

Abstract
Aberrant activation of Wnt/β‐catenin signaling causes tumorigenesis and promotes the proliferation of colorectal cancer cells. Porcupine inhibitors, which block secretion of Wnt ligands, may have only limited clinical impact for the treatment of colorectal cancer, because most colorectal cancer is caused by loss‐of‐function mutations of the tumor suppressor adenomatous polyposis coli (APC) downstream of Wnt ligands. Tankyrase poly(ADP‐ribosyl)ates (PARylates) Axin, a negative regulator of β‐catenin. This post‐translational modification causes ubiquitin‐dependent degradation of Axin, resulting in β‐catenin accumulation. Tankyrase inhibitors downregulate β‐catenin and suppress the growth of APC‐mutated colorectal cancer cells. Herein, we report a novel tankyrase‐specific inhibitor RK‐287107, which inhibits tankyrase‐1 and ‐2 four‐ and eight‐fold more potently, respectively, than G007‐LK, a tankyrase inhibitor that has been previously reported as effective in mouse xenograft models. RK‐287107 causes Axin2 accumulation and downregulates β‐catenin, T‐cell factor/lymphoid enhancer factor reporter activity and the target gene expression in colorectal cancer cells harboring the shortly truncated APC mutations. Consistently, RK‐287107 inhibits the growth of APC‐mutated (β‐catenin‐dependent) colorectal cancer COLO‐320DM and SW403 cells but not the APC‐wild (β‐catenin‐independent) colorectal cancer RKO cells. Intraperitoneal or oral administration of RK‐287107 suppresses COLO‐320DM tumor growth in NOD‐SCID mice. Rates of tumor growth inhibition showed good correlation with the behavior of pharmacodynamic biomarkers, such as Axin2 accumulation and MYC downregulation. These observations indicate that RK‐287107 exerts a proof‐of‐concept antitumor effect, and thus may have potential for tankyrase‐directed molecular cancer therapy.
Keywords: colorectal cancer, mouse xenograft model, poly(ADP‐ribose) polymerase, tankyrase inhibitor, Wnt/β‐catenin signaling
1. INTRODUCTION
Wnt/β‐catenin signaling is activated in over 90% of human colorectal cancer. Especially, loss‐of‐function mutations of the tumor suppressor adenomatous polyposis coli (APC), a key negative regulator of Wnt/β‐catenin signaling, are found in 90% of patients.1 During colorectal carcinogenesis, APC mutations are found in precancerous lesions of the intestine and cause initial clonal evolution in the very early stages of the so‐called adenoma‐carcinoma sequence.2 Normally, Wnt/β‐catenin signaling is activated by Wnt ligands. In the absence of Wnt ligands, β‐catenin is scaffolded by the destruction complex, which is constituted by Axin, APC, casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β), and phosphorylated by CK1 and GSK3β. This phosphorylation within the destruction complex leads to β‐transducin repeat‐containing protein (βTrCP)‐mediated ubiquitination and proteasomal degradation of β‐catenin. When Wnt ligands bind to Frizzled, a Wnt receptor, Axin associates with Frizzled and βTrCP dissociates from the destruction complex and β‐catenin.3 Then, the accumulated β‐catenin binds to the T‐cell factor/lymphoid enhancer factor (TCF/LEF) transcription factor in the nucleus, resulting in upregulation of the target genes, such as MYC, AXIN2 and APCDD1. In most colorectal cancer, APC function is disrupted and Wnt/β‐catenin signaling is dysregulated.
Based on this background, it has been postulated that Wnt/β‐catenin signaling is a promising therapeutic target for colorectal cancer. However, there have been no clinical anticancer drugs that selectively target this pathway.4 LGK974,5 a porcupine inhibitor, which targets Wnt‐activated cancer, has been examined in a phase I clinical trial (ClinicalTrials.gov Identifier: NCT01351103). Because LGK974 inhibits palmitoylation, which is required for activation and secretion of Wnt ligands,5 it may be applicable to Wnt‐driven cancer without APC loss‐of‐function or CTNNB1 (gene for β‐catenin) gain‐of‐function mutations. However, 90% of colorectal cancer has APC mutations and upregulates Wnt/β‐catenin signaling downstream of porcupine.1 Therefore, porcupine inhibitors may not be mechanistically suitable as anticancer drugs for most colorectal cancer.
Tankyrases (tankyrase‐1/TNKS and tankyrase‐2/TNKS2, also known as PARP5a/ARTD5 and PARP5b/ARTD6) are members of the poly(ADP‐ribose) polymerase (PARP) family proteins, which have PARP catalytic domains.6, 7 Although single knockout mice of either TNKS or TNKS2 are viable, their double knockout mice are embryonic lethal, supporting that these two genes are functionally redundant.8 Accordingly, the functional differences of TNKS and TNKS2 remain unknown. Tankyrase poly(ADP‐ribosyl)ates (PARylates) target proteins, that is, tankyrases add multiple ADP‐ribose moieties sequentially to target proteins using NAD+ as a substrate. Tankyrase has multiple ANK repeat clusters (ARC), through which it binds various target proteins.9, 10, 11 One of them is Axin, which anchors β‐catenin to prevent its translocation into the nucleus.12 Tankyrase PARylates Axin and the PARylated Axin are recognized by the ubiquitin ligase RNF146, leading to proteasomal degradation of Axin.13, 14, 15, 16 Consequently, β‐catenin is dissociated from the destruction complex and binds to the TCF/LEF transcription factor, which upregulates expression of the β‐catenin target gene. Thus, tankyrases are positive regulators of the canonical Wnt/β‐catenin signaling.
Given that tankyrase has PARP activity, its inhibition by small compounds will provide a therapeutic opportunity for Wnt‐driven cancer. XAV939, the first reported tankyrase‐selective PARP inhibitor, induces Axin stabilization and suppresses Wnt/β‐catenin signaling in colorectal cancer SW480 cells harboring a truncated APC mutation.15 IWR‐1 is another tankyrase inhibitor, initially identified as an Axin stabilizer.17 Although XAV939 and IWR‐1 are not suitable for in vivo experiments owing to their pharmacokinetic problems, several tankyrase‐specific inhibitors for in vivo administration have been developed, including G007‐LK, NVP‐TNKS656, K‐756, AZ1366, G‐631 and JW55.18, 19, 20, 21, 22, 23 G007‐LK inhibits proliferation of the colorectal cancer cells COLO‐320DM and SW403, both harboring truncated APC mutations, but not HCT‐15 and DLD‐1 cells, which also have truncated APC mutants.18 G007‐LK suppresses tumor growth in mouse xenograft models with two APC‐mutant colorectal cancer cells.18 Recently, we have reported that colorectal cancer cells harboring “short” truncated APC mutations lacking all seven β‐catenin‐binding 20‐amino acid repeats (20‐AAR) (hereinafter, referred to as “short APC”) show sensitivity to tankyrase inhibitors using 25 colorectal cancer cells (9 established cell lines and 16 patient‐derived cancer cell lines), although several patient‐derived cancer cell lines also showed sensitivity to tankyrase inhibitors in spite of the partial retention of 20‐AAR in the mutant APC.24 So far, none of these tankyrase inhibitors have been the subject of clinical trials.
Herein, we have developed a novel, potent and selective tankyrase inhibitor, RK‐287107. RK‐287107 inhibits proliferation of colorectal cancer cells that harbor short APC mutations. Orally given RK‐287107 at tolerable doses suppressed tumor growth in a mouse xenograft model, suggesting that RK‐287107 is a lead compound for the development of clinical tankyrase inhibitors.
2. MATERIALS AND METHODS
2.1. Chemical compounds, screening and in vitro PARP assay
Yeast high‐throughput screening for compounds that repress the growth inhibitory effect of tankyrase‐1 overexpression on fission yeast was carried out as described.25 Hit compounds were subjected to in vitro PARP assays. Detailed description is given in Doc S1.
2.2. Cell culture and proliferation assays
Human colorectal cancer cell lines COLO‐320DM, HCC2998, HCT‐116 and DLD‐1 were maintained in RPMI‐1640 medium with 10% heat‐inactivated FBS as described previously.24, 26 Human colorectal cancer cell lines SW403 and RKO were maintained in DMEM medium with 10% FBS. Cell proliferation was evaluated by MTT and BrdU assays. Detailed description is given in Doc S1.
2.3. Luciferase reporter assay
Conditioned media derived from the cultures of L cells and L‐Wnt3A cells were obtained according to ATCC protocol. Reporter assay was carried out as described in Doc S1.
2.4. Western blot analysis
Cell lysates were prepared and western blot analysis was carried out as previously described.9 Detailed description is given in Doc S1.
2.5. Immunofluorescence staining
Immunofluorescence staining was carried out as described previously24 using anti‐non‐phosphorylated β‐catenin antibody (D13A1, 1:500; Cell Signaling Technology, Danvers, MA, USA). Fluorescence images were quantified with ImageJ (National Institutes of Health, Bethesda, MD, USA).
2.6. Quantitative reverse transcription (qRT)‐PCR and transcriptome analyses
Total RNAs were prepared with the RNeasy Mini Kit (Qiagen, Hilden, Germany). qRT‐PCR and microarray analyses were done as described in Doc S1. The gene expression data have been deposited in the Gene Expression Omnibus (GEO) (GSE113965).
2.7. In vivo xenograft experiments
All animal procedures were carried out in the animal experiment room at the Japanese Foundation for Cancer Research (JFCR) according to protocols approved by the JFCR Animal Care and Use Committee. COLO‐320DM cells were suspended in Matrigel (Corning, Corning, NY, USA) and HBSS in a 1:1 ratio (5.2 × 106 cells/100 μL/mouse) and were implanted s.c. in the rear right flank of 6‐week‐old female NOD.CB17‐Prkdc scid/J mice (Charles River Laboratories Japan, Inc., Kanagawa, Japan). The length (L) and width (W) of the tumor mass were measured, and the tumor volume (TV) was calculated using the equation: TV = (L × W2)/2. When tumor volumes reached approximately 59‐120 mm3 (59‐104 mm3 and 71‐120 mm3 for i.p. and i.p./p.o. experiments, respectively), mice were separated into groups of 10 and six animals, respectively, with similarly sized tumors, and treatment was started the day after grouping. For i.p. administration, dosing solution contained 15% DMSO, 17.5% Cremophor, 8.75% ethanol, 8.75% PEG‐40 hydrogenated castor oil and 50% PBS. For oral administration, RK‐287107 was milled and suspended in sterilized 0.5% (w/v) methyl cellulose in water. Tumor growth inhibition coefficient (% TGI) was calculated according to the following equation: % TGI = [(Vvehicle – Vexperiment)/Vvehicle] × 100, where V is relative tumor volume on the final day. Pharmacodynamic and pharmacokinetic analyses were carried out as described in Doc S1.
3. RESULTS
3.1. RK‐287107 inhibits tankyrase‐1 and ‐2 but not PARP1 enzyme activities
Using the high‐throughput screening of nearly 140 000 small molecule compounds that repress the growth‐suppressing effect of tankyrase‐1 overexpression on fission yeast cells25 and subsequent PARP assay, we identified a compound, RK‐140160, as a tankyrase inhibitor (Figure 1A). IC50 values of RK‐140160 for tankyrase‐1 and tankyrase‐2 PARP activities in vitro were 42.2 and 42.3 nmol/L, respectively, which were almost equivalent to or lower than those of G007‐LK, a previously reported tankyrase‐selective inhibitor18 (Figure 1B). RK‐140160 at concentrations of up to 20 μmol/L did not inhibit PARP1. Therefore, by using RK‐140160 as a lead compound, we synthesized a series of structurally related derivatives and developed a novel compound, RK‐287107, as a more potent, and selective tankyrase inhibitor (Figure 1B,C). RK‐287107 inhibited tankyrase‐1 and tankyrase‐2 in vitro with IC50 values of 14.3 and 10.6 nmol/L, respectively, whereas it had no effect on PARP1 enzyme activity at up to 20 μmol/L. As a negative control, RK‐140790 is another derivative that had a closely similar structure to RK‐140160 but retained much less ability to inhibit tankyrases. The IC50 values of RK‐140790 for tankyrase‐1 and tankyrase‐2 were 428‐fold and 414‐fold higher than those of RK‐287107, respectively. Detailed information on the yeast high‐throughput screening, synthetic routes and chemical properties of RK‐287107 and its derivatives will be described elsewhere (F. Shirai, T. Tsumura, Y. Yashiroda, H. Yuki, H. Niwa, S. Sato, T. Chikada, Y. Koda, K. Washizuka, N. Yoshimoto, M. Abe, T. Onuki, Y. Mazaki, C. Hirama, T. Fukami, H. Watanabe, T. Honma, T. Umehara, M. Shirouzu, M. Okue, Y. Kano, T. Watanabe, K. Kitamura, E. Shitara, Y. Muramatsu, H. Yoshida, A. Mizutani, H. Seimiya, M. Yoshida, H. Koyama, manuscript in preparation).
Figure 1.
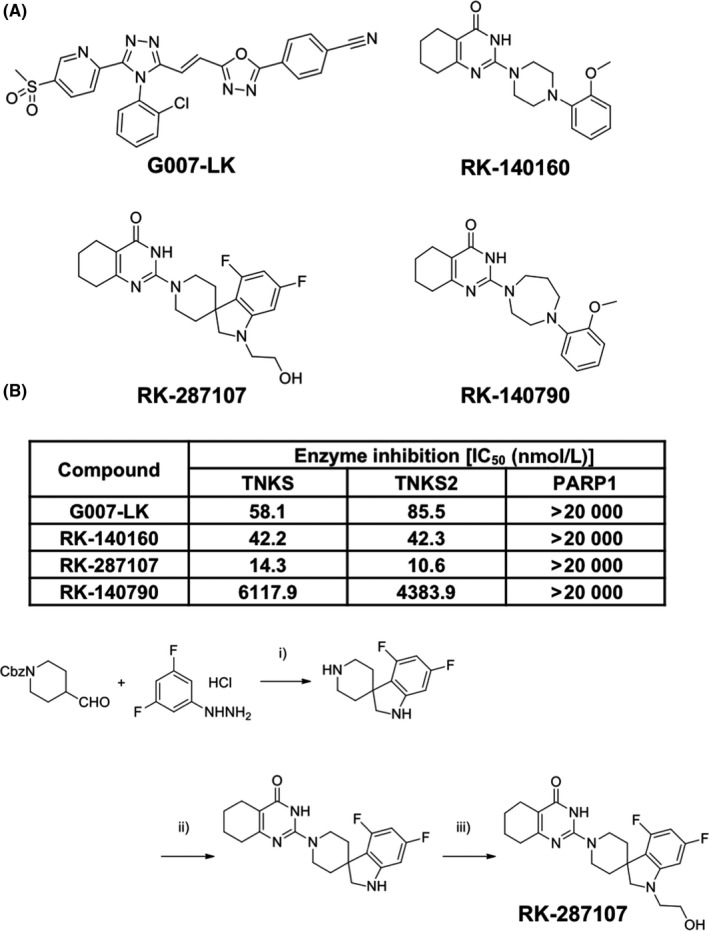
In vitro properties of newly identified tankyrase inhibitors. A, Chemical structures of tankyrase inhibitors and a related compound. G007‐LK, a previously reported tankyrase inhibitor;18 RK‐140160, tankyrase inhibitor identified by high‐throughput screening; RK‐287107, highly potent and specific tankyrase inhibitor synthesized by setting RK‐140160 as a lead compound; RK‐140790, a structurally related compound of RK‐140160 without potent activities to inhibit tankyrase. B, poly(ADP‐ribose) polymerase (PARP) inhibitory activities of the compounds. Each compound was subjected to PARP assay in vitro. IC 50 values are shown. TNKS, tankyrase‐1; TNKS2, tankyrase‐2. C, Synthetic routes of RK‐287107. (i) (1) TFA, CHCl3, reflux, 3 h; (2) NaBH(OAc)3, CHCl3‐MeOH, rt, 1 h; (3) TFA, reflux, 3 h; (ii) (1) 1‐Amidinopyrazole hydrochloride, Et3N, CHCl3, rt, 1 h; (2) Ethyl 2‐cyclohexanonecarboxylate, EtONa, EtOH, reflux, 3 h [(i) + (ii), 64% from aldehyde]; (iii) Glycolaldehyde dimer, NaBH(OAc)3, CHCl3‐AcOH, rt, 14 h (53%)
3.2. RK‐287107 shows an antiproliferative effect on colorectal cancer cells harboring short APC mutations
Short APC mutations predict the sensitivity of colorectal cancer cells to tankyrase inhibitors.24 To investigate the effect of RK‐287107 on the growth of six human colorectal cancer cell lines with various APC mutations, we carried out MTT assays. COLO‐320DM and SW403 cells, both of which had short APC, showed high sensitivity to RK‐287107 and G007‐LK (Figure 2A). RK‐140790, as an inactive negative control, showed only marginal effects, if any, on the growth of these cells. The 50% growth inhibition (GI50) values of RK‐287107 and G007‐LK on COLO‐320DM cells were 0.449 and 0.434 μmol/L, respectively. Meanwhile, these tankyrase inhibitors did not inhibit the growth of other cell lines, including RKO (APC wild‐type, β‐catenin‐independent), HCT‐116 (CTNNB1 gain‐of‐function mutation), HCC2998 and DLD‐1 (APC mutations with partial retention of 20‐AARs). To further examine the effect of RK‐287107 on cell proliferation, we carried out a BrdU assay in COLO‐320DM and RKO cells under treatment with RK‐287107, G007‐LK, RK‐140790 and olaparib,27, 28 a PARP1/2‐selective PARP inhibitor. As shown in Figure 2B (left), DNA synthesis in COLO‐320DM cells was reduced by RK‐287107 and G007‐LK in a dose‐dependent way. Inhibition of DNA synthesis at higher doses was larger in RK‐287107‐treated cells than in G007‐LK‐treated cells. As negative controls, olaparib and RK‐140790 showed no effects at concentrations up to 3 μmol/L, and marginally inhibited DNA synthesis at 10 μmol/L (Figure 2B left and right). RKO cells were resistant to these tankyrase inhibitors and olaparib, although 10 μmol/L olaparib inhibited the DNA synthesis to half that of the basal level (Figure 2B middle). These observations indicate that RK‐287107 preferentially inhibits the growth of tankyrase inhibitor‐sensitive cells, that is, short APC mutant colorectal cancer cells.
Figure 2.
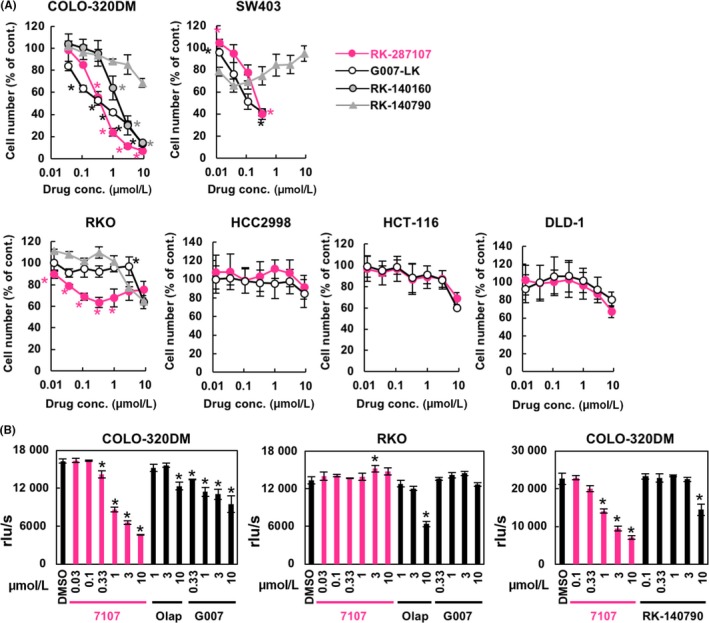
Antiproliferative effect of RK‐287107 on colorectal cancer cells. A, Effects of RK‐287107 and related compounds on cancer cell growth. Cells were treated with the compounds in triplicate for 120 h. Relative cell number was quantitated by MTT assays. Experiments were repeated at least twice and the average values were plotted. *P < 0.01 vs RK‐140790‐treated cells at the same concentration of compounds by Tukey‐Kramer test. B, Effects on DNA synthesis. BrdU assays were carried out using COLO‐320DM and RKO cells after 48‐h treatment with the compounds. 7107, RK‐287107; Olap, olaparib; G007, G007‐LK. Experiments were repeated at least twice and the representative results are shown. *P < 0.01 vs DMSO‐treated cells by Tukey‐Kramer test. Error bar indicates standard deviation
3.3. RK‐287107 stabilizes Axin and downregulates β‐catenin signaling
To examine the pharmacodynamic effects of RK‐287107 in COLO‐320DM cells, we carried out western blot analysis. Tankyrase PARylates itself and is consequently ubiquitinated by RNF146, resulting in degradation.13, 29, 30 Therefore, inhibition of tankyrase would block its PARylation/degradation, leading to protein accumulation. As shown in Figure 3A and Figure S1, RK‐287107 and G007‐LK caused accumulation of tankyrase and Axin1/2, which are the most direct pharmacodynamic biomarkers observed as a result of the reduced PARylation and subsequent stabilization. Levels of Axin2 accumulation were more evident than those of Axin1, consistent with a previous report.24 Of note, the extent of Axin2 accumulation was slightly reduced upon higher‐dose treatment with tankyrase inhibitors, because AXIN2 is a transcriptional target of β‐catenin (see below). Consequently, downregulation of active (ie, non‐phosphorylated) β‐catenin was observed both in the western blot analysis and indirect immunofluorescence microscopy (Figure 3B). The immunofluorescence intensities of active β‐catenin were 100%, 0.63%, 106%, and 26.4% in DMSO, RK‐287107, Olaparib, and G007‐LK‐treated cells, respectively. Although β‐catenin was localized in the nucleus in COLO‐320DM cells in a steady state, its fluorescence intensity was reduced by treatment with RK‐287107 or G007‐LK. Furthermore, these inhibitor‐treated cells showed several intracellular foci, which were considered to be degradasomes31 (Figure 3B, inset, arrowheads). Olaparib did not affect tankyrase, Axins or β‐catenin at all, excluding the possibility that the observed phenomena were caused by PARP1/2 inhibition.
Figure 3.
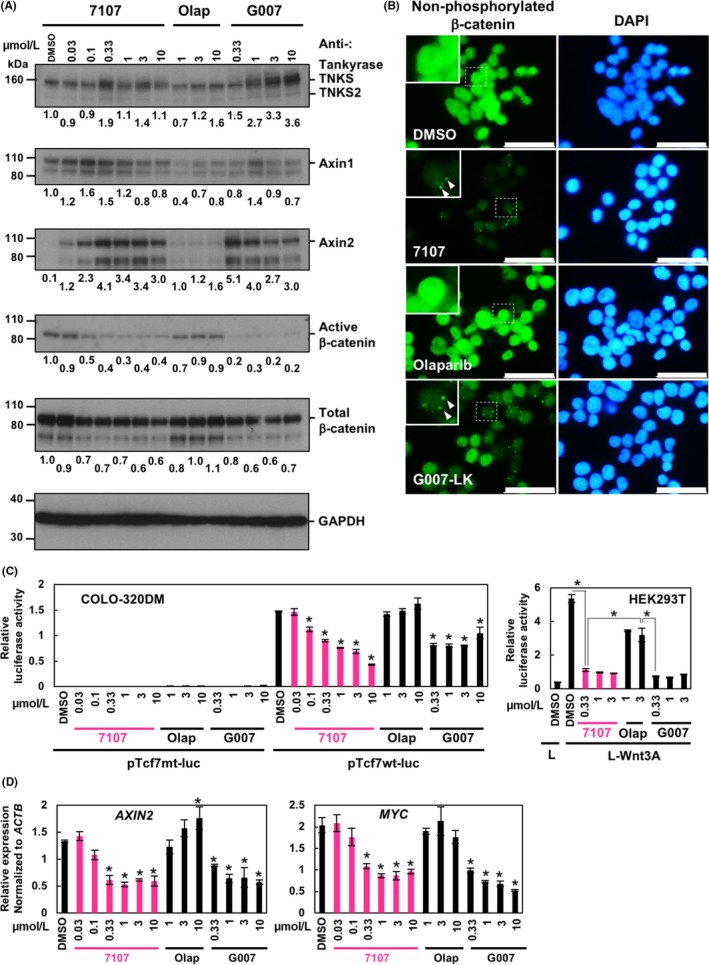
RK‐287107 downregulates β‐catenin signaling in cultured cells. A, Western blot analysis. COLO‐320DM cells were treated with RK‐287107, olaparib and G007‐LK for 16 h. Whole cell lysates were subjected to western blot analysis. Full‐length blots are presented in Figure S1. B, Indirect immunofluorescence staining. COLO‐320DM cells were treated with the compounds at 1 μmol/L for 16 h. Then the cells were subjected to immunofluorescence staining with anti‐non‐phosphorylated (active) β‐catenin antibody. Nuclear DNA was counterstained with DAPI. Scale bar, 20 μm. Inset, magnified view of the dotted area; arrowhead, degradosome focus. C, T‐cell factor (TCF) reporter assay. Cells were treated with the compounds for 30 h after transfection of the reporter vectors. In the case of HEK293T cells, L or L‐Wnt3A conditioned media were added to the cells simultaneously with the indicated inhibitors. Luciferase reporter assays were carried out. *P < .01 vs pTcf7wt‐luc‐transfected cells with DMSO or the indicated in the figure by Tukey‐Kramer test. D, qRT‐PCR assay of β‐catenin target genes. COLO‐320DM cells were treated with the inhibitors for 48 h. Relative expression levels of AXIN2 and MYC transcripts were determined by normalization with those of ACTB expression. Error bar indicates standard deviation. *P < 0.01 vs DMSO‐treated cells by Tukey‐Kramer test
To determine whether RK‐287107 suppresses the transcriptional activity of TCF/LEF, a downstream effector of the Wnt/β‐catenin pathway, we carried out TCF/LEF reporter luciferase assays. In COLO‐320DM cells, RK‐287107 suppressed the reporter activity in a dose‐dependent way (Figure 3C, left). Although G007‐LK less efficiently suppressed the reporter activity, olaparib had no effect. As HEK293T cells have an intact Wnt/β‐catenin pathway, high TCF/LEF luciferase activity was detected when conditioned medium of L‐Wnt3A cells containing Wnt3A ligands was added to HEK293T cells (Figure 3C, right). RK‐287107 and G007‐LK suppressed TCF/LEF luciferase activity in L‐Wnt3A‐stimulated HEK293T cells, whereas olaparib only marginally inhibited the activity. Next, we carried out quantitative RT‐PCR analyses on mRNA expression of AXIN2 and MYC, transcriptional target genes of Wnt/β‐catenin signaling. We found that RK‐287107 and G007‐LK, but not olaparib, suppressed the mRNA expression of AXIN2 and MYC in COLO‐320DM cells (Figure 3D). These observations indicate that RK‐287107 inhibits tankyrase, which leads to suppression of β‐catenin signaling.
3.4. RK‐287107 inhibits tumor growth in a mouse xenograft model
We next investigated the therapeutic effect of RK‐287107 in vivo by using immunodeficient mouse xenograft models. COLO‐320DM cells were s.c. injected into NOD‐SCID mice and tumor growth was monitored. As shown in Figure 4A (upper), i.p. administration of RK‐287107 at 100 and 300 mg/kg once per day resulted in 32.9% and 44.2% TGI, respectively. Under these treatment conditions, there was no detectable effect on body weight (Figure 4A, lower) or behavior of the mice. To determine the concentrations of RK‐287107 in plasma and tumor tissues 4 hours after drug administration, we carried out liquid chromatography‐mass/mass spectrometry (LC‐MS/MS) analyses. Plasma concentrations of RK‐287107 increased in a dose‐dependent way, whereas its concentration in tumor tissues was not significantly changed (Figure 4B).
Figure 4.
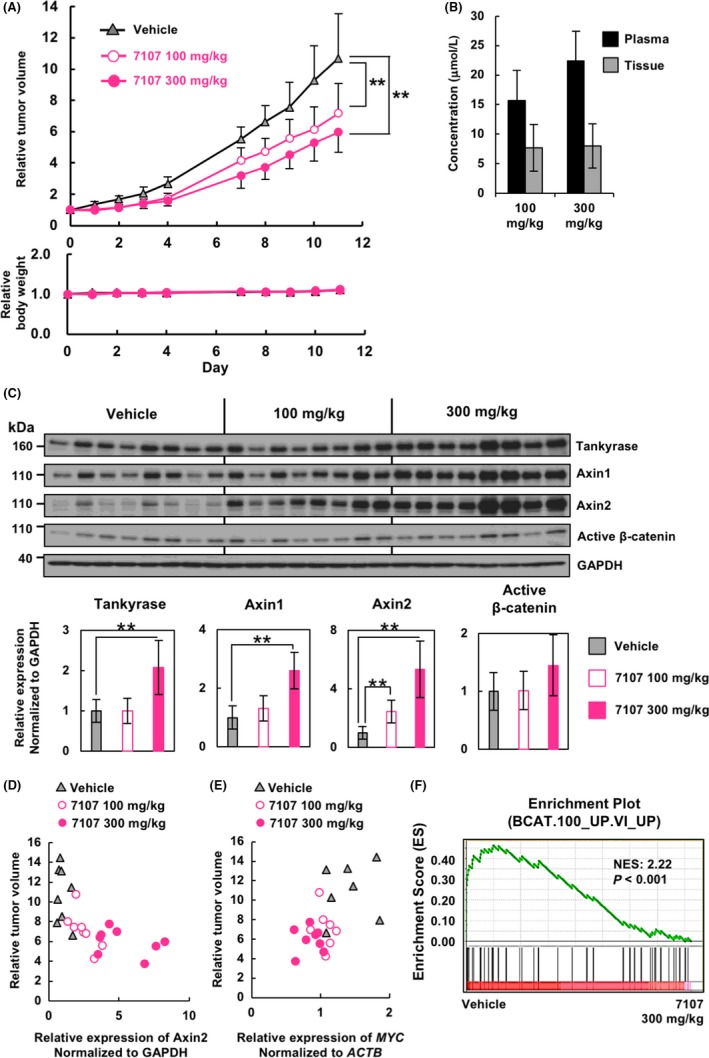
In vivo antitumor effect of RK‐287107 through repression of β‐catenin signaling. A, Tumor growth inhibition by RK‐287107 in vivo. NOD‐SCID mice were injected s.c. with COLO‐320DM cells. RK‐287107 was given i.p. under a 5‐days on/ 2‐days off schedule for 2 weeks at a dose of 100 mg/kg or 300 mg/kg once per day. Error bar indicates standard deviation. **P < 0.01 by Tukey‐Kramer test. B, Concentration of RK‐287107 in plasma and tumor tissue at the sampling point at 4 h from the final dosage of the compound. C, Xenograft tumors in (A) were collected at 4 h from the final administration, and subjected to western blot analysis. Each lane indicates a tumor derived from an independent mouse. Full‐length blots are presented in Figure S2. Bottom panels are quantitative representations. **P < 0.01 by two‐sided Student's paired t test. D,E, Scatter plots of Axin2 protein expression normalized to GAPDH (D) or MYC gene expression normalized to ACTB (E) and relative tumor volume of each sample. Samples for qRT‐PCR analysis were collected at 4 h from the final dosage. F, Gene Set Enrichment Analysis (GSEA) showing enrichment of the β‐catenin pathway‐related gene signature in the tumor tissues of the vehicle‐treated group vs that of the RK‐287107‐treated group (300 mg/kg)
To evaluate the pharmacodynamic effects of RK‐287107 at the protein level, we carried out western blot analysis at 4 hours after the final administration of the compound. As shown in Figure 4C and Figure S2, Axin1 and Axin2 accumulated in RK‐287107‐treated tumor tissues. In particular, the Axin2 level was increased in a dose‐dependent way and inversely correlated with relative tumor size (Pearson's correlation r = −0.65) (Figure 4D). We could not detect downregulation of β‐catenin presumably because the whole cell extracts, instead of the nuclear extracts, were subjected to the analysis (Figure 4C, see Section 4). RNA expression of MYC, one of the β‐catenin target genes, in the tumor tissues was downregulated by RK‐287107 and showed a moderate correlation with relative tumor size (Pearson's correlation r = 0.57) (Figure 4E). Transcriptome analysis with GeneChip microarrays and gene set enrichment analysis (GSEA) showed that β‐catenin target gene expression was higher in the vehicle‐treated samples than in RK‐287107‐treated samples, indicating that RK‐287107 suppressed the expression of β‐catenin target genes (Figure 4F). Together, these observations indicate that RK‐287107 suppresses the Wnt/β‐catenin pathway and tumor growth of the xenografted COLO‐320DM cells in vivo.
3.5. RK‐287107 is an orally effective tankyrase inhibitor in mice
The above‐mentioned antitumor efficacy of RK‐287107 was observed under the conditions of repetitive dosage of the compound. This suggests that oral dosage might be the most preferable administration route in a practical therapeutic setting. Therefore, we next carried out a xenograft experiment to determine whether RK‐287107 is available for oral dosage. Figure 5A (upper) shows that intraperitoneal (150 mg/kg, twice per day) and oral (300 mg/kg, twice per day) dosage of RK‐287107 resulted in 47.2% and 51.9% TGI, respectively. Relative body weights were slightly lower in the oral groups than in the i.p. group, presumably because the former groups were subjected to the mechanical stress of feeding needles (Figure 5A, lower). There was no detectable difference in body weight between the oral, vehicle and RK‐287107‐treated groups. Pharmacokinetic analysis of plasma and tissue samples indicated that RK‐287107 with oral administration showed a higher concentration of the compound than the i.p. administration under these dosage conditions (Figure 5B).
Figure 5.
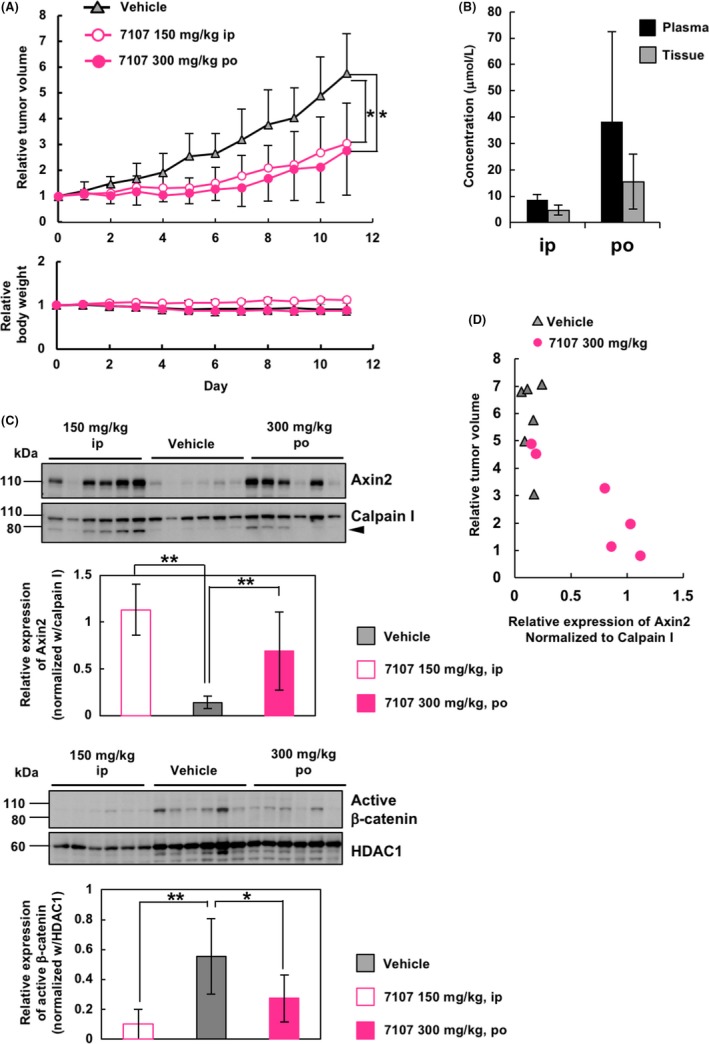
Orally given RK‐287107 exerts an antitumor effect. A, Tumor growth inhibition by orally given RK‐287107. NOD‐SCID mice were injected s.c. with COLO‐320DM cells before giving RK‐287107. RK‐287107 was given for 11 days at a dose of 150 mg/kg i.p. or 300 mg/kg p.o. twice a day. Error bar indicates standard deviation. *P < 0.05 by Tukey‐Kramer test. B, Concentration of RK‐287107 in plasma and tumor tissue at the sampling point at 4 h from the final dosage of the compound. Error bar indicates standard deviation. C, Western blot analyses of pharmacodynamic biomarkers in the same xenograft tumors as in (A) collected at 4 h from the final dosage. Cytoplasmic (upper panels) and nuclear (lower panels) extracts from the tumor tissues were subjected to western blot analysis. Full‐length blots are presented in Figure S3. Error bar indicates standard deviation. **P < 0.01 and *P < 0.05 by two‐sided Student's paired t test. D, Scatter plot of Axin2 protein expression normalized to Calpain I and relative tumor volume of each sample
Western blot analysis of the tumor tissues at 4 hours after the final dosage showed that Axin2 was upregulated in the cytoplasmic extracts of the RK‐287107‐treated groups with i.p. and oral administration (Figure 5C, upper, and Figure S3). Again, the relative tumor volumes showed good inverse correlation with the expression levels of Axin2 protein (Pearson's correlation r = −0.84) (Figure 5D). Furthermore, active (ie, non‐phosphorylated) β‐catenin was downregulated by either oral or i.p. administration of RK‐287107 (Figure 5C, lower). These observations indicate that RK‐287107 is an orally effective tankyrase inhibitor.
4. DISCUSSION
In the present study, we showed that a novel small‐molecule compound, RK‐287107, potently inhibits tankyrases but not PARP1 enzyme activity. Through blockade of the β‐catenin signaling pathway, RK‐287107 suppressed tumor growth of human colorectal cancer cells in mouse xenograft models with either i.p. or oral administration. RK‐287107 was optimized from a high‐throughput screening hit compound, RK‐140160. RK‐140160 is structurally similar to XAV939, the first described tankyrase inhibitor, and binds to the nicotinamide subsite of the donor NAD+ binding site. Optimization of the spiroindoline substituent, which was predicted to be exposed to the cytosolic space, resulted in improvement of enzymatic and cellular activities, drug metabolism and pharmacokinetics profile of the compounds. Detailed information on the chemical optimization will be described elsewhere elsewhere (F. Shirai, T. Tsumura, Y. Yashiroda, H. Yuki, H. Niwa, S. Sato, T. Chikada, Y. Koda, K. Washizuka, N. Yoshimoto, M. Abe, T. Onuki, Y. Mazaki, C. Hirama, T. Fukami, H. Watanabe, T. Honma, T. Umehara, M. Shirouzu, M. Okue, Y. Kano, T. Watanabe, K. Kitamura, E. Shitara, Y. Muramatsu, H. Yoshida, A. Mizutani, H. Seimiya, M. Yoshida, H. Koyama, manuscript in preparation).
We previously reported that the status of the APC mutation is a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer.24 RK‐287107 suppressed proliferation of COLO‐320DM and SW403 cells with short APC mutations, which lack 20‐AARs, but not that of RKO, HCC2998 and DLD‐1 with more than two 20‐AARs, indicating that sensitivity to tankyrase inhibitors is predictable by APC mutations (Figure 2A). HCT‐116 cells have a constitutively active mutation in the CTNNB1 gene caused by deletion of Ser45, which suggests that HCT‐116 has less sensitivity to tankyrase inhibitors. Therefore, it is reasonable that HCT‐116 cells did not show sensitivity to tankyrase inhibitors (Figure 2A). This result also indicates that RK‐287107 is a tankyrase‐specific inhibitor and has fewer off‐target effects. RK‐287107 reduced the level of active β‐catenin in the nuclei of COLO‐320DM cells (Figure 3B). We also carried out immunofluorescence staining of β‐catenin using SW403 cells. In these cells, however, β‐catenin was detected in the cytoplasm, nuclei, and at the cell‐cell junctions. Therefore, it is unclear from our results whether RK‐287107 reduces β‐catenin in the nuclei in SW403 cells (A. Mizutani, H. Yoshida, H. Seimiya, unpublished data).
In HEK293T cells, we needed to stimulate the cells with external Wnt3A ligand for activation of Wnt signaling (Figure 3C). Because HEK293T cells have WT APC, Axins accumulated upon tankyrase inhibition can cooperate with APC to degrade β‐catenin. This observation is consistent with a previous report on another tankyrase inhibitor, XAV939.15 In colorectal cancer cells, RKO cells retain WT APC and have no abnormalities in the Wnt signaling pathway. In fact, RKO cells do not accumulate β‐catenin,24 which explains why these cells are resistant to tankyrase inhibitors, including RK‐287107 (Figure 2A). Meanwhile, HCC2998 and DLD‐1 cells express the mutant APC with partial 20‐AAR deletions. These APC mutants are hypomorphic and exert a dominant‐negative effect on Axin‐dependent degradation of β‐catenin.24 Therefore, in these cells, RK‐287107‐induced Axin accumulation would not lead to β‐catenin degradation, which explains the resistance to tankyrase inhibitors. By contrast, COLO‐320DM cells have a “short” APC, which lacks all of the seven 20‐AARs. This mutant APC does not contribute to β‐catenin degradation, causing hyperactivation of β‐catenin signaling.24 Therefore, COLO‐320DM cells are highly dependent on β‐catenin, and tankyrase inhibitors efficiently downregulate β‐catenin and block cell growth.
Over the past decade, various tankyrase inhibitors have been developed,15, 17, 18, 19, 20, 21, 22, 23, 32 some of which have been adapted to in vivo administration in preclinical models.18, 19, 20, 21, 22, 23 Among such inhibitors, G007‐LK is one of the most studied tankyrase inhibitors in vivo, although it has only been given i.p.18 It has been reported that G007‐LK given at high doses in mice causes body weight loss and lesions in the small intestine, which are considered severe side‐effects.18 It has been reported that another tankyrase inhibitor, G‐631, also has severe side‐effects on the small intestine.23 Meanwhile, no body weight alterations or histological changes in the small intestine in mice have been observed on 21‐day oral dosage of JW55, a tankyrase‐specific inhibitor.22 In our present study, we did not observe any body weight loss in mice with i.p. administration of RK‐287107 (Figure 4A). When RK‐287107 was given orally, body weight loss was observed; however, the control group, in which the vehicle was given orally, showed equivalent body weight loss to the RK‐287107‐administered group. These results indicate that the body weight loss observed in this study was due to the stress of oral dosage and vehicle toxicity. RK‐287107 administration did not show obvious body weight loss in mice, whereas G007‐LK causes severe body weight loss in mice.18 One possible reason for this discrepancy would be that the metabolic stability of RK‐287107 is much lower than that of G007‐LK. After incubation with murine microsomes, percentages of the remaining G007‐LK and RK‐287107 were 96% and 18%, respectively (our unpublished observations). Because we used rather high doses of RK‐287107 (up to 300 mg/kg, Figures 4 and 5), it was mechanistically difficult to further increase the doses. We did not carry out pathological analyses on intestinal toxicity because RK‐287107 did not induce body weight loss as compared with the vehicle‐treated group.
Knockdown of Axin2, but not Axin1, abolishes tankyrase inhibitor‐mediated β‐catenin degradation in COLO‐320DM cells,24 suggesting that β‐catenin downregulation by RK‐287107 would mainly be caused by accumulated Axin2. In HCC2998 cells, which have APC mutations that partially retain 20‐AARs, tankyrase inhibitor‐mediated β‐catenin degradation depends on Axin2 but not on Axin1 when mutant APC is depleted by siRNA‐mediated knockdown.24 Meanwhile, not only Axin2, but also Axin1, accumulated in RK‐287107‐treated xenograft tumors (Figure 4C), suggesting that β‐catenin degradation caused by tankyrase inhibitors also depends on Axin1 in vivo.
When we used whole cell extracts instead of nuclear extracts, it was technically difficult to clearly detect downregulation of β‐catenin in RK‐287107‐treated xenograft tumors. This might be as a result of the fact that the signal of active β‐catenin decreased in a time‐dependent way during lysate preparation. Because accumulation of Axin2 was easily detected at 4 hours from the final drug administration in vivo, we routinely evaluated the pharmacodynamic biomarkers at this time point. Meanwhile, degradation of active β‐catenin emerged more slowly than Axin2 accumulation (our unpublished observations).
Some melanoma patients respond to immune checkpoint inhibitors (eg, anti‐programmed‐cell‐death 1 antibodies, such as nivolumab and pembrolizumab), whereas others do not respond. In those drug‐resistant patients, melanoma‐intrinsic β‐catenin signaling may often avoid T‐cell infiltration in melanoma by means of inhibition of CCL4 secretion.33 In tumors with activated β‐catenin signaling, 14% of cases have an active mutation of CTNNB1, whereas 61% of cases are caused by overexpression of Wnt ligand (WNT7B), receptor (FZD3) or β‐catenin itself.33 These observations suggest that tankyrase inhibitors could provide a novel opportunity for combination therapy with immune checkpoint inhibitors for non‐responder patients.
CONFLICTS OF INTEREST
TC, TT and MO are employees of Meiji Seika Pharma Co., Ltd. Other authors have no competing interests to declare.
Supporting information
ACKNOWLEDGMENTS
We would like to thank Tomokazu Ohishi, Mika Kuroiwa, Mao Miyake (Cancer Chemotherapy Center, JFCR), Mika Takagi, Chizuko Hirama, Yui Mazaki (Drug Discovery Seed Compounds Exploratory Unit, RIKEN Center for Sustainable Resource Science [CSRS]), Shin Sato, Takashi Umehara (Laboratory for Epigenetics Drug Discovery, RIKEN Center for Biosystems Dynamics Research), Hiroo Koyama (Drug Discovery Chemistry Platform Unit, RIKEN CSRS), Toshio Goto (RIKEN Program for Drug Discovery and Medical Technology Platforms) and Tetsuo Noda (Cancer Institute, JFCR) for their expertise support and advise. This study was carried out as a research program of RIKEN Program for Drug Discovery and Medical Technology Platforms, the Project for Development of Innovative Research on Cancer Therapeutics and Project for Cancer Research and Therapeutic Evolution, Japan Agency for Medical Research and Development (AMED). This work was supported in part by AMED (JP15cm0106030, JP18cm0106102 to HS and YY; G2011‐011 and M15114 to MY), Grants‐in‐Aid for Scientific Research, Japan Society for the Promotion of Science (JSPS) (Scientific Research [B] 16H04716 to HS and Scientific Research [C] 17K07186 to AM), a research grant from Princess Takamatsu Cancer Research Fund (to HS), and the Platform Project for Supporting Drug Discovery and Life Science Research from AMED under Grant Number JP17am0101086. Funding for open access charge: (Japan Society for the Promotion of Science/16H04716).
Mizutani A, Yashiroda Y, Muramatsu Y, et al. RK‐287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018;109:4003–4014. 10.1111/cas.13805
Funding information
Japan Agency for Medical Research and Development (JP15cm0106030, JP18cm0106102, G2011‐011, JP17am0101086, M15114). Princess Takamatsu Cancer Research Fund (Grant/Award Number: ‘N/A’), Japan Society for the Promotion of Science, (16H04716, 17K07186).
REFERENCES
- 1. The Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55‐67. [DOI] [PubMed] [Google Scholar]
- 3. Li VS, Ng SS, Boersema PJ, et al. Wnt signaling through inhibition of beta‐catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245‐1256. [DOI] [PubMed] [Google Scholar]
- 4. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu J, Pan S, Hsieh MH, et al. Targeting Wnt‐driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci USA. 2013;110:20224‐20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hsiao SJ, Smith S. Tankyrase function at telomeres, spindle poles, and beyond. Biochimie. 2008;90:83‐92. [DOI] [PubMed] [Google Scholar]
- 7. Seimiya H. The telomeric PARP, tankyrases, as targets for cancer therapy. Br J Cancer. 2006;94:341‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chiang YJ, Hsiao SJ, Yver D, et al. Tankyrase 1 and tankyrase 2 are essential but redundant for mouse embryonic development. PLoS ONE. 2008;3:e2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seimiya H, Smith S. The telomeric poly(ADP‐ribose) polymerase, tankyrase 1, contains multiple binding sites for telomeric repeat binding factor 1 (TRF1) and a novel acceptor, 182‐kDa tankyrase‐binding protein (TAB 182). J Biol Chem. 2002;277:14116‐14126. [DOI] [PubMed] [Google Scholar]
- 10. Seimiya H, Muramatsu Y, Smith S, Tsuruo T. Functional subdomain in the ankyrin domain of tankyrase 1 required for poly(ADP‐ribosyl)ation of TRF1 and telomere elongation. Mol Cell Biol. 2004;24:1944‐1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guettler S, LaRose J, Petsalaki E, et al. Structural basis and sequence rules for substrate recognition by Tankyrase explain the basis for cherubism disease. Cell. 2011;147:1340‐1354. [DOI] [PubMed] [Google Scholar]
- 12. Cong F, Varmus H. Nuclear‐cytoplasmic shuttling of Axin regulates subcellular localization of beta‐catenin. Proc Natl Acad Sci USA. 2004;101:2882‐2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Callow MG, Tran H, Phu L, et al. Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS ONE. 2011;6:e22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DaRosa PA, Wang Z, Jiang X, et al. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP‐ribosyl)ation signal. Nature. 2015;517:223‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang SM, Mishina YM, Liu S, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614‐620. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y, Liu S, Mickanin C, et al. RNF146 is a poly(ADP‐ribose)‐directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol. 2011;13:623‐629. [DOI] [PubMed] [Google Scholar]
- 17. Lu J, Ma Z, Hsieh JC, et al. Structure‐activity relationship studies of small‐molecule inhibitors of Wnt response. Bioorg Med Chem Lett. 2009;19:3825‐3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lau T, Chan E, Callow M, et al. A novel tankyrase small‐molecule inhibitor suppresses APC mutation‐driven colorectal tumor growth. Cancer Res. 2013;73:3132‐3144. [DOI] [PubMed] [Google Scholar]
- 19. Okada‐Iwasaki R, Takahashi Y, Watanabe Y, et al. The discovery and characterization of K‐756, a novel Wnt/beta‐catenin pathway inhibitor targeting tankyrase. Mol Cancer Ther. 2016;15:1525‐1534. [DOI] [PubMed] [Google Scholar]
- 20. Quackenbush KS, Bagby S, Tai WM, et al. The novel tankyrase inhibitor (AZ1366) enhances irinotecan activity in tumors that exhibit elevated tankyrase and irinotecan resistance. Oncotarget. 2016;7:28273‐28285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shultz MD, Cheung AK, Kirby CA, et al. Identification of NVP‐TNKS656: the use of structure‐efficiency relationships to generate a highly potent, selective, and orally active tankyrase inhibitor. J Med Chem. 2013;56:6495‐6511. [DOI] [PubMed] [Google Scholar]
- 22. Waaler J, Machon O, Tumova L, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72:2822‐2832. [DOI] [PubMed] [Google Scholar]
- 23. Zhong Y, Katavolos P, Nguyen T, et al. Tankyrase inhibition causes reversible intestinal toxicity in mice with a therapeutic index < 1. Toxicol Pathol. 2016;44:267‐278. [DOI] [PubMed] [Google Scholar]
- 24. Tanaka N, Mashima T, Mizutani A, et al. APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer. Mol Cancer Ther. 2017;16:752‐762. [DOI] [PubMed] [Google Scholar]
- 25. Yashiroda Y, Okamoto R, Hatsugai K, et al. A novel yeast cell‐based screen identifies flavone as a tankyrase inhibitor. Biochem Biophys Res Commun. 2010;394:569‐573. [DOI] [PubMed] [Google Scholar]
- 26. Mashima T, Taneda Y, Jang MK, et al. mTOR signaling mediates resistance to tankyrase inhibitors in Wnt‐driven colorectal cancer. Oncotarget. 2017;8:47902‐47915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evers B, Drost R, Schut E, et al. Selective inhibition of BRCA2‐deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14:3916‐3925. [DOI] [PubMed] [Google Scholar]
- 28. Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA. 2008;105:17079‐17084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sbodio JI, Lodish HF, Chi NW. Tankyrase‐2 oligomerizes with tankyrase‐1 and binds to both TRF1 (telomere‐repeat‐binding factor 1) and IRAP (insulin‐responsive aminopeptidase). Biochem J. 2002;361:451‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith S, Giriat I, Schmitt A, de Lange T. Tankyrase, a poly(ADP‐ribose) polymerase at human telomeres. Science. 1998;282:1484‐1487. [DOI] [PubMed] [Google Scholar]
- 31. Thorvaldsen TE, Pedersen NM, Wenzel EM, et al. Structure, dynamics, and functionality of tankyrase inhibitor‐induced degradasomes. Mol Cancer Res. 2015;13:1487‐1501. [DOI] [PubMed] [Google Scholar]
- 32. James RG, Davidson KC, Bosch KA, et al. WIKI4, a novel inhibitor of tankyrase and Wnt/ss‐catenin signaling. PLoS ONE. 2012;7:e50457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic beta‐catenin signalling prevents anti‐tumour immunity. Nature. 2015;523:231‐235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials