

Abstract
Mitochondria‐eating protein (Mieap), encoded by a p53‐target gene, plays an important role in mitochondrial quality control (MQC). Mieap has been reported to have a critical role in tumor suppression in colorectal cancer. Here, we investigated its role as a tumor suppressor in breast cancer. The enforced expression of exogenous Mieap in breast cancer cells induced caspase‐dependent apoptosis, with activation of both caspase‐3/7 and caspase‐9. Immunohistochemistry revealed endogenous Mieap in the cytoplasm in 24/75 (32%) invasive ductal carcinomas (IDC), 15/27 (55.6%) cases of ductal carcinoma in situ (DCIS) and 16/18 (88.9%) fibroadenomas (FA) (IDC vs DCIS; P = 0.0389, DCIS vs FA; P = 0.0234, IDC vs FA; P < 0.0001). In IDC, the Mieap promoter was methylated in 6/46 (13%) cases, whereas p53 was mutated in 6/46 (13%) cases. Therefore, the p53/Mieap‐regulated MQC pathway was inactivated in 12/46 IDC (26.1%). Interestingly, all tumors derived from the 12 patients with Mieap promoter methylation or p53 mutations pathologically exhibited more aggressive and malignant breast cancer phenotypes. Impairment of p53/Mieap‐regulated MQC pathway resulted in significantly shorter disease‐free survival (DFS) (P = 0.021), although p53 status is more prognostic in DFS than Mieap promoter methylation. These results indicate that p53/Mieap‐regulated MQC has a critical role in tumor suppression in breast cancer, possibly in part through mitochondrial apoptotic pathway.
Keywords: apoptosis, breast cancer, caspase, Mieap, promoter methylation
1. INTRODUCTION
Breast cancer is the most frequently diagnosed cancer and the leading cause of female cancer‐associated death worldwide, accounting for 25% of all cancer cases and 15% of all cancer deaths among females.1, 2 Although the causes of breast cancer are not fully understood, some risk factors, including age, genetics, a family history of breast cancer and estrogen exposure, are known to affect its development.3, 4 Most hereditary breast cancers are associated with alterations in either BRCA1 or BRCA2 (breast cancer susceptibility genes).5 However, in sporadic breast cancers, the most important gene is p53, given that it is a tumor suppressor gene that is mutated in >50% of human cancers.6 According to previous reports, p53 is also mutated in approximately 20%‐40% of breast cancers.7, 8 Recent data from The Cancer Genome Atlas revealed that 37% of breast cancer specimens had alterations in p53 (72% in [human epidermal growth factor] HER2‐rich and 80% in basal‐like breast cancer cases), indicating that it is a critical driver of tumor development even in breast cancer.9 p53 is clinically very important not only because of its high mutation rate but also because mutation is associated with more aggressive disease and worse overall survival.10
p53 is a transcription factor that activates the expression of various downstream genes in response to DNA damage.11 The central functions of this protein in tumor suppression are cell cycle arrest, apoptosis, DNA repair and anti‐angiogenesis.12, 13, 14, 15, 16 In particular, apoptosis is so important a function for p53‐related tumor suppression that p53 activates target genes, including Bax, Noxa, Puma, Apaf‐1 and p53AIP1, in response to DNA damage by radiation, UV and oxidative stress.11, 13, 17 Although the mechanisms of apoptosis induced by DNA damage have been clarified, mitochondria are a pivotal organ for apoptosis, where these apoptosis‐related proteins localize and play an important role in mitochondria through caspase activation.18 Recently, mitochondrial quality control has been revealed to be a novel function of p53. This function is regulated by a novel p53‐inducible protein called mitochondria‐eating protein (Mieap).19, 20 Mieap plays an important role in mitochondrial quality control (MQC) by repairing or eliminating unhealthy mitochondria. Mieap carries out its repair function by inducing the accumulation of intramitochondrial lysosomal proteins to eliminate oxidized mitochondrial proteins in response to mitochondrial damage, in a process called Mieap‐induced accumulation of lysosome‐like organelles within mitochondria (MALM). This leads to a decrease in reactive oxygen species (ROS) generation and an increase in mitochondrial ATP synthesis. When MALM is inhibited, Mieap induces the formation of a vacuole‐like structure known as the MIV. This engulfs the damaged mitochondria and results in the accumulation of lysosomes, leading to the degradation of unhealthy mitochondria.20 Further in vitro study revealed that BNIP3 and NIX co‐localized with Mieap in mitochondria and reduced ROS. The physical interaction of Mieap, BNIP3 and NIX at the mitochondrial outer membrane may play a critical role in the translocation of lysosomal proteins from the cytoplasm to the mitochondrial matrix.21
Although Mieap has been shown to be a key player in mitochondrial quality control, emerging evidence suggests that it also plays an important role in tumor suppression. In a mouse model, Mieap‐deficient (adenomatous polyposis coli) ApcMin/+ mice had a much shorter lifespan and increased numbers and sizes of intestinal tumors compared to those in Mieap‐wild type ApcMin/+ mice, suggesting that loss of Mieap increases ROS production in the intestinal mucosa and accelerates tumor progression.22 Furthermore, Mieap‐regulated mitochondrial quality control is frequently inactivated in human colorectal cancer by Mieap promoter methylation, BNIP3 promoter methylation or p53 mutation.23 Considering that Mieap is a downstream target of p53, this novel mechanism for mitochondrial quality control is a new function of the p53 tumor suppressor. These results strongly prompted us to speculate that p53/Mieap‐regulated mitochondrial quality control might be involved in tumor suppression in human breast cancer. In this study, we investigated this role.
2. MATERIALS AND METHODS
2.1. Cell lines, cell culture, and drug
Breast cancer cell lines MCF‐7 (luminal type), SK‐BR‐3 (HER2 type) and MDA‐MB‐231 (triple negative) were purchased from the ATCC (Manassas, VA, USA). All cell lines were grown in DMEM supplemented with 10% FBS (Sigma‐Aldrich, St. Louis, MO, USA), antibiotics (Sigma‐Aldrich) and non‐essential amino acids in a humidified atmosphere of 5% CO2 at 37°C. Etoposide was obtained from Sigma‐Aldrich.
2.2. Adenovirus
Mieap‐expressing adenovirus (Ad‐Mieap) was constructed as previously described.19, 20 As controls, we used Ad‐EGFP, Ad‐BNIP3 and Ad‐α1‐273 (Δ817), which express enhanced green fluorescent protein (EGFP), BNIP3 and a Mieap deletion mutant lacking the C‐terminal region (amino acids:1‐273) of the Mieap protein (273 amino acids), respectively.20, 21 BNIP3 is a BH3‐only protein that belongs to the Bcl‐2 family and is suggested to induce apoptosis by inhibiting anti‐apoptotic proteins.24 Our group recently demonstrated that BNIP3 is a co‐factor of Mieap for MQC.21
2.3. Fluorescence activated cell sorting analysis
We overexpressed Mieap using the constructed adenovirus in breast cancer cell lines. In addition, we treated these cells with etoposide (10 μmol/L). MCF‐7, SK‐BR‐3 and MDA‐MB‐231 cells were infected with the adenovirus at a moi of 30 and 60, respectively; cells were then trypsinized and washed with PBS. Subsequently, the cells were fixed in cold 70% ethanol at 4°C. Fixed cells were washed and then incubated for 30 minutes at 37°C with PBS containing 0.2 mg/mL RNase, and then incubated for a further 30 minutes at approximately 22°C with PBS containing 20 μg/mL propidium iodide. Cells were analyzed using the EC800 Flow Cytometry Analyzer (Sony, Tokyo, Japan). For each analysis, 5000‐20 000 cells were recorded, and the percentages of cells in different cell cycle phases (subG1, G1, S and G2/M) were determined using EC800 analysis software. The subG1 fraction was regarded as the apoptotic cell population.25
2.4. Caspase activity assay
Breast cancer cell lines were treated using the same conditions as described previously herein. Caspase activities were measured as described previously.25 The cell pellets were resuspended in cell lysis buffer (50 mmol/L HEPES, 100 mmol/L NaCl, 0.1% CHAPS, 1 mmol/L EDTA, 10% glycerol, 0.1% NP40, pH 7.4) for 15‐30 minutes on ice. Total protein (10‐30 μg) was added to the reaction buffer (the same as the cell lysis buffer (except for the addition of 0.1% NP40) containing NAc‐Asp‐Glu‐Val‐Asp‐pNA (Ac‐DEVE‐pNA, a substrate of caspase‐3/7; Sigma‐Aldrich), followed by incubation for 1‐2 hours at 37°C. The free pNA cleaved from its precursor was quantified using a spectrometer at 405 nm (Bio‐Rad, Hercules, CA, USA). The Caspase‐Glo 9 Assay reagent (Promega, Madison, MI, USA) was used as the substrate for activated caspase‐9. An equal volume of cell lysate and caspase‐Glo 9 reagent was added to each well, mixed, and incubated in the dark for 1 hour at room temperature. The resultant luminescence, which is proportional to caspase‐9 activity, was measured using a luminometer (Bio‐Rad). Absorbance was determined using an Envision 2104 multilabel reader (Perkin‐Elmer, Waltham, MA, USA). Data were analyzed using Student's t‐test. A P‐value <0.05 was considered statistically significant.
2.5. Western blotting
Caspase plays a central role in apoptosis, and the cleavage of poly ADP‐ribose polymerase (PARP) facilitates cellular disassembly. Cleaved PARP is a marker of cells undergoing apoptosis.26 Breast cancer cell lines were treated using the same conditions as those described previously herein. Cells were resuspended in radioimmunoprecipitation assay (RIPA) buffer (Sigma‐Aldrich) with protease inhibitors for 15‐30 minutes on ice. Protein (8 μg) was subjected to SDS‐PAGE and then transferred to PVDF membranes. The membranes were blocked for 1 hour and then incubated with respective primary antibodies overnight at 4°C. The anti‐Mieap antibody (rabbit, 1:1000) was prepared as described previously.19 Anti‐caspase‐3 antibody (rabbit, 1:1000), anti‐caspase‐7 antibody (rabbit, 1:1000), anti‐caspase‐9 (rabbit, 1:1000), anti‐cleaved PARP: poly (ADP‐ribose) polymerase (rabbit, 1:2000), anti‐Puma (rabbit, 1:1000), anti‐Bax(rabbit, 1:1000), anti‐Noxa (rabbit, 1:1000), anti‐Bak (rabbit, 1:1000) (from Cell Signaling Technology, Danvers, MA, USA), anti‐Apaf‐1(rat,1:100), anti‐caspase‐8 (mouse, 1:200) (from Santa Cruz, Santa Cruz, CA, USA), anti‐NIX (mouse, 1:250; Abnova, Taipei City, Taiwan) and anti‐BNIP3 (mouse, 1:1000; Abcam, Cambridge, UK). The blots were stripped and re‐probed with a β‐actin antibody (1:5000; Cell Signaling Technology) to ensure equal protein loading. After washing, the membrane was incubated with secondary antibody (1:20000; ProteinTech, Chicago, IL, USA) for 1 hour at room temperature. The immunoreactive proteins were visualized and captured using the LAS‐4000 system (Fujifilm, Tokyo, Japan).
2.6. Immunohistochemistry
We examined the expression of Mieap in vivo using surgical specimens dissected at the Gifu University Hospital (invasive ductal carcinomas [IDC]: 75, ductal carcinoma in situ [DCIS]: 27, fibroadenomas [FA]: 18). Immunohistochemistry (IHC) was performed using Dako EnVision+ Dual Link System‐HRP (Dako, Glostrup, Denmark). For antigen retrieval, sections were pretreated with 0.2% trypsin in 10 mmol/L tris‐HCI (pH 7.6) containing 0.1% CaCl2, for 30 minutes at 37°C. Sections were incubated overnight at 4°C with the rabbit polyclonal anti‐human Mieap antibody (diluted 1:200). Then, sections were incubated with EnVision reagents for 1 hour at room temperature.22 IHC evaluation was performed by experienced pathologists. When we started IHC for human cancer tissues in vivo, we repeated preliminary experiments for appropriate conditions using pre‐immune serum. For subsequent runs, we did not use this due to its shortage. However, as we found endothelial cells and smooth muscle of blood vessels to be good positive controls, we show Mieap‐expressing endothelial cells in Figure S1.
Informed consent was obtained from all patients who agreed to provide a surgical specimen. This study was approved by the central ethics committee of Gifu University.
2.7. Methylation‐specific PCR and p53 mutation search
Methylation‐specific PCR was performed as described previously.19, 23 We used cryopreserved surgical specimens obtained from May 2011 to May 2012 at the Gifu University Hospital, most of which were also used for IHC, described previously. We extracted genomic DNA from 46 samples that were cryopreserved, among 75 IDC cases examined by immunohistochemistry, using DNeasy (Qiagen, Hilden, Germany). Bisulfite‐treated genomic DNA was subjected to MSP. The MSP for Mieap, NIX and BNIP3 promoters was carried out using specific primers under the following conditions: 1 cycle at 94°C for 5 minutes; 35 cycles at 94°C for 30 seconds, 57°C for 30 seconds and 72°C for 1 minute; and a final extension step at 72°C for 7 minutes. The p53 mutation search was performed as described previously.23 Genomic PCR was performed, followed by DNA sequence analysis.
2.8. Statistical analysis
Immunohistochemistry for Mieap was analyzed using the χ2‐test. Disease‐free survival (DFS) was estimated using the Kaplan‐Meier method, and comparisons between groups were performed using a 2‐sided log‐rank test. Results were considered significant at P < 0.05.
3. RESULTS
3.1. Mieap induces cell death in breast cancer cells
First, we investigated the expression level of Mieap protein in breast cancer cells (MCF‐7, SK‐BR‐3 and MDA‐MB‐231) infected with Ad‐Mieap (at a moi of 30‐60). Twenty‐four hours after adenovirus infection, exogenous Mieap protein was observed in each cell line by western blotting, and this was compared to that in control cells infected with EGFP‐expressing adenovirus (Figure S2A). When Mieap was expressed in breast cancer cell lines, vacuole‐like structures (Mieap‐induced vacuoles [MIV]) were observed in the cytoplasm after 24 hours as previously reported (Figure S2B).20
MCF‐7 cells were infected with Ad‐Mieap at a moi of 5, 10, 20, 30 and 60. After 72 hours, we observed morphological changes microscopically and performed FACS analysis. MCF‐7 cells were morphologically smaller, fragmented, and floated freely in the medium, and this phenomenon occurred in a dose‐dependent manner (Figure 1A). FACS analysis demonstrated an increase in the subG1 fraction, suggesting nuclear DNA degradation in a moi‐dependent manner (Figure 1B). We also examined the effect of Mieap on various breast cancer cells. After 48‐72 hours of infection with Ad‐Mieap (30‐60 moi), each cell line was harvested for FACS analysis. Most cells were smaller and floating, particularly MCF‐7 and SK‐BR‐3 cells, when compared to Ad‐EGFP‐infected cells (Figure 1C). The subG1 fractions were 56.94% for MCF‐7 (30 moi, 72 hours) and 43.27% for SK‐BR‐3 (30 moi, 48 hours). In contrast, most MDA‐MB‐231 cells remained attached, with a subG1 fraction of 28.21% (60 moi, 72 hours; Figure 1D).
Figure 1.
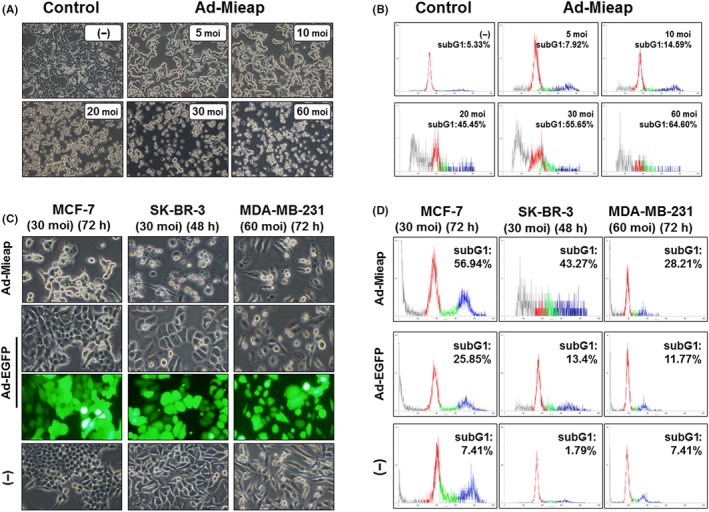
Mieap‐induced cell death in breast cancer cells occurs in a multiplicity of infection (moi)‐dependent manner. A, After 72 h of Ad‐Mieap infection, morphological changes were captured (100× magnification). Cell nuclei were fragmented, and the cell number was decreased in a moi‐dependent manner. B, Adenovirus‐infected cells (MCF‐7) were collected and subjected to FACS analysis. Moi number and percentage of the subG1 fraction (apoptotic cells) are shown in the upper right of each diagram. C, Morphological changes in 3 breast cancer cell lines (MCF‐7, SK‐BR‐3 and MDA‐MB‐231) 48‐72 h after infection at a moi of 30‐60 with Ad‐Mieap. Ad‐Mieap‐infected cells were decreased in number and size. D, After incubation for the indicated time, cells were evaluated by flow cytometry. The subG1 fraction was elevated after Ad‐Mieap infection. The subG1 fractions were 56.94% for MCF‐7 (30 moi, 72 h) and 43.27% for SK‐BR‐3 (30 moi, 48 h). In contrast, most MDA‐MB‐231 cells remained attached, with a subG1 fraction of 28.21%
3.2. Mieap‐induced cell death is apoptosis
As a preliminary experiment, we treated 3 cell lines with etoposide. We found cleaved caspase‐3/7/9 with activation of mitochondrial apoptotic factors particularly in SK‐BR‐3 and MDA‐MB‐231 (Figure S3).17, 27 To investigate the association between Mieap‐induced cell death and caspases, we determined the expression levels and activation of caspase‐3/7 and caspase‐9 by western blotting and caspase activity assays. We found that SK‐BR‐3 was the most sensitive cell line to Mieap‐induced cell death (Figure 1D). Forty‐eight hours after Ad‐Mieap infection (30 moi), not only caspase‐3/7 but also caspase‐9 was significantly elevated in SK‐BR‐3 cells (caspase‐3/7: P = 0.000003, caspase‐9: P = 0.0003). In MCF‐7 cells (Ad‐Mieap: 30 moi), caspase‐3/7 was significantly activated (P = 0.023). However, in MDA‐MB‐231 cells (Ad‐Mieap: 60 moi), no caspase activation was observed (Figure 2A). To confirm the activation of caspases, we performed western blotting for caspase‐3/7 and PARP. In SK‐BR‐3 cells, caspase‐3 was cleaved 48 hours after Ad‐Mieap infection. In MCF‐7 cells, caspase‐7, but not caspase‐3, was cleaved under the same conditions because caspase‐3 is deficient in MCF‐7 cells.28 Cleaved PARP, which is produced through cleavage by activated caspases,29 was detected 48‐72 hours after Ad‐Mieap infection in both cell lines. These results indicate that activation of caspase is a major cause of Mieap‐induced cell death. In contrast, in MDA‐MB‐231 cells, we could detect neither the activation of these caspases nor cleaved PARP, indicating that MDA‐MB‐231 cells might be resistant to Mieap‐induced cell death (Figure 2B).
Figure 2.
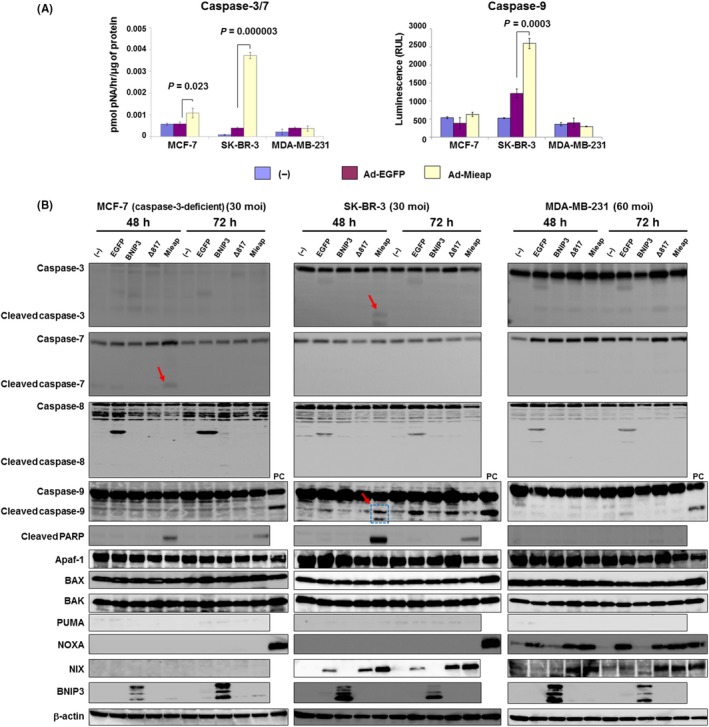
Mieap activates mitochondrial apoptotic pathways in human breast cancer cells in a caspase‐dependent manner. A, After 48 h (SK‐BR‐3) and 72 h (MCF‐7, MDA‐MB‐231) treatments with adenovirus (30 multiplicity of infection (moi) for MCF‐7 and SK‐BR‐3 and 60 moi for MDA‐MB‐231), caspase‐3/7 (left) and caspase‐9 (right) activities were measured. Caspases were significantly elevated in SK‐BR‐3 and MCF‐7 cells but not in MDA‐MB‐231 cells. B, Western blotting data for cleaved caspase‐3/7 (red arrows) and cleaved poly (ADP‐ribose) polymerase are indicated. Cell lysates from 3 breast cancer cell lines were harvested under the indicated conditions (48‐72 h, 30‐60 moi). Caspase‐3‐deficient MCF‐7 cells showed cleavage of caspase 7. No cleaved caspase‐8 was detected in any of the cells and caspase‐9 was cleaved in SK‐BR‐3 cells (arrow and dotted box). No mitochondria‐associated apoptotic factors were altered in MCF‐7 cells. In SK‐BR‐3 cells, NIX was elevated, resulting in caspase‐9 activation. Despite activation of both NOXA and NIX, caspases including caspase‐9 were not activated in MDA‐MB‐231 cells. Based on these data, Mieap can activate the mitochondria apoptotic pathway. However, the occurrence of Mieap‐induced cell death depends on caspase activation. PC indicates positive control. No infection (−), Ad‐EGFP, Ad‐BNIP3 and Ad‐Δ817 (deletion mutant [273 amino acids] of Mieap) were used as negative controls
Interestingly, in SK‐BR‐3 cells, cleaved caspase‐9/3 was detected after 48 hours of Ad‐Mieap treatment and was associated with NIX elevation. Although in MDA‐MB‐231 cells NOXA and NIX were elevated, neither caspases nor PARP was cleaved. In MCF‐7 cells, which are caspase‐3‐deficient cells, only caspase‐7 was cleaved, with no upregulation of mitochondrial apoptotic factors. We further investigated the expression of BNIP3/NIX in each cell line. Both proteins were not expressed in steady‐state conditions; however, NIX was elevated in SK‐BR‐3 and MDA‐MB‐231 after Ad‐Mieap infection. BNIP3 was not expressed irrespective of enforced Mieap expression. These results indicated that Mieap is involved in the mitochondrial apoptotic pathway in a caspase‐dependent manner. Based on these results, we concluded that Mieap‐induced cell death is apoptosis and that it is mediated by the mitochondrial apoptotic pathway.
3.3. Mieap expression is downregulated in tumors from breast cancer patients with an aggressive and malignant phenotype
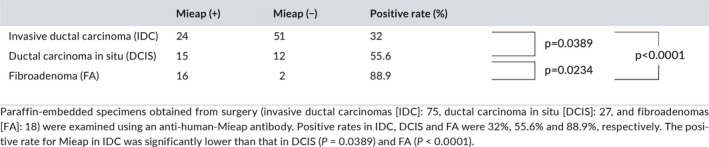
We next performed IHC using surgical specimens obtained from 120 patients (IDC: 75, DCIS: 27, and FA: 18) to examine the expression of Mieap in vivo. Mieap was detected in the cytoplasm, and representative cases (both positive and negative cases, respectively) are shown in Figure 3. Mieap was positive in 32% (24/75) of IDC, 55.6% (15/27) of DCIS and 88.9% (16/18) of FA. Its expression was also significantly higher in FA compared to that in DCIS (P = 0.0234), in DCIS compared to that in IDC (P = 0.0389), and in FA compared to that in IDC (P < 0.0001), indicating that its expression decreases with malignant potential (Table 1).
Figure 3.
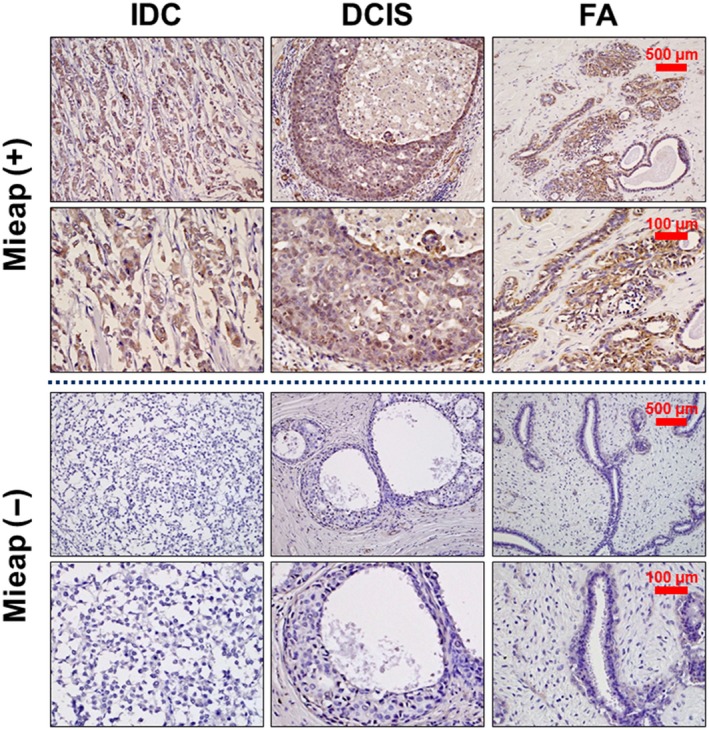
Immunohistochemistry for Mieap in breast tumors in vivo. Immunohistochemical staining of Mieap in tissue specimens obtained from patients with invasive ductal carcinomas (IDC), ductal carcinoma in situ (DCIS) and fibroadenomas (FA). Representative cases (Mieap‐positive [+], Mieap‐negative [−]) are indicated. Mieap is expressed in the cytoplasm. The scale bars represent 500 μm (upper row) and 100 μm (lower row) photomicrographs
Table 1.
Immunohistochemistry for Mieap in vivo using human breast cancer tissues
3.4. The p53/Mieap‐regulated mitochondrial quality control pathway is genetically and epigenetically inactivated in aggressive and malignant breast cancer
To investigate inactivation of the p53/Mieap‐regulated MQC pathway, we performed MSP for Mieap, NIX and BNIP3 promoters, as well as a p53 mutation search, because NIX and BNIP3 have been shown to be co‐factors for MQC.21 We investigated 46 IDC, which had been stored as frozen tissue for DNA extraction. MSP revealed methylation of the Mieap promoter in 6 of 46 IDC (13%). However, there was no methylation of the NIX or BNIP3 promoters (Figure 4). Genetic alterations of p53 were found in 6 of 46 IDC (13%). The p53/Mieap‐regulated MQC pathway was inactivated in 12 of 46 IDC (26.1%; Table 2). Of 12 cases, 6 with methylated Mieap promoter were luminal B and the other 6 with mutant p53 were triple negative as follows: 4, HER2‐rich; 1, luminal B; 1, whose subtypes were clinically more aggressive and malignant phenotypes in breast cancer (Table 3).30, 31
Figure 4.
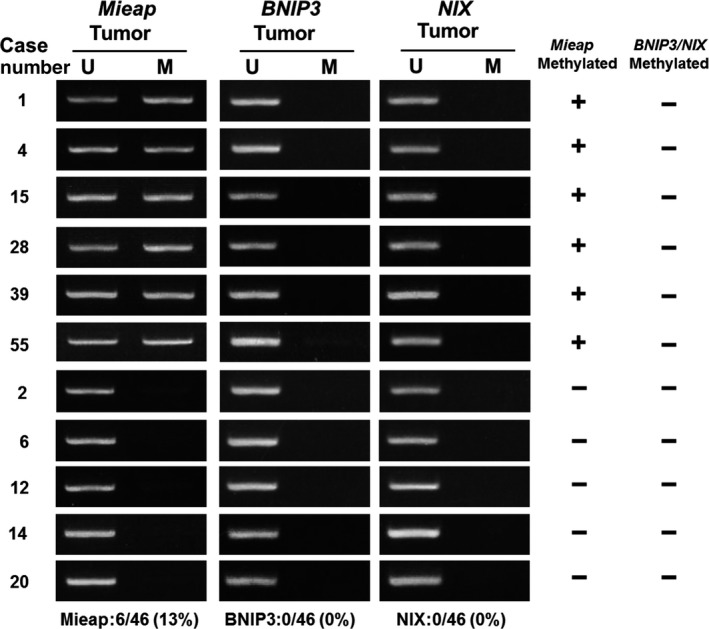
Methylation‐specific PCR (MSP) and breast cancer clinical outcome based on inactivation of Mieap‐regulated mitochondrial quality control (MQC). MSP analysis of the Mieap,NIX and BNIP3 promoters. Genomic DNA was extracted from surgically dissected invasive breast cancer and then treated with bisulfite treatment and subjected to MSP for Mieap/NIX/BNIP3 promoters. The results of MSP from 11 representative cases are shown. Mieap promoter methylation was detected in 6 cases. However, no samples showed NIX/BNIP3 methylation. M, methylated; U, unmethylated
Table 2.
Association between regulatory factors of Mieap expression and subtypes
| Case | Mieap methylated | NIX methylated | BNIP3 methylated | p53 mutation | Mieap expression | Subtype |
|---|---|---|---|---|---|---|
| 1 | + | − | − | − | − | Luminal B |
| 4 | + | − | − | − | − | Luminal B |
| 15 | + | − | − | − | − | Luminal B |
| 28 | + | − | − | − | − | Luminal B |
| 39 | + | − | − | − | − | Luminal B |
| 55 | + | − | − | − | − | Luminal B |
| 10 | − | − | − | Deletion in exon 4 | − | Triple negative |
| 32 | − | − | − | Deletion in exon 4 | − | Triple negative |
| 35 | − | − | − | Deletion in exon 7 | − | Triple negative |
| 42 | − | − | − | TGC→TGG(176C→W) | + | Triple negative |
| 52 | − | − | − | CGT→TGT (273R→C) | − | Luminal B |
| 56 | − | − | − | GAC→CAC (281D→H) | NE | HER2 |
| 13 | − | − | − | − | + | Luminal A |
| 33 | − | − | − | − | + | Triple negative |
| 40 | − | − | − | − | + | Luminal B |
| 44 | − | − | − | − | + | Luminal A |
| 49 | − | − | − | − | + | Luminal B |
| 50 | − | − | − | − | + | Luminal B |
| 2 | − | − | − | − | − | Luminal A |
| 6 | − | − | − | − | − | HER2 |
| 11 | − | − | − | − | − | Luminal/HER2 |
| 12 | − | − | − | − | NE | Luminal/HER2 |
| 16 | − | − | − | − | − | Luminal A |
| 17 | − | − | − | − | − | Luminal A |
| 18 | − | − | − | − | − | Luminal B |
| 19 | − | − | − | − | − | Luminal A |
| 20 | − | − | − | − | − | Luminal A |
| 22 | − | − | − | − | − | Luminal A |
| 23 | − | − | − | − | − | Luminal B |
| 24 | − | − | − | − | − | Luminal A |
| 25 | − | − | − | − | − | Luminal A |
| 26 | − | − | − | − | − | Luminal B |
| 27 | − | − | − | − | − | Luminal B |
| 29 | − | − | − | − | NE | Luminal B |
| 30 | − | − | − | − | − | Luminal A |
| 31 | − | − | − | − | − | Luminal A |
| 34 | − | − | − | − | NE | Luminal A |
| 37 | − | − | − | − | − | Triple negative |
| 38 | − | − | − | − | − | Luminal A |
| 41 | − | − | − | − | − | Luminal B |
| 43 | − | − | − | − | − | Triple negative |
| 46 | − | − | − | − | − | Luminal A |
| 47 | − | − | − | − | − | Luminal A |
| 48 | − | − | − | − | NE | Luminal A |
| 53 | − | − | − | − | − | Luminal B |
| 54 | − | − | − | − | − | Luminal A |
| 6/46 (13%) | 0/46 (0%) | 0/46 (0%) | 6/46 (13%) | 7/41 (17%) |
NE, not examined.
Results of MSP for Mieap/NIX/BNIP3 promoters and p53‐mutation search (exons 4‐8) are summarized. Methylation of the Mieap promoter was positive in 6/46 (13%) cases, whereas NIX/BNIP3 promoters were negative. Genetic alterations in p53 were detected in 6/46 (13%) cases. Finally, inactivation of the p53/Mieap/NIX/BNIP3‐regulated mitochondrial quality control (MQC) pathway was observed in 12/46 (26.1%) cases. These cases were aggressive phenotypes such as Luminal B, triple negative and HER2‐enriched. Among them, ten cases of 11 subjected to IHC lost Mieap expression.
Table 3.
Summary of clinicopathological parameters in human breast cancers
| Case | Age | Gender | Subtype | TN | Stage | Recurrence | Histopathological diagnosis |
|---|---|---|---|---|---|---|---|
| 1 | 58 | Female | Luminal B | T4bN1 | IIIB | − | Scirrhous carcinoma |
| 4 | 46 | Female | Luminal B | T2N0 | IIA | − | Scirrhous carcinoma |
| 15 | 76 | Female | Luminal B | T1N0 | I | − | Invasive lobular carcinoma |
| 28 | 48 | Female | Luminal B | T2N1 | IIB | + | Solid‐tubular carcinoma |
| 39 | 51 | Female | Luminal B | T3N1 | IIIA | − | Scirrhous carcinoma |
| 55 | 72 | Female | Luminal B | T2N0 | IIA | − | Scirrhous carcinoma |
| 2 | 74 | Female | Luminal A | T2N0 | IIA | − | Solid‐tubular carcinoma |
| 6 | 36 | Female | HER2 | T2N1 | IIB | + | Scirrhous carcinoma |
| 10 | 69 | Female | Triple negative | T1N1 | IIA | − | Solid‐tubular carcinoma |
| 11 | 45 | Female | Luminal/HER2 | T1N0 | I | − | Scirrhous carcinoma |
| 12 | 75 | Female | Luminal/HER2 | T1N2 | IIB | − | Solid‐tubular carcinoma |
| 13 | 72 | Female | Luminal A | T1N0 | I | − | Scirrhous carcinoma |
| 16 | 51 | Female | Luminal A | T1N1 | IIA | − | Papillotubular carcinoma |
| 17 | 60 | Female | Luminal A | T2N0 | IIA | − | Papillotubular carcinoma |
| 18 | 43 | Female | Luminal B | T2N0 | IIA | − | Papillotubular carcinoma |
| 19 | 66 | Female | Luminal A | T1N1 | IIA | − | Scirrhous carcinoma |
| 20 | 79 | Female | Luminal A | T1N0 | I | − | Solid‐tubular carcinoma |
| 22 | 58 | Female | Luminal A | T1N1 | IIA | − | Scirrhous carcinoma |
| 23 | 84 | Female | Luminal B | T1N0 | I | − | Scirrhous carcinoma |
| 24 | 62 | Female | Luminal A | T2N0 | IIA | − | Scirrhous carcinoma |
| 25 | 63 | Female | Luminal A | T2N0 | IIA | + | Scirrhous carcinoma |
| 26 | 49 | Female | Luminal B | T1N1 | IIA | − | Solid‐tubular carcinoma |
| 27 | 48 | Female | Luminal B | T2N0 | IIA | − | Scirrhous carcinoma |
| 29 | 57 | Female | Luminal B | T2N1 | IIB | − | Mucinous carcinoma |
| 30 | 61 | Female | Luminal A | T2N1 | IIB | − | Scirrhous carcinoma |
| 31 | 64 | Female | Luminal A | T2N1 | IIB | − | Scirrhous carcinoma |
| 32 | 52 | Female | Triple negative | T2N0 | IIA | − | Solid‐tubular carcinoma |
| 33 | 69 | Female | Triple negative | T2N0 | IIA | − | Papillotubular carcinoma |
| 34 | 51 | Female | Luminal A | T1N0 | I | − | Papillotubular carcinoma |
| 35 | 46 | Female | Triple negative | T2N0 | IIA | + | Solid‐tubular carcinoma |
| 37 | 62 | Female | Triple negative | T1N0 | I | − | Scirrhous carcinoma |
| 38 | 53 | Female | Luminal A | T1N0 | I | − | Papillotubular carcinoma |
| 40 | 42 | Female | Luminal B | T2N1 | IIB | − | Papillotubular carcinoma |
| 41 | 68 | Female | Luminal B | T1N0 | I | − | Solid‐tubular carcinoma |
| 42 | 57 | Female | Triple negative | T2N1 | IIB | + | Scirrhous carcinoma |
| 43 | 79 | Female | Triple negative | T2N0 | IIA | − | Papillotubular carcinoma |
| 44 | 51 | Female | Luminal A | T1N0 | I | − | Papillotubular carcinoma |
| 46 | 64 | Female | Luminal A | T1N0 | I | − | Papillotubular carcinoma |
| 47 | 63 | Female | Luminal A | T1N0 | I | − | Scirrhous carcinoma |
| 48 | 73 | Female | Luminal A | T1N0 | I | − | Papillotubular carcinoma |
| 49 | 33 | Female | Luminal B | T1N0 | I | − | Scirrhous carcinoma |
| 50 | 58 | Female | Luminal B | T2N0 | IIA | − | Solid‐tubular carcinoma |
| 52 | 74 | Female | Luminal B | T4bN1 | IIIB | + | Scirrhous carcinoma |
| 53 | 46 | Female | Luminal B | T2N0 | IIA | − | Papillotubular carcinoma |
| 54 | 70 | Female | Luminal A | T1N0 | I | − | Papillotubular carcinoma |
| 56 | 68 | Female | HER2 | T2N0 | IIA | − | Mucinous carcinoma |
Clinicopathological parameters of 46 invasive breast cancer patients are shown followed by Table 2.
Interestingly, the group with inactivation of the p53/Mieap‐regulated MQC pathway showed shorter DFS than that without these genetic alterations (P = 0.021, Figure 5). Four of six recurrent cases showed inactivation in the p53/Mieap‐regulated MQC pathway. Of 46 cases, we separately showed DFS curves for individuals with or without Mieap promoter methylation or those with or without p53 alterations (Figure S4). As shown in Figure S4A, there was no significant difference between those with Mieap promoter methylation (n = 6) and those without (n = 40). Six cases with a methylated Mieap promoter showed no expression of Mieap by IHC. However, as shown in Figure S4B, there was a significant difference between those with p53 mutations (n = 6) and those without (n = 40; P = 0.00035). Considering p53‐mutated cases, 4 of 5 revealed no Mieap expression. Mieap expression was completely undetectable in 10 of 11 cases with either methylated Mieap promoter or p53 alterations. In addition, as shown in Figure S4C, among 75 IDC evaluated by IHC (in Figure 3), there were no significant differences between 24 Mieap‐positive IDC cases and 51 Mieap‐negative cases. These results suggest that Mieap might not be prognostic, at least to the same extent as p53. Based on the previous reports that Mieap expression is regulated by either p53 status or methylation of Mieap, BNIP3 and NIX promoters, we suggest that it is important to evaluate MQC based on these factors (i.e. MQC pathway through p53 and BNIP3/NIX to Mieap).
Figure 5.
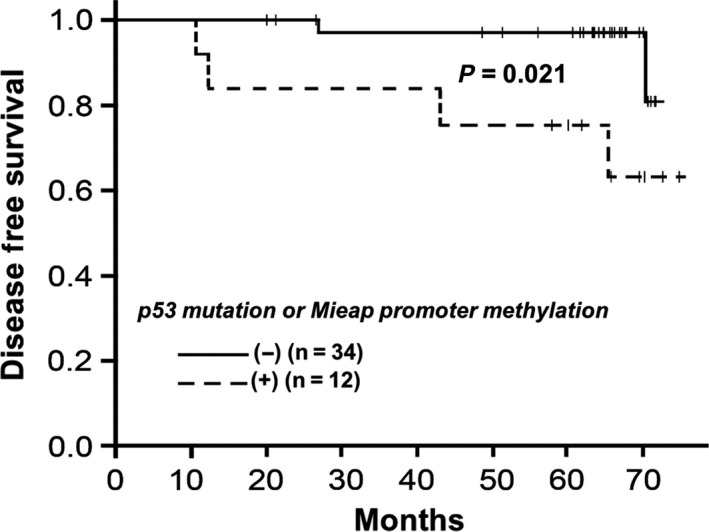
Kaplan‐Meier curve showing disease‐free survival (DFS) in breast cancer patients based on Mieap‐regulated mitochondrial control (MQC) expression. The group with inactivated Mieap‐regulated MQC (dotted line; n = 12) exhibited shorter DFS than that without genetic alterations (solid line; n = 34; P = 0.021)
4. DISCUSSION
According to a previous report, we demonstrated the physiological functions related to MQC, which were classified mainly into 2 mechanisms. One is referred to as MALM, whereas the other is MIV, which occurs when salvage of the damaged mitochondria appears to be impossible for recovery and survival.20 These mechanisms might be used depending on the degree of mitochondrial damage. In this study, we focused on the MIV, followed by Mieap‐induced cell death, and found strong evidence supporting the role of Mieap in breast cancer tumor suppression.
First, Mieap induced caspase‐dependent apoptosis in breast cancer cells. We infected adenoviruses at a moi of 30‐60 into each cell (for MIV), for which Ad‐Mieap mois were 5‐10‐fold greater compared to that for MALM. This Mieap‐induced apoptosis was likely consistent with the mitochondrial intrinsic pathway because Mieap activated caspase‐9/3 as observed in SK‐BR‐3 cells. However, given that MCF‐7 cells are known to be a caspase‐3‐deficient cell line,28 caspase‐7 was suggested to function instead, as Ac‐DEVE‐pNA is a substrate for both caspase‐3 and caspase‐7. Lakhani et al26 report that both caspase‐3 and caspase‐7 regulate mitochondrial events in the apoptotic pathway. These results suggest that apoptosis induced by Mieap occurs via a caspase‐dependent intrinsic pathway through the mitochondria. Through our experiments using 3 cell lines, we identified at least 3 patterns against Mieap: (i) good response: in SK‐BR‐3 cells, Mieap activates apoptotic factors in the mitochondria in a caspase (9/3)‐dependent manner; (ii) fair response: in MCF7 cells, Mieap activates apoptotic factors in mitochondria; however, cells do not undergo enough apoptosis, exhibiting activation of caspase‐7, but not caspase‐9/3 and (iii) poor response: in MDA‐MB‐231 cells, Mieap activates apoptotic factors in the mitochondria (however, no caspases are activated, resulting in resistance to Mieap‐induced cell death).
Second, Mieap protein was downregulated in tumors with more aggressive and malignant phenotypes of human breast cancer. In FA, which consists of both glandular breast tissue and stromal tissue, Mieap was expressed in the cytoplasm of glandular tissue in 16/18 (88.9%) cases. Interestingly, Mieap expression tended to decrease with malignant transformation (DCIS; 15/27 [55.6%], IDC; 24/75 [32%]; Table 1). These data suggest that Mieap plays an important role in preventing the malignant transformation and/or progression of breast tumors. Given that Mieap is under the control of p53, it is understandable that benign glandular tissues might express Mieap to suppress malignant transformation.
Third, p53/Mieap‐regulated MQC is genetically and epigenetically inactivated in tumors with more aggressive and malignant human breast cancer phenotypes. The p53/Mieap/BNIP3‐regulated MQC pathway is inactivated in >70% of colorectal cancers, suggesting that Mieap‐regulated MQC has a critical role in colorectal cancer suppression in vivo.23 In this study, we found that this pathway was inactivated in 12/46 (26.1%) invasive breast cancers via the promoter methylation of Mieap or p53 mutations, which occurred at a lower frequency compared to that observed for colorectal cancer. This might be due to the BNIP3 status in breast cancer. The promoter methylation of BNIP3 was not detected in any breast cancer samples, whereas it occurred in nearly 50% colorectal cancer specimens. Interestingly, among 12 cases exhibiting inactivation of the pathway, 6 cases with methylation of the Mieap promoter were luminal B type, which comprises aggressive and fast‐growing estrogen receptor‐positive breast cancers. The other 6 cases with a p53 mutation were also aggressive types (triple negative, 4; HER2‐rich, 1; luminal B, 1).30, 31 Furthermore, as shown in Table 2, there were 6 cases with p53 alterations, in which 5 were subjected to IHC for Mieap. Four of five cases did not express Mieap. Specifically, Mieap expression completely disappeared in 10 of 11 cases with either Mieap promoter methylation or p53 alterations. In contrast, only 1 expressed Mieap, despite the presence of a p53 mutation. This might be explained by the possibility that not only Mieap but some other p53‐target genes are often induced in a p53‐independent manner.
Fourth, the group with p53 mutation or Mieap promoter methylation showed shorter DFS, suggesting that inactivation of the p53/Mieap‐regulated MQC pathway might lead to failed cell death induction, resulting in enhanced malignant potential and aggressiveness in vivo.
In summary, Mieap induces caspase‐dependent apoptosis in breast cancer cells in vitro. Moreover, p53/Mieap‐regulated MQC is inactivated in breast cancers with more aggressive and malignant phenotypes in vivo. Therefore, p53/Mieap‐regulated MQC plays a critical role as a tumor suppressor in human breast cancer. Although there were limitations to our present work, this study forms the basis for future research projects to develop novel strategies to treat cancer.
DISCLOSURE STATEMENT
KY has received grants and personal fees from Taiho Pharmaceutical, Pfizer, Chugai Pharmaceutical and Yakult Honsha, grants from Bristol‐Myers Squibb and Kyowa Hakko Kirin, and honoraria from Taiho Pharmaceutical, Pfizer, Chugai Pharmaceutical, Kyowa Hakko Kirin and Yakult Honsha, and had a consultant and advisory relationship with Taiho Pharmaceutical and La Roche. All remaining authors declare that they have no conflict of interests.
Supporting information
ACKNOWLEDGMENTS
We thank Shizu K for technical support and Enya K, Hirata M, Iwata A, Mori K and Takano K for administrative assistance.
Gaowa S, Futamura M, Tsuneki M, et al. Possible role of p53/Mieap‐regulated mitochondrial quality control as a tumor suppressor in human breast cancer. Cancer Sci. 2018;109:3910–3920. 10.1111/cas.13824
Contributor Information
Manabu Futamura, Email: mfutamur@gifu-u.ac.jp.
Hirofumi Arakawa, Email: harakawa@ncc.go.jp.
REFERENCES
- 1. International Agency for Research on Cancer . GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. http://globocan.iarc.fr/Default.aspx
- 2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 3. Collaborative Group on Hormonal Factors in Breast Cancer . Breast cancer and breastfeeding: collaborative reanalysis of individual data from 47 epidemiological studies in 30 countries, including 50 302 women with breast cancer and 96 973 women without the disease. Lancet. 2002;360:187‐195. [DOI] [PubMed] [Google Scholar]
- 4. American Cancer Society . Breast Cancer Facts & Figures 2015–2016. Atlanta: American Cancer Society; 2015. https://www.cancer.org/research/cancer-facts-statistics/breast-cancer-facts-figures.html [Google Scholar]
- 5. Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72:1117‐1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49‐53. [DOI] [PubMed] [Google Scholar]
- 7. Coles C, Condie A, Chetty U, Steel CM, Evans HJ, Prosser J. p53 mutations in breast cancer. Cancer Res. 1992;52:5291‐5298. [PubMed] [Google Scholar]
- 8. Borresen‐Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21:292‐300. [DOI] [PubMed] [Google Scholar]
- 9. The Cancer Genome Atlas Network . Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang P, Du CW, Kwan M, Liang SX, Zhang GJ. The impact of p53 in predicting clinical outcome of breast cancer patients with visceral metastasis. Sci Rep. 2013;3:2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307‐310. [DOI] [PubMed] [Google Scholar]
- 12. El‐Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817‐825. [DOI] [PubMed] [Google Scholar]
- 13. Oda K, Arakawa H, Tanaka T, et al. p53AIP1, a potential mediator of p53‐dependent apoptosis, and its regulation by Ser‐46‐phosphorylated p53. Cell. 2000;102:849‐862. [DOI] [PubMed] [Google Scholar]
- 14. Tanaka H, Arakawa H, Yamaguchi T, et al. A ribonucleotide reductase gene involved in a p53‐dependent cell‐cycle checkpoint for DNA damage. Nature. 2000;404:42‐49. [DOI] [PubMed] [Google Scholar]
- 15. Futamura M, Kamino H, Miyamoto Y, et al. Possible role of semaphorin 3F, a candidate tumor suppressor gene at 3p21.3, in p53‐regulated tumor angiogenesis suppression. Cancer Res 2007;67:1451‐1460. [DOI] [PubMed] [Google Scholar]
- 16. Nakamura Y, Arakawa H. Discovery of Mieap‐regulated mitochondrial quality control as a new function of tumor suppressor p53. Cancer Sci. 2017;108:809‐817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arakawa H. p53, apoptosis and axon‐guidance molecules. Cell Death Differ. 2005;12:1057‐1065. [DOI] [PubMed] [Google Scholar]
- 18. Susan E. Apoptosis: A review of programed cell death. Toxicol Pathol. 2007;35:495‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyamoto Y, Kitamura N, Nakamura Y, et al. Possible existence of lysosome‐like organella within mitochondria and its role in mitochondrial quality control. PLoS ONE. 2011;6:e16054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kitamura N, Nakamura Y, Miyamoto Y, et al. Mieap, a p53‐inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PLoS ONE. 2011;6:e16060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nakamura Y, Kitamura N, Shinogi D, et al. BNIP3 and NIX mediate Mieap‐induced accumulation of lysosomal proteins within mitochondria. PLoS ONE. 2012;7:e30767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsuneki M, Nakamura Y, Kinjo T, Nakanishi R, Arakawa H. Mieap suppresses murine intestinal tumor via its mitochondrial quality control. Sci Rep. 2015;5:12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kamino H, Nakamura Y, Tsuneki M, et al. Mieap‐regulated mitochondrial quality control is frequently inactivated in human colorectal cancer. Oncogenesis. 2016;4:e181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ohnishi S, Futamura M, Kamino H, et al. Identification of NEEP21, encoding neuron‐enriched endosomal protein of 21 kDa, as a transcriptional target of tumor suppressor p53. Int J Oncol. 2010;37:1133‐1141. [DOI] [PubMed] [Google Scholar]
- 26. Lakhani SA, Masud A, Kuida K, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zang SH, Huang Q. Etoposide induces apoptosis via the mitochondrial‐ and caspase‐dependent pathways and in non‐cancer stem cells in Panc‐1 pancreatic cancer cells. Oncol Rep. 2013;30:2765‐2770. [DOI] [PubMed] [Google Scholar]
- 28. Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase‐3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 1998;273:9357‐9360. [DOI] [PubMed] [Google Scholar]
- 29. Oliver FJ, de la Rubia G, Rolli V, Ruiz‐Ruiz MC, de Murcia G, Murcia JM. Importance of poly (ADP‐ribose) polymerase and its cleavage in apoptosis. J Biol Chem. 1998;273:33533‐33539. [DOI] [PubMed] [Google Scholar]
- 30. Goldhirsch A, Winer EP, Coates AS, et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013;24:2206‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hennigs A, Riedel F, Gondos A, et al. Prognosis of breast cancer molecular subtypes in routine clinical care: A large prospective cohort study. BMC Cancer. 2016;16:734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials