

Abstract
Eight new lactones (δ-chloro-, δ-bromo- and δ-iodo-γ-lactones), each with a methylcyclohexane ring, were obtained by chemical means from (4-methylcyclohex-2-en-1-yl) acetic acid or (6-methylcyclohex-2-en-1-yl) acetic acid. Whole cells of ten fungal strains (Fusarium species, Syncephalastrum racemosum and Botrytis cinerea) were tested on their ability to convert these lactones into other products. Some of the tested fungal strains transformed chloro-, bromo- and iodolactone with a methyl group at C-5 into 2-hydroxy-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one during hydrolytic dehalogenation. When the same lactones had the methyl group at C-3, no structural modifications of halolactones were observed. In most cases, the optical purity of the product was low or medium, with the highest rate for chlorolactone (45.4%) and iodolactone (45.2% and 47.6%). All of the obtained compounds were tested with reference to their smell. Seven halolactones and the hydroxylactone obtained via biotransformation of halolactones with 5-methylcyclohexane ring were examined for their antimicrobial activity. These compounds were capable of inhibiting growth of some bacteria, yeasts and fungi.
Keywords: lactones, biotransformations, hydrolytic dehalogenation, Fusarium species, antimicrobial activity
1. Introduction
Modern life sciences are focused on the search for new bioactive compounds of natural origin. It is reported that many plants used in traditional medicine systems contain a variety of compounds responsible for their healing properties. A large part of these compounds are sesquiterpenoid hydroxylactones [1,2,3,4,5,6], substances of high biological activity. Hydroxylactones exhibit a number of biological properties, showing e.g., hepaprotective [7], cytotoxic [8,9,10,11,12,13,14], antiplasmodial [15], antifungal [16], antibacterial [17], antitumor and anti-inflammatory activity [18], acting as phytotoxic agents [19], and having an inhibitory effect on NO production [20] and against activity of some enzymes [21].
However, the isolation of such compounds from plants is often complicated, because their content is usually not impressive. Therefore, it is necessary to look for alternative methods of the preparation of more potentially effective bioactive molecules. One such method is the use of microorganisms capable of introducing a hydroxy group into a molecule of, for example, a lactone. Halogenolactones or saturated lactones are useful compounds for this purpose [22,23,24,25,26,27,28,29].
As part of our ongoing research on the biotransformation of halolactones with a methyl-substituted cyclohexane rings [30,31,32,33,34,35], in this paper we present further examples of a biohydroxylation of chloro-, bromo- and iodolactones containing a methylcyclohexane ring. We examined the effect of the methyl group position in the cyclohexane ring on the ability of microorganisms to replace the halogen atom with a hydroxy group.
2. Results and Discussion
2.1. Synthesis of Substrates
In our research on compounds with a methylcyclohexane ring two intermediate products—γ,δ-unsaturated esters 1a and 1b—were obtained during earlier syntheses [36]. Following basic hydrolysis, these esters gave γ,δ-unsaturated acids 2a [37] or 2b, which were substrates for the next step. New chlorolactones 3a,b, known bromolactone 4a [38], and a new bromolactone 4b were synthesized by chloro- or bromolactonization in THF [31]. Moreover, new iodolactones 5a,b and 6a,b were obtained by iodolactonization of acid 2, according to the previously described procedure [39] (Scheme 1).
Scheme 1.

Synthesis of halolactones 3–6.
Chloro-3a,b, bromo-4a,b and iodolactones-5a,b were subjected to a screening biotransformation. Iodolactone 6a was generated in small quantities, insufficient to carry out the biotransformation, so we decided to investigate only its biological activity. As for iodolactone 6b, the amount obtained was only sufficient to determine its structure.
2.2. Biotransformation of Halolactones
One of our aims was to identify fungal strains capable of conducting a hydrolytic dehalogenation of these halolactones. The following strains were selected: Fusarium culmorum AM10, Fusarium avenaceum AM11, Fusarium oxysporum AM13, Fusarium tricinctum AM16, Fusarium semitectum AM20, Fusarium equiseti AM22, Fusarium scirpi AM1199, Fusarium solani AM203, Syncephalastrum racemosum AM105, and Botrytis cinerea AM235. Progress of all the screening transformations was monitored by using standard techniques (TLC and GC). The results of this step are displayed in Table 1.
Table 1.
Hydrolytic dehalogenation of halolactones 3a,b–5a,b after 7 days of incubation.
| Entry | Microorganism | Chlorolactone | Bromolactone | Iodolactone | |||
|---|---|---|---|---|---|---|---|
| 3a | 3b | 4a | 4b | 5a | 5b | ||
| 1 | Fusarium culmorum AM10 | (++) | (‒) | (++) | (‒) | (+++) | (‒) |
| 2 | Fusarium avenaceum AM11 | (‒) | (‒) | (+++) | (‒) | (++++) | (‒) |
| 3 | Fusarium oxysporum AM13 | (‒) | (‒) | (+) | (‒) | (+++) | (‒) |
| 4 | Fusarium tricinctum AM16 | (‒) | (‒) | (‒) | (‒) | (‒) | (‒) |
| 5 | Fusarium semitectum AM20 | (‒) | (‒) | (+) | (‒) | (+) | (‒) |
| 6 | Fusarium equiseti AM22 | (+++) | (‒) | (++++) | (‒) | (++++) | (‒) |
| 7 | Syncephalastrum racemosum AM105 | (‒) | (‒) | (‒) | (‒) | (+++) | (‒) |
| 8 | Fusarium scirpi AM199 | (‒) | (‒) | (+++) | (‒) | (+++) | (‒) |
| 9 | Fusarium solani AM203 | (++) | (‒) | (++) | (‒) | (+++) | (‒) |
| 10 | Botrytis cinerea AM235 | (‒) | (‒) | (‒) | (‒) | (++) | (‒) |
Conversions: (−): 0%–35%; (+): 45%–51%; (++): 53%–68%; (+++): 73%–89%; (++++): 90%–96%.
Halolactones 3a–5a were transformed into a single product in all the biotransformations. The highest degree of conversion (over 90%) was observed for bromolactone 4a (entry 6) and iodolactone 5a (entry 2, 6). Some of the other microorganisms (entries 1, 8, 9 and 10 for 4a, entries 1, 3, 7, 8, 9 and 10 for 5a) transformed these two substrates with a good yield (over 53%). Chlorolactone 3a was converted in good yield (over 53%) by only three microorganisms (entries 1, 6, 9). Iodolactone 6a and 6b were not subjected to the biotransformation, as they were produced in insufficient amounts for this step. The screening biotransformation was only used for a selection of fungal strains capable of converting the substrates into a product with a yield exceeding 50%. During the screening transformations, only the degree of conversion was evaluated.
The same ten fungal strains were investigated regarding their ability to transform halolactones 3b–5b into other derivatives. Unfortunately, the tested strains were not capable of converting these substrates into any products.
Tests on the stability of lactones 3a, b–5a, b were also conducted. The control was carried out by adding each of the lactones to a sterile medium containing 3 g of glucose and 1 g of peptobac in 100 mL of water and shaking for a week. Studied samples were prepared similarly as in the case of the screening procedure. We found that only the substrate was present in the reaction mixture, indicating that the hydrolytic dehalogenation of lactones 3a, b–5a, b did not occur without the microbial participation.
For the next step, e.g., the preparative scale biotransformations, fungal strains that had been selected during the screening procedure were used. The results of these transformations are presented in Figure 1.
Figure 1.

Results (by GC) of preparative biotransformations of lactones 3a–5a.
As expected, the best results were obtained when F. equiseti AM22 (degree of conversion 89.2% for 3a, 90.7% for 4a, and 97.8% for 5a) was used as a biocatalyst. Very good results were also observed for F. avenaceum AM11 (degree of conversion 78.0% for 4a and 93.6% for 5a), F. oxysporum AM13 (degree of conversion 82.1% for 5a) and S. racemosum AM105 (degree of conversion 87.4% for 5a). From the microbial perspective, the best substrate for the biotransformation was iodolactone 5a, converted into hydroxylactone 7a by eight strains, and the worst was chlorolactone 3a, transformed by only three strains. Data presented above pointed to the conclusion that the microorganisms used for the biotransformation of halolactones were characterized by substrate specificity.
During all of the preparative biotransformations, a single product, hydroxylactone 7a, was obtained (Scheme 2).
Scheme 2.

Biotransformations of halolactones 3a–5a.
One of the aims of the described experiments was to obtain a product with high optical purity. Because of that we evaluated both the enantiomeric excess and optical rotation for every hydroxylactone obtained during the preparative biotransformations of halolactones 3a–5a. The enantiomeric excess was determined using GC with a chiral column (Beta Dex, 30 m × 0.25 mm × 0.25 μm), and the optical rotation was measured in chloroform solutions. More details on the quantities of hydroxylactone 7a and the data on its optical purity are presented in Table 2, Table 3 and Table 4.
Table 2.
Data on hydroxylactone 7a obtained during preparative biotransformations of lactone 3a.
| Entry | Strain | Yield (g)/(%) | ee | |
|---|---|---|---|---|
| 1 | F. culmorum AM10 | 0.017/18.3 | 45.4 | ‒33.344 (c = 0.69, CHCl3) |
| 2 | F. equiseti AM22 | 0.038/41.8 | 34.3 | ‒23.175 (c = 0.88, CHCl3) |
| 3 | F. solani AM203 | 0.012/13.3 | 17.9 | +14.509 (c = 0.46, CHCl3) |
Table 3.
Data on hydroxylactone 7a obtained during preparative biotransformations of lactone 4a.
| Entry | Strain | Yield (g)/(%) | ee | |
|---|---|---|---|---|
| 1 | F. culmorum AM10 | 0.024/32.4 | 32.4 | ‒22.477 (c = 0.85, CHCl3) |
| 2 | F. avenaceum AM11 | 0.032/10.4 | 10.4 | +9.765 (c = 1.65, CHCl3) |
| 3 | F. equiseti AM22 | 0.024/18.0 | 32.5 | ‒17.042 (c = 0.61, CHCl3) |
| 4 | F. scirpi AM199 | 0.028/41.2 | 38.5 | ‒27.339 (c = 0.87, CHCl3) |
| 5 | F. solani AM203 | 0.034/5.6 | 46.6 | +4.234 (c = 0.85, CHCl3) |
| 6 | B. cinerea AM235 | 0.021/20.2 | 28.9 | ‒19.213 (c = 0.94, CHCl3) |
Table 4.
Data on hydroxylactone 7a obtained during preparative biotransformations of lactone 5a.
| Entry | Strain | yield (g)/(%) | ee | |
|---|---|---|---|---|
| 1 | F. culmorum AM10 | 0.016/26.7 | 45.2 | ‒32.244 (c = 0.89, CHCl3) |
| 2 | F. avenaceum AM11 | 0.018/29.7 | 5.7 | ‒6.598 (c = 0.80, CHCl3) |
| 3 | F. oxysporum AM13 | 0.035/57.2 | 35.3 | +37.211 (c = 0.59, CHCl3) |
| 4 | F. equiseti AM22 | 0.036/58.5 | 15.1 | ‒17.740 (c = 0.97, CHCl3) |
| 5 | S. racemosum AM105 | 0.031/50.7 | 24.0 | ‒19.714 (c = 0.48, CHCl3) |
| 6 | F. scirpi AM199 | 0.030/49.6 | 47.6 | ‒34.134 (c = 0.65, CHCl3) |
| 7 | F. solani AM203 | 0.013/21.4 | 3.7 | 0 (c = 0.72, CHCl3) |
| 8 | B. cinerea AM235 | 0.014/23.2 | 45.2 | ‒32.196 (c = 0.49, CHCl3) |
As a result of microbial dehydrodehalogenation, hydroxylactone 7a was obtained with medium or low enantiomeric excess (ee). The best results were achieved using iodolactone 5a as a substrate. Three microorganisms, F. culmorum AM10 (Table 4, entry 1), F. scirpi AM199 (Table 4, entry 6) and B. cinerea AM235 (Table 4, entry 8) were capable of transforming this compound with moderate enantiomeric excess (45.2% or 47.6%). Similar results were obtained for chlorolactone 3a transformed by F. culmorum AM10 (ee = 45.4%) (Table 2, entry 1) and bromolactone converted by F. solani AM203 (ee = 46.6%) (Table 3, entry 5).
The majority of the microorganisms formed the (‒)-enantiomer of hydroxylactone 7a. Interestingly, some of them, such as F. avenaceum AM11 for bromolactone 4a (Table 3, entry 2), F. oxysporum AM13 for iodolactone 5a (Table 4, entry 3) and F. solani AM203 for chlorolactone 3a (Table 2, entry 3) and bromolactone 4a (Table 3, entry 5) were able to form (+)-enantiomer.
2.3. Biological Tests
The synthetic halolactones 3a, b–5a, b and 6a and the microbiologically obtained hydroxylactone 7a were subjected to biological tests. These tests were designed to identify the lactones capable of inhibiting or reducing growth of the selected microorganisms. Those microorganisms included an indicator bacterium E. coli, potentially pathogenic bacteria S. aureus and B. subtilis, a pathogenic bacterium C. albicans and Fusarium linii and Aspergillus niger that are capable of producing mycotoxins. The effects of the halolactones 3a–6a on the growth of bacteria, yeast and fungi are presented in Table 5 and Table 6.
Table 5.
The effects of the halolactones 3a–6a on the growth of bacteria, yeast and fungi.
| Entry | Strain | OD Control | max.OD of 3a | max.OD of 4a | max.OD of 5a | max.OD of 6a | max.OD of 7a |
|---|---|---|---|---|---|---|---|
| 1 | E. coli | 1.56 | 1.53 a | 1.57 a | 1.46 a | 1.50 a | 1.50 |
| 2 | S. aureus | 1.55 | 1.20 | 1.56 | 1.58 | 1.22 | 1.45 |
| 3 | B. subtilis | 1.37 | 1.39 b | 1.38 b | 1.40 b | 1.40 | 1.56 |
| 4 | C. albicans | 1.96 | 1.56 c | 0.41 | 0.95 | 1.80 c | 1.90 |
| 5 | S. cerevisiae | 2.30 | 0.80 | 0.70 | 1.37 | 1.67 | 0.37 |
| 6 | Y. lipolytica | 1.40 | 0.38 | 0.49 | 0.89 | 1.05 c | 1.41 |
| 7 | F. linii | 1.96 | 0.32 | 0.36 | 0.45 | 1.51 | 2.45 |
| 8 | A. niger | 3.0 | 2.00 d | 0.81 | 1.22 | 1.16 | 2.71 |
Notes: a rapid elimination of bacterial cells (reduced OD after 15–20 h of the culture); b longer lag phase (up to 3 h) and biphasic growth; c great elongation of the lag-phase, up to 25–28 h; d longer lag-phase, up to 10 h.
Table 6.
The effects of the halolactones 3b–5b on the growth of bacteria, yeast and fungi.
| Entry | Strain | OD Control | Max.OD of 3b | Max.OD of 4b | Max.OD of 5b |
|---|---|---|---|---|---|
| 1 | E. coli | 1.56 | 1.56 | 0.72 | 1.46 |
| 2 | S. aureus | 1.55 | 1.00 | 0.84 | 1.30 |
| 3 | B. subtilis | 1.4 | 1.00 a | 1.20 a | 1.20 |
| 4 | C. albicans | 1.96 | 1.82 | 1.85 | 0.58 |
| 5 | S. cerevisiae | 2.30 | 0.70 | 0.70 | 1.11 |
| 6 | Y. lipolytica | 1.39 | 0.39 | 0.36 | 1.00 b |
| 7 | F. linii | 2.05 | 0.35 | 0.37 | 0.31 |
| 8 | A. niger | 3.0 | 0.48 | 0.36 | 2.41 c |
Notes: a biphasic growth; b great elongation of the lag-phase; c longer lag phase up to 10 h.
The results, presented in Table 5, indicate that some of the tested lactones showed complete growth inhibition mainly against Y. lipolytica (Entry 6) and F. linii (Entry 7) strains. Rapid elimination of bacterial cells was observed for the E. coli strain (Entry 1). Some of the lactones were also capable of extending the lag-phase of microorganisms—B. subtilis (Entry 3) and C. albicans (Entry 4). In the case of compounds 3a–5a biphasic growth for B. subtilis (Entry 3) was observed.
Data presented in Table 6 indicates that some of the tested lactones showed complete growth inhibition, mainly against Y. lipolytica (Entry 6) and A. niger (Entry 8) strains. Iodolactone 5a was able to elongate the lag-phase of Y. lipolytica (Entry 6), F. linii (Entry 7) and A. niger (Entry 8). Similarly as in the case of lactones 3a–5a (Table 5) biphasic growth for B. subtilis (Entry 3) was also observed.
2.4. Discussion
The performed syntheses yielded eight racemic halo-γ-halolactones 3a, b–6a, b, consisting of a methylcyclohexane ring connected to a lactone ring. Their structures were determined based on their spectral data (1H-NMR, 13C-NMR, COSY, HMQC and IR), confirmed by GC-MS and elemental analysis.
2.4.1. Characterization of Compounds 3a–6a with 5-Methylcyclohexane Rings
The spectroscopic data indicated that acid 2a was a diastereoisomeric cis-trans (82%:18%) mixture. The coupling constant between the H-6 proton and the CH3-9 methyl group protons was equal to 6.6 Hz, which suggested an equatorial orientation of this methyl group. Wide multiplets coming from the H-6 proton and H-1 proton indicated their axial orientation. When this acid was subjected to chloro- or bromolactonization the creation of only one lactone (3a or 4a) was observed, while the iodolactone was created as a mixture of compounds 5a and 5b. Such result can be explained by different mechanisms of chloro-, bromo- and iodolactonization. According to Snider [40] and Jayaraman [41] during chloro- and bromolactonization the double bond is attacked by bromine leading to the formation of a bromonium ion. In the process of iodolactonization the complex of iodine and double bond is attacked by the carboxylate anion.
The absorption bands at 1778, 1784, 1784 and 1784 cm−1 in the IR spectra for these lactones indicated that the lactone ring present in their molecules was a γ-lactone. As claimed in the earlier works [31,33,34], in similar compounds the cyclohexane ring exists in the chair conformation. The analysis of the 1H-NMR spectra of lactones 3a, 4a and 5a indicated that in these lactones the cyclohexane ring also adopted the chair conformation. Signals from the H-1 proton (4.62, 4.63, 4.76 ppm) and H-2 proton (4.61, 4.61, 4.69 ppm) looking like singlets suggested that these protons were located in a trans-diequatorial positions. The wide multiplet of the H-6 proton suggested its axial position. This also implied that the halogen atom and C-O bond were in trans-diaxial positions. In the case of iodolactone 6a, the cyclohexane ring was also in a chair conformation, but the coupling constants of the H-1 proton (dd, J = 9.9 and 7.3 Hz) and H-2 proton (ddd, J = 13.7, 9.9 and 4.4 Hz) suggested their trans-diaxial positions. It means that in this case, the halogen atom and C-O bond were in trans diequatorial positions. Doublet of doublet of doublet (J = 13.7, 7.4 and 7.4 Hz), coming from H-6 proton, also indicated its axial position. The wide multiplet corresponding to the H-5 proton suggested its axial orientation, which indicated that the methyl group CH3-9 was in an equatorial position in the moleculesof halolactones 3a–6a (Figure 2).
Figure 2.

Structures of iodolactones 5a and 6a.
Only one product was obtained during all the preparative scale biotransformations performed on lactones 3a, 4a and 5a. Their structures were established based on the spectral data. The IR spectra showed that the γ-lactone ring (absorption band at 1769 cm−1) was retained in the product during the biotransformation. The presence of a hydroxy group in the molecule was suggested by the strong and broad band found at 3305 cm−1. Analysis of NMR spectra (1H, 13C, COSY and HMQC) of halolactones 3a–5a and the microbiologically obtained hydroxylactone 7a was helpful in determining the differences between these molecules. Major difference was observed for the H-1 and H-2 protons. The signal of the H-1 proton changed from a singlet (in the substrates) into a triplet (J = 3.8 Hz), while the signal of the H-2 proton became doublet of doublets of doublets with coupling constants J = 11.6, 4.7 and 3.8 Hz. This means that the H-1 proton retained its equatorial position (as in the substrates), but the H-2 proton changed its position from equatorial in the substrates into axial in the products (Figure 3).
Figure 3.
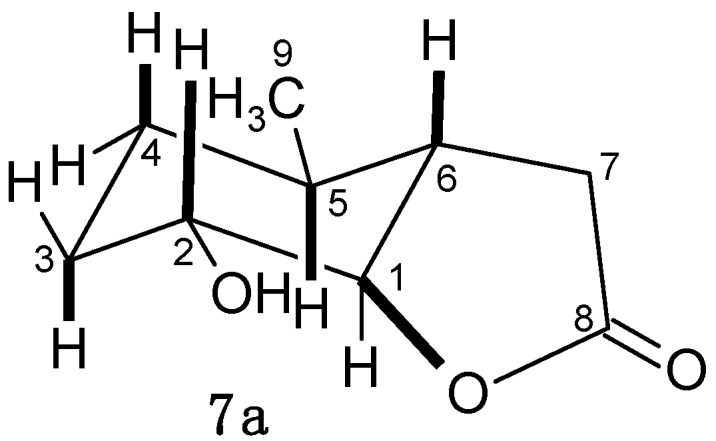
Structure of hydroxylactones 7a.
The data presented above suggests that the halogen atom present in the substrate molecule was replaced by a hydroxy group during a hydrolytic dehalogenation similar to a SN2 mechanism. Such a mechanism was observed earlier for similar halolactones subjected to hydrolytic dehalogenation by the same microorganisms [31,33,34], and also for enzymes from Absidia cylindrospora culture that catalyze hydrolytic dehalogenations in iodolactones [30]. We assume that the hydroxylactone 7a was formed as a result of the hydrolytic dehalogenation which mechanism is similar to the one described for haloalkane dehalogenase [42]. According to this mechanism one of the oxygens of the aspartate carboxylate group of the enzyme attacked the carbon C-2 to which the halogen was bound and replaced it by a SN2 substitution mechanism. During the next step the covalent alkyl-enzyme intermediate, to which the halide was still bound, was hydrolyzed by a molecule of water. This hydrolysis afforded the final product with an equatorial hydroxy group at C-2 (Scheme 3).
Scheme 3.

Equatorial location of the hydroxy group in lactone 7a as the stereochemical consequence of microbial hydrolytic dehalogenation (similar to a SN2 mechanism) of chloro-3a, bromo-4a and iodolactone 5a proceeding with inversion at C-2.
Knowing from the earlier experiments [33,34] that the microbiologically obtained hydroxylactones would be characterized by significant enantiomeric purity, the enantiomeric excess and optical rotation for hydroxylactones obtained during all preparative biotransformation were checked. As shown in Table 3, the enantiomeric excess was usually low or moderate. The best results (enantiomeric excess over 45%) were obtained for hydroxylactone 7a obtained from chlorolactone 3a, when F. culmorum AM10 was used as a biocatalyst and iodolactone 5a was transformed by F. culmorum AM10, S. racemosum AM105 and B. cinerea AM235. Most of the tested microorganisms preferably formed the (‒)-isomer of the hydroxylactone. Only F. avenaceum AM11 for lactone 4a, F. oxysporum AM13 for lactone 5a and F. solani AM203 for all three lactones used as substrates formed the (+)-isomer of hydroxylactone 7a.
2.4.2. Characterization of Compounds with 3-Methylcyclohexane Ring (3b–6b)
Acid 2b was a diastereoisomeric 79:21 cis-trans mixture similar to acid 2a. In this case we also observed formation of only one pure compound during chloro- or bromolactonization-3b and 4b, and the mixture of two iodolactones 5b and 6b. This situation also can be explained by the different mechanisms of chloro-, bromo- and iodolactonization described above for compounds 3a–5a. Analysis of the 1H-NMR spectra of lactones 3b–5b revealed similarity between these lactones and the previous ones 3a–5a. For these lactones, the cyclohexane ring also adopts the chair conformation. The signals of the H-1 proton (4.58, 4.71, 4.84 ppm), which appears as a multiplet with a small coupling constant, and the H-2 proton (4.39, 4.53, 4.73 ppm) being doublet of doublets with small coupling constants (3.3 and 3.2 Hz, 3.6 and 3.2 Hz, 3.6 and 3.0 Hz), indicated that these protons were located in the trans-diequatorial position. This also suggested that the halogen atom and C-O bond were in trans-diaxial positions. The structure of iodolactone 6b was similar to the structure of its 6a analog. In this case, the cyclohexane ring was in the chair conformation with H-1 (dd, J = 9.0 and 7.0 Hz) and H-2 (dd, J = 10.8 and 9.0 Hz) protons, located in the trans-diaxial position. The wide multiplet from the H-6 proton suggested its axial position (Figure 4).
Figure 4.

Structure of iodolactones 5b and 6b.
As presented in Table 1, none of the tested microorganisms were capable of introducing a hydroxy group in the place of the halogen atom. This was probably due to the presence of a methyl group located at the C-3 carbon. It is well known that the mechanism of the hydrolytic dehalogenation is similar to a SN2 one, therefore, the presence of a methyl group at C-3 probably prevents activation of this mechanism.
2.4.3. The Assessment of the Effects of Lactones on the Growth of Tested Microorganisms
Analysis of the results obtained during the biological tests leads to the conclusion that filamentous fungi and yeast were the most susceptible species, and bacteria the most resistant, to the tested lactones. Chlorolactones 3a, 3b, bromolactone 4a and iodolactones 5a, 5b were able to completely inhibit the growth of some yeast and fungi, and the other halolactones also exhibited a limiting effect. Surprisingly hydroxylactone 7a was the least active against the tested strains. It showed activity only against the S. aureus and S. cerevisiae strains. In the presence of lactone 7a the population of S. aureus was similar to that of the control cells, but after some time the bacterial cells began to die off. In the second case, the complete inhibition of S. cerevisiae growth was observed. During the tests, biphasic growth of B. subtilis was noticed in the presence of most of the lactones. This could mean that these bacteria were capable of using the lactones as a growth-inducing factor. It is supported by a known ability of B. subtilis to degrade N-acyl-l-homoserine lactone [43].
The greatest inhibitory effect on bacterial growth was shown by halolactones with a methyl group located at the C-3 carbon (compounds 3b, 4b, 5b). The same compounds showed a weak effect on yeast growth. In this case better results were obtained for halolactones with a methyl group located at the C-5 carbon (3a, 4a, 5a). The tests on fungal strains demonstrated that the position of the methyl group did not matter. In general, the compounds which were able to inhibit the growth of microorganisms to the greatest extent were the chlorolactones. It was also interesting that lactones with a halogen atom in an axial position showed stronger inhibitory effects against microorganisms than these with a halogen located in an equatorial position. Examples of the effects of selected lactones on the chosen microorganisms are presented in Figure 5 and Figure 6.
Figure 5.

Examples of the effects of selected lactones on the chosen bacteria strains.
Figure 6.

Examples of the effects of selected lactones on the chosen yeast strains.
2.4.4. An olfactory Analysis of Lactones
Knowing from previous experiments [36] that some derivatives obtained during the synthesis of hydroxylactones with the methylcyclohexane ring are characterized by specific smells, we decided to check the odor of the compounds described in this paper. It was found that an overwhelming majority of them had slight, uninteresting or unpleasant (decayed leftovers in the case of bromolactone 4b) odors. Only three compounds gave off an interesting smell:
acid 2a—medium intensive, sweet-sour, dry pine scent;
chlorolactone 3b—slight dried fruit scent;
iodolactone 5b—slight scent of dust, reminiscent of an old pharmacy.
3. Experimental Section
3.1. General
Progress of all chemical reactions, biotransformations and purity of isolated products were checked by TLC on silica gel-coated aluminium plates (DC-Alufolien Kieselgel 60 F254, Merck, Dramstadt, Germany) and also by GC analysis performed on a CP-3380 instrument (Varian, Agilent Technologies, Santa Clara, CA, USA) using an DB-17 column (cross-linked methyl silicone gum, 30 m × 0.32 mm × 0.25 µm). Temperatures during GC analysis were as follows: injector 150 °C, detector (FID) 280 °C, column temperature: 100 °C (hold 1 min), 100–200 °C (rate 10 °C/min), 200–280 (rate 50 °C/min), 300 °C (hold 1 min). The structures of obtained compounds were also confirmed by gas chromatography-mass spectrometry (GC-MS) analysis using Varian SATURN 2000 instrument (EI ionization) with HP-1 column (cross-linked methyl silicone gum, 25 m × 0.32 mm × 0.52 µm) under the following conditions: injector 200 °C, detector 300 °C, column temperature: 120 °C (hold 2 min), 120–300 °C (rate 20 °C/min), 300 °C (hold 3 min). The enantiomeric excess of the products obtained during biotransformation were determined by GC analysis using chiral column CP-cyclodextrin-B-325 (30 m × 0.25 mm × 0.25 µm) under the following conditions: injector 200 °C, detector (FID) 220 °C, column temperature: 140 °C (hold 45 min), 140–200 °C (rate 20 °C/min), 200 °C (hold 1 min). All products were purified by using of preparative column chromatography on silica gel (Kieselgel 60, 230–400 mesh). Melting points were determined on a Boetius apparatus. Refractive index was measured on Carl Zeiss (Jena, Germany) Abbe and Pulfrich refractometers. Elemental analysis was done using a Vario EL III CHNS automatic analyzer from Elemental Analyzersysteme (Wigan, UK). 1H-NMR, 13C-NMR, 1H-13C COSY (HMQC) and 1H-1H COSY spectra were recorded in a CDCl3 solution on an Avance DRX 300 spectrometer (Bruker, Billerica, MA, USA). IR spectra were determined using FTIR on a Thermo-Mattson IR 300 spectrometer (Waltham, MA, USA). Optical rotations were measured on P-2000 polarimeter (Jasco, Easton, PA, USA).
3.2. Synthesis of Substrates
3.2.1. (6–Methylcyclohex-2-en-1-yl)acetic Acid (2a)
Basic hydrolysis of ester 1a (9.2 g, 0.046 mol), according to the known procedure [31] yielded 6.0 g (77%) of acid 2a as a 82:18 diastereoisomeric cis-trans mixture with the following physical and spectral properties: nD = 1.4694, 1H-NMR (CDCl3): 0.89 (d, J = 6.9 Hz, 3H, CH3-9 trans), 1.00 (d, J = 6.6 Hz, 3H, CH3-9 cis), 1.31–1.50 (m, 4H, CH2-5), 1.56–1.62 (m, 1H, H-6 trans), 1.64–1.72 (m, 1H, H-6 cis), 1.98–2.05 (m, 4H, CH2-4), 2.18–2.23 (m, 4H, one of CH2-7 and H-1), 2.53–2.62 (dd, J = 18.9 and 8.9 Hz, 2H, one of CH2-7), 5.53–5.59 (m, 2H, H-3), 5.67–5.74 (m, 2H, H-2), 13C-NMR (CDCl3): 19.85 (C-9), 24.19 (C-4), 29.27 (C-5), 32.96 (C-6), 38.95 (C-1), 39.20 (C-7), 127.71 (C-3), 129.15 (C-2), 178.43 (C-8), IR (KBr, cm−1): 2921 (s), 1709 (s), 1411 (s), 1284 (s). EI-MS m/z (%) = 154 (8) [M]+, 108 (11), 94 (100), 79 (81), 67 (46), 43 (34), 39 (45). EA for C9H14O2 (154.21) calculated 70.10% C, 9.15% H, found 70.19% C, 9.10% H.
3.2.2. 2-Chloro-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one (3a)
Chlorolactonization of a diastereoisomeric 82:18 cis-trans mixture of acid 2a (2.5 g, 0.016 mol), according to the procedure of Grabarczyk and Białońska [31] gave 1.6 g (76%) of chlorolactone 3a with the following physical and spectral properties: nD = 1.4993, 1H-NMR (CDCl3): 1.01 (d, J = 6.5 Hz, 3H, CH3-9), 1.30 (m, 1H, H-5), 1.56 (m, 2H, CH2-4), 1.85 (m, 1H, one of CH2-3), 1.99 (m, 1H, CH2-3), 2.47 (m, 1H, H-6), 2.43 (d, J = 17.0 Hz, 1H, one of CH2-7), 2.67 (dd, J = 17.0 and 6.7 Hz, 1H, one of CH2-7), 4.53 (m, 2H, H-1 and H-2), 13C-NMR (CDCl3): 19.84 (C-9), 25.43 (C-4), 27.92 (C-3), 31.72 (C-5), 36.30 (C-7), 39.19 (C-6), 54.91 (C-2), 80.96 (C-1), 175.91 (C-8), IR (KBr, cm−1): 2952 (s), 1778 (s), 1454 (s), 1147 (s), 972 (s). EI-MS m/z (%): 153 (66) [M−HCl], 126 (17), 112 (68), 84 (68), 69 (100), 56 (18), 43 (50), 39 (33). EA for C9H13ClO2 (188.66), 57.30% C, 6.95% H, found 57.20% C, 6.90% H.
3.2.3. 2-Bromo-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one (4a)
After bromolactonization acid 2a (1.7 g, 0.011 mol of an 82:18 diastereoisomeric cis-trans mixture), according to the known method [31], 1.6 g (61%) of bromolactone 3 was obtained. The physical and spectral data of this product are as follows: m.p. = 47–48 °C, 1H-NMR (CDCl3): 1.03 (d, J = 6.5 Hz, 3H, CH3-9), 1.32 (m, 1H, H-5), 1.61 (m, 2H, CH2-4), 2.06 (m, 2H, CH2-3), 2.32 (m, 1H, H-6), 2.45 (d, J = 17.0 Hz,1H, one of CH2-7), 2.66 (dd, J = 17.0 and 6.6 Hz,1H, one of CH2-7), 4.64 (s, 1H, H-2), 4.65 (s, 1H, H-1), 13C-NMR (CDCl3): 19.85 (C-9), 26.48 (C-4), 28.33 (C-3), 31.82 (C-5), 36.95 (C-7), 39.19 (C-6), 47.64 (C-2), 81.30 (C-1), 176.00 (C-8), IR (KBr, cm−1): 2957 (s), 1784 (s), 1427 (s), 1153 (s), 966 (s). EI-MS m/z (%): 153 (11) [M−HBr], 111 (39), 95 (22), 85 (100), 67 (33), 55 (42), 41 (26), 39 (46). EA for C9H13BrO2 (233.11) calculated 46.37% C, 5.62% H, found 46.29% C, 5.66% H.
3.2.4. 2-Iodo-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one (5a) and 2-Iodo-5-methyl-9-oxabicyclo[4.3.0]-nonan-8-one (6a)
Iodolactonization of 1.8 g (0.012 mol) of acid 2a (82:18 diastereoisomeric cis-trans mixture), according to the known procedure [39] gave iodolactone 5a (1.9 g, 58%) and iodolactone 6a (0.3 g, 9.4%) with the following physical and spectral properties: 5a: m.p.=67–68 °C, 1H-NMR (CDCl3): 1.04 (d, J = 6.5 Hz, 3H, CH3-9), 1.31 (m, 1H, H-5), 1.61 (m, 2H, CH2-4), 1.85 (m, 1H, one of CH2-3), 2.00 (dd, J = 15.3 and 3.1 Hz, 1H, one of CH2-3), 2.40 (m, 1H, H-6), 2.45 (d, J = 16.9 Hz, 1H, one of CH2-7), 2.65 (dd, J = 16.9 and 6.6 Hz, 1H, one of CH2-7), 4.74 (s, 1H, H-1), 4.82 (m, 1H, H-2), 13C-NMR (CDCl3): 19.88 (C-9), 27.66 (C-2), 28.56 (C-4), 29.43 (C-3), 32.03 (C-5), 37.08 (C-7), 39.26 (C-6), 83.09 (C-1), 176.27 (C-8), IR (KBr, cm−1): 2924 (s), 1786 (s), 1457 (s), 1148 (s), 960 (s). EI-MS m/z (%): 153 (100) [M-HI], 112 (26), 84 (25), 69 (37), 43 (21), 39 (13). EA for C9H13IO2 (280.11) calculated 38.59% C, 4.68% H, found 38.48% C, 4.57% H. 6a: m.p. = 48–49 °C, 1H-NMR (CDCl3): 0.98 (d, J = 7.2 Hz, 3H, CH3-9), 1.22 (m, 1H, one of CH2-4), 1.47 (m, 1H, one of CH2-4), 1.95 (m, 2H, one of CH2-3 and H-5), 2.25 (dd, J = 17.2 and 8.2 Hz, 1H, one of CH2-7), 2.33 (dd, J = 17.2 and 13.7 Hz, 1H, one of CH2-7), 2.38 (m, 1H, one of CH2-3), 2.70 (ddd, J = 13.7, 7.4 and 7.4 Hz, 1H, H-6), 3.80 (ddd, J = 13.7, 9.9 and 4.4 Hz, 1H, H-2), 4.70 (dd, J = 9.9 and 7.3 Hz, 1H, H-1), 13C-NMR (CDCl3): 19.49 (C-9), 27.17 (C-2), 27.40 (C-7), 30.48 (C-4), 31.01 (C-5), 36.59 (C-3), 42.80 (C-6), 86.24 (C-1), 175.13 (C-8), IR (KBr, cm−1): 2936 (s), 1784 (s), 1454 (s), 1177 (s), 1016 (s). EI-MS m/z (%): 153 (100) [M−HI], 135 (26), 127 (13), 107 (20), 93 (28), 81 (36), 67 (21), 55 (34), 39 (31). EA for C9H13IO2 (280.11) calculated 38.59% C, 4.68% H, found 38.51% C, 4.60% H.
3.2.5. (4–Methylcyclohex-2-en-1-yl)acetic Acid (2b)
After basic hydrolysis of ester 1b (1.5 g, 0.0075 mol), according to the known procedure [31], acid 2b (1.1 g, 95%) was obtained as a diastereoisomeric 79:21 cis-trans mixture. The physical and spectral data of this product are as follows: nD = 1.4654, 1H-NMR (CDCl3): 0.96 (d, J = 6.9 Hz, 3H, CH3-9 cis), 0.97 (d, J = 6.9 Hz, 3H, CH3-9 trans), 1.16-1.23 (m, 4H, CH2-6), 1.69–1.72 (m, 2H, CH2-5 cis), 1.81–1.93 (m, 2H, CH2-5 trans), 2.12–2.17 (m, 2H, H-4), 2.29–2.37 (m, 4H, CH2-7), 2.50–2.60 (m, 2H, H-1), 5.48–5.62 (m, 4H, H-2 and H-3), 13C-NMR (CDCl3): 21.31 and 21.64 (C-9), 27.88 and 28.89 (C-6), 29.69 and 30.44 (C-4), 30.86 and 31.50 (C-5), 32.51 and 33.04 (C-1), 40.19 and 40.67 (C-7), 128.82 and 128.97 (C-3), 134.65 and 134.72 (C-2), 178.64 (C-8), IR (KBr, cm−1): 2928 (sb), 1709 (s), 1411 (s), 1292 (s). EI-MS m/z (%): 154 (8) [M]+, 136 (5), 108 (9), 94 (100), 79 (61), 67 (39), 53 (8), 39 (31). EA for C9H14O2 (154.21) calculated 70.10% C, 9.15% H. Found: 70.04% C, 9.10% H.
3.2.6. 2-Chloro-3-methyl-9-oxabicyclo[4.3.0]nonan-8-one (3b)
Chlorolactonization of a 79:21 diastereoisomeric cis-trans mixture of acid 2b (0.5 g, 0.0032 mol) according to the known procedure [31] yielded chlorolactone 3b (0.32 g, 53%) with the following physical and spectral properties: nD = 1.4914, 1H-NMR (CDCl3): 1.09 (d, J = 6.7 Hz, 3H, CH3-9), 1.24 (dd, J = 11.7 and 3.6 Hz, 1H, one of CH2-4), 1.39 (dd, J = 13.7 and 3.4 Hz, 1H, one of CH2-5), 1.56 (m, 1H, one of CH2-5), 1.85 (m, 1H, one of CH2-4), 2.05 (m, 1H, H-5), 2.26 (d, J = 15.9 Hz,1H, one of CH2-7), 2.66–2.71 (m, 2H, H-6 and one of CH2-7), 4.43 (m, 1H, H-2), 4.61 (dd, J = 3.3 and 3.2 Hz, 1H, H-1), 13C-NMR (CDCl3): 18.50 (C-9), 24.72 (C-5), 27.24 (C-4), 30.80 (C-3), 30.99 (C-6), 37.92 (C-7), 61.81 (C-2), 81.47 (C-1), 176.00 (C-8), IR (KBr, cm−1): 2936 (s), 1783 (s), 1455 (s), 1144 (s), 981 (s). EI-MS m/z (%): 189 (27) [M]+, 167 (12), 159 (22), 153 (68), 109 (45), 93 (89), 81 (43), 67 (62), 55 (53), 39 (100). EA for C9H13ClO2 (188.66), 57.30% C, 6.95% H, found 57.24% C, 6.88% H.
3.2.7. 2-Bromo-3-methyl-9-oxabicyclo[4.3.0]nonan-8-one (4b)
Using the previously described method [31] 0.28 g (62%) of bromolactone 4b was obtained from 0.3 g (0.0019 mol) of 79:21 diastereoisomeric cis-trans mixture of acid 2b. The physical and spectral data of the product are as follows: nD = 1.5181, 1H-NMR (CDCl3): 1.03 (d, J = 6.6 Hz, 3H, CH3-9), 1.21 (m, 1H, one of CH2-4), 1.37 (m, 1H, one of CH2-5), 1.52 (m, 1H, one of CH2-5), 1.80 (m, 2H, H-3 and one of CH2-4), 2.23 (d, J = 16.1 Hz,1H, one of CH2-7), 2.65 (dd, J = 16.1 and 6.4 Hz,1H, one of CH2-7), 2.75 (m, 1H, H-6), 4.53 (m, 1H, H-2), 4.71 (dd, J = 3.6 and 3.2 Hz, 1H, H-1), 13C-NMR (CDCl3): 20.26 (C-9), 25.83 (C-5), 27.37 (C-4), 30.66 (C-3), 31.00 (C-6), 38.23 (C-7), 57.08 (C-2), 81.88 (C-1), 176.13 (C-8), IR (KBr, cm−1): 2930 (s), 1788 (s), 1423 (s), 1138 (s), 950 (s). EI-MS m/z (%): 153 (100) [M−HBr], 135 (23), 109 (43), 93 (37), 81 (16), 67 (39), 55 (17), 39 (35). EA for C9H13BrO2 (233.11) calculated 46.37% C, 5.62% H, found 46.42% C, 5.70% H.
3.2.8. 2-Iodo-3-methyl-9-oxabicyclo[4.3.0]nonan-8-one (5b) and 2-Iodo-3-methyl-9-oxabicyclo-[4.3.0]nonan-8-one (6b)
Iodolactonization of acid 2b (0.3 g, 0.0019 mol of 79:21 diastereoisomeric cis-trans mixture), according to a known procedure [39] gave iodolactones 5b (0.29 g, 53%) and 6b (0.05 g, 9%) with the following physical and spectral properties: 5a: nD = 1.5525, 1H-NMR (CDCl3): 0.96 (d, J = 5.0 Hz, 3H, CH3-9), 1.22 (m, 2H, CH2-4), 1.37 (m, 1H, H-3), 1.83 (ddd, J = 10.1, 6.4 and 3.3 Hz, 1H, H-5), 2.27 (d, J = 16.7 Hz, 1H, one of CH2-7), 2.66 (dd, J = 16.7 and 6.4 Hz, 1H, one of CH2-7), 2.82 (m, 1H, H-6), 4.73 (m, 1H, H-2), 4.84 (dd, J = 3.6 and 3.0 Hz, 1H, H-1), 13C-NMR (CDCl3): 23.54 (C-9), 27.61 (C-5), 28.10 (C-3), 30.41 (C-4), 31.10 (C-6), 38.78 (C-7), 41.82 (C-2), 83.83 (C-1), 176.44 (C-8), IR (KBr, cm−1): 2925 (s), 1784 (s), 1454 (s), 1146 (s), 974 (s). EI-MS m/z (%): 153 (100) [M−HI], 135 (27), 107 (28), 93 (50), 79 (10), 67 (9), 55 (11), 39 (25). EA for C9H13IO2 (280.11) calculated 38.59% C, 4.68% H, found 38.71% C, 4.61% H. 5b: oil, 1H-NMR (CDCl3): 1.12 (d, J = 6.5 Hz, 3H, CH3-9), 1.72–1.78 (m, 5H, H-3, CH2-4, CH2-5), 2.31 (dd, J = 17.2 and 8.4 Hz, 1H, one of CH2-7), 2.45 (dd, J = 17.2 and 11.8 Hz, 1H, one of CH2-7), 2.67 (m, 1H, H-6), 3.70 (dd, J = 10.8 and 9.0 Hz, 1H, H-2), 4.78 (dd, J = 9.0 and 7.0 Hz, 1H, H-1), 13C-NMR (CDCl3): 23.77 (C-9), 27.78 (C-5), 27.90 (C-4), 31.59 (C-7), 35.58 (C-6), 38.02 (C-3), 39.44 (C-2), 86.25 (C-1), 175.32 (C-8), IR (KBr, cm−1): 2954 (s), 1772 (s), 1464 (s), 1178 (s), 952 (s). EA for C9H13IO2 (280.11) calculated 38.59% C, 4.68% H, found 38.53% C, 4.63% H.
3.3. Biotransformations
3.3.1. Microorganisms
The fungal strains used in all biotransformations came from the collection of the Institute of Biology and Botany, Medical University, Wrocław, Poland (Fusarium culmorum AM10, Fusarium avenaceum AM11, Fusarium oxysporum AM13, Fusarium tricinctum AM16, Fusarium semitectum AM20, Fusarium equiseti AM22, Fusarium scirpi AM1199, Fusarium solani AM203, Syncephalastrum racemosum AM105 and Botrytis cinerea AM235). These strains were cultivated on Sabouraud’s agar containing 0.5% of aminobac, 0.5% of peptone, 4% of glucose and 1.5% of agar dissolved in distilled water at 28 °C and stored in refrigerator at 4 °C. Screening and preparative biotransformations were performed as described before [34]. The physical and spectral data of 2-hydroxy-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one (7a) are as follows: m.p. = 89–90 °C, 1H-NMR (CDCl3): 0.93 (d, J = 6.4 Hz, 3H, CH3-9), 1.06 (m, 1H, one of CH2-4), 1.26 (m, 1H, H-5), 1.53 (m, 1H, one of CH2-3), 1.73 (m, 1H, one of CH2-4), 1.88 (m, 1H, one of CH2-3), 2.00 (m, 1H, H-6), 2.10 (m, 1H, OH), 2.40 (d, J = 16.9 Hz, 1H, one of CH2-7), 2.68 (dd, J = 16.9 and 6.6 Hz, 1H, one of CH2-7), 3.72 (ddd, J = 11.6, 4.7 and 3.8 Hz, 1H, H-2), 4.57 (t, J = 3.8 Hz, 1H, H-1), 13C-NMR (CDCl3): 19.60 (C-9), 28.86 (C-3), 30.78 (C-4), 31.88 (C-5), 37.02 (C-7), 42.98 (C-6), 70.08 (C-1), 81.83 (C-2), 176.33 (C-8), IR (KBr, cm−1): 3305 (sb), 2927 (s), 1769 (s), 1160 (s). EI-MS m/z (%):170 (6) [M]+, 170, 153 (11), 123 (8), 111 (39), 85 (100), 67 (33), 39 (42). EA for C9H14O3 (170.21) calculated 63.51% C, 8.29% H, found 63.43% C, 8.18% H
3.3.2. Bioassays
The tests were conducted on certain bacteria strains: Escherichia coli, Staphylococcus aureus, Bacillus subtilis, yeast strains: Candida albicans, Saccharomyces cerevisiae, Yarrowia lipolytica and also filamentous fungi strains: Aspergillus niger and Fusarium linii. The bacterial cultures were grown for 48 h in a liquid broth consisting of 15 g of dry bullion (Biocorp, Warszawa, Poland) and 10 g of glucose dissolved in 1 L of distilled water. Yeast and fungi cultures were growing in YPG medium which was composed of 10 g of yeast extract, 10 g of bacteriological peptone and 10 g of glucose dissolved in 1 L of distilled water for 48 and 96 h, respectively. The test were performed in the automated Bioscreen C system (Automated Growth Curve Analysis System, Lab systems, Helsinki, Finland).
Working volume in the wells of the Bioscreen plate was 300 µL, comprised of 280 µL of culture medium, 10 µL of cells or spore solution (final concentration). Tested lactones were dissolved in DMSO and used at the final concentration of 0.1% (w/v). Temperature was controlled at 30 °C (bacteria, yeasts) and 25 °C (filamentous fungi). The optical density of the cell suspensions was measured automatically at 560 nm in regular intervals of 30 min, for 3–4 days. The cell cultures were continuously shaken. Each culture was performed in three replicates. Data was analyzed by using spreadsheet software (Excel 97) and were calculated as the averages from the tree replicate analysis for each type of culture medium. The averages were used to generate the growth curves for each strain studied, constructed as a function of the incubation time and the absorbancy of the culture medium. The resulting microbial growth curves were compared to control cultures in medium supplemented with DMSO.
3.3.3. Odour Evaluation
The odour evaluation was performed for ethanolic solutions (10%) of samples with the use of a strip blotter.
4. Conclusions
Two-step chemical synthesis yielded eight halolactones with methylcyclohexane rings. Six of them were subjected to a screening biotransformation by ten fungal strains. Most of these strains were capable of transforming the halolactones with the methyl group at the C-4 position into the corresponding hydroxylactone. The selected microorganisms showed very high regioselectivity, because the hydroxy group was introduced at C-2 in the equatorial position due to the operative SN2 mechanism. The tested strains were also characterized by substrate specificity—iodolactone 5a was transformed at the greatest rate (eight strains) and chlorolactone 3a at the lowest rate (only three strains). The best results were obtained when F. equiseti AM22, F. avenaceum AM11, F. oxysporum AM13 and S. racemosum AM105 were used as biocatalysts. Degrees of conversion of the substrates 3a–5a were over 80%. Most of the microorganisms formed the (‒)-isomer of the hydroxylactone and the (+)-isomer was produced only by two microrganisms. The halolactones 3a–6a and 3b–5b and hydroxylactone 7a were tested for their ability to inhibit the growth of some bacteria, yeast and fungal strains. The biological tests indicated the ability of these compounds to inhibit or limit growth of most microorganisms. Odor evaluation of all the described compounds revealed that most of them were characterized by uninteresting smells, except for acid 2a, chlorolactone 3b and iodolactone 5b. The smell of acid 2a (sweet sour, dry pine) was particularly interesting. Chlorolactone 3b and iodolactone 5b were characterized by a slight dried fruit scent and a slight dusty scent, respectively.
Acknowledgments
Publication supported by Wroclaw Centre of Biotechnology, programme The Leading National Research Centre (KNOW) for years 2014–2018.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/02/3335/s1.
Author Contributions
Małgorzata Grabarczyk suggested the research work, performed all biotransformations, contributed for the discussion of results and wrote the paper. Katarzyna Wińska and Anna K. Żołnierczyk synthesized halolactones. Katarzyna Wińska performed the olfactory analysis. Wanda Mączka performed the GC-MS and polarimetry analyses. Barbara Żarowska performed the biological analysis on microorganisms. Mirosław Anioł contributed to the discussion of results. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 1a, b–6a, b are available from the authors.
References
- 1.Chen H., Xu C., Liu D.Q., An S.Q., Tan R.X. Buddlin, a new compound from Buddleja asiatica. Fitoterapia. 2005;76:588–589. doi: 10.1016/j.fitote.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Pujar P.P., Sawaikar D.D., Rojatkar S.R., Nagasampagi B.A. A new germacranolide from Artemisia pallens. Fitoterapia. 2000;71:590–592. doi: 10.1016/S0367-326X(00)00168-4. [DOI] [PubMed] [Google Scholar]
- 3.Ahmed A.A., Khattab A.M., Grace M.H., Sahl M.M. A new eudesmanolide from Crataegus flava fruits. Fitoterapia. 2001;72:756–759. doi: 10.1016/S0367-326X(01)00315-X. [DOI] [PubMed] [Google Scholar]
- 4.Ortet R., Prado S., Mouray E., Thomas O.P. Sesquiterpene lactones from the endemic Cape Verdean Artemisia gorgonom. Phytochemistry. 2008;69:2961–2965. doi: 10.1016/j.phytochem.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 5.Marco J.A., Sanz-Cervera J.F., Yuste A., Sancenon F., Carda M. Sesquiterpenes from Centaurea aspera. Phytochemistry. 2005;66:1644–1650. doi: 10.1016/j.phytochem.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Huang H.L., Xu Y.J., Liu H.L., Liu X.Q., Shang J.N., Han G.T., Yao M.J., Yuan C.S. Eremophilane-type sesquiterpene lactones from Ligularia hodgsonii Hook. Phytochemistry. 2011;72:514–517. doi: 10.1016/j.phytochem.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Gan L.S., Zheng Y.L., Mo J.X., Liu X., Li X.H., Zhou C.X. Sesquiterpene lactones from the root tubers of Lindera agregata. J. Nat. Prod. 2009;72:1497–1501. doi: 10.1021/np900354q. [DOI] [PubMed] [Google Scholar]
- 8.Buskuhl H., de Oliveira F.L., Blind L.Z., de Freitas R.A., Barison A., Campos F.R., Corilo Y.E., Eberlin M.N., Caramori G.F., Biavatti M.W. Sesquiterpene lactones from Vernonia scorpioides and their in vitro cytotoxicity. Phytochemistry. 2010;71:1539–1544. doi: 10.1016/j.phytochem.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Fortuna A.M., Juarez Z.N., Bach H., Nematallah A., Av-Gay Y., Sanchez-Arreola E., Catalan C.A.N., Turbay S., Hernandez L.R. Antimicrobial activities of sesquiterpene lactones and inositol derivatives from Hymenoxys robusta. Phytochemistry. 2011;72:2413–2418. doi: 10.1016/j.phytochem.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Pan L., Lantvit D.D., Riswan S., Kardono L.B.S., Chai H.B., de Blanco E.J.C., Farnsworth N.R., Soejarto D.D., Swanson S., Kinghorn A.D. Bioactivity-guided isolation of cytotoxic sesquiterpenes of Rolandra fruticosa. Phytochemistry. 2010;71:635–640. doi: 10.1016/j.phytochem.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J.Q., Zhang M., Zhang C.F., Qi H.Y., Bashall A., Bligh S.W.A., Wang Z.T. Cytotoxic sesquiterpenes from Ligularia platyglossa. Phytochemistry. 2008;69:2231–2236. doi: 10.1016/j.phytochem.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 12.Kim K.H., Noh H.J., Choi S.U., Park K.M., Seok S.J., Lee K.R. Lactarane sesquiterpenoids from Lactarius subvellereus and their cytotoxicity. Bioorg. Med. Chem. Lett. 2010;20:5385–5388. doi: 10.1016/j.bmcl.2010.07.119. [DOI] [PubMed] [Google Scholar]
- 13.Liu Q., Ahn J.H., Kim S.B., Lee C., Hwang B.Y., Lee M.K. Sesquiterpene lactones from the roots of Lindera strychnifolia. Phytochemistry. 2013;87:112–118. doi: 10.1016/j.phytochem.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Komala I., Ito T., Nagashima F., Yagi Y., Kawahata M., Yamaguchi K., Asakawa Y. Zierane sesquiterpene lactone, cembrane and fusicoccane diterpenoids, from the Tahitian liverwort Chandonanthus hirtellus. Phytochemistry. 2010;71:1387–1394. doi: 10.1016/j.phytochem.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 15.Pillay P., Vleggaar R., Maharaj V.J., Smith P.J., Lategan C.A. Isolation and identification of antiplasmodial sesquiterpene lactones from Oncosiphon piluliferum. J. Ethnopharmacol. 2007;112:71–76. doi: 10.1016/j.jep.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigues A.M.S., Theodoro P.N.E.T., Eparvier V., Basset C., Silva M.R.R., Beauchene J., Espindola L.S., Stien D. Search for antifungal compounds from the wood of durable tropical trees. J. Nat. Prod. 2010;73:1706–1707. doi: 10.1021/np1001412. [DOI] [PubMed] [Google Scholar]
- 17.Yesilada E., Gurbuz I., Bedir E., Tatli I., Khan I.A. Isolation of anti-ulcerogenic sesquiterpene lactones from Centaurea solstitialis L. ssp. solstitialis through bioassay-guided fractionation procedures in rats. J. Ethnopharmacol. 2004;95:213–219. doi: 10.1016/j.jep.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 18.Xiao W., Li X., Li N., Bolati M., Wang X., Jia X., Zhao Y. Sesquiterpene lactones from Saussurea involucrate. Fitoterapia. 2011;82:983–987. doi: 10.1016/j.fitote.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Evidente A., Masi M., Linaldeddu B.T., Franceschini A., Scanu B., Cimmino A., Andolfi A., Motta A., Maddau L. Afritoxinones A and B, dihydrofuropyran-2-ones produced by Diplodia africana the causal agent of branch dieback on Juniperus phoenicea. Phytochemistry. 2012;77:245–250. doi: 10.1016/j.phytochem.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 20.Sumioka H., Harinantenaina L., Matsunami K., Otsuka H., Kawahata M., Yamaguchi K. Linderolides A–F, eudesmane-type sesquiterpene lactones and linderoline, a germacrane-type sesquiterpene from the roots of Lindera strychnifolia and their inhibitory activity on NO production in RAW 264.7 cells in vitro. Phytochemistry. 2011;72:2165–2171. doi: 10.1016/j.phytochem.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 21.Wube A.A., Wenzig E.M., Gibbons S., Asres K., Bauer R., Bucar F. Constituents of the stem bark of Discopodium penninervium and their LTB4 and COX-1 and -2 inhibitory activities. Phytochemistry. 2008;69:982–987. doi: 10.1016/j.phytochem.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Koshimura M., Utsukihara T., Kawamoto M., Saito M., Horiuchi C.A., Kuniyoshi M. Biotransformation of bromosesquiterpenes by marine fungi. Phytochemistry. 2009;70:2023–2026. doi: 10.1016/j.phytochem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 23.Cheng C.H., Chung H.M., Hwang T.L., Lu M.C., Wen Z.H., Kuo Y.H., Wang W.H., Sung P.J. Echinoclerodane A: A new bioactive clerodane-type diterpenoid from a gorgonian coral Echinomuricea sp. Molecules. 2012;17:9443–9450. doi: 10.3390/molecules17089443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H.L., Geng C.A., Ma Y.B., Zhang X.M., Chen J.J. Three new secoiridoids, swermacrolactones A–C and anti-hepatitis B virus activity from Swertia macrosperma. Fitoterapia. 2013;89:183–187. doi: 10.1016/j.fitote.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Li S., Li J., Guan X.L., Li J., Deng S.P., Li L.Q., Tang M.T., Huang J.G., Chen Z.Z., Yang R.Y. Hypoglycemic effects and constituents of the barks of Cyclocarya paliurus, and their inhibiting activities to glucosidase and glycogen phosphorylase. Fitoterapia. 2011;82:1081–1085. doi: 10.1016/j.fitote.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Li Y., Yang X.W. Five new eudesmane-type sesquiterpenoid lactones biotransformed from atractylenolide I by rat hepatic microsomes. Fitoterapia. 2013;85:95–100. doi: 10.1016/j.fitote.2012.12.033. [DOI] [PubMed] [Google Scholar]
- 27.Wedge D.E., Galindo J.C.G., Macias F.A. Fungicidal activity of natural and synthetic sesquiterpene lactone analogs. Phytochemistry. 2000;53:747–757. doi: 10.1016/S0031-9422(00)00008-X. [DOI] [PubMed] [Google Scholar]
- 28.Aranda G., Moreno L., Cortes M., Prange T., Maurs M., Azerad D. A new example of 1a-hydroxylation of drimanic terpenes through combined microbial and chemical processes. Tetrahedron. 2001;57:6051–6056. doi: 10.1016/S0040-4020(01)00595-6. [DOI] [Google Scholar]
- 29.Maurs M., Azerad R., Cortes M., Aranda G., Delahaye M.B., Ricard L. Microbial hydroxylation of natural drimenic lactones. Phytochemistry. 1999;52:291–296. doi: 10.1016/S0031-9422(99)00199-5. [DOI] [PubMed] [Google Scholar]
- 30.Gładkowski W., Mazur M., Białońska A., Wawrzeńczyk C. Lactones 35. Metabolism of iodolactones with cyclohexane ring in Absidia cylindrospora culture. Enzyme Microb. Technol. 2011;48:326–333. doi: 10.1016/j.enzmictec.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 31.Grabarczyk M., Białońska A. Biotransformations of chloro-, bromo- and iodolactone with trimethylcyclohexane system using fungal strains. Biocatal. Biotransform. 2010;28:408–414. doi: 10.3109/10242422.2010.538688. [DOI] [Google Scholar]
- 32.Grotowska A.K., Wawrzeńczyk C. Lactones 13. Biotransformation of iodolactones. J. Mol. Catal. B. 2002;19–20:203–208. doi: 10.1016/S1381-1177(02)00168-6. [DOI] [Google Scholar]
- 33.Grabarczyk M. Fungal strains as catalysts for the biotransformation of halolactones by hydrolytic dehalogenation with the dimethylcyclohexane system. Molecules. 2012;17:9741–9753. doi: 10.3390/molecules17089741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grabarczyk M., Mączka W., Wińska K., Żarowska B., Anioł M. Antimicrobial activity of hydroxylactone obtained by biotransformation of bromo- and iodolactone with gem-dimethylcyclohexane ring. J. Braz. Chem. Soc. 2013;24:1913–1919. [Google Scholar]
- 35.Żołnierczyk A.K., Anioł M., Wawrzeńczyk C. Microbial dehalogenation of chloro- and bromolactones. Przem. Chem. 2013;92:802–805. [Google Scholar]
- 36.Grabarczyk M., Wińska K., Mączka W., Anioł M. Synthesis and odour characteristics of hydroxylactones with methylcyclohexane system. Przem. Chem. 2014;93:1000–1003. [Google Scholar]
- 37.Lebrun M.E., Pfeiffer J.Y., Beauchemin A.M. Synthesis of 2-epi-pumiliotoxin C via a challenging intramolecular hydroamination key step. Synlett. 2009;7:1087–1090. [Google Scholar]
- 38.Semmelhack M.F., Epa W.R., Cheung A.W.H., Gu Y., Kim C., Zhang N., Lew W. Palladium-promoted synthesis of ionophore antibiotics. Strategy and assembly of the homochiral tetrahydrofuran and tetrahydropyran portions of tetronomycin. J. Am. Chem. Soc. 1994;116:7455–7456. doi: 10.1021/ja00095a078. [DOI] [Google Scholar]
- 39.Grabarczyk M., Szumny A., Gładkowski W., Białońska A., Ciunik Z., Wawrzeńczyk C. Lactones 18. Synthesis of bicyclic lactones with methyl-, di- and trimethyl substituted cyclohexane system. Pol. J. Chem. 2005;79:1763–1771. [Google Scholar]
- 40.Snider B.B., Johnston M.I. Regioselectivity of the halolactonization of γ,δ-unsaturated acids. Tetrahedron Lett. 1985;26:5497–5500. doi: 10.1016/S0040-4039(01)80869-8. [DOI] [Google Scholar]
- 41.Ranganathan S., Muraleedharan K.M., Vaish N.K., Jayaraman N. Halo- and selenolactonisation: The two major strategies for cyclofunctionalisation. Tetrahedron. 2004;60:5273–5308. doi: 10.1016/j.tet.2004.04.014. [DOI] [Google Scholar]
- 42.Janssen D.B. Evolving haloalkane dehalogenases. Curr. Opin. Chem. Biol. 2004;8:150–159. doi: 10.1016/j.cbpa.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 43.Medina-Martínez M.S., Uyttendaele M., Rajkovic A., Nadal P., Debevere J. Degradation of N-acyl-l-homoserine lactones by Bacillus cereus in culture media and pork extract. Appl. Environ. Microbiol. 2007;73:2329–2332. doi: 10.1128/AEM.01993-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.