

Abstract
Endocrine therapy and radiotherapy are the main treatments for luminal A breast cancer. However, drug and radiotherapy resistance could occur during long‐term treatment, leading to local recurrence and distant metastasis. Some studies have found that drug resistance might be related to human epidermal growth factor receptor‐3 (HER3) overexpression. However, whether HER3 plays a role in radiotherapy resistance is unknown. The purpose of this study is to elucidate the effect of HER3 in radiotherapy and to assess whether HER3 could be a potential target for radiosensitivity. We used retroviruses to construct stable low expression of HER3 in MCF‐7 and ZR75‐1cells. The CCK‐8 assay was used to observe proliferation. Colony‐forming assay was used to detect radiosensitivity. Flow cytometry was used to observe the cell cycle and apoptosis. Immunofluorescence assay was used to detect the number of γH2AX foci in the nucleus with or without ionizing radiation (IR). Western blot analysis was used to verify the change of relative proteins. Nude mice were used to observe tumor growth in vivo. In our study, silencing HER3 reduced cell proliferation and clone formation ability after IR, so silencing HER3 increased the sensitivity of luminal A breast cancer cells to radiotherapy. In terms of radiosensitivity mechanisms, it is suggested that the silencing of HER3 enhanced IR‐induced DNA damage, reduced DNA repair, and increased apoptosis and G2/M arrest. In addition, silencing HER3 combined with IR clearly inhibited the transplanted tumor growth in vivo. Therefore, we concluded that HER3 played a role in radiotherapy resistance. Silencing HER3 increased the radiosensitivity of luminal A breast cancer cells and HER3 could be a potential target for radiosensitivity.
Keywords: HER3, ionizing radiation, luminal A breast cancer, mechanism, radiosensitivity
1. INTRODUCTION
The 13th St. Gallen International Breast Cancer Conference (2013) reached a new consensus on the molecular subtypes of breast cancer. Among them, the luminal A molecular phenotype is estrogen receptor‐positive, progesterone receptor‐positive, human epidermal growth factor receptor‐2 (HER2)‐negative, and Ki‐67 <20%. The consensus also points out that luminal A breast cancer is sensitive to endocrine therapy and radiotherapy (RT) is necessary for mastectomy and breast‐conserving therapy.1 However, some patients have developed resistance to endocrine therapy and distant metastasis during long‐term treatment. Previous studies have shown that the occurrence of appeal was related to the high expression of HER3 and downstream RAS/RAF/MAPK and PI3K/AKT signaling pathways.2, 3, 4, 5, 6, 7, 8
Human epidermal growth factor receptor‐3 is a member of the human epidermal growth factor receptor (EGFR) family (EGFR/HER1, HER2, HER3, and HER4). It consists of three major domains, namely the extracellular ligand binding domain, the transmembrane region, and the intracellular tyrosine kinase‐binding domain. Due to its weak tyrosine kinase activity, HER3 mainly forms heterodimers with other members of the EGFR family, particularly with EGFR and HER2, resulting in the phosphorylation of the carboxyl‐terminal tyrosine residue of HER3, thereby activating RAS/RAF/MAPK and PI3K/AKT signaling pathway, playing an important role in cell proliferation and survival.8, 9, 10
Radiotherapy is one of the major means for breast cancer treatment. It plays an important role in the local control of breast cancer. It can significantly reduce the local recurrence rate and mortality in breast‐conserving surgery and mastectomy patients.10, 11 However, local recurrence and distant metastasis might also occur during long‐term treatment, resulting in RT resistance. Increasing the sensitivity of breast cancer patients to RT is one of the effective ways to solve RT resistance. Given the important role of HER3 in endocrine therapy resistance, we therefore wonder whether HER3 also plays an important role in RT resistance.
In the present study, we used human luminal A breast cancer cells MCF‐7 and ZR75‐1 as experimental subjects, in which the expression of HER3 is higher than that of normal breast epithelial cells.12, 13 We constructed cell lines with low expression of HER3, then we designed experiments to observe whether knockdown of HER3 increased radiosensitivity and explored the mechanisms of radiosensitization. We found that silencing HER3 increased the sensitivity of luminal A breast cancer cells to RT. Possible mechanisms are that silencing HER3 enhanced IR‐induced DNA damage, reduced DNA repair, increased apoptosis and G2/M arrest. Our data indicated that HER3 is a potential target for radiosensitization.
2. MATERIALS AND METHODS
2.1. Cell culture and irradiated methods
Human breast cancer cell lines MCF‐7 and ZR75‐1 were provided by the Shanghai Institute of Cell Biology (Shanghai, China) and cultured in DMEM. All culture media were supplemented with 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. Both cell lines grew at 37°C in a humidified atmosphere containing 5% CO2. Cells in the IR groups were subjected to 2, 4, 6, or 8 Gy X‐ray irradiation from a medical linear accelerator (Precise accelerator, Elekta, Sweden) at room temperature.
2.2. Small interfering RNA and shRNA‐mediated HER3 knockdown
Small interfering RNAs and retroviral vectors expressing shRNA were obtained from (Shanghai Gene Pharma Pharmaceutical Technology, Shanghai, China). The sequences for siRNAs targeting HER3 were 5′‐GGACUCGAGCAACAUUGAUTT‐3′, 5′‐GCGACUAGACAUCAAGCAUTT‐3′, and 5′‐GCUUGUCCUGUCGAAAUUATT‐3′. The sequence for control siRNA was 5′‐UUCUCCGAACGUGUCACGUTT‐3′. After verification, the final selected sequence for shRNA targeting HER3 was 5′‐GGACUCGAGCAACAUUGAUTT‐3′. According to the manufacturer's instructions, at 72 hours post‐transfection the transfected cells were plugged with puromycin. One week after drug selection, stably transfected cell lines were obtained.
2.3. Western blot analysis
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors (Kaygen, Nanjing, Jiangsu Province, China). The protein concentration was quantified by a BCA kit (Beyotime Biotechnology, Shanghai, China). Equal amounts of the proteins were separated by SDS‐PAGE and transferred to PVDF membranes (Millipore, Bas‐Rhin, France). The membranes were soaked with 5% skim milk, incubated with primary Abs against GAPDH, HER3, P‐HER3, AKT, P‐AKT, poly (ADP‐ribose) polymerase (PARP), γH2AX, Bcl‐2, Bax, and Bad at 4°C for 1 night and incubated with secondary Abs (BioWorld, Saint Louis, MN, USA) at room temperature for 1 hour. Immunoblotted proteins were detected by the ChemiDoc XRS imaging system (Quantity One Quantitation software; Bio‐Rad Laboratories, Hercules, CA, USA).
2.4. Cell viability assay
Cell proliferation was measured by a CCK‐8 assay. Cells were digested and plated at a concentration of 5 × 103 cells/well in 96‐well plates at 37°C. A CCK8 cell proliferation and cytotoxicity assay kit (Beyotime Biotechnology) was used for an additional 2 hours after 24, 48, and 72 hours of culture. The absorbance was measured at a wavelength of 450 nm in a microplate reader (ELx800, BioTek, Winooski, VT, USA).
2.5. Clonogenic survival assay
Cells were seeded in 6‐well plates. After overnight culture, cells were then irradiated with 6 MV X‐rays at doses of 2, 4, 6 and 8 Gy at room temperature. The cells were then cultured in a 5% CO2 incubator at 37°C for 10 to 14 days. The colonies were fixed and stained with crystal violet to count the number of colonies (>50 cells/colony).
2.6. Flow cytometry analysis
Cells were seeded at a concentration of 2 × 105 cells/well in 6‐well plates. After incubation for 24 hours, the cells were exposed to 8 Gy X‐rays. After 24 hours, an FITC Annexin V Apoptosis Detection Kit (BD Biosciences, Oxford, UK) was used to detect the apoptotic cells and PI/RNase Staining Buffer (BD Biosciences) was used to detect cell cycle by flow cytometry.
2.7. Immunofluorescence assay
Cells were seeded a concentration of 5 × 104 into confocal laser small dishes and harvested at 0.5, 1, 6, 24 hours post IR. Cells were subsequently fixed in 4% paraformaldehyde at room temperature for 30 minutes and permeabilized in 0.1% Triton X‐100 (Sigma, Santa Clara, CA, USA) for 15 minutes. The cells were then blocked with 5% BSA (Gibco, NY, USA) for 1 hour and incubated with primary antibody γH2AX (1 μg/mL; Abcam, Cambridge, Cambridgeshire, UK) overnight at 4°C. Cells were washed in TBST 3 times every 5 minutes before incubating with a secondary Ab (Beyotime Biotechnology) for 1 hour. Cells were treated with 2 μg/mL DAPI (Beyotime Biotechnology) for 5 minutes and then visualized using confocal fluorescence microscopy (Leica, Frankfurt, Germany).
2.8. Xenograft mouse model
Female BALB/c nude mice (5‐6 weeks of age) were purchased from the Nanjing Medical University Animal Centre (Nanjing, China). The mice were randomly assigned to 4 groups (n = 6): (i) sh‐Control; (ii) sh‐Control and IR (8 Gy); (iii) sh‐HER3; and (iv) sh‐HER3 and IR (8 Gy). Nude mice were injected with 0.2 mL PBS containing 5 × 106 cells into the right armpit to establish the xenografts. When the tumor size reached 50‐100 mm3 (day 1), tumors were irradiated. Mice were killed on day 9 after IR (8 Gy). Body weights and tumor volumes were measured every other day. Tumor volumes were calculated by measuring the length [L] and width [W] of tumors using callipers. The formula tumor volume = (L × W2)/2 was used to calculate the tumor volume. The Ethics Committee of Nanjing Medical University approved animal experiments.
2.9. Data analysis
Mean ± SD from triplicate assays were calculated and the differences between treatment groups were determined using t tests, one‐way ANOVA, and two‐way ANOVA. Statistical analysis was carried out by using GraphPad Prism 5.0 (GraphPad SoftWare, San Diego, Ca, USA) and a P value <0.05 was considered statistically significant (*P < .05, **P < .01).
3. RESULTS
3.1. Silencing HER3 reduces cell proliferation and increases radiosensitivity
First, we silenced HER3 protein with three siRNAs. As shown in Figure 1A, we have chosen the best 1305 sequences in the following experiments. We developed MCF‐7 and ZR75‐1 cells with low expression of HER3 by shRNA (Figure 1B). Then we validated the cell proliferation using the CCK‐8 assay and cell survival fraction by clone formation assay in control groups and silenced HER3 groups. Experimental results showed that the proliferation rate significantly reduced in HER3 knockdown cells with the extension of cell culture time (P < .05) (Figure 1C). The clone formation assay for cell survival fraction analysis showed that silencing HER3 resulted in weakened clonal formation ability compared with controls (Figure 1D). The survival fraction (SF) of MCF‐7 and ZR75‐1 cells by silencing HER3 with 2 Gy was 0.28 and 0.40, respectively (Table 1). The single‐hit multitarget model formula [SF = 1−(1−e−D/D0) ^n] was used to calculate the SF value. The sensitization enhancement ratio in shHER3‐MCF‐7 and shHER3‐ZR75‐1 cells was 1.49 and 1.34, respectively (Table 1). These results suggested that silencing HER3 significantly enhanced radiosensitivity in luminal A breast cancer cells.
Figure 1.
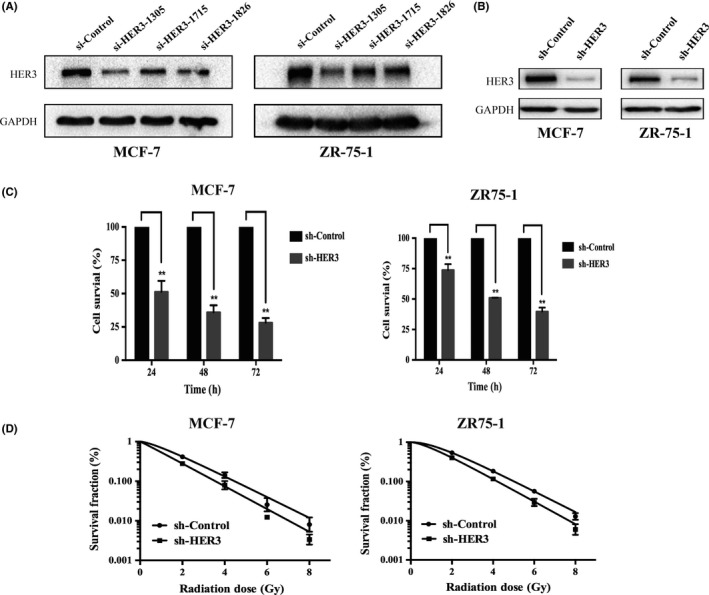
Silencing human epidermal growth factor receptor‐3 (HER3) reduces cell proliferation and clone formation ability with ionizing radiation (IR). sh‐Control, stably transduced with lentivirus vector; sh‐HER3, stably transduced with lentivirus‐mediated HER3 shRNA. A., Silencing HER3 by three different siRNAs in MCF‐7 and ZR75‐1 cells. HER3 protein was determined by western blot. B, Stable knockdown of HER3 was successfully established in both cells by shRNA. C, Cell proliferation was detected by CCK‐8 at different times. *P < .05, **P < .01. D, Survival curve was fitted according to the multitarget single‐hit model
Table 1.
Radiosensitization activity of MCF‐7 and ZR75‐1 breast cancer cells stably transduced with lentivirus‐mediated human epidermal growth factor receptor‐3 shRNA (sh‐HER3) or control
| K | N | D0 | Dq | SF2 | SER | |
|---|---|---|---|---|---|---|
| MCF‐7 | ||||||
| sh‐Control | 0.61 | 1.53 | 1.65 | 0.30 | 0.42 | |
| sh‐HER3 | 0.66 | 1.06 | 1.51 | 0.04 | 0.28 | 1.49 |
| ZR75‐1 | ||||||
| sh‐Control | 0.61 | 2.21 | 1.64 | 0.56 | 0.54 | |
| sh‐HER3 | 0.67 | 1.68 | 1.50 | 0.34 | 0.40 | 1.34 |
D0, mean lethal dose; Dq, quasithreshold dose; K, a passivation constant, derived directly from the fitting equation; N, extrapolation number, derived directly from the fitting equation; SER, sensitization enhancement ratio; SF2, survival fraction (2 Gy).
3.2. Silencing HER3 increases IR‐induced DNA damage and reduces DNA repair
In order to explore whether silencing HER3 could regulate IR‐induced DNA damage and repair, we used immunofluorescence to detect the number of γ‐H2AX foci after IR at different times. The average number of γH2AX foci per cell was calculated as a marker, which was thought to reflect the degree of DNA damage and repair.14, 15, 16, 17 The increased number of γH2AX foci means an increase of DNA damage, and the disappearance of γH2AX foci means the completion of DNA repair.18, 19, 20 As we expected, silencing HER3 increased the number of γ‐H2AX foci in the nucleus without IR. After 6 Gy IR, the number of γH2AX foci in both groups peaked at 30 minutes, and in the silenced HER3 group, the number increased significantly compared to the control group. Next, we continued to observe the number of disappeared γH2AX foci at 1 hour, 6 hours, and 24 hours after IR. Our study showed that, as time progressed, the number of disappeared γH2AX foci in the control group was higher compared with the silenced HER3 group at the same time point (P < .05) (Figure 2A,B). Furthermore, we verified related proteins, such as γH2AX and PARP. Poly (ADP‐ribose) polymerase rapidly recognizes and binds to DNA breaks to facilitate DNA repair.21 Our results showed that, after silencing HER3 with IR, the expression of γH2AX was clearly increased compared with silencing HER3 or IR alone (Figure 2C,D). The level of PARP increased in IR alone but decreased in the silenced HER3 group, especially in the combined silenced HER3 and IR groups (Figure 2C,D). These data suggested that silencing HER3 resulted in an increase of DNA damage and reduction of DNA repair when combined with IR.
Figure 2.
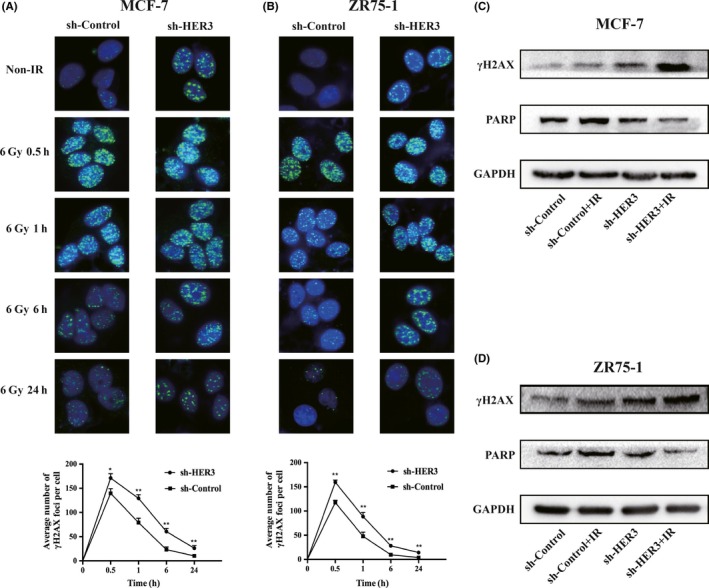
Silencing human epidermal growth factor receptor‐3 (HER3) increases ionizing radiation (IR)‐induced DNA damage and reduces DNA repair. A,B, DNA damage was revealed by immunofluorescence detection of γH2AX foci with or without 6 Gy IR for different times (0.5, 1, 6, and 24 hours) in MCF‐7 and ZR75‐1 cells. *P < .05, **P < .01. C,D, γH2AX and poly (ADP‐ribose) polymerase (PARP) proteins were detected after 24 hours of IR by western blot. GAPDH was an internal reference
3.3. Silencing HER3 prolongs IR‐induced G2/M arrest
Sensitivity to IR is different in different cell cycles. The G2/M phase is the most radiosensitive.22 We used flow cytometry analysis to clarify the cell cycle progression. We found that IR blocked cells in the G2/M phase after 24 hours and the result was the same as silencing HER3 alone (P < .05) (Figure 3A,B, Table S1). Silencing HER3 with IR could significantly increase the proportion of the G2/M period compared with single treatment (P < .05) (Figure 3A,B, Table S1). It was determined that silencing HER3 increased IR‐induced G2/M arrest. Therefore, we could have inferred that silencing HER3 increased radiosensitivity by inducing G2/M arrest.
Figure 3.
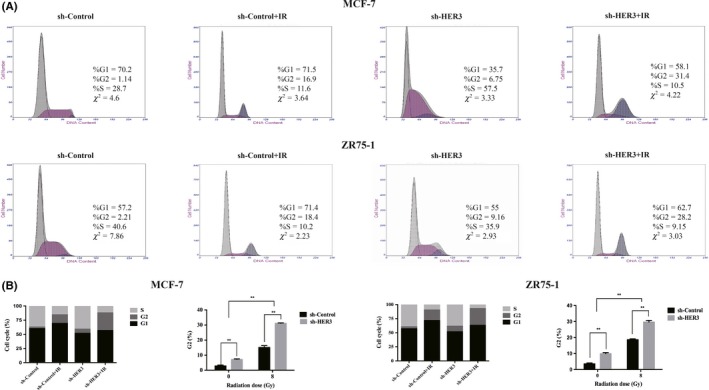
Silencing human epidermal growth factor receptor‐3 (HER3) prolongs ionizing radiation (IR)‐induced G2/M arrest. A, Cell cycle was detected by flow cytometry after 8 Gy IR after 24 hours. B, Proportion of G2/M phase in entire cell cycle. *P < .05, **P < .01
3.4. Silencing HER3 increases IR‐induced apoptosis
To further illustrate the role of silencing HER3 and IR in apoptosis, we evaluated apoptosis by flow cytometry and western blot analyses. The total apoptosis rate, including early and late apoptosis populations, was calculated. In our research, IR alone‐induced apoptosis did not increase significantly (P > .05); however, silencing of HER3 combined with IR can significantly increase the rate of apoptosis in MCF‐7 cells (P < .05) (Figure 4A,B, Table S2). By contrast, in ZR75‐1 cells, IR alone‐induced apoptosis increased (P < .05), when combined with silenced HER3, the rate of apoptosis further increased (P < .05) (Figure 4A,B). To further validate our findings, we examined the upstream HER/PI3K/AKT pathway and downstream apoptosis‐related proteins: Bax (pro‐apoptotic protein), Bad (pro‐apoptotic protein), and Bcl‐2 (anti‐apoptotic protein). The results showed that the effect was not obvious on the expression of HER3 or phosphorylation of HER3 in either cell line after 24 hours of IR. Silencing HER3 could significantly reduce HER3 and phosphorylation of HER3 expression. When combined with IR, the downstream phosphorylation of AKT expression significantly reduced. In agreement, Bax and Bad protein levels were upregulated and the Bcl‐2 protein level was downregulated when IR and HER3 silencing were combined, compared with IR or HER3 silencing alone (Figure 4C).
Figure 4.
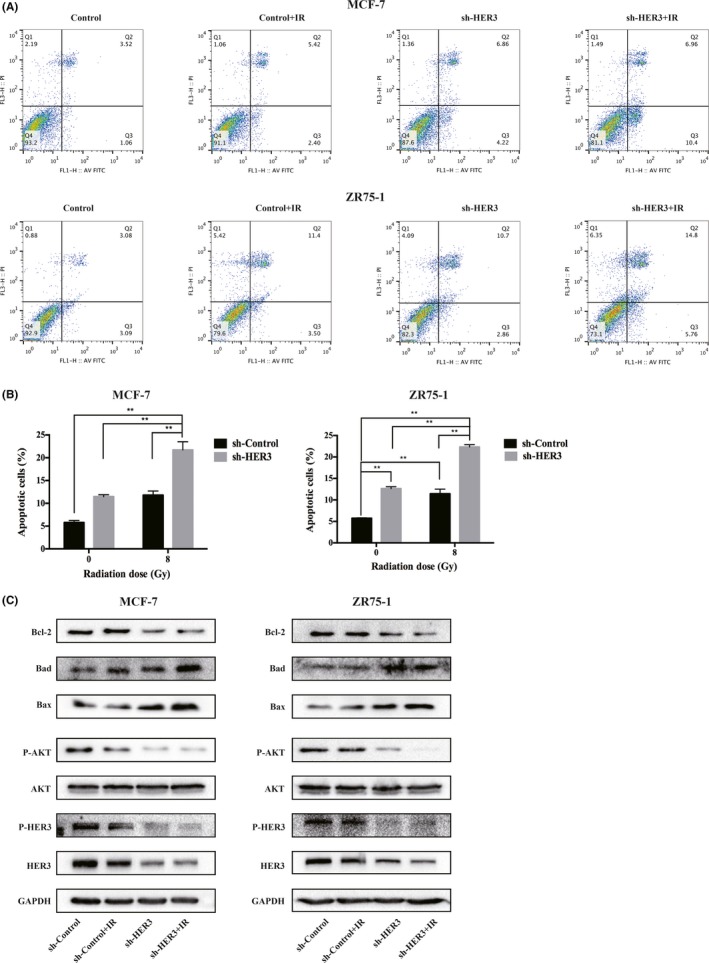
Silencing human epidermal growth factor receptor‐3 (HER3) increases ionizing radiation (IR)‐induced apoptosis. A,B, Flow cytometry was used to analyze apoptosis in MCF‐7 and ZR75‐1 cells after 8 Gy IR for 24 hours. *P < .05, **P < .01. C, Western blot was used to detected HER3/PI3K/AKT signaling and apoptosis‐related proteins Bax, Bad, and Bcl‐2 after 8 Gy IR for 24 hours
3.5. Silencing HER3 inhibits tumor growth in breast cancer xenograft nude mice
As shown in Figure 5, the combination of HER3 silencing and IR significantly decreased tumor growth. Tumor volume increased at a much slower rate in nude mice in the sh‐HER3 and IR group compared with the control group (P < .05) (Figure 5B), while tumor weight was significantly lower in the sh‐HER3 and IR group (P < .05) (Figure 5C). The relative tumor proliferation rate (T/C [%]) was measured to evaluate antitumor activity. The T/C (%) of the sh‐Control and IR, sh‐HER3, and sh‐HER3 and IR groups were 60.4%, 90.5%, and 24.1%, respectively (Figure 5D), and tumor inhibition rates reached 39.7%, 9.5% and 75.9%, respectively.
Figure 5.
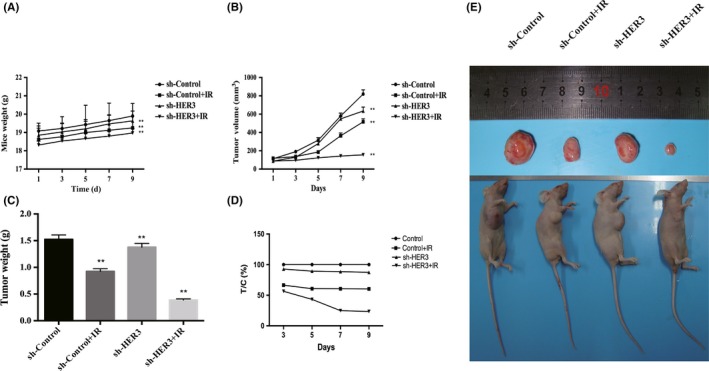
Silencing human epidermal growth factor receptor‐3 (HER3) inhibits tumor growth in breast cancer xenograft nude mice. MCF‐7 (sh‐Control and sh‐HER3) cells were used to form transplant tumor. A. Weight of nude mice was measured every other day after 8 Gy ionizing radiation (IR) in different groups. *P < .05, **P < .01. B,C, Volume and weight were measured every other day after 8 Gy IR in different groups. *P < .05, **P < .01. D, Relative tumor proliferation rate T/C (%) was calculated in different groups. E, Sizes of nude mice and xenografts were photographed and compared in different groups
4. DISCUSSION
Radiotherapy is a primary treatment for local control of breast cancer, and many patients who receive RT have excellent long‐term outcomes. However, different molecular subsets of patients with breast cancer experience different recurrence rates following RT. More than 20%‐40% of 5‐year locoregional recurrence rates have been reported following treatment with RT. One approach to improving the efficacy of RT for breast cancer patients is to improve the radiosensitivity.23, 24 Radiosensitivity of tumor cells largely depends on their ability to repair IR‐induced DNA damage. 25 The phosphorylated form of H2AX (γH2AX) is recognized as a histone involved in the event of DNA damage, and there is a one‐to‐one relationship between the numbers of γH2AX foci with DNA breaks caused by IR. γH2AX has been well used as a marker of DNA double‐strand breaks.26 Poly (ADP‐ribose) polymerase is a target for RT. It binds to DNA breaks rapidly leading to DNA repair and some PARP inhibitions have shown sensitivity to RT in vitro.27, 28 We have found that silencing HER3 increased IR‐induced DNA damage and reduced DNA repair. In response to DNA damage, the cell cycle checkpoint is activated and cell cycle progression is arrested, thereby allowing time to repair DNA breaks before they are passed on to the next generation.29, 30 Our study also found that silencing HER3 prolonged IR‐induced G2/M arrest, and we obtained the same conclusion from a previous study.28 Studies have shown that PARP activation is related to the EGFR‐ERK signaling pathway and IR‐induced G2/M arrest is associated with ERK1/2 activation.28, 31 Interestingly, HER3 could form heterodimers with other members of the EGFR family, such as EGFR and HER2, activating the MAPK/ERK signaling pathway.32, 33 This provided the basis for our further study. Therefore, in the next study we need to find the key molecules that HER3 uses to regulate DNA damage and repair and G2/M arrest.
Previous studies have shown that activation of HER3 signaling enhanced cell proliferation,29 and we obtained the same experimental result in luminal A breast cancer cells. Human epidermal growth factor receptor‐3 binds to PI3K and activates downstream AKT, causing a series of responses, including anti‐apoptotic effects.34, 35 Evidence indicates that the rate of IR‐induced apoptosis closely correlates with the double‐strand breaks.36 Our study found that silencing HER3 with IR inhibited the activation of the PI3K/AKT pathway to inhibit apoptosis. It was a relatively clear pathway in our experiment.
Using a xenograft nude mouse model, we showed that silencing HER3 with IR enhanced the radiosensitivity of luminal A breast cancer cells in vivo. The combination of HER3 silencing and IR notably suppressed tumor weight compared with the other treatment groups.
In conclusion, we found that silencing HER3 enhanced the radiosensitivity of luminal A breast cancer cells in vitro and in vivo. The mechanisms of radiosensitization might be aggravating DNA damage, weakening DNA repair, and inducing G2/M arrest and apoptosis. Human epidermal growth factor receptor‐3 is a potential molecular target of radiosensitization.
CONFLICT OF INTEREST
The authors declare no conflict of interest in this article.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by funds from the National Natural Science Foundation of China (Grant Nos. 81472809, 81502653, and 81672983), Young Medical Talents Project of Jiangsu Province (Grant No. QNRC2016571), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) (Grant No. JX10231801).
He G, Di X, Yan J, Zhu C, Sun X, Zhang S. Silencing human epidermal growth factor receptor‐3 radiosensitizes human luminal A breast cancer cells. Cancer Sci. 2018;109:3774–3782. 10.1111/cas.13810
Guofeng He, Xiaoke Di, and Jingjing Yan contributed equally to this work.
References
- 1. Goldhirsch A, Winer EP, Coates AS, et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen international expert consensus on the primary therapy of early breast cancer 2013. Ann Oncol. 2013;24:2206‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sutherland RL. Endocrine resistance in breast cancer: new roles for ErbB3 and ErbB4. Breast Cancer Res. 2011;13:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maurer C, Martel S, Zardavas D, Ignatiadis M. New agents for endocrine resistance in breast cancer. Breast. 2017;34:1‐11. [DOI] [PubMed] [Google Scholar]
- 4. Knowlden JM, Hutcheson IR, Jones HE, et al. Elevated levels of epidermal growth factor receptor/c‐erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen‐resistant MCF‐7 cells. Endocrinology. 2003;144:1032‐44. [DOI] [PubMed] [Google Scholar]
- 5. Hutcheson IR, Goddard L, Barrow D, et al. Fulvestrant‐induced expression of ErbB3 and ErbB4 receptors sensitizes oestrogen receptor‐positive breast cancer cells to heregulin beta1. Breast Cancer Res. 2011;13:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frogne T, Benjaminsen RV, Sonne‐Hansen K, et al. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 2009;114:263‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keegan NM, Gleeson JP, Hennessy BT, Morris PG. PI3K inhibition to overcome endocrine resistance in breast cancer. Expert Opin Investig Drugs. 2018;27:1‐15. [DOI] [PubMed] [Google Scholar]
- 8. Liu B, Ordonez‐Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD. Downregulation of erbB3 abrogates erbB2‐mediated tamoxifen resistance in breast cancer cells. Int J Cancer. 2007;120:1874‐82. [DOI] [PubMed] [Google Scholar]
- 9. Bae SY, La Choi Y, Kim S, et al. HER3 status by immunohistochemistry is correlated with poor prognosis in hormone receptor‐negative breast cancer patients. Breast Cancer Res Treat. 2013;139:741‐50. [DOI] [PubMed] [Google Scholar]
- 10. Koutras AK, Fountzilas G, Kalogeras KT, Starakis I, Iconomou G, Kalofonos HP. The upgraded role of HER3 and HER4 receptors in breast cancer. Crit Rev Oncol Hematol. 2010;74:73‐8. [DOI] [PubMed] [Google Scholar]
- 11. van der Leij F, Elkhuizen PH, Bartelink H, van de Vijver MJ. Predictive factors for local recurrence in breast cancer. Semin Radiat Oncol. 2012;22:100‐7. [DOI] [PubMed] [Google Scholar]
- 12. Subik K, Lee JF, Baxter L, et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki‐67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Can. 2010;4:35‐41. [PMC free article] [PubMed] [Google Scholar]
- 13. Momeny M, Saunus JM, Marturana F, et al. Heregulin‐HER3‐HER2 signaling promotes matrix metalloproteinase‐dependent blood‐brain‐barrier transendothelial migration of human breast cancer cell lines. Oncotarget. 2015;6:3932‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double‐strand breaks in vivo. J Cell Biol. 1999;146:905‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rothkamm K, Lobrich M. Evidence for a lack of DNA double‐strand break repair in human cells exposed to very low x‐ray doses. Proc Natl Acad Sci USA. 2003;100:5057‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Banath JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double‐strand breaks. Can Res. 2003;63:4347‐50. [PubMed] [Google Scholar]
- 17. Schmid TE, Dollinger G, Beisker W, et al. Differences in the kinetics of gamma‐H2AX fluorescence decay after exposure to low and high LET radiation. Int J Radiat Biol. 2010;86:682‐91. [DOI] [PubMed] [Google Scholar]
- 18. Bourton EC, Plowman PN, Smith D, Arlett CF, Parris CN. Prolonged expression of the gamma‐H2AX DNA repair biomarker correlates with excess acute and chronic toxicity from radiotherapy treatment. Int J Cancer. 2011;129:2928‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mariotti LG, Pirovano G, Savage KI, et al. Use of the gamma‐H2AX assay to investigate DNA repair dynamics following multiple radiation exposures. PLoS One. 2013;8:e79541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Svetlova M, Solovjeva L, Nishi K, Nazarov I, Siino J, Tomilin N. Elimination of radiation‐induced gamma‐H2AX foci in mammalian nucleus can occur by histone exchange. Biochem Biophys Res Comm. 2007;358:650‐4. [DOI] [PubMed] [Google Scholar]
- 21. Liu SK, Coackley C, Krause M, Jalali F, Chan N, Bristow RG. A novel poly(ADP‐ribose) polymerase inhibitor, ABT‐888, radiosensitizes malignant human cell lines under hypoxia. Radiother Oncol. 2008;88:258‐68. [DOI] [PubMed] [Google Scholar]
- 22. Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928‐42. [DOI] [PubMed] [Google Scholar]
- 23. Feng FY, Speers C, Liu M, et al. Targeted radiosensitization with PARP1 inhibition: optimization of therapy and identification of biomarkers of response in breast cancer. Breast Cancer Res Treat. 2014;147:81‐94. [DOI] [PubMed] [Google Scholar]
- 24. Rehman S, Reddy CA, Tendulkar RD. Modern outcomes of inflammatory breast cancer. Int J Radiat Oncol Biol Phys. 2012;84:619‐24. [DOI] [PubMed] [Google Scholar]
- 25. Zhou ZR, Yang ZZ, Wang SJ, et al. The Chk1 inhibitor MK‐8776 increases the radiosensitivity of human triple‐negative breast cancer by inhibiting autophagy. Acta Pharmacol Sin. 2017;38:513‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang N, Chang Y, Rios A, An Z. HER3/ErbB3, an emerging cancer therapeutic target. Acta Biochim Biophys Sin. 2016;48:39‐48. [DOI] [PubMed] [Google Scholar]
- 27. Veuger SJ, Hunter JE, Durkacz BW. Ionizing radiation‐induced NF‐kappaB activation requires PARP‐1 function to confer radioresistance. Oncogene. 2009;28:832‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chalmers AJ, Lakshman M, Chan N, Bristow RG. Poly(ADP‐ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin Radiat Oncol. 2010;20:274‐81. [DOI] [PubMed] [Google Scholar]
- 29. Sanchez Y, Wong C, Thoma RS, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497‐501. [DOI] [PubMed] [Google Scholar]
- 30. Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177‐96. [DOI] [PubMed] [Google Scholar]
- 31. Hagan MP, Yacoub A, Dent P. Radiation‐induced PARP activation is enhanced through EGFR‐ERK signaling. J Cell Biochem. 2007;101:1384‐93. [DOI] [PubMed] [Google Scholar]
- 32. Yan Y, Hein AL, Greer PM, et al. A novel function of HER2/Neu in the activation of G2/M checkpoint in response to gamma‐irradiation. Oncogene. 2015;34:2215‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yan Y, Black CP, Cowan KH. Irradiation‐induced G2/M checkpoint response requires ERK1/2 activation. Oncogene. 2007;26:4689‐98. [DOI] [PubMed] [Google Scholar]
- 34. Dey N, Williams C, Leyland‐Jones B, De P. A critical role for HER3 in HER2‐amplified and non‐amplified breast cancers: function of a kinase‐dead RTK. Am J Transl Res. 2015;7:733‐50. [PMC free article] [PubMed] [Google Scholar]
- 35. Kawakami H, Yonesaka K. HER3 and its Ligand, Heregulin, as Targets for Cancer Therapy. Recent Pat Anti‐Cancer Drug Discov. 2016;11:267‐74. [DOI] [PubMed] [Google Scholar]
- 36. Raleigh DR, Haas‐Kogan DA. Molecular targets and mechanisms of radiosensitization using DNA damage response pathways. Future Oncol. 2013;9:219‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials