

Abstract
Transcriptome-wide expression profiling of neurons has provided important insights into the underlying molecular mechanisms and gene expression patterns that transpire during learning and memory formation. However, there is a paucity of tools for profiling stimulus-induced RNA within specific neuronal cell populations. A bioorthogonal method to chemically label nascent (i.e. newly transcribed) RNA in a cell-type-specific and temporally controlled manner, which is also amenable to bioconjugation via click chemistry, was recently developed and optimized within conventional immortalized cell lines. However, its value within a more fragile and complicated cellular system, such as neurons, as well as for transcriptome-wide expression profiling, has yet to be demonstrated. Here, we report the visualization and sequencing of activity-dependent nascent RNA derived from neurons using this labeling method. This work has important implications for improving transcriptome-wide expression profiling and visualization of nascent RNA in neurons, which has the potential to provide valuable insights into the mechanisms underlying neural plasticity, learning, and memory.
Keywords: Nascent RNA, transcriptome-wide profiling, neuron, CuAAC, UPRT
Graphical Abstract
INTRODUCTION
Activity-dependent gene expression and its regulation by epigenetic mechanisms are critical for manifesting experience-dependent changes in cellular behavior that underlie neural plasticity, learning, and memory.1–4 In order to understand the processes that underlie the ability of a cell to alter its cellular behavior and encode memory, it is necessary to be able to profile the transcriptome and the dynamic changes that it undergoes. Moreover, given our growing appreciation for the behavioral and molecular heterogeneity present between different cell types and individual cells within the brain, transcriptome-wide profiling studies are now shifting towards utilizing cell-type-specific or single-cell sequencing technology.5, 6
Although significant advances have been made in sequencing homogeneous cell populations and single cells, current techniques are still hindered by an inability to separate nascent (i.e. stimulus-induced) RNA from steady-state (i.e. prestimulus) RNA populations (Figure 1). This lack of specificity and control over the investigation of RNA species that are produced during stimulus exposure has meant that many studies suffer from unnecessary noise in their data, which can make it challenging to identify pertinent targets in a transcriptome-wide manner. Therefore, methods that selectively label nascent RNA should prove invaluable for studies examining RNA biology and function.
Figure 1.

Illustrative graph depicting the production of stimulus-induced nascent RNA versus pre-stimulus steady-state RNA over time. Stimulus exposure (i.e. cellular activation) occurs during a set time period indicated by the red dotted lines.
The classic approach towards investigating nascent RNA involves introducing cells to a chemically-modified nucleoside, such as 4-thiouridine (4TUridine)7 or 5-ethynyl-uridine (5EUridine)8, that can be enzymatically incorporated into actively transcribed RNA by both endogenous and artificially-introduced phosphoribosyltransferases. Streptavidin enrichment of tagged nascent RNA can then be performed following biotinylation; 4TUridine-labeled RNA requires sulfhydryl-reactive biotinylation reagents whereas 5EUridine-labeled RNA bioconjugates to azide-biotin via the Cu(I)-catalysed alkyne-azide cycloaddition (CuAAC)9 reaction. However, a major disadvantage of both of these nucleoside analogs is that they are not cell-type-specific, and will therefore incorporate into the nascent RNA of any cell that they penetrate.
To address this limitation, a chemogenetic strategy for cell-type-specific nascent RNA labeling in mammalian cells was developed. Overexpression of the Toxoplasma gondii enzyme uracil phophoribosyltransferase (UPRT) within a discrete cell population is able to selectively drive the incorporation of a modified nucleobase 4-thiouracil (4TUracil) into nascent RNA.10 However, despite its success in controlling the spatial and temporal aspects of nascent RNA labeling, enrichment of 4TUracil-labeled RNA with sulfhydryl-reactive biotinylation reagents (e.g. HPDP-biotin) suffers from poor labeling and enrichment bias due to the transient nature of disulfide bonds.11 Moreover, due to the ubiquity of sulfurs within cells, the ability to utilize 4TUracil for imaging purposes is severely limited. Fortunately, recent work by our group has demon-strated that the alkyne-modified nucleobase analog, 5-ethynyl-uracil (5EUracil), is also a substrate for UPRT-driven cell-type-specific labeling of nascent RNA (Figure 2).12 However, chemical labeling of nascent RNA using UPRT and 5EUracil together (hereafter referred to as the UPRT-5EUracil system) has only been demonstrated within stably transfected UPRT-HeLa cells. Often times, optimized methods within immortal cell lines do not always efficiently translate across to primary cell cultures, which are a substantially more fragile and complicated cellular system. Furthermore, a demonstration of improved transcriptome-wide expression profiling using nascent RNA labeled with the UPRT-5EUracil system needs to be fully explored to understand the limitations and utility of the system.
Figure 2.

Overview of cell-type-specific bioorthogonal meta-bolic labeling of nascent RNA in mammalian cells using the UPRT-5EUracil system.
Here, we demonstrate robust nascent RNA labeling in primary cortical neurons in vitro, which can be used to improve enrichment of stimulus-responsive transcripts for transcriptome-wide expression profiling as well as imaging of nascent RNA localization using standard confocal microscopy. We expect that nascent RNA labeling in neurons will become a widespread tool for future studies investigating neural plas-ticity, learning, and memory.
RESULTS AND DISCUSSION
The first step in establishing the UPRT-5EUracil system for use in specific neuronal cell populations was to verify that UPRT overexpression in combination with 5EUracil exposure would be able to effectively label nascent RNA in primary cortical neurons (PCNs) in vitro. We used potassium chloride (KCl) treatment as a means of mimicking neural activity in vitro, in order to optimize our protocols for the visualization and identification of nascent RNA transcripts. In order to achieve neuron-specific labeling, we packaged a synapsin I-driven UPRT overexpression lentiviral plasmid that was generated by replacing the GFP sequence in the modified FUGW vector pFSy(1.1)GW (Addgene#2723213) with the Toxoplasma gondii UPRT sequence modified with a HA-tag (HA-UPRT, Addgene #4711010). The human synapsin 1 (hSYN1) gene promoter within the pFSy(1.1)GW vector contains a silencing element that represses gene transcription in non-neuronal cells.14, 15 As a control, we packaged the original pFSy(1.1)GW vector containing GFP. For nascent RNA labeling, we used the active intermediate 5EUridine analog as a positive control whilst RNA from UPRT+ cells without 5EUracil exposure was used as a negative control.
Robust overexpression of UPRT within neurons derived from embryonic C57BL/6J mice was evident after 7 days in culture (Figure 3A), and nascent RNA was successfully captured and visualized after adding 5EUracil for 3 hours to UPRT+ cells (Figure 3B,C). Visualization of alkyne-functionalized nascent RNA showed no significant labeling within UPRT- cells (Supplementary Figure 1); however, a dot blot analysis revealed some minor labeling (Figure 3B, Supplementary Figure 2). There are two possibilities regarding the background signal within UPRT-cells. Previous studies have shown that human and mouse cells are unable to convert uracil to uridine monophosphate (UMP); this has been attributed to a lack of uracil-binding capacity by UPRT orthologs from higher eukaryotes, which lack two residues in the uracil-binding domain that are highly conserved in the protozoan, yeast, and bacterial enzymes.16, 17 However, there is conservation in the active site of these UPRT orthologs, which suggests that they may be enzymatically active. Hence, it is possible that other co-enzymes or uracil-binding proteins could be acting to bring uracil to the mammalian UPRT, thereby leading to minimal levels of incorporation. Furthermore, other biochemical pathways are able to convert uracil to UMP in mammalian cells (e.g. orotate phosphoribosyl-transferase (OPRT)18 or a combination of uridine phosphory-lase19 and uridine kinase20).
Figure 3.

Robust nascent RNA labeling in neurons using the UPRT-5EUracil system. (A) UPRT is highly overexpressed in mouse PCNs using a synapsin I-driven lentiviral construct. UPRT lentivirus (+), GFP lentivirus (−), un-paired Student’s t-test (α=0.05, n=3) ****P<0.0001. (B) Visualization of alkyne-labeled biotinylated RNA on a dot blot (n=1/condition), which demonstrates effective nascent RNA capture in UPRT+ cells after treatment for 3h with 5EUracil (UPRT+5EUracil). (C) Visualization of nascent RNA labeled with 5EUracil (green) and HA-UPRT(red) in UPRT+ neurons (n=1). Scalebar: 50μm.
However, these pathways are substantially less efficient and their activity is rare.21 Thus, although there could be slight background incorporation of 5EUracil into nascent RNA without UPRT overexpression, the data still supports the use of the UPRT-5EUracil labeling method as a tool for the investigation of nascent RNA expression patterns within mammali-an cells, particularly given the large improvement in signal obtained during differential gene expression analysis post-sequencing (see Figures 4 & 5).
Figure 4.

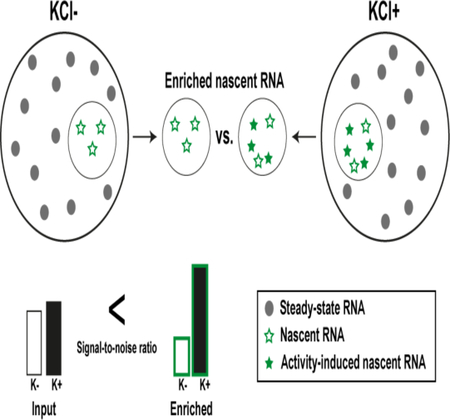
Pipeline for processing RNA from activated (KCl+) and non-activated (KCl-) neurons, which are then subdivided into two different groups for sequencing: input and enriched.
Figure 5.

Nascent RNA sequencing has improved sensitivity compared to standard RNA-seq. A Venn diagram illustrates the overlap of differentially expressed genes (KCl- Vs KCl+) detected using standard RNA-seq (input, left gray circle) and nascent RNA sequencing (enriched, right green circle). The four experimental groups shown are as follows: input KCl-, input KCl+, enriched KCl, enriched KCl+ (4 biological replicates per group). For gene target lists that were specifically identified in either the input or enriched differential gene expression analysis, the FDR value had to be both <0.05 within the selected comparison and >0.1 in the other so as to exclude any targets that were close to being significant in both (overlap).
Following validation of the UPRT-5EUracil system in neurons as well as optimization of the visualization protocol, we next moved towards developing a pipeline for sequencing (Figure 4). In order to demonstrate the advantages of nascent RNA sequencing using the UPRT-5EUracil system, we wanted to be able to sequence and directly compare enriched samples to non-enriched samples. For our study, we chose to divide each treatment condition (KCl- vs KCl+) into two sample streams: input and enriched (see Figure 4). Input samples represent an internal biological control that are derived from the same biological origin but are not subjected to enrichment, thereby allowing us to directly compare our nascent RNA sequencing approach to standard RNA-seq. For the sample processing pipeline, we needed to first validate/optimize the CuAAC biotinylation reaction, enrichment method, low-input ribosomal RNA (rRNA) removal, RNA fragmentation, double-stranded cDNA (dscDNA) synthesis, and library preparation procedure for sequencing activity-dependent nascent RNA from neurons (see Methods and Supplementary Information).
First, we found that shortening the CuAAC biotinylation reaction time to 10 mins (compared to 30 mins as previously used12) provided a better compromise between the amount of biotinylation and RNA quality, which is important for downstream sequencing applications (Supplementary Figure 3). In order to enrich nascent RNA, we evaluated two different approaches: 1) cDNA elution and 2) RNA elution (Supplementary Figure 4). Given the unquantifiable nature and expectedly low yield of cDNA transcripts following cDNA elution, we adopted the more straightforward RNA elution protocol (Supplementary Figure 5). Another consideration was that performing reverse transcription prior to enrichment could be biasing our yields towards certain transcript lengths and not accurately capturing the breadth of the nascent RNA transcriptome. Hence, we chose to minimize processing prior to enrichment. Improving the proportion of useful reads during sequencing requires rRNA depletion, as rRNA comprises >80% of total RNA. We found that rRNA removal was still possible on relatively low amounts of enriched nascent RNA (Supplementary Figure 6). Finally, we needed to optimize RNA fragmentation prior to dscDNA synthesis in order to obtain appropriately sized libraries for sequencing; we found that heating for 4–6 min at 94°C yielded a reasonable size distribution and therefore used 5 min for all subsequent samples (Supplementary Figure 7). Finally, we prepared the 16 samples for sequencing on the Illumina HiSeq 4000 platform using the Kapa HyperPrep Kit with 8 single-indexed adapters to uniquely barcode 8 samples per lane (2× lanes for a 2×150 cycle run, Supplementary Table 1).
We found a significant improvement in the identification of activity-regulated gene targets using nascent RNA sequencing compared with standard RNA-seq (Figure 5, see Section 3.10 for a description of the bioinformatics analysis). In particular, the former enabled the identification of 3347 activity-regulated genes (1662 up-regulated, 1685 down-regulated) that could not be detected by standard RNA-seq. This approach was also able to identify 94% of activity-regulated targets uncovered using standard RNA-seq, with the majority of these overlapping genes being more statistically robust in the nascent RNA sequencing analysis (False Discovery Rate (FDR) enriched < input in 97.7% up-regulated targets and 89.2% down-regulated targets). Enrichment analysis showed general differences in the gene ontology (GO) terms for significant gene targets obtained from each of the comparisons shown in Figure 5, for example, “poly(A) RNA binding” is only amongst the top 10 GO terms for up-regulated gene targets detected using enrichment (Supplementary Figure 8). Of especial interest, we observe a significant improvement in the detection of several immediate early genes (e.g. Arc, Nr4a2, Npas4), which are responsible for activating various specialized signaling cascades that result in changes to synaptic plasticity (Supplementary Figure 9A).22 Moreover, several genes implicated in synaptic remodeling, learning, and memory were only uncovered by nascent RNA sequencing. For example, Sox11 is involved in neurite growth,23 Kif1b is a molecular motor for ribonucleoprotein complexes from neuronal dendrites,24 and Tet3 is involved in the hydroxylation of 5-methylcytosine to 5-hydroxymethylcytosine, which is a highly dynamic epigenetic mark that is redistributed after fear extinction learning and is required for rapid behavioral adaptation25 (Supplementary Figure 9B). Furthermore, in addition to reducing the number of constitutively-expressed gene fragments (e.g. Pgk1, Actb, Gapdh), enrichment also allowed the detection of activity-dependent changes in these transcripts (Supplementary Figure 9C), which has previously been shown before in targeted qPCR studies.26 Therefore, standard RNA-seq appears to lack sufficient power to detect subtle changes in activity-dependent gene expression and has led to the misuse of several housekeeping genes. Taken together, our data indicates that enriching for nascent RNA prior to sequencing significantly improves the sensitivity of downstream analysis compared to traditional approaches that sequence the total RNA pool, and can provide important insights into the dynamics of gene regulation at both the steady-state and nascent RNA level.
Overall, we demonstrate the value of the UPRT-5EUracil system for analyzing transcriptome-wide expression patterns in a neuron-specific and temporally controlled manner. We have optimized protocols for visualizing and identifying nascent RNA within primary cortical neurons. As anticipated, we were able to detect a significantly greater number of activity-regulated genes following enrichment of nascent RNA compared to standard RNA-seq. By adjusting the timing of 5EUracil exposure, the transcriptional turnover of various genes can be probed within a user-defined time window for any neuronal cell population of interest. Furthermore, nascent RNA can also be visualized by conventional fluorescence imaging, which in combination with other RNA labeling techniques (e.g. clampFISH27) could allow RNA localization studies to distinguish between redistribution of steady-state versus nascent RNA transcripts during stimulation.
Chemical approaches have served as the foundation for investigating nascent RNA biology for many years. Previous work used thiol-modified analogs, such as 4TUridine and 4TUracil, or alkyne-modified nucleoside analogs, such as 5EUridine, to label nascent RNA. However, these approaches have the disadvantages of reliance on disulfide bond formation and a lack of cell-type specificity. One means of circumventing biases associated with disulfide enrichment is to chemically modify28 or recode29 the 4TUridine base to mimic a U-to-C conversion that can later be decoded using bioinformatics. However, this approach does not facilitate imaging of nascent RNA. Importantly, our work provides the first demonstration of nascent RNA labeling in mammalian neurons that is amenable to both visualization and sequencing methodologies.
Interestingly, recent work in HeLa cells and the invertebrate Drosophila melanogaster has extended the cell-type specificity of the UPRT system by using a two-step analog conversion process: 5-ethynyl-cytosine (5ECytosine) is converted into 5EUracil by the enzyme cytosine deaminase (CD) prior to conversion of 5EUracil into 5EUridine monophosphate by UPRT.30 This process is referred to as the CD-UPRT pathway. The strength of the CD-UPRT pathway is that CD itself has no orthologs in mammalian species, which improves the cell-type specificity of nascent RNA labeling. However, as a result of the two-step conversion process, the labeling efficiency is greatly reduced and requires a longer incubation time (>3 hours). Hence, the UPRT-5EUracil system is more suitable for studies that require shorter time windows of investigation, for example, during behavioral training, transient stimulus exposure, and time-course gene expression studies.
We anticipate that the UPRT-5EUracil nascent RNA labeling method will become a widely adopted chemical tool within the neuroscience community for investigating cell-type- and stimulus-specific changes in transcriptome-wide gene expression patterns. Furthermore, our method can also be used to extend cell-type specificity to already existing methods that utilize 5EUridine; for example, investigating RNA-binding proteins that interact with nascent RNA during stimulus exposure.31 The next challenge will be to combine the UPRT-5EUracil labeling approach with methods that investigate other aspects of RNA biology (e.g. RNA structure and modification). Other improvements will include providing tighter temporal control over nascent RNA labeling by using a photoinducible 5EUracil analog.32 Ultimately, we envision that future studies investigating neural plasticity, learning, and memory formation will greatly benefit from the spatial and temporal specificity conferred by the cell-type-specific UPRT-5EUracil system.
METHODS
Primary cortical neuron culture
Pregnant female C57BL6/J mice were euthanized at embryonic day 17 and cortical tissue was immediately dissected from the embryonic litter. Neurons were dissociated by finely chopping the tissue and then digested for 20 minutes at 37°C with papain (20 units, Sigma Aldrich #P4762). Benzonase nuclease (250 units, Sigma Aldrich #E1014–5KU) was added to the papain digestion solution to prevent cell clumping. Papain-digested tissue was passed through a 40 μm cell strainer (BD Falcon) to produce a single-cell suspension before plating onto either coverslips coated with poly-D-lysine or plates/dishes coated with poly-ornithine. Neurobasal medium (GIBCO #2110349) supplemented with 1% Penicillin/Streptomycin (GIBCO #15140122), 1% GlutaMAX (GIBCO #35050061), and 2% B-27 supplement (GIBCO #17504044) was used during plating and for maintaining cells afterwards (full media change every 2 – 3 days). First media change was always performed 12 – 24 hours following plating. Lentiviral treatment was performed following the first media change (i.e. days in vitro (DIV) 2) and then removed 2 days later. Labeling of nascent RNA with 5EUracil was performed 6 – 8 days following addition of virus. For collection of nascent RNA for sequencing: each embryonic litter was considered a biological sample and plated onto 10× 100mm dishes at a density of 5 million cells/dish (5× KCl-, 5× KCl+) before labeling nascent RNA a week later and pooling the extracted total RNA from each condition together. Total RNA was extracted immediately following the stimulation period either with or without KCl treatment (20 mM, KCl- group was given equivalent volume of Neurobasal media). All animal procedures were approved by The University of Queensland Animal Ethics Committee and conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Lentiviral packaging
For UPRT overexpression within neurons, the Toxoplasma gondii UPRT enzyme sequence capped with a HA-tag was placed downstream of a synapsin I promoter on a modified FUGW vector pFSy(1.1)GW (deposited by Pavel Osten13 into Addgene, plasmid #27232). The HA-tagged UPRT sequence was obtainedand PCR amplified from the pBS-HA::UPRT plasmid (deposited by Mike Cleary & Chris Doe10 into Addgene, plasmid #47110). Sequential PCR amplification was used to add BamHI and XbaI cutting sites onto either end of the HA::UPRT sequence (see Supplementary Table 2 for primers) before digestion and cloning into the BamHI and XbaI sites of the transfer vector pFSy(1.1)GW, replacing the original EGFP sequence (see Supplementary Figure 10). For packaging UPRT overexpression lentivirus, 80% confluent HEK293T cells were transfected for 4 hours with the UPRT transfer vector (13.3 μg), pMD2.G (6.7 μg, Addgene plasmid #12259), pRSV-rev (20 μg, Addgene plasmid #12253), and pMDLg/pRRE (20 μg, Addgene plasmid #12251) in Opti-MEM media (GIBCO #31985070) using 23.3 μL of Lipofectamine-2000 per 175 cm2 single-layer flask surface. Following transfection, HEK293T cells were maintained in DMEM media with sodium pyruvate (GIBCO #11995065), 1% Penicillin/Streptomycin (GIBCO #15140122), 10% fetal bovine serum (GIBCO #16000069), and 10 mM sodium butyrate (Sigma Aldrich #B5887–5G) for 40–48 hours prior to collection of culture supernatant. Virus particles were concentrated by ultracentrifugation at 21000 rpm for 90 min in a Beckman Type 50.2 Ti rotor, resuspended in 1× PBS, and stored at −80°C until use. At the same time, the original pFSy(1.1)GW vector containing EGFP was packaged as a virus negative control.
Validating UPRT overexpression
cDNA synthesis was performed on total RNA (500 ng) using the Quantitect Reverse Transcription Kit (Qiagen). ARotorGeneQreal-time PCR cycler with 2× SYBR Master Mix (Qiagen) was used to perform quantitative PCR. Phosphoglycerate kinase 1 (Pgk1) was used as an internal housekeeping control gene (see Supplementary Table 3 for primer pairs). For each PCR reaction, mRNA levels were normalized relative to the Pgk1 gene using the 2-ΔΔCT method. Each PCR reaction was run in duplicate for each sample and repeated at least twice. mRNA levels were compared between conditions using an un-paired Student’s t-test (α= 0.05) with Welch’s correction applied where appropriate.
Verifying ribosomal RNA removal
cDNA synthesis was performed on 2 ng of pre- and post-rRNA removal RNA samples (Input and Enriched) using the Quan-titect Reverse Transcription Kit (Qiagen). Quantitative PCR was performed on a RotorGeneQ real-time PCR cycler with 2×××× SYBR Master mix (Qiagen). Phosphoglycerate kinase 1 (Pgk1) was used as an internal housekeeping control gene (see Supplementary Table 4 for primer pairs). For each PCR reaction, mRNA levels were normalized relative to the Pgk1 gene using the 2-ΔΔCT method with the fold change of each rRNA-depleted sample calculated relative to the sample before ribosomal RNA depletion. Fold change values were then multiplied by 100 to obtain the % remaining of each rRNA type investigated (5S, 5.8S, 18S, 28S rRNA).
Labeling nascent RNA
5EUracil and 5EUridine nucleic acid analogs were added to neuron culture medium from 200 mM and 100 mM DMSO stocks with a final concentration of 200μM and 100 μM respectively (<1% DMSO in media). Analogs were incubated with cells for 10 min prior to and during a 3-hour stimulation period with either KCl (20 mM) or no KCl (equivalent amount of Neurobasal media added).
CuAAC biotinylation
Following chemical stimulation (KCl– vs KCl+) and incubation with nucleic acid analogs, total cellular RNA was harvested using NucleoZOL (Macherey-Nagel) according to the manufacturer’s instructions. For biotinylation reaction conditions, we adopted the protocol used by the Spitale group (UCI).12 In summary, 10 μg of total RNA was mixed with 1 mM azide-biotin, 12 mM CuSO4 to a final concentration of 2 mM, 4.6 mM THPTA to a final concentration of 0.76 mM, and fresh 10.6 mM NaASC to a final concentration of 1.76 mM. CuAAC reactions were incubated on shaker at room temperature for 10 min before immediately purifying using the Zymo Research RNA Clean & Concentrator-5 kit according to the manufacturer’s instructions with the in-column DNAse I incubation (#R1014).
CuAAC imaging
Following chemical stimulation (KCl- vs KCl+) and incubation with nucleic acid analogs, cells plated on coverslips were washed twice in PBS (5 min/wash) prior to fixation at room temperature with 4% formaldehyde in 0.1% Triton X-100 in PBS for 10 minutes. Cellular fixation was followed by two washes in cold PBS (10 min/wash) on a shaker before blocking at room temperature for 2 hours with 1× casein blocking buffer (Sigma Aldrich, #B6429). Cells were then rinsed briefly in PBS to remove excess protein before a 1 hour incubation at 37°C with a CuAAC reaction mixture containing 1 mM CuSO4, 5 mM THPTA, 2.5 mM NaASC, and 25 μM azide-AlexaFluor488 in nuclease-free H2O. After the CuAAC reaction, cells were washed three times in cold PBS (10 minutes/wash). 4’,6-diamidino-2-phenylidole (DAPI, 1:1000 in PBS) was added to cells for 10 minutes and then rinsed off with a 5-minute wash in 0.1% Triton X-100 in PBS and a final wash of PBS for at least 10 minutes before mounting coverslips onto microscope slides using DAKO fluorescent mounting media (#S3023).
Dot blot
Prior to RNA loading, a Hybond-N+ membrane (GE Healthcare) was equilibrated in 10X SSC for 15 min and then dried completely. Equal amounts of post-CuAAC biotinylated RNA were individually applied onto the equilibrated membrane using either a standard vacuum blotter (Bio-Rad, Bio-Dot Apparatus #1706545) or hand-held pipette. Next, the RNA-blotted membrane was UV crosslinked (120000 μJ, 1 min) in a standard UV crosslinking oven (Boekel, UV Cross-linker AH #234100–2). Following UV crosslinking, membranes were blocked for 30 min (120mM NaCl, 16mM Na2HPO4, 8mM NaH2PO4, 170mM SDS) and then incubated with a streptavidin-IR800CW fluorescent dye (1:10000, LI-COR, #926–3220) for 1 hour. The membrane was then washed overnight in 1:10 blocking solution and imaged on a LI-COR Odyssey Fc imaging system with 2 min exposure at 800 nm.
Streptavidin enrichment
Following CuAAC biotinylation of alkyne-modified nascent RNA, post-CuAAC biotinylated RNA was subjected to streptavidin enrichment. 5 μL of Dynabeads MyOne Streptavidin C1 beads (Invitrogen, #65002) was prepared for every μg of post-CuAAC biotinylated RNA by washing four times in Bead Binding Buffer (BBB; 1M NaCl, 100mM Tris-HCl pH 7.0, 10mM EDTA, 0.2% (vol/vol) Tween-20) before resuspending in 75 μL BBB. Two strategies for streptavidin enrichment were performed during this study and are outlined below: 1) cDNA elution, and 2) RNA elution.
cDNA elution
For cDNA elution, cDNA/RNA hybrids were generated using the Superscript III First-Strand Synthesis System (Invitrogen, #18080051) on 5 μg of post-CuAAC biotinylated RNA excluding the digestion step with RNAseH. Afterwards, the cDNA/RNA hybrids were diluted to 75 μL and added to the washed streptavidin beads with 20 units of RNAse inhibitor and allowed to bind at room temperature on rotation for one hour. Following binding of biotinylated RNA/cDNA species to the streptavidin beads, beads were washed 3× with Bead Washing Buffer (BWB; 4M NaCl, 100mM Tris-HCl pH 7.0, 10mM EDTA, 0.2% (vol/vol) Tween-20) and then twice in 1× PBS. cDNA complementary to biotinylated nascent RNA (still bound to the beads) was eluted by incubating in 50 μL of Elution Buffer (EB: 75mM NaCl, 50mM HEPES, 3 mM MgCl2, 0.125% (wt/vol) N-lauroylsarcosine sodium salt, 0.025% NaDOC, 5mM DTT, 12.5mM D-Biotin, RNAseA/H/T1 cocktail) for 30 min at 37°C in a thermoshaker rotating at 1000 rpm before addition of 1 μL of 100% DMSO with heating at 95°C for 4 – 8 min. The eluted cDNA was immediately subjected to purification using the Zymo Research DNA Clean and Concentrator-5 kit with the variation of adding an equal amount of 100% ethanol as DNA binding buffer (1:1) before transfer to the spin column. The remainder of column purification was performed according to the manufacturer’s instructions with elution of cDNA fragments into 20 μL of nuclease-free water.
RNA elution
2× batches of 25 μg of post-CuAAC biotinylated RNA were diluted to 75 μL and then added to the pre-washed streptavidin beads with 20 units of RNAse inhibitor and allowed to bind at room temperature on rotation for one hour. Following binding of biotinylated RNA species to the streptavidin beads, beads were washed 3× with Bead Washing Buffer (BWB; 4M NaCl, 100mM Tris-HCl pH 7.0, 10mM EDTA, 0.2% (vol/vol) Tween-20) and then twice in 1× PBS. Nascent RNA was eluted in 50 μL of ß-Mercaptoethanol Elution Buffer (ß-MEB; 1M NaCl, 50mM MOPS, 5mM EDTA, 2M ß-Mercaptoethanol) for 2 min at 95°C and then immediately purified using the Zymo Research RNA Clean and Concentrator-5 kit according to the manufacturer’s instructions with a final elution volume of 20 μL nuclease-free water.
Ribosomal RNA removal
Ribosomal RNA (rRNA) was removed from total RNA (5 μg reaction) and enriched nascent RNA (500 ng reaction) using the Illumina Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) according to the manufacturer’s instructions. For total RNA and enriched nascent RNA reactions, samples were purified immediately following rRNA removal using RNACleanXP beads (Beckman Coulter) in a 1.8:1 and 3.7:1 ratio, respectively.
Preparation of libraries for sequencing
RNA samples for sequencing were converted to dscDNA using the NEBNext® Ultra™ II RNA First Strand Synthesis Module (#E7771) and NEBNext® Ultra™ II Non-Directional RNA Second Strand Synthesis Module (#E6111). RNA fragmentation was performed at 94°C for 5 min (recommended time for samples of RIN 2–6: 7–8 min). Following conversion to dscDNA, sample libraries were prepared using the KAPA Hyper Prep Kit (#KK8504) using a 300nM adapter stock concentration (Set A) and 12-cycle amplification step (see Supplementary Table 4 for adapters used for each sample). Sample libraries were size-selected to be between ~200 – 1000 bp (median: 400 bp) using 0.6× and 1.6× sequential bead cleanups (AMPureXP beads, Beckman Coulter). All samples then underwent a 2×150 run using two lanes of an Illumina HiSeq 4000 (8 samples/lane) at GENEWIZ (Suzhou, China).
Bioinformatics analysis of sequencing data
Cutadapt (version 1.8.1) (http://cutadapt.readthedocs.io) was used to cut the adaptor sequences as well as low quality nucleotides at both ends. When a processed read is shorter than 36 bp, the read was discarded by cutadapt, with the parameter setting of “-q 20,20 --minimum-length=36”. Processed reads were aligned to the mouse reference genome (mm10) using HISAT2 (version 2.0.5),33 with the parameter setting of “--no-unal --fr --dta --known-splicesite-infile splicesites.txt” (see Supplementary Table 5 for counts of total and mappable reads). This setting is to i) suppress SAM records for reads that failed to align (“--no-unal”), ii) specify the Illumina’s paired-end sequencing assay (“--fr”), iii) require longer anchor lengths for the detection of high-confidence novel exon-exon junctions, and iv) provide a list of known splice sites in mouse (“--known-splicesite-infile splicesites.txt”). Samtools (version 1.3)34 was then used to convert “SAM” files to “BAM” files, sort and index the “BAM” files. The “htseq-count” script in HTSeq package (v0.7.1) (http://www-huber.embl.de/HTSeq) was used to quantitate the gene expression level by generating a raw count table for each sample (i.e. counting reads in gene features for each sample). Based on these raw count tables, edgeR (version 3.16.5)35 was adopted to perform the differential expression analysis between treatment groups and controls. EdgeR used a trimmed mean of M-values to compute scale factors for library size normalization.36 It used the quantile-adjusted conditional maximum likelihood (qCML) method to estimate dispersions37 and the quasi-likelihood F-test to determine differential expression.38 Extremely lowly expressed genes were removed before the differential expression analysis (i.e. only keep genes with non-zero counts in at least four samples). Differentially expressed (DE) genes between two groups were identified with FDR < 0.05. The nascent RNA-seq (i.e. enriched) specific DE genes were defined when genes with FDR<0.05 in the enriched KCl- vs. enriched KCl+ comparison and FDR > 0.1 in the input KCl- vs. input KCl+ comparison, and vice versa for input-specific DE genes. Gene ontology enrichment analysis for DE genes was performed using the functional annotation tool in DAVID Bioinformatics Resources (version 6.8).39, 40
Image acquisition and analysis
Images of alkyne-modified nascent RNA conjugated to azide-AlexaFluor488 were acquired on a ZIS LSM 510 META confocal microscope using a 63× oil objective with numerical aperture 1.4. All images were converted into tiffs using ImageJ and then processed further in Adobe Photoshop to threshold for background fluorescence staining using the negative analog control sample (UPRT treated cells without addition of 5EUracil) as a baseline.
Assessment of nucleic acid quality and fragmentation
RNA/DNA concentration was measured using an NanoPhotometer N60 (Implen) unless below the minimum concentration range whereby a Qubit assay (Invitrogen, RNA HS or DNA HS) was performed instead. Capillary electrophoresis was performed using the 2100 Agilent Bioanalyzer (Agilent Technologies, Palo Alto, California) to assess RNA/DNA quality and fragmentation (i.e. size distribution). RNA samples were analyzed on RNA Pico II and RNA Nano chips whilst DNA samples were analyzed on HS DNA chips. For RNA samples, the ratio of ribosomal RNAs (18S/28S) and the RNA integrity number (RIN, 1 – 10) was also automatically calculated. For DNA samples, the median fragment size (bp) was calculated by the Agilent software after outlining a region of interest.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Bredy Lab and Rowan Tweedale for careful reading and critique of the manuscript. E.L.Z. is supported by a UQ Graduate School Scholarship, X.L. (UQ Development Award), P.R.M (NSERC Postgraduate Award), and L.J.L. (Westpac Future Leaders Scholarship). This work was also supported by DP180102998 (T.W.B.) and 1R21MH113062 (R.C.S.).
Funding Sources
This work was supported by an ARC Discovery Project grant (DP180102998) and a National Institute of Mental Health R21 grant (1R21MH113062).
ABBREVIATIONS
- 4TUracil
4-thiouracil
- 4TUridine
4-thiouridine
- 5ECytosone
5-ethynyl-cytosine
- 5EUracil
5-ethynyl-uracil
- 5EUridine
5-ethynyluridine
- BBB
bead binding buffer
- BWB
bead washing buffer
- ß-MEB
ß-Mercaptoethanol elution buffer
- cDNA
complementary DNA
- CD
cytosine deaminase
- CuAAC
Cu(I)-catalysed alkyneazide cycloaddition
- CuSO4
copper sulphate
- DAPI
4’,6-diamidino-2-phenylindole
- DE
differentially expressed
- DIV
days in vitro
- DMSO
dimethylsulfoxide
- dscDNA
double-stranded cDNA
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- FDR
false discovery rate
- FISH
fluorescent in situ hybridization
- GFP
green fluorescent protein
- GO
gene ontology
- HA
human influenza hemagglutinin
- HCl
hydrochloric acid
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- KCl
potassium chloride
- MgCl2
magnesium chloride
- MOPS
3-morpholinopropane-1-sulfonic acid
- mRNA
messenger RNA
- NaASC
sodium ascorbate
- NaDOC
sodium deoxycholate
- NaCl
sodium chloride
- Na2HPO4
disodium phosphate
- NaH2PO4
monosodium phosphate
- OPRT
orotate phosphoribosyltransferase
- PBS
phosphate buffered saline
- PCN
primary cortical neurons
- PCR
polymerase chain reaction
- PRPP
phosphoribosyl pyrophosphate
- qPCR
quantitative PCR
- RNA
ribonucleic acid
- RNA-seq
RNA sequencing
- rRNA
ribosomal RNA
- SDS
sodium dodecyl sulfate
- THPTA
Tris(3-hydroxypropyltriazolylmethyl)amine
- UMP
uridine monophosphate
- UPRT
uracil phosphoribosyltransferase
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Baker-Andresen D, Ratnu VS, and Bredy TW (2013) Dynamic DNA methylation: a prime candidate for genomic meta-plasticity and behavioral adaptation, Trends Neurosci. 36, 3–13. [DOI] [PubMed] [Google Scholar]
- 2.Marshall P, and Bredy TW (2016) Cognitive neuroepigenetics: the next evolution in our understanding of the molecular mechanisms underlying learning and memory?, NPJ Sci Learn 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nainar S, Marshall PR, Tyler CR, Spitale RC, and Bredy TW (2016) Evolving insights into RNA modifications and their functional diversity in the brain, Nat Neurosci 19, 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leighton LJ, Ke K, Zajaczkowski EL, Edmunds J, Spitale RC, and Bredy TW (2018) Experience-dependent neural plasticity, learning, and memory in the era of epitranscriptomics, Genes Brain Behav. 17, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okaty BW, Sugino K, and Nelson SB (2011) Cell Type-Specific Transcriptomics in the Brain, J. Neurosci 31, 6939–6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duran RCD, Wei HC, and Wu JQ (2017) Single-cell RNA-sequencing of the brain, Clin. Transl. Med 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melvin WT, Milne HB, Slater AA, Allen HJ, and Keir HM (1978) INCORPORATION OF 6-THIOGUANOSINE AND 4-THIOURIDINE INTO RNA - APPLICATION TO ISOLATION OF NEWLY SYNTHESIZED RNA BY AFFINITY CHROMATOGRAPHY, Eur. J. Bio-chem 92, 373–379. [DOI] [PubMed] [Google Scholar]
- 8.Jao CY, and Salic A (2008) Exploring RNA transcription and turnover in vivo by using click chemistry, Proc. Natl. Acad. Sci. U. S. A 105, 15779–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolb HC, Finn MG, and Sharpless KB (2001) Click chemistry: Diverse chemical function from a few good reactions, Angew. Chem.-Int. Edit 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- 10.Miller MR, Robinson KJ, Cleary MD, and Doe CQ (2009) TU-tagging: cell type-specific RNA isolation from intact complex tissues, Nat. Methods 6, 439–U457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duffy EE, Rutenberg-Schoenberg M, Stark CD, Kitchen RR, Gerstein MB, and Simon MD (2015) Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry, Mol Cell 59, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen K, Fazio M, Kubota M, Nainar S, Feng C, Li X, Atwood SX, Bredy TW, and Spitale RC (2017) Cell-Selective Bioorthogonal Metabolic Labeling of RNA, J Am Chem Soc 139, 2148–2151. [DOI] [PubMed] [Google Scholar]
- 13.Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, and Osten P (2004) Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo, Proc. Natl. Acad. Sci. + 101, 18206–18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li LA, Suzuki T, Mori N, and Greengard P (1993) Identification of a Functional Silencer Element Involved in Neuron-Specific Expression of the Synapsin-I Gene, Proc. Natl. Acad. Sci. U. S. A 90, 1460–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLean JR, Smith GA, Rocha EM, Hayes MA, Beagan JA, Hallett PJ, and Isacson O (2014) Widespread neuron-specific transgene expression in brain and spinal cord following synapsin promoter-driven AAV9 neonatal intracerebro-ventricular injection, Neurosci Lett 576, 73–78. [DOI] [PubMed] [Google Scholar]
- 16.Pfefferkorn ER, and Pfefferkorn LC (1977) Specific Labeling of Intracellular Toxoplasma-Gondii with Uracil, J Protozool 24, 449–453. [DOI] [PubMed] [Google Scholar]
- 17.Li JX, Huang SD, Chen JZ, Yang ZX, Fei XW, Zheng M, Ji CN, Yi X, and Mao YM (2007) Identification and characterization of human uracil phosphoribosyltransferase (UPRTase), J Hum Genet 52, 415–422. [DOI] [PubMed] [Google Scholar]
- 18.Reyes P, and Guganig ME (1975) Studies on a Pyrimidine Phosphoribosyltransferase from Murine Leukemia P1534j - Partial-Purification, Substrate-Specificity, and Evidence for Its Existence as a Bifunctional Complex with Orotidine 5’-Phosphate Decarboxylase, J Biol Chem 250, 5097–5108. [PubMed] [Google Scholar]
- 19.Watanabe SI, Hino A, Wada K, Eliason JF, and Uchida T (1995) Purification, Cloning, and Expression of Murine Uridine Phosphorylase, J Biol Chem 270, 12191–12196. [DOI] [PubMed] [Google Scholar]
- 20.Cheng N, Payne RC, and Traut TW (1986) Regulation of Uridine Kinase - Evidence for a Regulatory Site, J Biol Chem 261, 3006–3012. [PubMed] [Google Scholar]
- 21.Carter D, Donald RGK, Roos D, and Ullman B (1997) Expression, purification, and characterization of uracil phosphoribosyltransferase from Toxoplasma gondii, Mol Biochem Parasit 87, 137–144. [DOI] [PubMed] [Google Scholar]
- 22.West AE, and Greenberg ME (2011) Neuronal Activity-Regulated Gene Transcription in Synapse Development and Cognitive Function, Csh Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jankowski MP, Cornuet PK, McILwrath S, Koerber HR, and Albers KM (2006) SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth, Neuroscience 143, 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charalambous DC, Pasciuto E, Mercaldo V, Boyl PP, Munck S, Bagni C, and Santama N (2013) KIF1B beta transports dendritically localized mRNPs in neurons and is recruited to synapses in an activity-dependent manner, Cell Mol Life Sci 70, 335–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, D’Alessio A, Zhang Y, and Bredy TW (2014) Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation, Proc. Natl. Acad. Sci. U. S. A. 111, 7120–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rydbirk R, Folke J, Winge K, Aznar S, Pakkenberg B,and Brudek T (2016) Assessment of brain reference genes for RT-qPCR studies in neurodegenerative diseases, Sci Rep-Uk 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rouhanifard SH, Mellis IA, Dunagin M, Bayatpour S,Symmons O, Cote A, and Raj A (2018) Exponential fluorescent amplification of individual RNAs using clampFISH probes, bioRxiv.
- 28.Herzog VA, Reichholf B, Neumann T, Rescheneder P,Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, and Ameres SL (2017) Thiol-linked alkylation of RNA to assess expression dynamics, Nat. Methods 14, 1198–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schofield JA, Duffy EE, Kiefer L, Sullivan MC, and Simon MD (2018) TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding, Nat. Methods 15, 221–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hida N, Aboukilila MY, Burow DA, Paul R, Greenberg MM, Fazio M, Beasley S, Spitale RC, and Cleary MD (2017) EC-tagging allows cell type-specific RNA analysis, Nucleic Acids Research 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bao XC, Guo XP, Yin MH, Tariq M, Lai YW, Kanwal S, Zhou JJ, Li N, Lv Y, Pulido-Quetglas C, Wang XW, Ji L, Khan MJ, Zhu XH, Luo ZW, Shao CW, Lim DH, Liu X, Li N, Wang W, He MH, Liu YL, Ward C, Wang T, Zhang G, Wang DY, Yang JH, Chen YW, Zhang CL, Jauch R, Yang YG, Wang YM, Qin BM, Anko ML, Hutchins AP, Sun H, Wang HT, Fu XD, Zhang BL, and Esteban MA (2018) Capturing the interactome of newly transcribed RNA, Nat. Methods 15, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng C, Li Y, and Spitale RC (2017) Photo-controlled cell-specific metabolic labeling of RNA, Org Biomol Chem 15, 5117–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim D, Langmead B, and Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements, Nat Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009) The Sequence Alignment/Map format and SAMtools, Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson MD, McCarthy DJ, and Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson MD, and Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data, Genome Biol 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCarthy DJ, Chen Y, and Smyth GK (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation, Nucleic Acids Res 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lun AT, Chen Y, and Smyth GK (2016) It’s DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR, Methods Mol Biol 1418, 391–416. [DOI] [PubMed] [Google Scholar]
- 39.Huang da W, Sherman BT, and Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nature protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 40.Huang da W, Sherman BT, and Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists, Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.