

Abstract
Recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) has been a longstanding challenge for head and neck oncologists, and current treatments still have limited efficacy. ERK is aberrantly overexpressed and activated in HNSCC. Herein, we aimed to investigate the cause of the limited therapeutic effect of selumetinib, a selective inhibitor of MEK in HNSCC, as MEK/ERK reactivation inevitably occurs. We assessed the effects of combining selumetinib with fibroblast growth factor receptor 3 (FGFR3) inhibitor (PD173074) on tumor growth. Selumetinib transiently inhibited MAPK signaling and reactivated ERK signaling in HNSCC cells. Rebound in the ERK and Akt pathways in HNSCC cells was accompanied by increased FGFR3 signaling after selumetinib treatment. Feedback activation of FGFR3 was a result of autocrine secretion of the FGF2 ligand. The FGFR3 inhibitor PD173074 prevented MAPK rebound and sensitized the response of HNSCC cells to selumetinib. These results provided rational therapeutic strategies for clinical studies of this subtype of patients that show a poor prognosis with selumetinib. Our data provide a rationale for combining a MEK inhibitor with inhibitors of feedback activation of FGFR3 signaling in HNSCC cells. ERK rebound as a result of the upregulation of FGFR3 and the ligand FGF2 diminished the antitumor effects of selumetinib, which was overcome by combination treatment with the FGFR3 inhibitor.
Keywords: fibroblast growth factor receptor, head and neck cancer, MAP kinase signaling system, MEK inhibition, receptor cross‐talk
Abbreviations
- AZD6244
selumetinib, a MEK1/2 inhibitor
- BRAF
serine/threonine‐protein kinase
- EGFR
epidermal growth factor receptor
- FGFR3
fibroblast growth factor receptor 3
- HNSCC
head and neck squamous cell carcinoma
- KRAS
Kristen ras
- NSCLC
non‐small cell lung cancer
- PD173074
fibroblast growth factor receptor 3 (FGFR3) inhibitor
- RAF
named after the rapidly accelerated fibrosarcoma, MAP kinase kinase kinase
- RAS
named after the transforming oncogene found in rat sarcoma, monomeric guanosine triphosphatase
- RTK
receptor tyrosine kinase
1. INTRODUCTION
Head and neck squamous cell carcinoma is the sixth leading cancer by incidence worldwide.1 Despite high cure rates in early‐stage HNSCC with either surgery or radiotherapy, nearly 60% of HNSCC patients still present with locally advanced‐stage disease.2
In HNSCC, multiple mutations or aberrant overactivation of the epidermal growth factor receptor (EGFR)‐ MEK signaling pathway has been frequently observed.3, 4 EGFR expression is detected in >90% of HNSCC and frequently triggers the RAS/RAF/MEK/ERK cascade. Clinical and biological evidence has suggested that the ERK pathway may play a pivotal role in tumor progression. ERK hyperactivation was investigated in general for cancers harboring RTK dysregulation, or KRAS or BRAF mutations.5 In HNSCC, ERK overexpression was observed in tumor cells as well as in infiltrating lymphoid cells.5 ERK expression and activation was reported to be associated with clinicopathological parameters and cell proliferation in oral tongue squamous carcinoma.5, 6
ERK inhibition could be an attractive strategy for anticancer therapies against thyroid cancer or advanced melanoma. Indeed, MEK and RAF inhibitors are compatible therapeutic agents for tumors addicted to ERK signaling. However, acquired resistance to these inhibitors and other targeted therapeutics can emerge in tumor cells. Several groups have investigated the mechanism of intrinsic (that is, present before treatment) or acquired resistance (acquired during treatment by various therapy‐induced adaptive responses) against ERK blockades in many cancers. Selumetinib (AZD6244), a MEK1/2 inhibitor, provoked intrinsic resistance by PI3K signaling in colorectal cancer cells with KRAS or BRAF mutations.7, 8 Some studies have generated evidence that acquired resistance to MEK inhibition induces feedback activation of several RTK called “kinome reprogramming” through the inhibition of a physiological negative loop.9, 10 Currently, adaptive resistance to MEK inhibition is thought to be driven by feedback activation of fibroblast growth factor receptor 1 (FGFR1) among the RTK in KRAS‐mutant lung cancer.11, 12 However, the mechanism associated with acquired resistance to MEK inhibition has not been clearly determined.
Data from numerous studies have shown that ERK activation was intimately associated with acquired‐resistance mechanisms. ERK‐activity rebound appeared at an early phase after treatment with the RAF inhibitor vemurafenib in BRAF‐mutant thyroid cancer cell lines resistant to vemurafenib, and this resistance was overcome using combination treatment with the HER kinase inhibitor lapatinib.13 Rapid recovery of ERK signaling has also been shown in melanoma cells and papillary thyroid cancer cells in the presence of a BRAF inhibitor.14, 15
Based on MAPK activation in HNSCC, many strategies have been developed to suppress MAPK activity. Small‐molecule MEK inhibitors represent the most specific and effective strategy tested to date in several cancers. Although preclinical data are extensive, objective response rates in these studies have been modest.16 Herein, we investigated the acquired resistance of a MEK inhibitor in several oral tongue cancer cell samples expressing high levels of ERK. MEK inhibition promoted an ERK rebound during early phase treatment, which was due to feedback‐induced, ligand‐dependent activation of FGFR3 signaling. RTK FGFR3 is another target for which researchers are currently developing inhibitors. The current study was designed to explore whether feedback activation of FGFR3 occurs after MEK inhibition and whether combined inhibition of FGFR3 and MEK is a promising treatment strategy for HNSCC.
2. MATERIALS AND METHODS
2.1. Ethics statement
All animal experiment protocols were approved by the committee of ethics on animal research of Yonsei University College of Medicine (Accession No. 2014‐0366). All mice were treated in accordance with the guidelines for the care and use of laboratory animals of the institutional animal care and use committee at our institute.
2.2. Cell lines and reagents
Cal27 human tongue squamous cell carcinoma cell line and FADU human hypopharyngeal squamous cell carcinoma cell line were purchased from the ATCC (Manassas, VA, USA). HN6 human tongue squamous cell carcinoma cell line was kindly provided by Dr Kim (Ajou University, Suwon, Korea). Cal27 and HN6 cells were maintained in DMEM (Lonza) supplemented with 10% FBS and 1% penicillin/streptomycin. FADU cells were cultured in RPMI‐1640 (Lonza, Walkersville, MD, USA) supplemented with 10% FBS and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA). All cell lines were cultured in a humidified atmosphere of 5% CO2 at 37°C. AZD6244 was purchased from Selleck Chemicals, and PD173074 was purchased from Sigma‐Aldrich (St Louis, MO, USA).
2.3. Receptor tyrosine kinase array
Phospho‐RTK array assay was carried out according to the manufacturer's instructions (PathScan Antibody Array Kit, Fluorescent Readout; Cell Signaling Technology, Danvers, MA, USA). Cells were lysed in cell lysis buffer (Cell Signaling Technology). Briefly, 30‐150 μg of each lysate was incubated with blocked arrays overnight at 4°C in an orbital shaker. After washing with the provided buffer, the arrays were incubated for 2 hours at room temperature with a 1× detection antibody cocktail and subsequently with streptavidin‐conjugated DyLight680 (Thermo Fisher Scientific, Rockford, IL, USA) for 30 minutes at room temperature. After washing with array wash buffer, fluorescent signals were detected using a fluorescent digital imaging system capable of excitation at 680 nm and detection at 700 nm.
2.4. Clonogenic assay
Cells were seeded at 2‐5 × 104 cells/well in six‐well plates and exposed to the indicated drugs for 24 hours. The culture medium was replaced every 3 days. After 14‐21 days, the cells were fixed in 4% paraformaldehyde (PFA) and stained with 0.1% crystal violet. Colonies containing at least 50 individual cells were counted using a stereomicroscope.
2.5. Immunoblotting
Cells were washed twice with ice‐cold PBS and lysed in ice‐cold Tris buffer (10 mmol/L Tris‐HCl [pH 7.4], 100 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L NaF, 20 mmol/L Na4P2O7, 2 mmol/L Na3VO4, 1% Triton X‐100, 10% glycerol, 0.1% SDS, and 0.5% deoxycholate; Invitrogen) containing 1 mmol/L PMSF and a protease inhibitor cocktail (Sigma). Cell lysates were centrifuged and then mixed with SDS‐containing sample buffer followed by boiling for 5 minutes. Whole‐cell extracts (25 μg/lane) were separated by 10% SDS‐PAGE and electrotransferred to a PVDF membrane (Millipore, Burlington, MA, USA). Antibodies against anti‐phospho FGFR3 (1: 500), ‐FGFR3 (1: 500), ‐phospho Akt (1: 500), ‐Akt (1: 1000), ‐phospho ERK (1: 1000), ‐ERK (1: 1000), ‐Caspase 3 (1: 1000), and GAPDH (1: 2000) were used for immunoblotting. All of these antibodies were purchased from Cell Signaling Technology, Inc.
2.6. Cell viability assay
Cell Counting Kit‐8 (Dojindo Molecular Technologies, Inc., Rockville, MD, USA) was used to determine cell viabilities, according to the manufacturer's instructions. Cells were plated in a 96‐well plate at a density of 104 cells/well. After serum starvation, cells were treated with the indicated drugs for the indicated times or concentrations before adding CCK‐8 solution (a water‐soluble tetrazolium salt). Cells were incubated for 1‐2 hours, and the absorbance at 450 nm was measured using a microplate reader (Molecular Devices, San Jose, CA, USA).
2.7. Silencing of FGFR3 and FGF2
Cells were transfected with 100 nmol/L FGFR3 siRNA and 50 nmol/L FGF2 siRNA or a control siRNA (Santa Cruz Biotechnology, Dallas, TX, USA) using Lipofectamine RNAiMAX (Invitrogen) and incubated for 24 hours at 37°C in a humidified 5% CO2 incubator. FGFR3 siRNA and FGF2 siRNA were designed and synthesized by Bioneer (Deajeon, Korea; sequences of FGFR3 siRNA and FGF2 siRNA can be seen in Table S1, Data S1).
2.8. Enzyme‐linked immunosorbent assay
Cells were collected for the measurement of FGF2 concentration by FGF2 ELISA kit (Catalog no. ab99979, Abcam, Cambridge, UK). Assays were carried out according to the manufacturer's instructions in a 96‐well plate.
2.9. Cell cycle analysis
Cell cycle progression was analyzed by propidium iodide (PI; Sigma) staining and flow cytometry. After drug treatment, the cells were trypsinized and washed in ice‐cold PBS. After fixation in 70% ethanol, the cells were suspended in PI solution (50 μg/mL) containing DNase‐free RNase (1 μg/mL) and incubated for 30 minutes at 37°C. Measurement of the cellular DNA content was carried out using a BD FACSVerse flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) and analyzed using FlowJo v10 software (FlowJo, Ashland, OR).
2.10. Apoptosis assay
Apoptosis was assessed by Annexin V‐FITC (BD Biosciences Pharmingen, San Diego, CA, USA) dual staining. After drug treatment, cells were harvested by trypsinization. The cells were washed with ice‐cold PBS and then washed with 1× binding buffer. Cells were then labeled with Annexin V‐FITC according to the manufacturer's instructions. Apoptotic cells were analyzed using a BD FACSVerse flow cytometer (Becton Dickinson Immunocytometry Systems). For TUNEL assays, cells were seeded on glass coverslips in six‐well plates and treated with drugs. After treatment, cells were fixed with 4% PFA and analyzed using the In Situ Cell Death Detection Kit (Roche Diagnostics, Mannheim, Germany). Apoptotic cells were detected according to the manufacturer's instructions. Slides were visualized with a Zeiss AxioVert 200 inverted epifluorescence microscope. Images were captured with an AxioCam digital microscope camera and analyzed with AxioVision software (Carl Zeiss Vision, Oberkochen, Germany).
2.11. Orthotopic xenograft mouse model
Cal27 cells were harvested from subconfluent cultures by trypsinization and washed with serum‐free medium. Cal27 cells (5 × 105/30 μL) were inoculated directly onto the anterior tongues of 6‐week‐old male nude mice (Orientbio Inc., Seongnam‐si, Korea) using a Hamilton syringe (Hamilton Company, Reno, NV, USA). Mice were then randomized into four groups (4 mice/group). Tumors developed for 14 days. The mice were treated twice per week for 28 days by i.p. injection with DMSO (vehicle control), 20 mg/(kg day) AZD6244, 10 mg/(kg day) PD173074, or 10 mg/(kg day) PD173074 at 1 day post‐injection of 20 mg/(kg day) AZD6244. Tumor volumes were calculated as (length × width2) × 0.5. The mice were weighed once a week, and tumor growth was measured weekly using a digimatic caliper (Mitutoyo Co., Kawasaki‐shi, Japan). The mice were killed 52 days post‐injection, and tumors were harvested to carry out histopathology and biochemical assays. Samples were fixed overnight in neutral buffered formalin, and immunohistochemical staining was carried out using tumor tissues and primary antibodies against Ki67, p‐FGFR3, p‐Akt, p‐ERK, and cleaved caspase 3. Immunostained sections were analyzed by Allred score under a Nikon light microscope (Nikon, Tokyo, Japan) (Data S1).17 Microscopy images were captured using AxioCam digital microscope cameras and AxioVision image processing (Carl Zeiss Vision).
2.12. In vivo fluorescence imaging
Mice were injected with MMPsense 680 (PerkinElmer, Waltham, MA, USA) by tail vein injection. At 24 hours post‐injection, the mice were imaged using an IVIS Spectrum instrument (Caliper LifeSciences, Waltham, MA, USA) while under anesthesia using isoflurane in oxygen. Fluorescence images were recorded with a 1‐second exposure using a 700/10 nm filter after excitation at 680 nm. Autofluorescence was removed using IVIS Spectrum Living Imaging Software.
2.13. Statistical analysis
All data are represented as mean ± SD from three experiments carried out in triplicate. Comparative statistical analyses were done with Student's t test and one‐way analysis of variance using SPSS 20.0 statistical software (SPSS, Chicago, IL, USA). P‐values <0.05 were considered statistically significant (*P < .05; **P < .01; ***P < .001).
3. RESULTS
3.1. Extracellular signal‐regulated kinase reactivation in MEK inhibitor‐treated HNSCC cells
Recent reports have indicated that ERK activation is frequently dysregulated in cancer cells and is associated with anticancer‐drug resistance. Therefore, we investigated whether the ERK pathway is related to resistance in HNSCC using AZD6244 as a selective MEK inhibitor to inhibit the ERK pathway. We used three cell lines: Cal27 cells and HN6 cells (established from human tongue carcinomas) and FADU cells (established from a human hypopharyngeal carcinoma). The cells were treated with AZD6244 for the indicated durations, after which the medium was replaced with fresh medium lacking AZD6244 (Figure 1A). Results showed that ERK activation rebounded transiently within a few hours after AZD6244 treatment in HNSCC cell lines. ERK activity disappeared shortly after treatment, but resurged over time, even though the Cal27 and HN6 cell lines showed differences in the time period before the ERK rebound occurred (Figure 1A). FADU cells did not show an ERK rebound within 24 hours.
Figure 1.
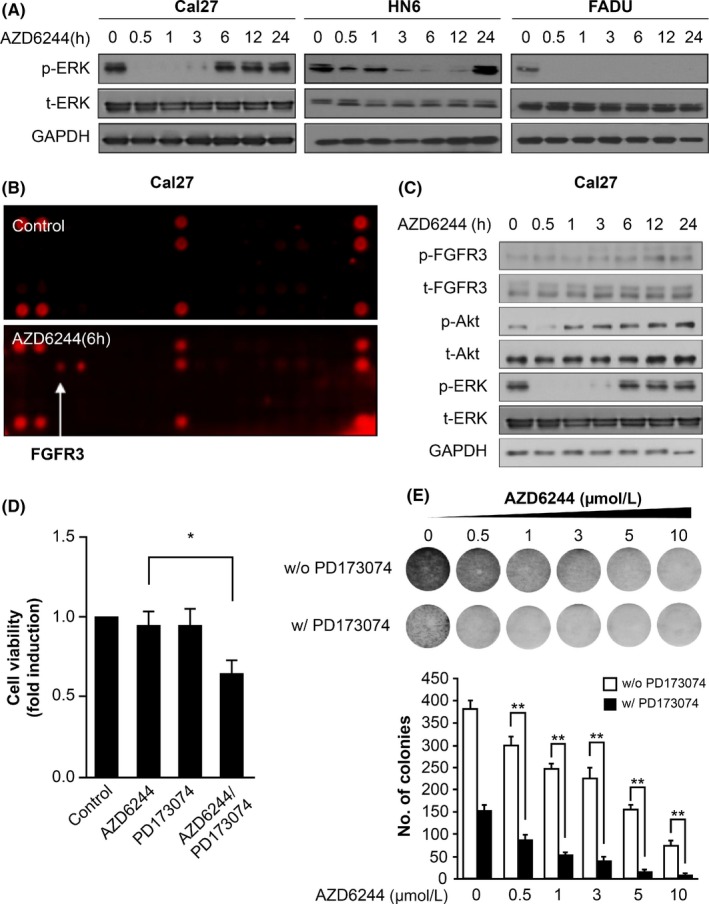
MEK inhibitor induced an ERK‐activity rebound and fibroblast growth factor receptor 3 (FGFR3) activation. A, Phosphorylated ERK and total ERK protein expression are shown in a representative western blot. Head and neck squamous cell carcinoma (HNSCC) cell lines were treated with 0.5 μmol/L AZD6244 or 0.1% DMSO as a vehicle control for different durations. AZD6244 was replaced with fresh media at the indicated times. GAPDH was detected as a loading control. B, Phospho‐RTK assay in Cal27 cells treated with AZD6244 for 6 h. Cal27 cells incubated with 0.1% DMSO for 6 h served as a control. C, Representative western blot analysis of FGFR3, Akt, and ERK expression in Cal27 cells after treatment with 0.5 μmol/L AZD6244 for different time periods. Media containing AZD6244 was replaced with fresh media (lacking AZD6244) at the indicated times. GAPDH was detected as a loading control. D, Cell growth was measured in Cal27 cells treated with AZD6244 or PD173074 as an FGFR inhibitor in cell‐viability assays. Cells were treated for 48 h with 0.1% DMSO, 0.5 μmol/L AZD6244 alone, 1 μmol/L PD173074, or 0.5 μmol/L AZD6244 with 1 μmol/L PD173074. Bars represent means ± SEM between replicates (n = 3). Significant differences compared to the corresponding controls, *P < .05. E, Clone‐formation ability of Cal27 cells treated with AZD6244 was evaluated in clonogenic assays. Cal27 cells were treated with a dose gradient of AZD6244 in the absence or presence of 1 μmol/L PD173074 for 14 d and were studied in clonogenic assays. Bars represent means ± SEM between replicates (n = 3). Significant differences compared to the corresponding controls, **P < .01
Next, to determine whether RTK are related to the ERK rebound after MEK inhibition, a phospho‐RTK array was carried out in Cal27 cells after a 6‐hour treatment with AZD6244 (Figure 1B). In cells treated with AZD6244, FGFR3 expression was elevated among RTK and some factors involved in downstream signal‐transduction pathways. Our western blot results showed that ERK reactivation was accompanied by increased FGFR3 activity under MEK inhibition, where phosphorylated FGFR3 levels increased, but total FGFR3 levels did not show any change after AZD6244 treatment (Figure 1C, results of HN6 and FADU cells can be seen in Figure S1). Using PD173074 as an FGFR3 inhibitor, we evaluated the effect of FGFR3 activity on cell proliferation when cells were exposed to the MEK inhibitor. Combined treatment with AZD6244 and PD173074 attenuated cell proliferation significantly more than treatment with AZD6244 or PD173074 alone (P < .05). Treatment with AZD6244 or PD173074 alone did not influence cell growth itself (Figure 1D). After a dose gradient exposure to AZD6244, combination treatment with PD173074 clearly reduced the number of colonies more so than did AZD6244 treatment alone (Figure 1E). These results showed that coinhibition of MEK and FGFR3 may synergistically increase the sensitivity of HNSCC cells to AZD6244.
3.2. MEK inhibition upregulated FGFR3
To further investigate whether FGFR3 activation is effective against the AZD6244‐induced ERK‐activity rebound, siRNA against FGFR3 was used in Cal27 cells. We showed that the ERK‐activity rebound arose under MEK inhibition and that the p‐FGFR3 level increased gradually in Cal27 cells. When FGFR3 expression weakened, the ERK‐activity rebound began to diminish at 6 hours post‐AZD6244 treatment in Cal27 cells (Figure 2A, results of HN6 cells can be seen in Figure S2). As a result of the AZD6244 half‐maximal inhibitory concentration (IC50) in HNSCC cells, Cal27 cells (AZD6244 IC50, 1.33 μmol/L) showed a higher IC50 than observed in HN6 cells (AZD6244 IC50, 0.58 μmol/L) or FADU cells (AZD6244 IC50, 1.16 μmol/L), but these differences were not significant (Figure 2B). After siFGFR3 transfection, proliferation of Cal27 cells was significantly reduced compared to that of control transfectants treated with scrambled siRNA (Figure 2C). Next, we explored FGF2 expression under MEK inhibition to determine the role of FGF2 in FGFR3 activation. FGF2, referred to as basic FGF, is a major ligand of FGFR3. In Cal27 cells, FGF2 level in the media increased time‐dependently after treatment with AZD6244 (Figure 2D). In FGF2 siRNA transfectants, the p‐FGFR3 level decreased and the ERK rebound also reduced, suggesting that FGF2 mediated FGFR3 feedback activation during MEK inhibition (Figure 2E). To rule out the off‐target effects, two different siFGFR3 and siFGF2 were transfected to Cal27 cells as shown in Figure S3 and the representative result is shown in Figure 2.
Figure 2.
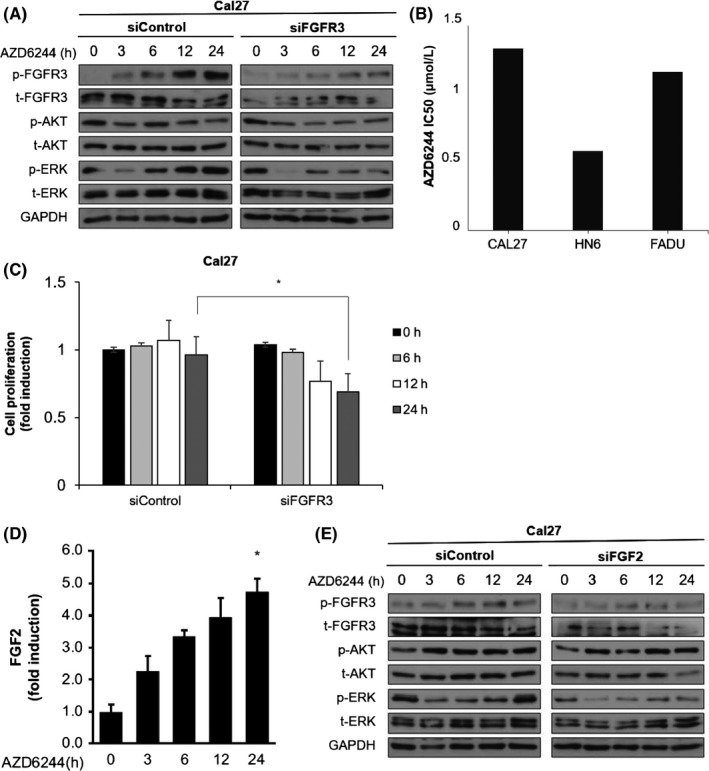
Knockdown of fibroblast growth factor receptor 3 (FGFR3) abrogated ERK rebound and cell growth in head and neck squamous cell carcinoma (HNSCC) cells treated with AZD6244. A, Relative FGFR3, Akt, and ERK expression levels in Cal27 cells transfected with siFGFR3 or a scrambled oligo control (siControl) for 48 h, and treated with AZD6244 are shown by representative western blot analysis. HNSCC cell lines were treated with 0.5 μmol/L AZD6244 or 0.1% DMSO for different time periods. Media containing AZD6244 was replaced with fresh media (lacking AZD6244) at the indicated times. GAPDH was detected as a loading control. B, IC 50 of AZD6244 in HNSCC cell lines. Concentrations of AZD6244 (μmol/L) that inhibited the viability of Cal27, HN6, and FADU cells by 50% (IC 50) after 72 h were determined by MTT assays. Each value represents mean ± SEM from three independent experiments, carried out in triplicate. C, Cell growth was measured by cell viability assays with Cal27 cells treated as described in (A). Bars represent means ± SEM between replicates (n = 3). Significant differences compared to the corresponding controls, *P < .05. D, FGF2 expression in Cal27 cells treated with AZD6244 was determined using an FGF2 ELISA kit, as described in Materials and Methods. Cal27 cells were treated with AZD6244 for the indicated times. Bars represent means ± SEM between replicates (n = 3). Significant difference compared to the control, *P < .05. E, FGFR3, Akt, and ERK expression in Cal27 cells transfected with siFGF2 or siControl for 48 h, and treated with AZD6244 at the indicated times are shown with a representative western blot
3.3. Fibroblast growth factor receptor 3 inhibition and MEK inhibition induced apoptotic cell death
To explore the mechanism by which FGFR3 inhibition retarded cell proliferation, we analyzed cell cycle progression in Cal27 cells by flow cytometry. Sub‐G1 distribution following combination treatment with AZD6244 and PD173074 increased, which represented delayed cell proliferation due to apoptotic cell death (Figure 3A, histogram can be seen in Figure S4). Next, we used the selective FGFR3 inhibitor PD173074 to investigate whether FGFR3 inhibition affected the ERK rebound and could inhibit cell growth in AZD6244‐treated Cal27 cells. After treatment with AZD6244 alone, the p‐FGFR3 level increased and the ERK rebound was evoked, but not after treatment with both AZD6244 and PD173074. Treatment with PD173074 and AZD6244 attenuated the ERK rebound in Cal27 cells. Level of cleaved caspase 3 did not increase following AZD6244 treatment alone, but increased significantly after combined treatment with PD173074. A trend of increased caspase 3 cleavage after combination treatment was observed in Cal27 cells (Figure 3B, results of HN6 cells can be seen in Figure S5). Next, apoptotic cells were quantified by Annexin V/PI staining. Combined treatment significantly increased the apoptotic index compared to treatment with AZD6244 alone. After treating cells with AZD6244, the proportion of apoptotic cells was 9.4 ± 1.5% in Cal27 cells. However, the proportion of apoptotic cells after combined treatment with AZD6244 and PD173074 was 21.2 ± 2.4% (Cal27 cells), suggesting that an additive effect was observed with AZD6244 and PD173074 (Figure 3C). Furthermore, we found that DNA fragmentation significantly increased after combined treatment compared with that observed after treatment with AZD6244 or PD173074 alone. DNA fragmentation was determined in TUNEL assays. Coinhibition of FGFR and MEK resulted in an increased number of TUNEL‐positive cells. Apoptosis was induced significantly by the combination treatment (Figure 3D).
Figure 3.
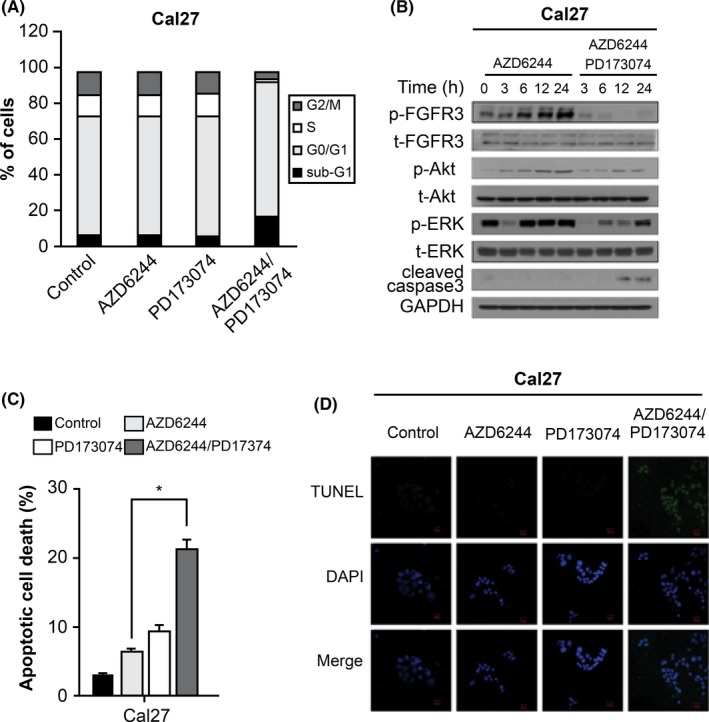
MEK inhibition and fibroblast growth factor receptor 3 (FGFR3) inhibition induced apoptotic cell death in head and neck squamous cell carcinoma (HNSCC) cells treated with AZD6244. A, Cell cycle distribution in Cal27 cells treated with AZD6244 or AZD6244 plus PD173074 for 24 h. Cell cycle progression was measured by flow cytometry using propidium iodide (PI) to stain DNA. Each value represents the mean ± SEM from three independent experiments carried out in triplicate. B, FGFR3, Akt, ERK, and caspase 3 expression in Cal27 cells treated with AZD6244 alone or with AZD6244 plus PD173074 for the indicated times are shown in representative western blot images. HNSCC cell lines were treated with 0.5 μmol/L AZD6244 alone or with 0.5 μmol/L AZD6244 plus 1 μmol/L PD173074 for the indicated times. GAPDH was detected as a loading control. C, Apoptosis of Cal27 cells treated with AZD6244 alone or with combination AZD6244 and PD173074 treatment for 24 h. Cells were analyzed by flow cytometry using Annexin V/PI double staining. Graph shows the percentage of annexin V‐positive, PI‐negative cells in terms of mean ± SEM (n = 3 replicates). Significant difference compared to the corresponding control, *P < .05. D, TUNEL staining in Cal27 cells exposed to AZD6244 or PD173074. Cells were treated with 0.1% DMSO, AZD6244 alone, PD173074 alone, or AZD6244 plus PD173074 for 24 h, after which TUNEL staining was carried out. Results shown represent data from experiments carried out in triplicate
3.4. Combination of FGFR inhibitor and MEK inhibitor leads to antitumor activity in vivo
We observed synergistic effects following combination treatment with the MEK inhibitor and FGFR inhibitor in vitro. To test whether FGFR inhibition could be an effective strategy for controlling resistance to MEK inhibition in vivo, we used an orthotopic xenograft mouse model of HNSCC. Cal27 cells were injected into the tongue of nude mice and tumor volume was evaluated on day 14. Mice were i.p. injected with vehicle, AZD6244, or both inhibitors in combination twice weekly (n = 4 mice/group). In Figure 4B, the presence of invasive orthotopic tumor was confirmed by an in vivo imaging system. Tumor volumes were monitored on the indicated days. As shown in Figure 4A,C, relative tumor volumes in the combination group were lower than those in the vehicle group or in the AZD6244 group. AZD6244 treatment alone did not show a significant effect on tumor growth, whereas cotreatment with AZD6244 and PD173074 led to tumor regression. H&E staining showed that vehicle and AZD6244‐treated tumors showed significantly greater tumor cellularities and sizes versus mice given combination treatment (Figure 4D). The molecular events were explored by immunohistochemical analysis. Enhanced levels of Ki67 and p‐FGFR3 were found in tumors of the vehicle‐control and AZD6244 groups, indicating strong cell proliferation in the tumor masses (Figure 4E, magnified images and Allred scores can be seen in Figure S6 and Table S2). In contrast, combination treatment with AZD6244 and PD173074 resulted in a significant decline in the levels of Ki67 and p‐FGFR3, which was the result of inhibited tumor cell proliferation. Prevalence of apoptotic cells in tumors in the combination‐treatment group was significantly higher than in tumors from vehicle‐control or AZD6244‐treated mice, as shown by cleaved caspase 3 staining. Treatment of mice with PD173074 only showed no effect on histology or apoptosis in a group of four treated mice. These results suggested that combination treatment with both the MEK inhibitor and the FGFR inhibitor had a stronger effect on tumor growth and metastasis than did monotherapy with either agent alone.
Figure 4.
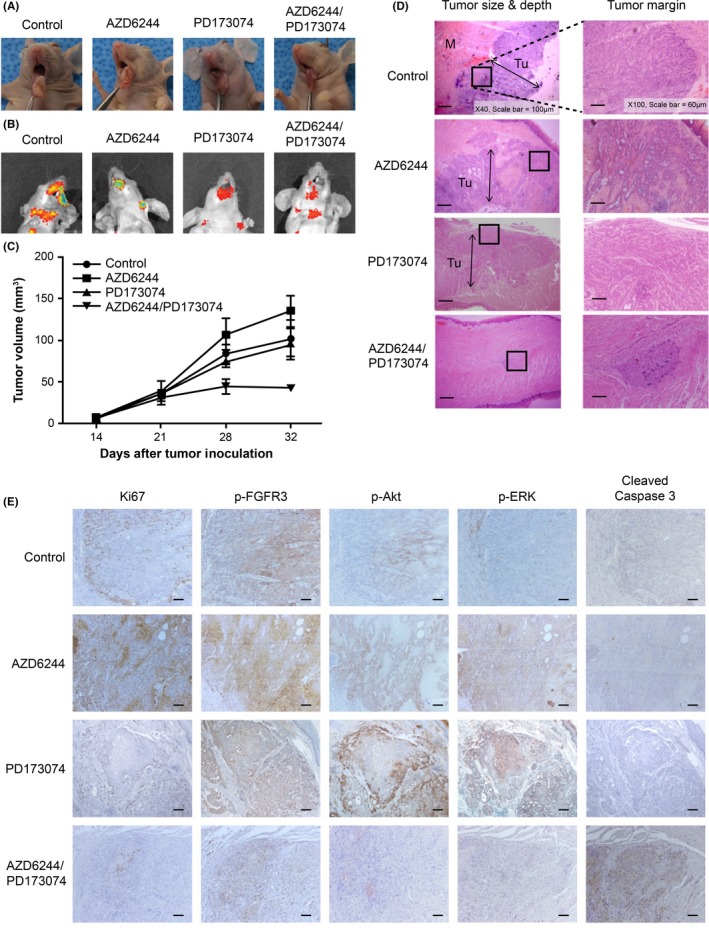
Fibroblast growth factor receptor 3 (FGFR3) inhibition cooperated with MEK inhibition to inhibit tumor growth in orthotopic xenograft mice. A, Primary tumor growth of Cal27 cells in xenograft mice injected with the indicated inhibitors. Cal27 cells were injected s.c. into the tongues of nude mice (n = 4/group). The mice were treated twice per week for 28 d by i.p. injection with DMSO (vehicle control), 20 mg/(kg day) AZD6244, 10 mg/(kg day) PD173074, or 10 mg/(kg day) PD173074 at 1 d post‐injection of 20 mg/(kg day) AZD6244. B, In vivo imaging system (IVIS) images of tumors in the tongue at 52 d post‐injection. The tumor was detected by in vivo fluorescence imaging analysis, as described in Materials and Methods. C, Tumor volumes of mice (n = 4/group) were measured with a caliper on the indicated days. Each value represents mean ± SEM between replicates (n = 4/group). D, H&E staining of tumor tissue in a mouse model. Magnification, 40× or 100×. Scale bar = 100 μm or 60 μm. E, Ki67, phospho‐FGFR3, phospho‐Akt, phospho‐ERK, and cleaved caspase‐3 expression in tumor tissues were measured by immunohistochemistry. Magnification, 100×. Scale bar = 60 μm.
4. DISCUSSION
Targeted single‐agent therapies that block highly oncogenic functions have a major limitation in that acquired resistance involves high levels of feedback activation of the related signaling network. The relative insensitivity of BRAF V600E‐mutated melanoma to BRAF inhibition emerges through an enhanced level of cyclin D1.18 Cyclin E overexpression confers insensitivity of trastuzumab in Her2+ breast cancer cells.19 The lack of responsiveness of BRAFV600E‐positive colorectal cancer cells to BRAF inhibition is associated with the feedback activation of EGFR.20 ERK 1/2‐pathway reactivation during acquired resistance has led to the identification of gatekeeper mutations in oncogenic kinases promoting the use of the alternative MEK pathway. Shi et al21 reported that the acquired resistance of melanoma cells to BRAF inhibition can result in amplification of V600E BRAF expression. Enhanced CRAF levels drive resistance to BRAF inhibitors in melanoma.22 COT expression also confers acquired resistance to RAF inhibition by triggering a MAP kinase switch.23 However, as mentioned above, cyclins D1, E, and G1 or the RTK EGFR have been correlated with acquired resistance and could emerge as common pathways involved in sustaining ERK 1/2 activation in tumor cells. Approximately 30% of acquired resistance cases emerged independently of remarkable mutations.24
In the present study, we observed that ERK reactivation was invoked by FGFR3 activation to counteract the MEK inhibitor in HNSCC cells. We used AZD6244 as a selective MEK inhibitor and observed an ERK‐activity rebound in AZD6244‐treated HNSCC cell lines. In this experiment, we found that MEK inhibition induced FGFR3 activation. Inhibition of MEK with the FGFR3 inhibitor relieved the reactivation of ERK and the insensitivity of cancer cells against the MEK inhibitor. This likely occurred in vivo in xenograft mice and increased the anticancer effect of the MEK inhibitor. Thus, FGFR3 activation in response to MEK inhibition caused feedback activation of the ERK 1/2 cascade when FGFR3 was phosphorylated and Akt was activated. When FGFR3 activation was suppressed by FGFR siRNA or PD173074, these results were not observed. Recently, it was reported that ERK reactivation in tumors induced extensive and complex feedback activation of RTK. Prahallad et al20 reported that ERK reactivation by a BRAF inhibitor caused induction of RTK and the EGFR pathway. These data also indicated that combined treatment with a BRAF inhibitor and cetuximab synergistically inhibited tumor growth in vivo.20
Recent findings showed that FGFR dysregulation plays an important role in many cancers. Recently, Marshall et al25 reported that FGF‐FGFR signaling frequently occurs in HNSCC and was dominant or codominant with EGFR in HNSCC. Bevacizumab‐resistant HNSCC showed upregulated FGF2 and FGFR3 by increased ERK signaling.26 FGF‐FGFR autocrine loop signaling was induced after chronic adaptation to gefitinib and, thus, FGFR1‐specific tyrosine kinase inhibitor (TKI) prevented acquired resistance to gefitinib.27 We also showed that induction of the FGF2 ligand and FGFR3 following MEK inhibition promotes ERK reactivation and insensitivity to MEK inhibition. These findings provide evidence that FGFR signaling could serve as an additional candidate for a bypass mechanism in cancer cell lines rendered resistant to anticancer drugs.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea government (grant number: NRF‐2017R1C1B5018124), awarded to Myung Jin Ban. This research was also supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C0179) awarded to Yoon Woo Koh.
Ban MJ, Byeon HK, Yang YJ, et al. Fibroblast growth factor receptor 3‐mediated reactivation of ERK signaling promotes head and neck squamous cancer cell insensitivity to MEK inhibition. Cancer Sci. 2018;109:3816–3825. 10.1111/cas.13839
Ban and Byeon equally contributed to this study.
Contributor Information
Hyunjung Kee, Email: hjkee@yuhs.ac.
Yoon Woo Koh, Email: YWKOHENT@yuhs.ac.
REFERENCES
- 1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893‐2917. [DOI] [PubMed] [Google Scholar]
- 2. Global Burden of Disease Cancer Collaboration , Fitzmaurice C, Dicker D, et al. The global burden of cancer 2013. JAMA Oncol. 2015;1:505‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee JW, Soung YH, Kim SY, et al. Somatic mutations of EGFR gene in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2005;11:2879‐2882. [DOI] [PubMed] [Google Scholar]
- 4. Stransky N, Egloff AM, Tward A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Theocharis S, Kotta‐Loizou I, Klijanienko J, et al. Extracellular signal‐regulated kinase (ERK) expression and activation in mobile tongue squamous cell carcinoma: associations with clinicopathological parameters and patients survival. Tumor Biol. 2014;35:6455‐6465. [DOI] [PubMed] [Google Scholar]
- 6. Wang L, Liu T, Nishioka M, Aguirre RL, Win SS, Okada N. Activation of ERK1/2 and cyclin D1 expression in oral tongue squamous cell carcinomas: relationship between clinicopathological appearances and cell proliferation. Oral Oncol. 2006;42:625‐631. [DOI] [PubMed] [Google Scholar]
- 7. Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY‐142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer. 2009;125:2332‐2341. [DOI] [PubMed] [Google Scholar]
- 8. Wee S, Jagani Z, Xiang KX, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286‐4293. [DOI] [PubMed] [Google Scholar]
- 9. Duncan JS, Whittle MC, Nakamura K, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple‐negative breast cancer. Cell. 2012;149:307‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun C, Hobor S, Bertotti A, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 2014;7:86‐93. [DOI] [PubMed] [Google Scholar]
- 11. Little AS, Balmanno K, Sale MJ, et al. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal. 2011;4:ra17. [DOI] [PubMed] [Google Scholar]
- 12. Kitai H, Ebi H, Tomida S, et al. Epithelial‐to‐mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS‐mutant lung cancer. Cancer Discov. 2016;6:754‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Montero‐Conde C, Ruiz‐Llorente S, Dominguez JM, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF‐mutant thyroid carcinomas. Cancer Discov. 2013;3:520‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paraiso KH, Fedorenko IV, Cantini LP, et al. Recovery of phospho‐ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br J Cancer. 2010;102:1724‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanly EK, Tuli NY, Bednarczyk RB, et al. Hyperactive ERK and persistent mTOR signaling characterize vemurafenib resistance in papillary thyroid cancer cells. Oncotarget. 2016;7:8676‐8687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang D, Boerner SA, Winkler JD, LoRusso PM. Clinical experience of MEK inhibitors in cancer therapy. Biochim Biophys Acta. 2007;1773:1248‐1255. [DOI] [PubMed] [Google Scholar]
- 17. Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155‐168. [PubMed] [Google Scholar]
- 18. Smalley KS, Lioni M, Dalla Palma M, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E‐mutated melanomas. Mol Cancer Ther. 2008;7:2876‐2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Scaltriti M, Eichhorn PJ, Cortes J, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci USA. 2011;108:3761‐3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100‐103. [DOI] [PubMed] [Google Scholar]
- 21. Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853‐4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sequist LV, Waltman BA, Dias‐Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marshall ME, Hinz TK, Kono SA, et al. Fibroblast growth factor receptors are components of autocrine signaling networks in head and neck squamous cell carcinoma cells. Clin Cancer Res. 2011;17:5016‐5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gyanchandani R, Ortega Alves MV, Myers JN, Kim S. A proangiogenic signature is revealed in FGF‐mediated bevacizumab‐resistant head and neck squamous cell carcinoma. Mol Cancer Res. 2013;11:1585‐1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ware KE, Hinz TK, Kleczko E, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2‐FGFR1 autocrine growth loop. Oncogenesis. 2013;2:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials