

Abstract
Modified nucleoside analogues are of great biological importance as antiviral and antitumoral agents. There is special interest in the preparation of C-aryl nucleosides with an aromatic ring in different positions of the glycone for their biological activity. Different chemical synthesis strategies for these targets are described in this review.
Keywords: C-arylnucleoside, nucleoside, carbohydrate, total synthesis, asymmetric catalysis
1. Introduction
Nucleoside analogues have shown high effectiveness as antiviral and antitumoral agents. In order to improve the pharmacologic activity, a variety of functionalities have been introduced into either the ribose moiety [1,2,3,4] or the heterocyclic moiety [4,5], particularly an aromatic core. This review is focused on the synthesis of C-aryl nucleoside analogues having C-C bonds between an aryl core and the glycone moiety. The particular C-C bond formations covered in this review are those in positions 1', 2', 3', 4' and 5' of the ribose ring. The well-known C-nucleosides in which the anomeric bond has been replaced by a C-C bond have been the focus of recent reviews [6,7] and are therefore not included in this review. In this regards, this review has been arranged to describe the different methodologies for the formation of C-aryl bond according to the type of organic reaction involved: addition to a carbonyl group, C-C cross-coupling, addition to epoxides and cyclization. One special section is dedicated to the formation of the glycone ring starting from an aromatic core.
2. Addition of an Aromatic Ring to a Carbonyl Group
Introduction of an aromatic core can occur via the attack of organometallic reagents such as a lithium, magnesium, aluminum or titanium reagents to both aldehydes or ketones. Starting from nucleoside and carbohydrate analogues possessing a ketone, and depending of the nature of the glycone part, the reaction can lead to diastereoselectivity.
2.1. Addition of Aromatic Organolithiums to Carbonyl Groups
Two strategies were developed. The first one was the direct addition of an aromatic ring to a carbonyl group starting from nucleoside analogues and the second one was the addition of an aromatic ring to the carbonyl group of carbohydrate as starting material, followed by introduction of a nucleobase.
In 1987, Miyasaka and co-workers reported the synthesis of 3' (S)-C-phenyl-β-d-xylofuranosyluracil (4) in good yield [8]. This family of modified nucleoside analogues has been known to have potent biological activity and to be useful for elucidation of enzyme recognition of substrates. Starting from 2',5'-bis-O-tert-butyldimethylsilyl-3'-ketouridine (2) obtained in two steps from uridine (1) [9], treatment with an excess of phenyllithium in THF for 3 h at below −70 °C furnished the corresponding alcohol 3 in 72% yield. Then, classical deprotection of 3 in presence of TBAF in THF gave the corresponding triol 4 (Scheme 1). The authors did not report the presence of a diastereoisomeric mixture during the addition of the aromatic ring to the carbonyl group. Application of this approach to the synthesis of the corresponding 2'-C- phenyl analogue did not afford the target aromatic derivative, probably due to the known instability of 2'-ketouridine.
Scheme 1.
Synthesis of 3'(S)-C-phenyl-β-D-xylofuranosyluracil (4).

Reagents and Conditions: (i) Reference [9]; (ii) PhLi, THF, −70 °C, 3 h, 72%; (iii) TBAF, THF.
A similar sequence was applied to the aldehyde 6 [10] which was obtained in four steps from thymidine (5) via subsequent protection of the 5'-OH group, silylation of the 3'-OH group, removal of the protection of the 5'-OH group and then Moffatt oxidation of the 5'-OH group. This strategy furnished the 5'-C-aryl derivatives 11 and 12 [11] as nucleotide analogues for a study on site-specific DNA cleavage [11,12]. Starting from 1-bromo-2-nitrobenzene in the presence of phenyl lithium, a metal-halide exchange in THF at −105 °C permitted obtaining an epimeric mixture of alcohols 7 and 8 (7 (5'S)/8 (5'R) (4.6:1) in 66% yield. The diastereoisomeric excess (de 64%) was not explained by the authors. Then, conversion of the mixture of isomers 7 and 8 gave, after flash column chromatography, the acetals 9 and 10 in 76% and 16% yields, respectively. A conventional deprotection step followed by transformation of the hydroxyl group in position 3' to a phosphoramidite afforded the intermediates 11 and 12 in 77% and 71% yields (over two steps), respectively (Scheme 2). The phosphoramidites 11 and 12 were incorporated into oligonucleotides by standard automated DNA synthesis.
Scheme 2.
Synthesis of 5'(S)- and 5'(R)-C-phenyluridine analogues 11 and 12.

Reagents and Conditions: (i) Reference [10]; (ii) 1-bromo-2-nitrobenzene, PhLi, THF, −105 °C, 5 h, 66%; (iii) ethylvinyl ether, PPTS, CH2Cl2, 18 h, 9: 76% and 10: 16%; (iv) (a) TBAF, THF, 2 h; (b) 2-cyanoethyl-N,N-diisopropylphosphorochloroamidite, (iPr)2EtN, CH2Cl2, 2 h, 11: 77%, 12: 71% for the two steps.
In parallel, addition of aromatic ring on a carbonyl group was realized on carbohydrate starting materials. In 2001, Sasaki and co-workers reported for the first time the synthesis of W-shape nucleic acid (WNA) designed for selective formation of anti-parallel triplexes formation [13]. WNAs are bicyclic nucleoside analogues bearing an aromatic moiety for stacking and a heterocyclic part as purine base for Hoogesteen hydrogen bonds. The strategy started from D-ribono-1,4-lactone 14 which was prepared in four steps from D-ribose (13) via protection of the 2,3-dihydroxy groups, acetylation of the residual hydroxyl groups, selective deacetylation and then oxidation of the anomeric position. Addition of phenyllithium in THF furnished the two anomers 15 in 53% yield [13] (Scheme 3). In the next steps, this sequence demanded protection of the primary hydroxyl group with a silyl group.
Scheme 3.
Synthesis of 1-C-phenyl-D-ribofuranosyl analogues 15.

Reagents and Conditions: (i) (a) Acetone, H+; (b) Ac2O, pyridine; (c) piperidine, THF, 55% for the three steps; (d) PCC, CH2Cl2; (ii) PhLi, THF, −70 °C, 3 h, 53%.
To complete this work, Sasaki and co-workers reported three years later a similar strategy by changing the protecting group in position 5 (silyl vs. acetyl) (Scheme 4) [13,14]. In this case, the C-C coupling between phenyllithium and the lactone 16 gave the two 1-C-phenyl lactol epimers 17 in 79% yield [14]. Allylation at the 1-position of compounds 17 gave a mixture of two anomers 18 (ratio of α/β 7:6) in 82% yield. An elegant chemical sequence for the bicyclo[3.3.0]octane derivative was reported by Sasaki and co-workers. Subsequent oxidative cleavage of the vinyl group of 18 gave the corresponding aldehyde and deprotection of the diol in position 2,3 spontaneously provided the two corresponding bicyclo[3.3.0]octane derivatives 19 in 28% yield (two steps). After acetylation of the two hydroxyl groups furnishing the two epimers 20 in 90% yield, conventional N-glycosidation with thymine was done to produce the target α- and β-isomers 22 and 21 in 37% and 42% yields, respectively. After flash column chromatography, each nucleoside analogues 21 and 22 were deprotected to give the corresponding diols 23 and 24 in 71% and 47% yields, respectively. After classical protection and activation steps, the corresponding phosphoramidites were incorporated to oligonucleotides by standard automated DNA synthesis.
Scheme 4.
Synthesis of thymidine analogues 23 and 24.

Reagents and Conditions: (i) (a) Acetone, H+; (b) TBDPSCl, TEA, DMAP, CH2Cl2; (c) PCC, CH2Cl2, 77% for the three steps; (ii) PhLi, THF, −78 °C, 2 h, 79%; (iii) CH2=CHCH2TMS, ZnBr2, CH3NO2, 0 °C then rt, 2 h, 82%; (iv) (a) aq. OsO4, NaIO4, pyridine, rt, 30 h; (b) H2SO4 (5%), THF, 60 °C, 6 h, 28% for the two steps; (v) Ac2O, pyridine, 0 °C, 39 h, 90%; (vi) HDMS, TMSCl, SnCl4, thymine, CH3CN, 50 °C, 4 h, 21: 42%; 22: 37%; (vii) (a) TBAF, THF, rt, 2 h; (b) NaOH, THF, MeOH, 0 °C, 1 h, 23: 71%; 24: 47% for the two steps.
At this stage, from the mixture of the key glycosyl donors 20, the strategy described provides straightforward access in an efficient fashion to the different nucleoside analogues 25–40 in a bicyclo[3.3.0]octane series as presented in Figure 1 [13,14,15]. As usual, N-glycosidation with a guanine derivative afforded a mixture of 7-N and 9-N alkylated isomers and α- and β-isomers 33, 34, 37 and 38. It is noteworthy that introduction of the nucleobase furnished in each case a mixture of two isomers, but the authors did not mention at any time the ratio of the α-isomer. In addition to the above-mentioned syntheses, Sasaki and co-workers reported the preparation of the halogeno- and amino-functionalized bicyclonucleoside analogues 41–50 (Figure 2) [16].
Figure 1.

Bicyclo[3.3.0]octane nucleoside analogues 25–40 having a phenyl group [13,14,15].
Figure 2.

Bicyclo[3.3.0]octane nucleoside analogues 41–50 having a substituted aromatic ring.
During this period, Sasaki and co-workers reported the synthesis of compounds 53 and 54 [17] using the same strategy described above [14]. In this case, acetylation of the hydroxyl group of 17 as pre-treatment for the N-glycosidation did not furnish the corresponding acetate but caused carbohydrate ring opening to yield the corresponding undesired acyclic derivative [18].
Due to this reactivity, the authors developed the direct N-glycosidation of the two epimeric alcohols 17. Thymine was mixed in presence of the silylating agent BSA and Lewis acid TMSOTf with the epimeric mixture of 17 at 0 °C to produce the β-nucleoside 51 (α-phenyl) in 31% yield. The same reaction at 50 °C furnished a mixture of two isomers (α-nucleoside/β-nucleoside, 52/51, 6:31) showing that the β-nucleoside 51 was formed by thermodynamic process. Then, classical deprotection of the primary hydroxyl group of 51 and 52 afforded the nucleoside analogues 53 and 54 in 49% and 63% yields, respectively (Scheme 5).
Scheme 5.
Synthesis of 1'(R)- and 1'(S)-C-phenyl-D-ribofuranosylthymine analogues 53 and 54.

Reagents and Conditions: (i) PhLi, THF, −78 °C then rt, 4 h, 71%; (ii) thymine, BSA, TMSOTf, 50 °C, 4.5 h, 51: 31%, 52: 6%; (iii) TBAF, THF, rt, 2 h, 53: 49%, 54: 63%.
Introduction of all four nucleobases were realized using similar strategy giving compounds 55–60 and two of them were selectively deprotected to obtain the β-isomers 61 and 62 (Figure 3). In 1982, Vasella and co-workers reported the synthesis of 4'-C-aryl-D-ribonucleosides as synthons for the synthesis of antibiotics [19]. Starting from the 1,4-lactone derivative 64 obtained from the ribonolactone 63 in two steps, addition of an excess of 2-methoxymethoxyphenyllithium at 10 °C afforded two isomeric lactones 65 and 66 in 65% yield with an excess of the L-lyxo 66 (54%). In order to have more D-ribose derivative, the authors described the conversion of the L-lyxo form 66 to the target D-ribo form 65 in 89% yield by treatment with piperidine and the addition of methanesulfonyl chloride and TEA. Reduction of the isolated lactone 65 with DiBAL-H afforded the two lactols 67 in 95% yield and then subsequent deprotection of the diol and acetylation of the free hydroxyl group gave the glycone derivatives 68 in 90% yield, respectively.
Figure 3.

1'(R)- and 1'(S)-C-phenyl-D-ribofuranosylnucleoside analogues 55–62.
Using the Vorbrüggen methodology, addition of N6-benzoyladenine to the mixture of anomers 68 in presence of TMSOTf and HDMS afforded selectively the β-isomer via a C2 acetyloxonium intermediate. Then, direct treatment of the nucleoside analogue with NH3 in methanol led to the target adenosine derivative 69 in 68% yield (two steps) (Scheme 6).
Scheme 6.
Synthesis of (4'(R)-C-phenyl-D-ribo-tetrofuranosyl)adenine analogue 69.

Reagents and Conditions: (i) (a) Cyclohexanone, FeCl3, Sikkon, 50 °C, 2.5 h, 89%; (b) NaOH, H2O, NaIO4, 0 °C then BaCl2.10 H2O, 4 °C, 10 min, 93%; (ii) CH3OCH2OPhLi, Et2O, 10 °C, 3 h, 65: 30%, 66: 35%; (iii) DIBAL-H, toluene −78 °C, 10 min, 95%; (iv) (a) aq. HCl (0.2 M), 50 °C, 7 h; (b) Ac2O, pyridine, 90% for the two steps; (v) (a) N6-benzoyladenine, HDMS, TMSOTf, CH3CN, 60 °C, 1 h; (b) NH3, MeOH, rt, 48 h, 68% for the two steps.
2.2. Addition of Aromatic Organomagnesium Reagents to Carbonyl Groups or Analogues
Using aromatic organomagnesium reagents, two strategies were developed starting from either a nucleoside analogue or from a carbohydrate derivative. Substitution of phenyllithium by the corresponding Grignard reagent was described by Miyasaka and co-workers for the synthesis of 3'(S)-C-phenyl-β-D-xylofuranosyluracil (4). Unfortunately the target compound was obtained in poor yield (30%) (see Scheme 1) [8].
Vasella and co-workers have also reported in the same paper described above the use of phenylmagnesium bromide instead 2-methoxymethoxyphenyllithium for the synthesis of 4'-C-aryl-D-ribonucleoside analogue [19]. Starting from the platform molecule 64, addition of an excess of phenylmagnesium bromide at 10 °C afforded two isomeric lactones 70 and 71 in 81% yield. Attempts to improve the diastereoselectivity of the Grignard reaction showed that the ratio varied between 58:42 (10 °C, normal addition) and 25:75 (−40 °C, inverse addition). Then following the same strategy, reduction of the lactone 70, deprotection and acetylation, N-glycosidation and treatment in basic media conducted to the target adenosine derivative 74 in 58% yield (four steps) (Scheme 7).
Scheme 7.
Synthesis of (4'(R)-C-phenyl-D-ribotetrofuranosyl)adenine analogue 74.

Reagents and Conditions: (i) (a) Cyclohexanone, FeCl3, Sikkon, 50 °C, 2.5 h, 89%; (b) NaOH, H2O, NaIO4, 0 °C then BaCl2.10 H2O, 4 °C, 10 min, 93%; (ii) PhMgBr, THF, 10 °C, 2 h, 70: 47%, 71: 34%; (iii) DIBAL-H, toluene, −78 °C, 10 min, 99%; (iv) aq. AcOH, 60 °C, 2 h, 89%; (v) (a) N6-benzoyl-N6,9-bis(trimethylsilyl)adenine, SnCl4, CH2Cl2, rt, 1 h; (b) NH3, MeOH, rt, 15 h, 66%.
In 2008, Enders and co-workers developed an elegant strategy for the preparation of 4'-C-arylnucleosides [20]. A versatile and efficient route for the selective synthesis of the platform molecule 78 having two asymmetric carbon atoms was described. Starting from the achiral 2,2-dimethyl-1,3-dioxan-5-one (75), α-alkylation using RAMP-hydrazone methodology furnished enantioselectively the corresponding ester 76 in 57% yield (three steps) [21]. Diastereoselective Grignard reaction afforded, after flash chromatography, the major syn diastereoisomer 77 in 88% yield (Scheme 8).
Scheme 8.
Synthesis of 5(R)-C-phenyltetrahydrofurane derivative 81.

Reagents and Conditions: (i) Reference [21]; (ii) PhMgBr, THF, −78 °C then flash chromatography, 88%; (iii) (a) HCl (3N), MeOH, rt; (b) TBDMSOTf, pyridine, THF, 0 °C, 89% for the two steps; (iv) (a) DIBAL-H, CH2Cl2, −78 °C; (b) Ac2O, pyridine, rt, 62% for the two steps; (v) TMSSPh, BF3.OEt2, hexane, −95 °C to rt, 89%; (vi) Bis-TMS-thymine, NBS, 4-A molecular sieve, CH2Cl2, −78 °C to −26 °C, 87%.
Conventional cleavage of the acetonide, subsequent cyclization giving the lactone and then protection of the residual two hydroxyl groups afforded the corresponding 5-phenyltetrahydrofuran analogue 78 in 89% yield (two steps). Reduction of the lactone 78 with DIBAL-H and subsequent acetylation of the lactol furnished selectively the acetal 79 in 62% yield. The authors reported that only the α-anomer was observed. Instead of directly using the acetal 79, Enders and co-workers preferred to convert compound 79 to the corresponding mixture of thioglycosides 80 in 89% yield. Then, a classical silyl-Hilbert-Johnson reaction was applied to give the thermodynamically more stable β-anomers 81 in 87% yield. No attempt to remove the protecting group on compound 81 was mentioned. Application of this strategy furnished the fluoro derivative 82 (Figure 4).
Figure 4.
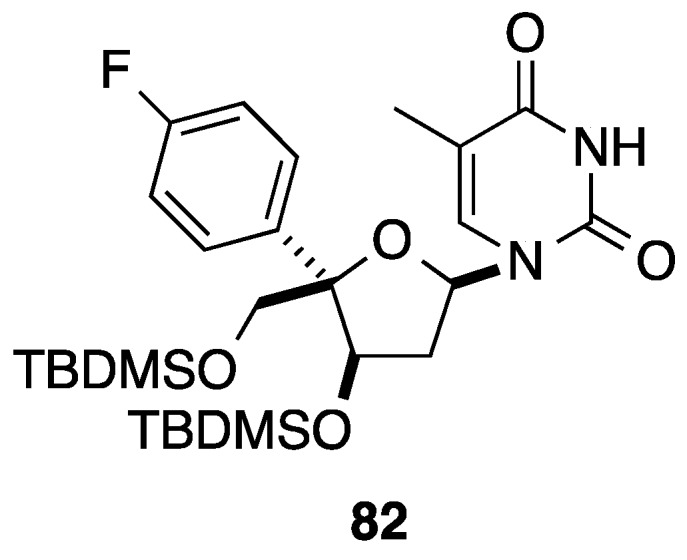
5(R)-(4-Fluorophenyl)tetrahydrofuran derivative 82.
2.3. Addition of Aromatic Organoaluminium Reagents to Carbonyl Groups
Addition of aromatic organoaluminium reagents was described starting from either nucleoside analogues or carbohydrate derivatives. Substitution of phenyllithium by the corresponding phenylaluminium reagent was described by Miyasaka and co-workers for the synthesis of 3'-C-phenyluridine analogue 4 (Scheme 9) [8]. The carbalumination of the ketone 2 was attempted in the presence of an excess of phenylaluminium in CH2Cl2 at −70 °C, but no reaction occurred. It needed to run at room temperature for one hour. The authors reported that even under reflux no decomposition was observed. This reaction led selectively to the nucleoside analogue 3 which could not be isolated in pure form. Subsequent deprotection of the alcohol 3 gave the target nucleoside analogue 4 in 26% yield (two steps). This sequence using Ph3Al permitted to prepare compound 4, but with a lower yield than that using PhLi.
Scheme 9.
Synthesis of 3'(S)-C-phenyl-β-D-xylofuranosyluracil (4).

Reagents and Conditions: (i) Reference [9]; (ii) Ph3Al, CH2Cl2, rt, 1 h; (iii) TBAF, THF, rt, 2 h, 26% for the two steps.
Application of this method from the 2'-keto derivative 83 [22] was realized to furnish the 2'(S)-C-phenyluridine analogue 84 in 30% yield (Scheme 10).
Scheme 10.
Synthesis of 2'(S)-C-phenyl-β-D-arabinofuranosyluracil (84).

Reagents and Conditions: (i) Reference [22]; (ii) (a) Ph3Al, CH2Cl2, rt, 1 h; (b) TBAF, THF, rt, 2 h, 30% for the two steps.
2.4. Addition of Aromatic Organotitanium Reagents to Carbonyl Groups or Analogues
Using chiral titanium complexes, addition to aldehydes led enantioselectively to the corresponding alcohol as a platform for the synthesis of 2(R)-C-phenyl carbohydrate derivatives. In 1992, Duthaler and co-workers reported the synthesis of 2-C-phenylribofuranosyl analogues [23]. Starting from aldehyde 85, an (R,R)-configured allyltitanium reagent was added to glyceraldehyde 85 to furnish the corresponding allyl derivative 86 in 75% yield. The diastereoselectivity was excellent and exclusive Si-face addition was observed. After successive benzoylation of the secondary hydroxyl group of 86, deprotection of the diol and silylation of the primary hydroxyl group gave the allylic compound 88. Ozonolysis of the vinyl bond of 88 afforded the resulting lactol 89 in 50% (four steps) [24,25]. Acetylation of the glycone 89 and N-glycosidation using the Vorbrüggen methodology furnished a mixture of the two anomers 91 and 92 (91–92, 3.5/1). After flash chromatography and classical deprotection of the primary hydroxyl group the target nucleoside analogue 93 was obtained (Scheme 11). Modification of compound 94 permitted its incorporation into oligonucleotides by standard automated DNA synthesis.
Scheme 11.
Synthesis of (2(R)-C-phenylribofuranosyl)thymine derivative 94.

Reagents and Conditions: (i) Reference [23]; (ii) BzCl, pyridine; (iii) (a) TFA, MeOH; (b) TBDMSOTf, TEA, CH2Cl2; (iv) O3, MeOH, −78 °C and then Me2S, −78 °C, rt, 50% for the five steps; (v) Ac2O, DMAP, pyridine, CH2Cl2, 0 °C-rt, 87%; (vi) (a) thymine, BSA C2H4Cl2, 83 °C; (b) TMSOTf, C2H4Cl2, rt, 91: 53% 92: 15%;(vii) TBAF, THF, rt; (viii) MeONa, MeOH, rt.
In this paper, the authors used the previous intermediates 90 to prepare the cytosine analogue 95 following a classical methodology of nucleobase insertion and hydroxyl deprotection (Figure 5).
Figure 5.
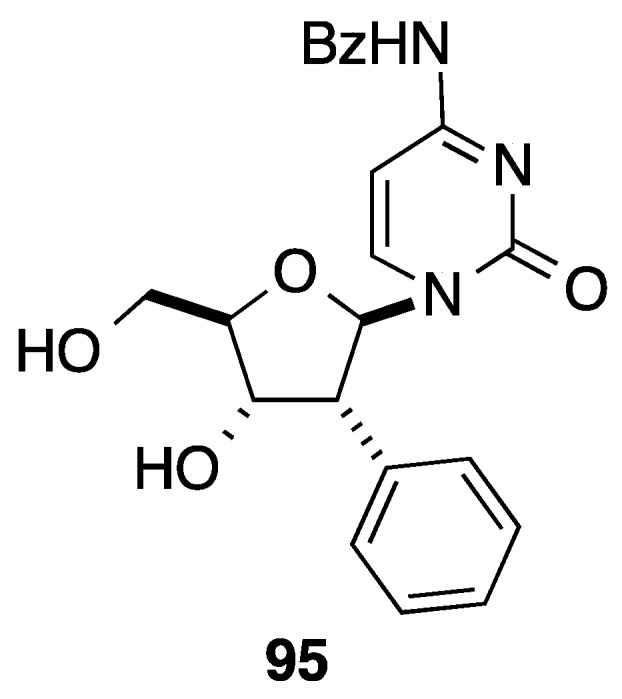
(2(R)-C-Phenylribofuranosyl)cytosine derivative 95.
3. Cross-Coupling Reactions
Palladium-catalyzed cross coupling reactions were studied to obtain mainly d4T analogues having 2',3'-didehydro-2',3'-dideoxy-D-ribose as the glycone part. Two main strategies were developed: the first one was the formation of a bromovinyl intermediate or analogue and the second one the formation of an unsaturated stannyl intermediate.
Tanaka and co-workers described the synthesis of 2'-C- and 3'-C-branched 2',3'-unsaturated nucleosides via palladium-catalysed cross-coupling of the bromovinyl intermediates [26,27]. The 3'-bromo derivative 103 was prepared starting from uridine (1) in a multi-step sequence. Treatment of the 3'-O-mesyl derivative 96 [28] with (PhSe)2 in presence of NaBH4 in refluxing THF-EtOH gave selectively the phenylseleno derivative 97 in 81% yield with inversion of configuration at the 3' position. After perdeacylation and selective silylation of the primary hydroxyl group, bromination in the presence of SOBr2 and imidazole in CCl4 afforded a mixture of β-bromoselenides 100 and 101. Then without purification, the crude mixture of the regioisomers 100 and 101 was submitted to a selenoxide elimination. Treatment of 100 and 101 with MCPBA in CH2Cl2 furnished the bromovinyl derivatives 102 and 103 in 38% and 42% yields, respectively. After flash chromatography, compound 103 was subjected to a Stille reaction using organotin reagents, as coupling partners, in presence of (Ph3P)2PdCl2 (10 mol %) in dioxane at 100 °C for 24 h to obtain the nucleoside analogue 104 in 39% yield (Scheme 12). The same group reported two years later that application to the Stille reaction starting from the bromovinyladenine nucleoside analogue did not give any phenyl derivative [27].
Scheme 12.
Synthesis of 3'-C-phenyl d4U analogue 104.

Reagents and Conditions: (i) Reference [28]; (ii) (PhSe)2, EtOH, NaBH4, THF, reflux, 48 h, 81%; (iii) aq NaOH (1N), EtOH, rt, 24 h; (iv) TBDMSCl, pyridine, rt, 24 h, 96% for the two steps; (v) imidazole, SOBr2, CCl4, rt, 6 h; (vi) MCPBA, rt, 12 h, 102: 38%, 103: 42% for the two steps; (vii) Ph4Sn, (Ph3P)2PdCl2 (10 mol %), TEA, DMF, 100 °C, 24 h, 39%.
Tanaka and co-workers described the synthesis of 3'-C-phenyl-d4A analogue 113 by radical-mediated desulfonylative stannylation [29]. Starting from the epoxide 106 obtained from adenine (105) [30], silylation of the hydroxymethyl group by a conventional method gave the epoxide 107 in 88% yield. To avoid the oxidation of the amino group of adenine, pivaloylation of 107 gave the protected adenine derivative 108 in 97%. Then, selective ring opening by addition of thiophenolate gave the thioether 109 in 90%. Compound 109 was submitted to MCPBA oxidation to give the β-hydroxysulfone product 110 in quantitative yield. Deprotection of the amino group of compound 100 and subsequent methylsulfonylation directly afforded the cis-elimination product 111 in 81% yield (two steps). Radical-mediated desulfonylative stannylation of 111 proceeded efficiently by reacting with Bu3SnH in the presence of AIBN and triethylamine in refluxing benzene to give the 3'-C-stannyl nucleoside 112 in 76% yield (Scheme 13). With the 3'-C-stannyl derivative 112 in hands, the 3'-C-phenyl analogue 113 was prepared by the Stille reaction in presence of PhI, Pd(PPh3)4 and CuI in DMF at room temperature for 28 h. The target aromatic derivative 113 was obtained in 66% yield.
Scheme 13.
Synthesis of 3'-C-phenyl d4A analogue 113.

Reagents and Conditions: (i) Reference [30]; (ii) TBDMSCl, imidazole, DMF, 0 °C, 25 min, 88%; (iii) PivCl, iPr2NEt, CH2Cl2, 0 °C, 1.5 h, 97%; (iv) PhSH, NaOH, MeOH, reflux, 2.5 h, 90%; (v) MCBPA, MeOH, 0 °C, 2.5 h, 100%; (vi) (a) NH3, MeOH, 4 °C, 12 h; (b) MsCl, DMAP, pyridine, 0 °C then rt, 12 h, 81% for the two steps; (vii) Bu3SnH, AIBN, TEA, benzene, 80 °C, 5.5 h, 76%; (viii) PhI, Pd(PPh3)4 (10 mol %), CuI, DMF, rt, 28 h, 66%.
The same year, Tanaka and co-workers developed a similar strategy for the preparation of 2'-C-phenyl d4U analogue 121 starting from uridine (1) [31] (Scheme 14). The main difference was the oxidation step of the phenylthio group to the benzenesulfonyl group which was realized at the end of the strategy (Scheme 14). Starting from uridine (1), the O2,2'-anhydrouridine 114 furnished selectively the 2'-phenylthio derivative 115 [32]. It is clear that only the anhydro strategy led to the desired selectivity. Then, selective protection of the primary hydroxyl group followed by mesylation of the 3'-OH group and elimination afforded the vinyl derivative 118 in 80% yield (three steps). Oxidation of the phenylthio derivative 118 was realized by treatment with MCPBA in methanol and gave the benzenesulfonyl derivative 119 in 79% yield. Classical radical reaction permitted to prepare the 2'-stannyl derivative 120 in 35% yield with a recovered material 119 (40%). The Stille coupling reaction between compound 120 and PhI in presence of Pd(Ph3P)4, CuI afforded the target 2'-C-phenyl d4U 121 in 80% yield.
Scheme 14.
Synthesis of 2'-C-phenyl d4U analogue 121.

Reagents and Conditions: (i and ii) Reference [32]; (iii) TBDMSCl, pyridine, rt, 15 h, 100%; (iv) MsCl, pyridine, rt, 12 h, 80%; (v) DBN, CH3CN, reflux, 2 h, 100%; (vi) MCPBA, MeOH, rt, 3 h, 79%; (vii) Bu3SnH, AIBN, benzene, 80 °C, 6 h, 35%; (viii) PhI, Pd(Ph3P)4, CuI, DMF, rt, 20 h, 80%.
In order to decrease the number of step for the preparation of 2'-C-phenyl- and 3'-C-phenyl-2',3'-didehydro-2',3'-dideoxynucleoside, Tanaka and co-workers developed the direct stannylation of D4T 122 (Scheme 15) [33]. Starting from unprotected d4T 122, stannylation was carried out using Bu3SnOMe at 90 °C for 90 min and furnished the bis-tributylstannyl d4T 123. Subsequently, compound 123 was mixed with a solution of LTMP containing TMEDA at −70 °C for 15 min.
Scheme 15.
Synthesis of 3'-C-phenyl d4T 126.

Reagents and Conditions: (i) Bu3SnOMe, 90 °C, 1.5 h; (ii) LTMP, TMEDA, THF, −70 °C, 15 min, 124: 60%, 125: 9% for the two steps; (iii) PhI, Pd(PPh3)4, CuI, DMF, rt, 12 h, 97%.
A mixture of two regioisomers were obtained, the target 3'-C-stannyl derivative 124 in 60% yield and the isomer 2'-C-phenyl derivative 125 in 9% yield. Starting from compound 124, conventional Stille cross-coupling with PhI in presence of Pd(PPh3)4 and CuI permitted to prepare the target nucleoside analogue 126 in 97% yield.
Few years later, Tanaka and co-workers reported the same strategy starting from d4U [34]. Application of the aforementioned strategy permitted the synthesis of the d4U analogues 127–132. Conversion of the uridine analogues 127 and 129–132 using Reese methodology furnished the corresponding 3'-C-aryl d4C analogues 133–137, respectively (Figure 6).
Figure 6.

3'-C-Aryl d4U 127–132 and 3'-C-aryl d4C 133–137.
Using the 3'-C-aryl d4C derivatives 133 and 135–137, Tanaka and co-workers described the synthesis of the corresponding ddC analogue 141–144 (Scheme 16 and Figure 7) [34]. After protection of the 5'-OH group of compound 127 with an acetate, catalytic hydrogenation occured stereoselectively to give the 3'-β-phenyl analogue 139 in 88% yield then subsequent conversion of the uracile moiety to the cytosine one using Reese methodology permitted to prepare the ddC structure 141 in 88% yield (5 steps) (Scheme 16). Application of the aforementioned strategy permitted the synthesis of the ddC analogues 142–144 with the same diastereoselectivity (Figure 7).
Scheme 16.
Synthesis of 3'(S)-C-phenyl ddC analogue 141.

Reagents and Conditions: (i) Reference [34]; (ii) H2, Pd/C (5%), EtOH, EtOAc, rt, 4 days; (iii) 2,4,6-triisopropylbenzenesulfonyl chloride, DMAP, Et3N, 0 °C, 3 h; (iv) (a) aq NH3 (28%), rt, 1.5 h; (b) Ac2O, DMAP, iPr2NEt, CH2Cl2, 0 °C, 30 min; (c) NH3, MeOH, 5 °C, 6 days, 81% for the five steps.
Figure 7.

3'(S)-C-aryl ddC 142–144.
4. Addition of Aromatic Rings to an Epoxide
Introduction of an aromatic core can occur via the attack of an aromatic organoaluminium reagent to a nucleoside analogue having an epoxide. In this regards, Haraguchi and co-workers reported the ring opening of a nucleoside 1',2'-epoxide with an organoaluminium reagent for the preparation of the 1'-C-phenyl uridine analogue 147 [35]. The strategy developed by the authors was to start with the 1',2'-unsaturated nucleoside analogue 145 obtained in three steps from uridine (1) (Scheme 17) [36].
Scheme 17.
Synthesis of 1'(R)-C-phenyl uridine analogue 147.

Reagents and Conditions: (i) Reference [36]; (ii) dimethyldioxirane, acetone, CH2Cl2; (iii) Ph3Al, CH2Cl2, −30 °C, 4.5 h, 55%.
Then, selective epoxidation of compound 145 was realized with an acetone solution of dimethyldioxirane and furnished only the 1',2'-α-epoxide 146. Nucleoside analogue 146 reacted with an excess of triphenylaluminium in CH2Cl2 at −30 °C for 4.5 h. In this case, preferential formation of the syn-ring-opened β-anomer 146 was seen giving only the α-phenyl derivative 147 in 55% yield.
The authors proposed a possible reaction pathway for this reaction (Scheme 18). With an excess of organoaluminium reagent, the epoxide 146 gave the trialuminium derivative A which formed the oxonium intermediate B. Finally the epoxide acted as a directing group in the presence of the triphenylaluminium reagent, then a nucleophilic attack of the phenyl ligand occurred on the α face of the glycone part and furnished only the syn-ring-opened product 147 [35].
Scheme 18.

Possible reaction pathway for the synthesis of 1'(R)-C-phenyl uridine analogue 147.
5. Cyclization
The formation of a highly functionalized aromatic core via catalytic [2+2+2]-alkyne cyclotrimerization has been well described [37]. This cyclization was reported on the glycone moiety either before the N-glycosidation or after.
In order to identify new therapeutic candidates, Ramana and co-workers reported the synthesis of tricyclonucleosides having a 3-O,4-C-(o-phenylenemethylene) moiety using a cyclotrimerization of the sugar part and then N-glycosidation [38,39]. Starting from the diol 149 obtained from 1,2-5,6-di-O-isopropylidene-α-D-glucose (148) [40], sodium metaperiodate mediated cleavage and subsequent Ohira-Bestmann alkynylation of the aldehyde furnished the corresponding diyne 150 in 78% yield (two steps). Compound 150 under an acetylene atmosphere in the presence of Wilkinson's catalyst in toluene was first mixed at −78 °C during 25 min to give after 4 h at 80 °C the desired xylotetrofuranose derivative 151 in 65% yield. After deprotection and acetylation of the diol, a mixture of the two anomers 152 was obtained in 87% yield. Due to the assistance of the acetyl group in position 2' under conventional nucleobase insertion conditions, the two isochroman derivatives 152 gave selectively the protected β-nucleoside analogue 153 in 79% yield. Then, classical deprotection of the residual secondary hydroxyl group furnished the target copound 154 in 95% yield (Scheme 19).
Scheme 19.
Synthesis of isochroman derivative 154.

Reagents and Conditions: (i) Reference [40]; (ii) (a) NaIO4, MeOH, rt, 30 min; (b) K2CO3, Ohira-Bestmann reagent, MeOH, rt, 6 h, 78% for the two steps; (iii) acetylene, toluene, RhCl(PPh3)3, toluene, −78 °C, 25 min then 80 °C, 4 h, 65%; (iv) (a) aq AcOH (60%), reflux, 2 h; (b) Ac2O, TEA, DMAP, CH2Cl2, 0 °C, 1 h then rt, 1 h, 87% for the two steps; (v) thymine, BSA, CH3CN, reflux, 15 min then TMSOTf, 50 °C, 2 h, 79%; (vi) MeONa, MeOH, rt, 20 min, 95%.
Application of the aforementioned procedure permitted to prepare the different analogues 155 and 156 (Figure 8).
Figure 8.

Isochroman derivatives 155 and 156.
The strategy developed by Ramana and co-workers did not permit preparation of the 3'-C-spiro analogue. In their hands, during the deprotection of the 1,2-diol and then the peracetylation only the pyranose glycone moiety was obtained. In order to obtain the target 3'-C-spiro nucleoside analogue 165, the authors reported a new route using the same key reactions: formation of the diyne, N-glycosidation and [2+2+2]-cyclotrimerization [41]. Starting from D-xylose (157), the propargyl derivative 158 was obtained in five steps [42]. Propargylation of the alcohol 158 followed by a sequence of deprotection/protection of the primary hydroxyl group furnished the pivaloyl ester 161 in 66% yield (three steps). Selective acetonide hydrolysis of 161 and peracetylation gave an anomeric mixture of diacetates 162 in 87% yield (two steps). Conventional Vorbrüggen methodology followed by Zemplen’s deacylation permitted to obtain selectively the corresponding nucleoside analogue 164 as platform molecule for the cyclotrimerization in 58% yield (two steps). Using a similar protocol described above [38,39], the substitution of the Wilkinson catalyst by Cp*RuCl(cod) (Ru vs. Rh) permitted to prepare the target 3'-C-spiro nucleoside analogue 165 in 79% yield (Scheme 20).
Scheme 20.
Synthesis of the 3'-C-spiro uridine analogue 165.

Reagents and Conditions: (i) Reference [42]; (ii) NaH, propargyl bromide, THF, 0 °C to rt, 3 h, 83%; (iii) TBAF, THF, rt, 8 h, 98%; (iv) PivCl, TEA, DMAP, CH2Cl2, 0 °C to rt, 6 h, 81%; (v) (a) Aq AcOH (60%), reflux, 2 h; (b) Ac2O, TEA, DMAP, CH2Cl2, 87% for the two steps; (vi) uracil, BSA, TMSOTf, CH3CN, 50 °C, 2 h, 75%; (vii) MeONa, MeOH, rt, 20 min, 78%; (viii) Cp*RuCl(cod) (5 mol %), C2H4Cl2, EtOH, rt, 4–6 h, 79%.
By using symmetric and unsymmetric alkynes, application of this strategy permitted different 3'-C-spiro nucleoside analogues 166–177 having substituted phenyl core to be obtained (Figure 9) [41].
Figure 9.

3'-C-Spiro uridine analogues 166–177.
6. Construction of the Glycone Part Starting from an Aromatic Moiety
The synthesis of nucleoside analogues having a C-C bond between an aromatic core and the glycone moiety can be realized starting from a benzene derivative via a multi-step strategy. In this regards, chloroacetophenone and benzaldehyde derivatives were used as starting materials.
In 2009, Lopp and co-workers described the enantioselective synthesis of 4'-aryl-2',3'-dideoxy-nucleoside analogues in nine steps (Scheme 21) [43]. Starting from benzaldehyde (178), addition of the 1-acetoxybut-3-en-2-one (179) furnished the corresponding ester 180 and then treatment in basic media gave the corresponding lactone 181 in 28% yield (two steps). Enantioselective oxidation of the enol tautomer 181 was realized in presence Ti(Oi-Pr)4, t-BuOOH and (+)-diethyl tartrate and permitted the preparation of the carboxylic acid 182 in 36% yield (ee 86%). It was notable that the formation of the keto acid was observed (16% yield) and a considerable amount of starting material 181 remained unreacted, permitting a recycling step [44]. With the chiral compound 182 having the D-configuration, Lopp and co-workers developed a conventional strategy for the preparation of the target nucleoside analogues 189. Subsequent reduction of the carboxyl group of 182 using a borane complex, protection of the resulting hydroxyl group and reduction of the lactone furnished the two diastereoisomeric lactols 185 in 79% yield (three step). Then, acetylation followed by N-glycosidation and deprotection of the primary hydroxyl group gave, after flash chromatography, the target β-D-isomer 189 and α-D-isomer 190 in 40% yield, respectively.
Scheme 21.
Synthesis of the 4'-C-phenyl thimidine analogues 189 and 190.

Reagents and Conditions: (i) 3-Benzyl-5-(2-hydroxyethyl)-4-methylthiazolium chloride, TEA, dioxane, 70–80 °C, 15 h, 71%; (ii) MeONa, MeOH, reflux, 40%; (iii) Ti(Oi-Pr)4, (+)-diethyl tartrate, t-BuOOH, CH2Cl2, −20 °C, 114 h, 36% (ee 86%); (iv) BH3.SMe2, THF, 86%; (v) TBDMSCl, imidazole, CH2Cl2, 100%; (vi) DIBAL-H, toluene, 92%; (vii) Ac2O, TEA, CH2Cl2, 90%; (viii) thymine, BSA, TMSOTf, CH3CN, 88% (187:44%, 188:44%); (ix) TBAF, THF, 100%.
Lopp and co-workers developed in the same paper the formation of substituted benzene derivatives 191–194 (Figure 10) [43].
Figure 10.

4'-C-Phenyl thimidine analogues 191–194.
In 2003, Trost and co-workers published a elegant synthesis of 4'-C-phenyl nucleoside analogues as depicted in Scheme 22. The authors reported a multi-step strategy using the Pd-catalyzed dynamic kinetic asymmetric transformation (Pd-DYKAT) of vinylepoxide, metathesis, isomerization and N-glycosidation [45]. Starting from the chloroacetophenone (195) addition of vinylmagnesium bromide furnished the phenyl substrate 196 in 91% yield. The racemic mixture of 196 reacted with allylic alcohol in presence of Pd2(dba)3.CHCl3 as pre-catalyst, (1S,2S)- diphosphine C as chiral ligand and triallyl borate as co-catalyst to give the 2-(S)-phenyl-2-allyloxybut-3-en-1-ol (197) in 33% yield (ee 87%). Ring-closing metathesis was realized with Grubbs I catalyst (2 mol %) and furnished the desired compound 198 in 86% yield. In order to allow the nucleoside base installation, protection of the primary hydroxyl group and then isomerization to convert 2,5-dihydrofuran 199 to 2,3-dihydrofuran 200 were realized. The authors obtained the glycal 200 in 90% yield using [H2Ru(CO)(PPh3)3] as ruthenium catalyst. The N-glycosidation was effected using the silylated uracil and PhSeCl in presence of InCl3 as Lewis acid and gave a mixture of the two anomeric products 201 and 202 in 67% yield. The major nucleoside analogue 201 had the β-L-configuration which is the enantiomeric form of the natural nucleoside. After classical treatment of 201 and 202 with TBAF and then purification by preparative HPLC, the deprotected 4'-C-phenyl β-L-isomer 203 and α-L-isomer 204 were obtained in 85% and 13% yields, respectively.
Scheme 22.
Synthesis of the 4'-C-phenyl L-uridine analogues 203 and 204.

Reagents and Conditions: (i) CH2CHMgBr, THF, 91%; (ii) [Pd2dba3].CHCl3, (1S,2S)- Ligand C, DMAP, dioxane, (CH2CHCH2)3B, rt, 19 h, 33% (ee 87%); (iii) Cl2(PCy3)2RuCHPh, CH2Cl2, rt, 11 h, 86%; (iv)TBDPSCl, imidazole, DMAP, DMF, rt, 45 mn, 97%; (v) [H2Ru(CO)-(PPh3)3], toluene, rt, 70 °C, 4 h and then 80 °C, 9 h, 90%; (vi) PhSeCl, dioxane, bis(trimethylsilyl)uracil, InCl3, rt, 3 h, 67%; (vii) TBAF, THF, rt, 1.5 h, 203:85%, 204:13%.
In the same paper and using the same methodology, Trost and co-workers described the synthesis of the purine analogue 205 (Figure 11) [45].
Figure 11.
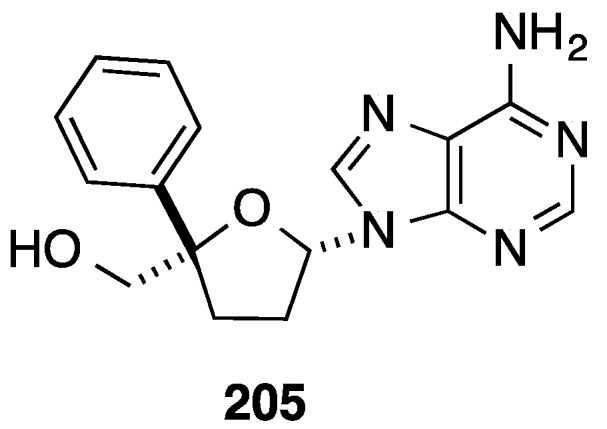
4'-C-Phenyl purine analogue 205.
From 1996 to 2006 Len and co-workers described the synthesis of d4T analogues having a benzo[c]furan core. Various strategies including racemic synthesis and asymmetric synthesis have been reported by his group, all of them starting from phthalaldehyde [46,47,48,49,50,51,52,53,54,55]. In parallel to this work, the Liu group reported the racemic synthesis of benzo[c]furan nucleoside analogues [56]. For simplicity, only the asymmetric synthesis of benzo[c]furan analogues reported by Len is described here [48]. After selective protection of phthalaldehyde 206, Wittig homologation of the remaining formyl group gave the corresponding styrene 208 in 58% yield (Scheme 23). Asymmetric dihydroxylation of the vinyl group using the commercial Sharpless reagent, AD-mix α afforded the corresponding dihydro derivative 209 in 85% yield (ee > 99%). The enantioselectivity of the dihydroxylation was important since only the new stereocenter having S-configuration can furnish the D-series. In order to avoid the isochroman formation, selective benzoylation of the primary hydroxyl group of 209 was necessary. Then classical treatment of the ester 210 in acidic methanol permitted the deprotection of the formyl group, the cyclization and methylation to afford a mixture of the two anomeric 1,3-dihydrobenzo[c]furan derivatives 211 in 82% yield. Without separation of the two epimers 211, standard Vorbüggen chemistry furnished the β-isomer 212 and α-isomer 213, due to the lack of neighboring group participation to direct stereoselectivity. After removal of the benzoyl protection and subsequent silica gel chromatography, the target nucleosides 214 and 215 were obtained enantiomerically pure in 19% and 9% overall yield, respectively. The related enantiomers analogous to L-nucleosides were synthesized using the same strategy but employing AD-mix β. Using the same strategy, Len and co-workers [46,47,48,49,50,51,52,53,54,55] reported the synthesis of d4T analogues 216–220 (Figure 12).
Scheme 23.
Synthesis of the benzo[c]furan analogues of d4U 214 and 215.

Reagents and Conditions: (i) propan-1,3-diol, PTSA, toluene, 110 °C, 5 h, 75%; (ii) Ph3PCH2Br, n-BuLi, THF, 0 °C to rt, 1 h, 77%; (iii) AD-mix α, aq. t-BuOH, −10 °C to 0 °C, 1 h, 85% (ee > 99%); (iv) PivCl, TEA, toluene, −10 °C, 12 h, 78%; (v) MeOH, HCl, rt, 1 h, 72%; (vi) silylated uracil, SnCl4, C2H4Cl2, 0 °C, 2 h, 212:35%, 213:38%; (vii) aq. NaOH (1N), rt, 2 h, 93%.
Figure 12.

Benzo[c]furan nucleoside analogues 216–220.
7. Conclusions
Different syntheses of C-aryl nucleoside analogues have been driven by attempts to improve upon the biological activities of commercial antiviral and antitumoral nucleosides, to provide structure–activity data and to offer a continuity of new drugs as alternatives to the previous generation to combat the rise of resistance. Formation of C-aryl nucleoside analogues using three main strategies depending of the starting materials—nucleoside, carbohydrate or aromatic compounds—was described.
Starting from nucleoside analogues, addition of an aromatic ring on a keto group or an epoxide and C-C cross coupling were reported. First, classical oxidation of the glycone part and then addition of phenyllithium or triphenylaluminium permitted preparation of 2'-C, 3'-C and 5'-C-phenyl derivatives [8,11,22]. Selective addition of triphenylaluminium on an nucleoside analogue having an epoxide in position 1',2' was also reported [35]. C-C cross coupling was described for the formation of 2'-C- and 3'-C-aryl-2',3'-didehydro-2',3'-dideoxynucleoside analogues using two multi-step strategies: palladium-catalyzed cross coupling via bromovinyl nucleoside analogues [26,27] and palladium-catalyzed cross coupling via butyltinvinyl nucleoside analogues [29]. A more efficient short strategy was reported by the same group starting from d4T and d4U with the formation of the 3'-C-phenyl derivative via the corresponding butyltinvinyl nucleoside analogues [33,34]. It was noteworthy that 3'-C-phenyl d4U derivatives were used as intermediate for the formation of the corresponding ddC analogues by conventional hydrogenation [34].
Starting from carbohydrate analogues, two strategies were reported: addition of aromatic ring on a keto group and [2+2+2]-cyclotrimerization. Starting from ribose, addition of phenyllithium on a 1,4-ribonolactone derivative followed by a N-glycosidation allowed the synthesis of the corresponding 1-C-phenyl nucleoside analogues and the corresponding bicyclo derivatives [13,14,15,16,17]. Another strategy was developed for the preparation of 4'-C-phenyl nucleoside analogues using addition of phenyllithium or phenylmagnesium bromide on a lactol followed by N-glycosidation [19]. Tricyclonucleoside analogues were obtained in a multi-steps strategy starting from D-glucose and D-xylose. The main key reactions were the formation of the corresponding diyne, the [2+2+2]-cyclotrimerization and then the N-glycosidation. This strategy permitted to prepare different isochromane analogues having a bridge between the 3'-C and 4'-C carbon atoms [38,39]. A modified strategy using the [2+2+2]-cyclotrimerization at the end of the protocol afforded the 3'-C-spiro-annulated nucleoside analogues [41].
Different approaches were reported starting from achiral compounds having an aromatic core or not. In these cases, asymmetric syntheses were studied. Most often the 4'-C-phenyl nucleoside analogues were obtained using this strategy. Starting from achiral 1,3-dihydroxyacetone derivative, asymmetric α-alkylation was studied and then diastereoselective addition of phenylmagnesium bromide permitted introduction of the aromatic ring on the skeleton [21]. Then classical glycone formation followed by N-glycosidation furnished the target nucleoside analogues. Starting from benzaldehyde, the enol intermediate was used for enantioselective oxidation and then the corresponding lactone afforded the 4'-C-phenyl nucleoside analogues using a classical protocol [43,44]. Chloroacetophenone was used as starting material for the synthesis of 4'-C-phenyl L-nucleosides as enantiomers of the natural nucleoside series [45]. The key reaction used was a Pd-catalyzed dynamic kinetic asymmetric transformation furnishing the enantiomeric diene. Then through classical ring closing metathesis, isomerization and N-glycosidation, the target nucleoside analogues were obtained. The 2'-C-phenyl nucleoside analogues were obtained starting from glyceraldehyde derivatives. Asymmetric Si-face addition of allyltitanium reagent furnished diastereoselectively the corresponding alcohol as key intermediate. Then conventional formation of the glycone moiety followed by N-glycosidation furnished the target nucleoside analogue. Finally, a series of 2'-C- and 3'-C-dibranched nucleosides with a benzo[c]furan core have been synthesised by a convergent route employing conventional Vorbruggen chemistry on a preformed benzo[c]furan system [48]. An interesting feature of the route to this unusual glycone system in nucleoside chemistry is the highly effective use of the stereoselective Sharpless hydroxylation to obtain compounds analogous to conventional nucleosides in the D- and L-series, accordingly.
Acknowledgments
Authors are grateful to the Région Picardie and Europe (FEDER) for their financial support.
Author Contributions
C.L. and G.E. contributed to the literature review (mostly G.E.) and manuscript writing (mostly C.L.). Both authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Len C., Mackenzie G. Synthesis of 2',3'-didehydro-2',3'-dideoxynucleosides having variations at either or both of the 2'- and 3'-positions. Tetrahedron. 2006;62:9085–9107. doi: 10.1016/j.tet.2006.07.050. [DOI] [Google Scholar]
- 2.Len C., Postel D. Synthesis of 2',3'-didehydro-2',3'-dideoxynucleosides via nucleoside route. Curr. Org. Synth. 2006;21:261–281. doi: 10.2174/157017906777934881. [DOI] [Google Scholar]
- 3.Lebreton J., Escudier J.M., Arzel L., Len C. Synthesis of bicyclonucleosides having a C-C bridge. Chem. Rev. 2010;110:3371–3418. doi: 10.1021/cr800465j. [DOI] [PubMed] [Google Scholar]
- 4.Len C., Mondon M., Lebreton J. Synthesis of cyclonucleosides having a C-C bridge. Tetrahedron. 2008;64:7453–7475. doi: 10.1016/j.tet.2008.04.095. [DOI] [Google Scholar]
- 5.Herve G., Sartori G., Enderlin G., Mackenzie G., Len C. Palladium-catalysed Suzuki reaction in aqueous solvents applied to unprotected nucleosides and nucleotides. RSC Adv. 2014;4:18558–18594. doi: 10.1039/c3ra47911k. [DOI] [Google Scholar]
- 6.Stambasky J., Hocek M., Kocovsky P. C-Nucleosides: Synthetic strategies and biological applications. Chem. Rev. 2009;109:6729–6764. doi: 10.1021/cr9002165. [DOI] [PubMed] [Google Scholar]
- 7.Ferrero M., Gotor V. Biocatalytic selective modifications of conventional nucleosides, carbocyclic nucleosides and C-nucleosides. Chem. Rev. 2000;100:4319–4348. doi: 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]
- 8.Hayakawa H., Tanaka H., Itoh N., Nakajima M., Miyasaka T., Yamaguchi K., Iitaka Y. Reaction of organometallic reagents with 2'- and 3'-ketouridine derivatives: Synthesis of uracil nucleosides branched at the 2'- and 3'-positions. Chem. Pharm. Bull. 1987;35:2605–2608. doi: 10.1248/cpb.35.2605. [DOI] [Google Scholar]
- 9.Hansske F., Robins M.J. Nucleic acid related compounds. 43. A convenient procedure for the synthesis of 2' and 3'-ketonucleosides. Tetrahedron Lett. 1983;24:1589–1583. doi: 10.1016/S0040-4039(00)81717-7. [DOI] [Google Scholar]
- 10.Yang C.O., Wu H.Y., Fraser-Smith E.B., Walker K.A.M. Synthesis of 4'-cyanothymidine and analogs as potent inhibitors of HIV. Tetrahedron Lett. 1992;33:37–40. doi: 10.1016/S0040-4039(00)77667-2. [DOI] [Google Scholar]
- 11.Dussy A., Meyer C., Quennet E., Bickle T.A., Giese B., Marx A. New light-sensitive nucleosides for caged DNA strand breaks. ChemBioChem. 2002;3:54–60. doi: 10.1002/1439-7633(20020104)3:1<54::AID-CBIC54>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 12.Thoeni S., Kressierer C.J., Giese B. Site-specific DNA cleavage on a solid support: A method for mismatch detection. Angew. Chem. Int. Ed. 2007;46:2112–2114. doi: 10.1002/anie.200603092. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki S., Yamauchi H., Nagatsugi F., Takahashi R., Taniguchi Y., Maeda M. W-shape nucleic acid (WNA) for selective formation of non-natural anti-parallel triplex including a TA interrupting site. Tetrahedron Lett. 2001;42:6915–6918. doi: 10.1016/S0040-4039(01)01446-0. [DOI] [Google Scholar]
- 14.Sasaki S., Taniguchi Y., Takahashi R., Senko Y., Kodama K., Nagatsugi F., Maeda M. Selective formation of stable triplexes including a TA or a CG interrupting site with new bicyclic nucleoside analogues (WNA) J. Am. Chem. Soc. 2004;126:516–528. doi: 10.1021/ja037211z. [DOI] [PubMed] [Google Scholar]
- 15.Taniguchi Y., Nakamura A., Senko Y., Nagatsuji F., Sasaki S. Effects of halogenated WNA derivatives on sequence dependency for expansion of recognition sequences in non-natural-type triplexes. J. Org. Chem. 2006;71:2115–2122. doi: 10.1021/jo052413u. [DOI] [PubMed] [Google Scholar]
- 16.Taniguchi Y., Togo M., Aoki E., Uchida Y., Sasaki S. Synthesis of p-amino-WNA derivatives to enhance the stability of the anti-parallel triplex. Tetrahedron. 2008;64:7184–7170. [Google Scholar]
- 17.Nasr T., Taniguchi Y., Sasaki S. Synthesis of 1'-phenyl substituted nucleoside analogs. Heterocycles. 2007;71:2659–2668. doi: 10.3987/COM-07-11150. [DOI] [Google Scholar]
- 18.Hayakawa H., Miyazawa M., Tanaka H., Miyasaka T. A ribonolactone-based approach to the synthesis of 1'-carbon-substituted thymine ribonucleosides. Nucleosides Nucleotides. 1994;12:297–308. doi: 10.1080/15257779408013242. [DOI] [Google Scholar]
- 19.Beer D., Mewly R., Vasella A. l-Erythruronic acid derivatives as building blocks for nucleoside analogs. Synthesis of 4'-C-aryl-D-ribonucleosides. Helv. Chim. Acta. 1982;65:2570–2582. doi: 10.1002/hlca.19820650828. [DOI] [Google Scholar]
- 20.Enders D., Voith M., Lenzen A. The dihydroxyacetone unit—A versatile C3 building bloc in organic synthesis. Angew. Chem. Int. Ed. 2005;44:1304–1325. doi: 10.1002/anie.200400659. [DOI] [PubMed] [Google Scholar]
- 21.Enders D., Hieronymi A., Raabe G. Asymmetric synthesis of 4'-quaternary 2'-deoxy-3'- and -4'-epi-β-C- and -N-nucleosides. Synthesis. 2008;10:1545–1558. doi: 10.1055/s-2008-1072577. [DOI] [Google Scholar]
- 22.Cook A.F., Moffatt J.G. Sulfoxide-carbodiimide reactions. VI. Synthesis of 2'- and 3'-ketouridines. J. Am. Chem. Soc. 1967;89:2697–2705. doi: 10.1021/ja00987a036. [DOI] [Google Scholar]
- 23.Duthaler R.O., Hafner A., Alsters P.L., Rothe-Streit P., Rihs G. Stetreoselective transformations mediated by chiral monocyclo-pentadienyl titanium, zirconium, and hafnium complexes. Pure Appl. Chem. 1992;64:1897–1910. doi: 10.1351/pac199264121897. [DOI] [Google Scholar]
- 24.Schmidt C. Efficient synthesis of 2'-deoxy-2'-α-C-substituted nucleosides. Synlett. 1994:238–240. doi: 10.1055/s-1994-22809. [DOI] [Google Scholar]
- 25.Schmidt C., Bevierre M.O., De Mesmaeker A., Altmann K.H. The effects of 2'- and 3'-alkyl substituents on oligonucleotide hybridization and stability. Bioorg. Med. Chem. Lett. 1994;4:1969–1974. doi: 10.1016/S0960-894X(01)80545-X. [DOI] [Google Scholar]
- 26.Haraguchi K., Itoh Y., Tanaka H., Miyasaka T. Preparation and reactions of 2'- and 3'-vinyl bromides of uracil-nucleosides: Versatile synthons for anti-HIV agents. Tetrahedron Lett. 1991;32:3391–3394. doi: 10.1016/S0040-4039(00)92715-1. [DOI] [Google Scholar]
- 27.Haraguchi K., Itoh Y., Tanaka H., Akita M., Miyasaka T. Uracil and adenine nucleosides having a 2',3'-bromovinyl structure: Highly versatile synthons for the synthesis of 2'-C- and 3'-C-branched 2',3'-unsaturated derivatives. Tetrahedron. 1993;49:1371–1390. doi: 10.1016/S0040-4020(01)90190-5. [DOI] [Google Scholar]
- 28.Kamaike K., Uemura F., Yamakage S., Nishino S., Ishido Y. Partial protection of carbohydrate derivatives. Part 23. Simple, efficient procedure for the preparation of 3'- and 2'-O-(tetrahydropyran-2-yl)ribonucleoside derivatives involving highly regioselective 2',5'-di-O-acylation or that followed by acyl migration on silica gel and subsequent O-(tetrahydropyran-2-yl)ation. Nucleosides Nucleotides. 1987;6:699–736. doi: 10.1080/15257778708073420. [DOI] [Google Scholar]
- 29.Onuma S., Kumamoto H., Kawato M., Tanaka H. A versatile intermediate for the synthesis of 3'-substituted 2',3'-didehydro-2',3'-dideoxyadenosine (d4A): Preparation of 3'-C-stannyl-d4A via radical-mediated desulfonylative stannylation. Tetrahedron. 2002;58:2497–2503. doi: 10.1016/S0040-4020(02)00153-9. [DOI] [Google Scholar]
- 30.Robins M.J., Fouron Y., Mengel R. Nucleic acid related compounds. II. Adenosine 2',3'-ribo-epoxide. Synthesis, intramolecular degradation, and transformation into 3'-substituted xylofuranosyl nucleosides and the lyxo-epoxide. J. Org. Chem. 1974;39:1564–1570. doi: 10.1021/jo00924a025. [DOI] [PubMed] [Google Scholar]
- 31.Kumamoto H., Onuma S., Tsuchiya K., Egusa Y., Tanaka H., Satoh T. Sulfoxide-metal exchange for the synthesis of the 2'-tributylstannyl derivative of the 2',3'-didehydro-2',3'-dideoxyuridine (d4U): A general entry to 2'-carbon-substituted analogues of d4U. Nucleosides Nucleotides Nucleic Acids. 2002;21:275–286. doi: 10.1081/NCN-120006826. [DOI] [PubMed] [Google Scholar]
- 32.Divakar K.J., Reese C.B. Reaction between 2,2'-anhydro-1-β-D-arabinofuranosyluracil and thiolate ions. J. Chem. Soc. Perkin Trans. 1. 1982:1625–1628. doi: 10.1039/p19820001625. [DOI] [Google Scholar]
- 33.Kumamoto H., Tanaka H. Simple entry to 3'-substituted analogues of anti-HIV agent Stavudine based on an anionic O-C stannyl migration. J. Org. Chem. 2002;67:3541–3547. doi: 10.1021/jo0107958. [DOI] [PubMed] [Google Scholar]
- 34.Kumamoto H., Onuma S., Tanaka H., Dutschman G.E., Cheng Y.C. 3'-Carbon-substituted pyrimidine nucleosides having a 2',3'-dideoxy and 2',3'-didehydro-2'-3'-dideoxy structure: Synthesis and antiviral evaluation. Antivir. Chem. Chemother. 2006;17:225–234. doi: 10.1177/095632020601700406. [DOI] [PubMed] [Google Scholar]
- 35.Haraguchi K., Kubota Y., Tanaka H. Ring opening of nucleoside 1',2'-epoxides with organoaluminium reagents: Stereoselective entry to ribonucleosides branched at the anomeric position. J. Org. Chem. 2004;69:1831–1836. doi: 10.1021/jo030262u. [DOI] [PubMed] [Google Scholar]
- 36.Haraguchi K., Itoh Y., Matsumoto K., Hashimoto K., Nakamuta K.T., Yanaka H. Stetreoselective synthesis of 1'-C-branched arabinofuranosyl nucleosides via anomeric radicals generated by 1,2-acyloxy migration. J. Org. Chem. 2003;68:2006–2009. doi: 10.1021/jo020620d. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto Y. Recent advances in intramolecular alkyne cyclotrimerization and its application. Curr. Org. Chem. 2005;9:503–519. doi: 10.2174/1385272053544399. [DOI] [Google Scholar]
- 38.Ramana C.V., Suryawanshi S.B. A [2+2+2]-cyclotrimerization approach for the synthesis of enantiopure isochromans using carbohydrate derived dialkyne template. Tetrahedron Lett. 2008;49:445–448. doi: 10.1016/j.tetlet.2007.11.103. [DOI] [Google Scholar]
- 39.Suryawanshi S.B., Dushing M.P., Gonnade R.G., Ramana C.V. The isochroman and 1,3-dihydroisobenzofuran-annulation on carbohydrate templates via [2+2+2]-cyclotrimerization and synthesis of some tricyclic nucleosides. Tetrahedron. 2010;66:6085–6096. doi: 10.1016/j.tet.2010.06.011. [DOI] [Google Scholar]
- 40.Adhavana M., Chari K.S. Polymer-supported ferric chloride as a heterogeneous catalyst for chemoselective deprotection of acetonides. Synthesis. 2005:708–710. [Google Scholar]
- 41.Dushing M.P., Ramana C.V. Target cum flexibility: synthesis of C(3')-spiroannulated nucleosides. Tetrahedron Lett. 2011;52:4627–4630. doi: 10.1016/j.tetlet.2011.06.100. [DOI] [Google Scholar]
- 42.Hattori H., Tanaka M., Fukushima M., Sasaki T., Matsuda A. J. Med. Chem. 1996;39:5005–5011. doi: 10.1021/jm960537g. [DOI] [PubMed] [Google Scholar]
- 43.Jogi A., Paju A., Pehk T., Kailas T., Muurisepp A.M., Lopp M. Synthesis of 4'-aryl-2',3'-dideoxynucleoside analogues. Tetrahedron. 2009;65:2959–2965. doi: 10.1016/j.tet.2009.02.010. [DOI] [Google Scholar]
- 44.Jogi A., Paju A., Pehk T., Kailas T., Muurisepp A.M., Kanger T., Lopp M. Asymmetric synthesis of 2-aryl-5-oxotetrahydrofuran-2-carboxylic acids. Synthesis. 2006;18:3031–3036. [Google Scholar]
- 45.Trost B.M., Brown B.S., McEachern E.J., Kuhn O. Asymmetric synthesis of oxygen heterocycles via Pd-catalyzed dynamic kinetic asymmetric transformations: Application to nucleosides. Chem. Eur. J. 2003;9:4442–4451. doi: 10.1002/chem.200304949. [DOI] [PubMed] [Google Scholar]
- 46.Ewing D.F., Fahmi N., Len C., Mackenzie G., Ronco G., Villa P., Shaw G. Nucleoside analogues: Glycones based on the benzo[c]furan core. Collect. Czechoslov. Chem. Commun. 1996;61:145–147. [Google Scholar]
- 47.Ewing D.F., Fahmi N., Len C., Mackenzie G., Ronco G., Villa P., Shaw G. Nucleoside analogues with a novel glycone based on the benzo[c]furan core. Nucleosides Nucleotides. 1999;18:2613–2630. doi: 10.1080/07328319908044630. [DOI] [PubMed] [Google Scholar]
- 48.Ewing D.F., Fahmi N., Len C., Mackenzie G., Pranzo A. Stereoisomeric pyrimidine nucleoside analogues based on the benzo[c]furane core. J. Chem. Soc. Perkin Trans. 1. 2000;21:3561–3565. doi: 10.1039/b006417n. [DOI] [Google Scholar]
- 49.Ewing D.F., Len C., Mackenzie G., Ronco G., Villa P. Asymmetric synthesis of 1,3-dihydrobenzo[c]furan derivatives using a d-xylose moiety as a chiral auxiliary. Tetrahedron Asymmetry. 2000;11:4995–5002. doi: 10.1016/S0957-4166(00)00488-2. [DOI] [Google Scholar]
- 50.Belloli E., Len C., Mackenzie G., Ronco G., Bonte J.P., Vaccher C. Diastereomeric resolution of nucleoside analogues, new potential antiviral agents, using high-performance liquid chromatography on polysaccharide-type chiral stationary phases. J. Chromatogr. A. 2001;943:91–100. doi: 10.1016/S0021-9673(01)01431-5. [DOI] [PubMed] [Google Scholar]
- 51.Pilard S., Riboul D., Glaçon V., Moitessier N., Chapleur Y., Postel D., Len C. Asymmetric dihydroxylation of chiral styrene derivatives: Development of an analytical strategy for the determination of the diastereomeric excess. Tetrahedron Asymmetry. 2002;13:529–537. doi: 10.1016/S0957-4166(02)00136-2. [DOI] [Google Scholar]
- 52.Len C., Sélouane A., Postel D., Villa P., Aubertin A.M., Egron D., Gosselin G., Périgaud C. Synthesis, stability and biological evaluation of 1,3-dihydrobenzo[c]furan analogues of d4T and its SATE pronucleotide. Nucleosides Nucleotides Nucleic Acids. 2003;5–8:943–946. doi: 10.1081/NCN-120022691. [DOI] [PubMed] [Google Scholar]
- 53.Egron D., Périgaud C., Gosselin G., Aubertin A.M., Faraj A., Sélouane A., Postel D., Len C. 1,3-Dihydrobenzo[c]furan nucleoside analogues: Additional studies of the thymine derivative. Bioorg. Med. Chem. Lett. 2003;13:4473–4475. doi: 10.1016/j.bmcl.2003.09.031. [DOI] [PubMed] [Google Scholar]
- 54.Len C., Mackenzie G., Ewing D.F., Sheppard G., Banoub J. Electrospray tandem mass spectrometric analysis of diastereo- and stereoisomeric pyrimidine nucleoside analogues based on the 1,3-dihydrobenzo[c]furan core. Carbohydr. Res. 2003;338:2311–2324. doi: 10.1016/j.carres.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 55.Lipka E., Sélouane A., Postel D., Len C., Vaccher M.P., Bonte J.P., Vaccher C. Enantioseparation of four cis and trans diastereoisomers of d4T analogs, by High Performance Liquid Chromaography and Capillary Electrophoresis. J. Chromatogr. A. 2004;1034:161–167. doi: 10.1016/j.chroma.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 56.Liu Z.P., Meng Z.L., Wang D.F. Synthesis of novel nucleoside analogues based on 1,3-dihydrobenzo[c]furan core. Chin. J. Chem. 2006;24:504–508. doi: 10.1002/cjoc.200690097. [DOI] [Google Scholar]