

Abstract
As part of our continuing research on canthin-6-one antimicrobial agents, a new series of ester derivatives of 10-hydroxycanthin-6-one were synthesized using a simple and effective synthetic route. The structure of each compound was characterized by NMR, ESI-MS, FT-IR, UV, and elemental analysis. The antimicrobial activity of these compounds against three phytopathogenic fungi (Alternaria solani, Fusarium graminearum, and Fusarium solani) and four bacteria (Bacillus cereus, Bacillus subtilis, Ralstonia solanacearum, and Pseudomonas syringae) were evaluated using the mycelium linear growth rate method and micro-broth dilution method, respectively. The structure-activity relationship is discussed. Of the tested compounds, 4 and 7s displayed significant antifungal activity against F. graminearum, with inhibition rates of 100% at a concentration of 50 μg/mL. Compounds 5, 7s, and 7t showed the best inhibitory activity against all the tested bacteria, with minimum inhibitory concentrations (MICs) between 3.91 and 31.25 μg/mL. Thus, 7s emerged as a promising lead compound for the development of novel canthine-6-one antimicrobial agents.
Keywords: 10-hydroxycanthin-6-one, ester derivatives, antibacterial activity, antifungal activity
1. Introduction
The canthine-6-one alkaloids represent a fairly large subclass of β-carboline. The first member (Figure 1) in the series was isolated in 1952 from Pentaceras australis [1]. Since then, more than 60 compounds with the characteristic tetracyclic core have been isolated from many other species, principally from the Rutaceae and Simaroubaceae families, and recently from fungi [2]. These compounds exhibited a broad spectrum of biological properties, including antiviral [3,4,5], anticancer [6,7,8], antiparasitic [9,10,11], anti-inflammatory [12,13], antibacterial [14,15,16], and antifungal [17,18] activity. Notably, canthine-6-one alkaloids have received considerable attention owing to their high antimicrobial potencies. Soriano-Agaton et al. [17] reported the antifungal activity of four naturally occurring canthin-6-ones and their derivatives against five pathogenic fungi. However, the structure-activity relationship (SAR) for the antifungal activity remained unclear. Lagoutte et al. [18] investigated the mechanism of action of the antifungal canthin-6-one series in Saccharomyces cerevisiae. They showed an affinity of canthine-6-ones for lipid droplets and an effect on lipid metabolism suggesting a stimulation of alkyl chain desaturase enzyme systems. In 2007, O’Donnell et al. [16] isolated two canthin-6-ones which displayed minimum inhibitory concentrations (MICs) in the range 8–32 µg/mL against a panel of fast-growing Mycobacterium species and 8–64 µg/mL against multidrug-resistant and methicillin-resistant strains of Staphylococcus aureus. In a program screening of antimicrobials from natural products and their derivatives, our group [14,19] synthesized a series of hydrophilic derivatives of canthin-6-one having a peak MIC of 0.98 μg/mL against S. aureus.
Figure 1.
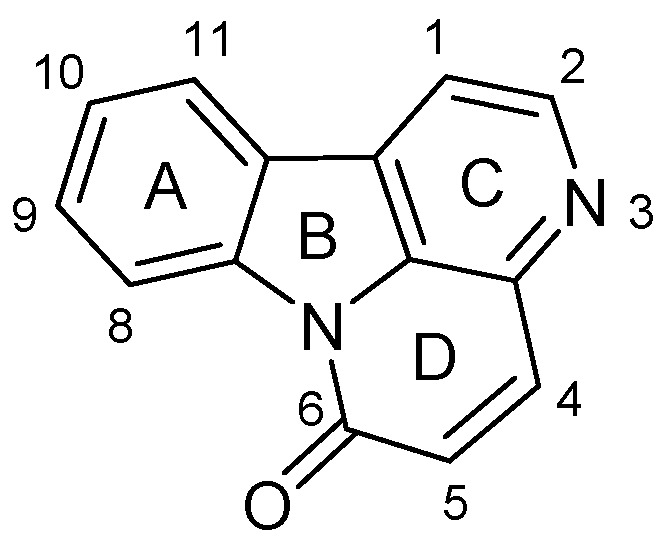
Structure of canthin-6-one.
Plant diseases caused by microorganisms have been recognized as a worldwide threat to crop production [20], and they are extremely difficult to manage in agricultural production. Moreover, the repeated use of conventional antimicrobials over many years has resulted in serious environmental and resistance problems. Libraries of natural products are expected to provide a rich resource for the discovery of new drugs [21]. In the past decades, natural product-based or -derived plant protectants have attracted lots of attention due to their low toxicity and good biocompatibility [22].
To exploit the potential antimicrobial activities of compounds having a canthin-6-one skeleton, a novel series of ester derivatives of 10-hydroxycanthin-6-one were synthesized and evaluated for their bioactivity against three phytopathogenic fungi and four bacterial strains. Furthermore, the structure-activity relationship was discussed.
2. Results and Discussion
2.1. Chemistry
The synthetic route used for the target compounds is outlined in Scheme 1. The natural product 10-methoxycanthin-6-one (5) was synthesized using a modification of previously reported methods [23], with 5-methoxytryptamine (1) as the starting material. Another key parent compound, 10-hydroxycanthin-6-one (6), was obtained by treating compound 5 with boron tribromide under an argon atmosphere, with anhydrous dichloromethane as solvent. At 0 °C, a new series of ester derivatives of 10-hydroxycanthin-6-one (7a–7x) was obtained in 48%–86% yield by the reaction of compound 6 with the corresponding acyl chloride in the presence of triethylamine.
Scheme 1.
Synthetic route and chemical structures of compounds 4–6 and 7a–7x. Reagents and conditions: (a) i: benzaldehyde, methanol, r.t.; ii: NaBH4, r.t., 98%; (b) α-ketoglutaric acid, p-TSA, dry toluene: dioxane = 3:2, DST, reflux, 83%; (c) HCOONH4, 5% Pd/C, methanol: toluene = 1:1, reflux, 75%; (d) 5% Pd/C, xylene, reflux, 90%; (e) BBr3, dry DCM, −78 °C to r.t., 63%; (f) acyl chloride, TEA, dry DCM, 0 °C, 48%–86%.

| 7b: R = 2′-pyridyl | 7h: R = 4′-ClC6H4 | 7n: R = 2′-CF3C6H4 | 7t: R = CH3CH2 |
| 7c: R = 2′-CH3C6H4 | 7i: R = 2′,4′-Cl2C6H3 | 70: R = 3′-CF3C6H4 | 7u: R = CH3(CH2)2 |
| 7d: R = 3′-CH3C6H4 | 7j: R = 4′-CH3OC6H4 | 7p: R = 4′-CF3C6H4 | 7v: R = CH3(CH2)3 |
| 7e: R = 4′-CH3C6H4 | 7k: R = 2′-FC6H4 | 7q: R = 1′-naphthyl | 7w: R = CH3(CH2)4 |
The structures of all compounds were elucidated by spectroscopic analysis, including ESI-MS, 1H-NMR spectra, 13C-NMR spectra, FT-IR, UV, and elemental analysis. All the spectral and analytical data obtained were in agreement with the assigned structures.
2.2. Biological Evaluation
2.2.1. Antifungal Activity
The in vitro antifungal properties of synthesized compounds 4–6 and 7a–7x were evaluated against three phytopathogenic fungi (Alternaria solani, Fusarium graminearum, and Fusarium solani) using the mycelium linear growth rate method [24]. Thiabendazole (TBZ), a commercially available agricultural fungicide, was used as a positive control. The results are summarized in Table 1.
Table 1.
Average inhibition rates of compounds 4–6 and 7a–7x against three phytopathogenic fungi.
| Compounds | Average Inhibition Rate ± SD (%) | |||
|---|---|---|---|---|
| No. | R | A. solani | F. graminearum | F. solani |
| 4 | - | 61.2 ± 0.4 g | 100.0 ± 0.0 a | 53.4 ± 0.7 n |
| 5 | - | 71.8 ± 0.5 b | 77.7 ± 0.4 d | 92.9 ± 0.1 a |
| 6 | - | 2.0 ± 0.5 q | 74.5 ± 0.5 e | 57.9 ± 0.6 k |
| 7a | C6H5 | 53.1 ± 0.7 i | 48.8 ± 0.4 j | 41.7 ± 0.7 q |
| 7b | 2′-pyridyl | 66.6 ± 0.0 de | 47.0 ± 0.7 k | 79.8 ± 0.5 d |
| 7c | 2′-CH3C6H4 | 2.7 ± 0.7 q | 35.6 ± 0.4 n | 65.3 ± 1.5 i |
| 7d | 3′-CH3C6H4 | 45.8 ± 0.9 k | 43.6 ± 0.6 l | 64.1 ± 0.4 j |
| 7e | 4′-CH3C6H4 | 28.7 ± 0.8 l | 23.6 ± 0.2 q | 77.0 ± 0.6 e |
| 7f | 2′-ClC6H4 | 2.1 ± 0.8 q | 31.8 ± 0.4 p | 68.1 ± 0.4 h |
| 7g | 3′-ClC6H4 | 21.4 ± 0.8 n | 3.4 ± 0.3 s | 86.1 ± 0.6 c |
| 7h | 4′-ClC6H4 | 5.7 ± 0.9 p | 3.1 ± 0.6 s | 72.2 ± 0.5 g |
| 7i | 2′,4′-Cl2C6H3 | 50.1 ± 0.8 j | 7.1 ± 0.5 r | 22.8 ± 0.6 u |
| 7j | 4′-CH3OC6H4 | 50.6 ± 0.3 j | 6.3 ± 0.4 r | 41.9 ± 0.3 q |
| 7k | 2′-FC6H4 | 68.4 ± 0.7 c | 37.6 ± 0.5 m | 40.5 ± 0.4 r |
| 7l | 3′-FC6H4 | 54.0 ± 0.9 i | 33.0 ± 0.7 o | 49.4 ± 0.2 p |
| 7m | 4′-FC6H4 | 58.9 ± 1.3 h | 51.1 ± 0.6 h | 52.1 ± 0.9 o |
| 7n | 2′-CF3C6H4 | 64.4 ± 0.9 f | 80.5 ± 0.4 c | 13.5 ± 1.0 w |
| 7o | 3′-CF3C6H4 | 25.7 ± 0.7 m | 32.6 ± 0.8 po | 54.6 ± 0.3 m |
| 7p | 4′-CF3C6H4 | 8.3 ± 0.3 o | 80.1 ± 1.0 c | 22.9 ± 0.4 u |
| 7q | 1′-naphthyl | 65.1 ± 1.1 ef | 49.9 ± 0.6 i | 25.2 ± 0.7 t |
| 7r | 2′-naphthyl | 25.8 ± 0.2 m | 50.2 ± 0.6 hi | 20.3 ± 0.1 v |
| 7s | CH3 | 68.4 ± 0.6 c | 100.0 ± 0.0 a | 86.0 ± 0.2 c |
| 7t | CH3CH2 | 59.1 ± 0.9 h | 70.0 ± 0.3 f | 80.1 ± 0.3 d |
| 7u | CH3(CH2)2 | 66.9 ± 0.9 cd | 44.4 ± 0.7 l | 69.2 ± 0.9 h |
| 7v | CH3(CH2)3 | 49.9 ± 0.8 j | 85.4 ± 0.4 b | 74.2 ± 0.2 f |
| 7w | CH3(CH2)4 | 66.5 ± 0.9 de | 55.1 ± 0.2 g | 56.6 ± 0.3 l |
| 7x | CH3(CH2)10 | 72.2 ± 0.2 b | 85.6 ± 0.4 b | 29.8 ± 0.4 s |
| TBZ | - | 89.1 ± 0.6 a | 100.0 ± 0.0 a | 89.3 ± 0.4 b |
All values are expressed as means ± SD (n = 3). Tested fungi were treated with each compound at a concentration of 50 μg/mL for 72 h. The differences between data with the different lower case letters within a column are significant for the same tested fungus (p < 0.05).
As shown in Table 1, all the compounds displayed activity to some degree against each of the tested fungi. In general, the compounds showed the highest activity against F. graminearum, followed by F. solani. At a concentration of 50 μg/mL, 58% of the test items (47/81) revealed good activity, with inhibition rates of 49.9%–100%. Compounds 4 and 7s had the highest activity against F. graminearum, with inhibition rates of 100%, equal to that of TBZ. Two compounds (5 and 7x) had inhibition rates exceeding 70% against A. solani. Six compounds (4, 7n, 7p, 7s, 7v, and 7x) displayed more than 80% inhibition activity against F. graminearum, and four compounds (5, 7g, 7s, and 7t) had over 80% inhibition activity against F. solani. Of these compounds, compounds 5, 7s, and 7t showed broad-spectrum fungicidal activity against all the phytopathogenic fungi tested. Compound 7g showed special activity against F. solani, displaying 86.1% inhibition activity, but had a low level of activity against F. graminearum and A. solani. Compound 7p exhibited much higher activity against F. graminearum than against F. solani and A. solani.
2.2.2. Antibacterial Activity
The antibacterial activity of the compounds was investigated using a previously reported method [14,25]. First, compounds 4–6 and 7a–7x (10 μg) were screened for their antibacterial activity against two Gram-positive (Bacillus cereus and Bacillus subtilis) and two Gram-negative bacterial strains (Ralstonia solanacearum and Pseudomonas syringae) using the disk diffusion method. Penicillin sodium was used as a positive control, and a dichloromethane poured disk as a negative control. The diameter of the inhibition zone was assayed to make a preliminary evaluation of the antibacterial potential of the tested compounds. The results are summarized in Table 2.
Table 2.
Inhibition zones (mm) of some synthesized compounds (10 μg).
| Compounds | Gram-Positive Bacteria | Gram-Negative Bacteria | ||
|---|---|---|---|---|
| B. cereus | B. subtilis | R. solanacearum | P. syringae | |
| 5 | 11.2 ± 0.5 | 10.9 ± 0.8 | 12.6 ± 0.4 | 10.9 ± 1.0 |
| 6 | - * | 9.8 ± 0.4 | - | 8.5 ± 0.1 |
| 7s | 12.2 ± 0.1 | 16.9 ± 0.7 | 12.3 ± 0.2 | 18.4 ± 0.2 |
| 7t | 16.6 ± 0.2 | 17.4 ± 0.1 | 13.0 ± 0.2 | 21.0 ± 0.4 |
| 7u | 13.5 ± 0.3 | 13.9 ± 0.4 | - | 13.0 ± 0.8 |
| 7v | 12.7 ± 0.2 | 10.3 ± 1.1 | - | 15.4 ± 0.2 |
| 7w | 9.4 ± 0.1 | 8.5 ± 0.3 | - | 12.5 ± 0.2 |
| Penicillin Sodium | - | 22.5 ± 0.2 | 16.2 ± 0.2 | 20.4 ± 0.8 |
All values are expressed as means ± SD (n = 3). * Inhibition zone < 8 mm.
The results revealed that compounds 4, 7a–7r, and 7x showed no antibacterial activity (growth inhibition zones < 8 mm), while compounds 5, 6, and 7s–7w exhibited moderate activity against both Gram-positive and Gram-negative bacteria. Compounds 7s and 7t showed higher antibacterial activities than the parent compounds 5 and 6. However, compounds 7u–7w exhibited equivalent or weaker antibacterial activity than compound 5.
The MICs of compounds 5, 6, and 7s–7w were determined in further studies using the micro-broth dilution method. The results are shown in Table 3.
Table 3.
The in vitro antibacterial activity as MIC (μg/mL) for compounds 5, 6, and 7s–7w.
| Compounds | Gram-Positive Bacteria | Gram-Negative Bacteria | ||
|---|---|---|---|---|
| B. cereus | B. subtilis | R. solanacearum | P. syringae | |
| 5 | 3.91 | 7.81 | 15.62 | 7.81 |
| 6 | 15.62 | >125 | >125 | >125 |
| 7s | 15.62 | 15.62 | 31.25 | 7.81 |
| 7t | 15.62 | 7.81 | 15.62 | 7.81 |
| 7u | 31.25 | 31.25 | ND * | 15.62 |
| 7v | >125 | 125 | ND | 31.25 |
| 7w | >125 | 62.5 | ND | >125 |
| Penicillin Sodium | 3.91 | 7.81 | 7.81 | 0.98 |
* Not determined.
Significant activity was observed for compounds 5, 7s, and 7t against all the tested bacterial strains, with MICs between 3.91 and 31.25 μg/mL. Compound 5 was active against the tested Gram-positive bacteria, with MICs equivalent to that of penicillin sodium. However, none of the investigated derivatives were found to be superior to the reference drug.
2.3. Structure-Activity Relationship
Based on the results of the present study, the structure–activity relationship (SAR) for the antifungal activity may be summarized as shown in Figure 2. Comparison of the antifungal activities of compound 4 and 10-methoxycanthin-6-one (5) suggests that the conjugated unsaturated bonds of rings C and D (see Figure 1) are non-essential. The aliphatic ester derivatives of 10-hydroxycanthin-6-one had much higher antifungal activity than did the aromatic ester derivatives.
Figure 2.

Structure-activity relationship for the antifungal activity.
The weak antibacterial activity of compound 4 suggests that the conjugated unsaturated bonds of rings C and D play an important role in antibacterial activity. The introduction of aromatic esters decreases the antibacterial activity. Other compounds with a shorter or longer aliphatic chain did not show any distinct effect, except for compound 7x (Figure 3).
Figure 3.

Structure-activity relationship for the antibacterial activity.
3. Materials and Methods
3.1. General Details
All the reagents and solvents were obtained locally and were of analytical grade or were purified according to standard methods before use. The tested fungi and bacteria (B. cereus, B. subtilis, R. solanacearum, P. syringae, A. solani, F. graminearum, and F. solani) were provided by the Institute of Pesticide Science, Northwest A&F University, China. Melting points were determined using a digital melting-point apparatus and were uncorrected. 1H-NMR (500 MHz) and 13C-NMR (125 MHz) spectra were recorded using a Bruker Avance III 500 MHz instrument (Bruker, Madison, WI, USA), with TMS as the internal standard and CDCl3 or dimethylsulfoxide (DMSO-d6) as the solvent. Chemical shifts (δ values) and coupling constants (J values) are given in this paper in ppm and Hz, respectively. ESI-MS was carried out using a Thermo Fisher LCQ Fleet instrument (Shimadzu, Beijing, China). Elemental analysis of the synthesized compounds was undertaken using a CE-440 elemental analyzer (EAI, North Chelmsford, MA, USA). Ultraviolet spectra (UV) were obtained using a Thermo Scientific Evolution 300 UV-vis spectrophotometer (Shimadzu, Beijing, China). Infrared spectra (IR) were recorded on a Bruker TENSOR 27 spectrometer (Bruker, Madison, WI, USA) with KBr disks.
3.2. Synthesis of Target Compounds
3.2.1. Synthesis of Compound 2
A previously described method [26] was used to synthesize compound 2. A solution of 5-methoxytryptamine (1) (4.76 g, 25 mmol) and benzaldehyde (3.06 mL, 30 mmol) in methanol was vigorously stirred at room temperature for 36 h. Solid NaBH4 (2.84 g, 75 mmol) was added to the resulting solution, and the mixture stirred at room temperature for 1 h. The reaction was quenched by adding saturated aqueous NaHCO3 (100 mL), and the mixture concentrated under vacuum until approximately 50 mL remained. The residue was filtered, and the filter cake was washed with CH2Cl2. The organic phase was separated, and the aqueous phase was extracted with CH2Cl2 (3 × 50 mL). The combined organic phases were washed with brine (3 × 50 mL) and dried over anhydrous Na2SO4. The organic solvent was removed under vacuum to give the title compound as a brown solid (6.87 g, yield 98%, m.p. 71.2–72.3 °C), which was used without further purification. 1H-NMR (500 MHz, CDCl3) δ: 8.06 (s, 1H), 7.37–7.28 (m, 2H), 7.26–7.18 (m, 4H), 7.02 (d, J = 2.5 Hz, 1H), 6.95 (d, J = 2.2 Hz, 1H), 6.84 (dd, J = 8.8, 2.4 Hz, 1H), 3.87–3.76 (m, 5H), 3.01–2.93 (m, 4H), 1.87(s, 1H). 13C-NMR (125 MHz, CDCl3) δ: 153.9, 140.1, 131.6, 128.4, 128.2, 127.9, 127.0, 122.8, 113.5, 112.3, 111.9, 100.8, 56.0, 53.9, 49.3, 25.8. ESI-MS m/z: [M + H]+(found) = 281.16, [M + H]+(calcd) = 281.37. Elemental analysis calculated for C18H20N2O (%): C, 77.11; H, 7.19; N, 9.99 (found: C, 77.02; H, 7.23; N, 10.05). 1H-NMR, 13C-NMR, and FT-IR spectra for some target compounds were showed in the Supplementary Materials.
3.2.2. Synthesis of Compound 3
A mixture of compound 2 (5.61 g, 20 mmol), α-ketoglutaric acid (3.51 g, 24 mmol), p-toluenesulfonic acid (0.69 g, 4 mmol), and a solution of toluene/dioxane (60 mL 3:2 v/v) was heated at 180 °C for 48 h, using a Dean-Stark trap for water removal. Upon completion, the reaction mixture was cooled to room temperature and the organic solvent removed. The residue was dissolved in a mixture of CH2Cl2 (60 mL) and saturated aqueous NaHCO3 (80 mL). The aqueous phase was separated and the product extracted with CH2Cl2 (3 × 50 mL). The combined organic phases were washed with brine (3 × 50 mL) and dried over anhydrous Na2SO4. The obtained crude product was chromatographed on silica gel eluting with dichloromethane to afford compound 3 as a white powder (5.75 g, yield 83%, m.p. 192.5–193.7 °C). 1H-NMR (500 MHz, CDCl3) δ: 8.26 (d, J = 8.8 Hz, 1H), 7.43–7.27 (m, 5H), 6.89 (dd, J = 8.8, 2.5 Hz, 1H), 6.86 (d, J = 2.5 Hz, 1H), 4.23 (d, J = 13.5 Hz, 1H), 3.85 (s, 3H), 3.56–3.52 (m, 1H), 3.39 (d, J = 13.5 Hz, 1H), 3.25–3.15 (m, 1H), 2.91–2.68 (m, 3H), 2.61–2.47 (m, 3H), 1.96–1.85 (m, 1H). 13C-NMR (125 MHz, CDCl3) δ: 167.7, 156.9, 138.5, 135.4, 130.5, 129.6, 128.9, 128.5, 127.3, 116.9, 113.4, 112.0, 101.8, 57.7, 57.3, 55.7, 50.1, 32.8, 27.6, 21.4. ESI-MS m/z: [M + H]+(found) = 347.23, [M + H]+(calcd) = 347.43. Elemental analysis calculated for C22H22N2O2 (%): C, 76.28; H, 6.40; N, 8.09 (found: C, 76.12; H, 6.51; N, 8.03).
3.2.3. Synthesis of Compound 4
To a flask was added compound 3 (5.75 g, 16.6 mmol), HCOONH4 (10.52 g, 167 mmol), Pd/C (5%, 1.5 g), methanol (25 mL), and toluene (25 mL). The mixture was stirred vigorously at 80 °C. After 4 h, the reaction was quenched by addition of saturated aqueous NaHCO3 until pH 8.5 was reached. The mixture was filtered, and the filter cake was washed with copious amounts of dichloromethane and methanol until TLC indicated complete removal of the product. The combined filtrates were concentrated under vacuum until approximately 50 mL remained, and then extracted with CH2Cl2 (3 × 80 mL). The combined organic phases were washed with brine (3 × 80 mL) and dried over anhydrous Na2SO4. The sample was concentrated and purified using silica gel column chromatography, eluting with ethyl acetate-methanol (v/v = 2:1) to give product 4 as a green solid (3.19 g, yield 75%, m.p. > 250 °C). 1H-NMR (500 MHz, CDCl3) δ 8.24 (d, J = 8.9, 1H), 6.92–6.85 (m, 2H), 4.02–3.95 (m, 1H), 3.86 (s, 3H), 3.56–3.49 (m, 1H), 3.18–3.10 (m, 1H), 2.86–2.67 (m, 3H), 2.66–2.58 (m, 1H), 2.33–2.24 (m, 1H), 1.85–1.74 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ: 167.8, 156.9, 136.9, 131.0, 129.2, 116.8, 112.8, 112.0, 101.9, 55.8, 50.7, 43.9, 33.1, 29.2, 21.7. ESI-MS m/z: [M + H]+(found) = 257.09, [M + H]+(calcd) = 257.31. Elemental analysis calculated for C15H16N2O2 (%): C, 70.29; H, 6.29; N, 10.93 (found: C, 70.33; H, 6.26; N, 10.89).
3.2.4. Synthesis of Compound 5
To a stirred solution of compound 4 (2.56 g, 10 mmol) in xylene was added Pd/C (5%, 1.8 g), and the mixture was heated at reflux for 4 days. The mixture was then cooled and filtered, and the filtered solid washed carefully with dichloromethane and methanol. The combined filtrates were concentrated under vacuum and purified using column chromatography on silica gel using dichloromethane–acetone (v/v = 5:1) as eluent to give compound 5 as a bright yellow solid (2.25 g, yield 90%, m.p. 202.9–204.1 °C). IR, υmax (KBr) cm−1: 1676 (C=O), 1450 (C=N). UV λmax (lg ε) (DCM) 380 (3.91) nm. 1H-NMR (500 MHz, DMSO-d6) δ: 8.84 (d, J = 5.0 Hz, 1H), 8.38 (d, J = 8.9 Hz, 1H), 8.30 (d, J = 5.0 Hz, 1H), 8.12 (d, J = 9.8 Hz, 1H), 7.99 (d, J = 2.6 Hz, 1H), 7.34 (dd, J = 8.9, 2.6 Hz, 1H), 6.99 (d, J = 9.8 Hz, 1H), 3.92 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ: 159.3, 157.9, 145.7, 139.3, 136.4, 133.8, 132.5, 130.3, 129.0, 125.7, 118.0, 118.0, 116.4, 106.5, 56.0. ESI-MS m/z: [M + H]+(found) = 251.25, [M + H]+(calcd) = 251.26. Elemental analysis calculated for C15H10N2O2 (%): C, 71.99; H, 4.03; N, 11.19 (found: C, 72.03; H, 4.12; N, 11.07).
3.2.5. Synthesis of Compound 6
To a solution of 10-methoxycanthin-6-one (2.25 g, 9 mmol) in anhydrous dichloromethane (100 mL) was added dropwise boron tribromide (8.50 mL, 90 mmol) under argon at −78 °C, and the mixture stirred overnight at room temperature. The reaction was cautiously quenched with iced water (80 mL). The solids were filtered and the filter cake washed with dichloromethane and then purified using silica gel column chromatography to give 10-hydroxycanthin-6-one (6) as a yellow powder (1.34 g, yield 63%, m.p. > 250 °C). IR, υmax (KBr) cm−1: 3424 (OH), 1691 (C=O), 1436 (C=N). UV λmax (lg ε) (DCM) 375 (3.89) nm. 1H-NMR (500 MHz, DMSO-d6) δ: 10.01 (s, 1H), 8.79 (d, J = 5.0 Hz, 1H), 8.30 (d, J = 8.8 Hz, 1H), 8.23 (d, J = 5.0 Hz, 1H), 8.08 (d, J = 9.8 Hz, 1H), 7.67 (d, J = 2.5 Hz, 1H), 7.19 (dd, J = 8.8, 2.5 Hz, 1H), 6.96 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, DMSO-d6) δ 158.9, 156.2, 146.1, 139.7, 136.2, 132.4, 132.2, 130.0, 129.0, 126.0, 119.0, 117.7, 117.4, 109.5.ESI-MS m/z: [M + H]+(found) = 237.21, [M + H]+(calcd) = 237.23. Elemental analysis calculated for C14H8N2O2 (%): C, 71.18; H, 3.41; N, 11.86 (found: C, 71.20; H, 3.45; N, 11.77).
3.2.6. General Procedure for the Synthesis of Compounds 7a–7x
According to the previously reported method [27], compounds 7a–7x were prepared through the direct condensation of 10-hydroxycanthin-6-one (6) with the corresponding acyl chloride. The general procedure was as follows. A solution of benzoyl chloride (0.05 mL, 0.45 mmol) in CH2Cl2 (5 mL) was added slowly to an ice-cooled mixture of 10-hydroxycanthin-6-one (0.71 g, 0.3 mmol) and triethylamine (0.21 mL, 1.5 mmol) in anhydrous CH2Cl2 (30 mL) over 15 min with stirring. The reaction mixture was stirred overnight at room temperature to provide a clear solution, and then quenched with saturated aqueous NaHCO3 (50 mL). The organic phase was separated, washed with brine (3 × 50 mL), and finally dried over anhydrous Na2SO4. The solvent was removed and the residue purified by column chromatography on silica gel, eluting with dichloromethane–acetone (v/v = 20:1) to afford 7a as a pale yellow powder (0.82 g, yield 80%, m.p. 220.4–221.2 °C). IR, υmax (KBr) cm−1: 1685 (C=O), 1669 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 364 (4.25) nm, 383 (4.24) nm. 1H-NMR (500 MHz, CDCl3) δ 8.87 (d, J = 5.0 Hz, 1H), 8.75 (d, J = 8.7 Hz, 1H), 8.33–8.25 (m, 2H), 8.09 (d, J = 9.8 Hz, 1H), 8.05 (d, J = 2.3 Hz, 1H), 7.97 (d, J = 5.0 Hz, 1H), 7.76–7.69 (m, 1H), 7.63–7.56 (m, 3H), 7.04 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ165.3, 159.3, 148.7, 145.9, 139.6, 137.0, 136.4, 134.0, 130.3, 130.0, 129.8, 129.1, 129.0, 128.7, 125.5, 124.5, 118.0, 116.7, 116.2. ESI-MS m/z: [M + H]+(found) = 341.35, [M + H]+(calcd) = 341.34; Elemental analysis calculated for C21H12N2O3 (%): C, 74.11; H, 3.55; N, 8.23 (found: C, 74.09; H, 3.45; N, 8.26).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl nicotinate (7b): Yield: 64%; yellow solid; m.p. 235.8–236.8 °C; IR, υmax (KBr) cm−1: 1739 (C=O), 1683 (ester), 1588 (C=N). UV λmax (lg ε) (DCM) 364 (4.04) nm, 382 (4.03) nm. 1H-NMR (500 MHz, CDCl3) δ 9.45 (d, J = 2.2 Hz, 1H), 8.90 (dd, J = 4.9, 1.7 Hz, 1H), 8.84 (d, J = 5.0 Hz, 1H), 8.73 (d, J = 8.8 Hz, 1H), 8.50 (dt, J = 7.9, 2.0 Hz, 1H), 8.06–8.00 (m, 2H), 7.93 (d, J = 5.0 Hz, 1H), 7.56 (dd, J = 8.8, 2.3 Hz, 1H), 7.51 (ddd, J = 8.0, 4.9, 0.9 Hz, 1H), 6.99 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 164.0, 159.3, 154.3, 151.5, 148.2, 146.0, 139.8, 137.7, 137.2, 136. 6, 132.6, 129.6, 129.0, 125.6, 125.3, 124.3, 123.6, 118.1, 116.6, 116.1. ESI-MS m/z: [M + H]+(found) = 342.34, [M + H]+(calcd) = 342.33; Elemental analysis calculated for C20H11N3O3 (%): C, 70.38; H, 3.25; N, 12.31 (found: C, 70.25; H, 3.13; N, 12.28).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 2-methylbenzoate (7c): Yield: 85%; yellow powder; m.p. 214.7–215.8 °C; IR, υmax (KBr) cm−1: 1715 (C=O), 1667 (ester), 1423 (C=N). UV λmax (lg ε) (DCM) 364 (4.26) nm, 382 (4.24) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 4.9 Hz, 1H), 8.72 (d, J = 8.8 Hz, 1H), 8.23 (d, J = 7.9 Hz, 1H), 8.04 (d, J = 9.8 Hz, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.94 (d, J = 5.0 Hz, 1H), 7.57–7.49 (m, 2H), 7.40–7.33 (m, 2H), 7.00 (d, J = 9.8 Hz, 1H), 2.72 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 165.8, 159.4, 148.7, 146.0, 141.6, 139.7, 137.0, 136.5, 133.1, 132.6, 132.1, 131.3, 129.8, 129.0, 128.1, 126.1, 125.6, 124.6, 118.0, 116.6, 116.3, 22.0. ESI-MS m/z: [M + H]+(found) = 355.33, [M + H]+(calcd) = 355.37; Elemental analysis calculated for C22H14N2O3 (%): C, 74.57; H, 3.98; N, 7.91 (found: C, 74.62; H, 4.03; N, 7.88).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 3-methylbenzoate (7d): Yield: 80%; pale yellow powder; m.p. 183.2–183.9 °C; IR, υmax (KBr) cm−1: 1738 (C=O), 1636 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 364 (4.26) nm, 382 (4.24) nm. 1H-NMR (500 MHz, CDCl3) δ 8.81 (dd, J = 6.6, 4.8 Hz, 1H), 8.69 (t, J = 8.9 Hz, 1H), 8.07–7.97 (m, 4H), 7.90 (dd, J = 8.9, 4.9 Hz, 1H), 7.54 (dd, J = 8.8, 2.3 Hz, 1H), 7.48 (d, J = 7.7 Hz, 1H), 7.42 (td, J = 7.5, 2.9 Hz, 1H), 6.98 (dd, J = 9.8, 5.9 Hz, 1H), 2.46 (d, J = 2.3 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 165.4, 159.3, 148.7, 145.9, 139.6, 138.6, 137.0, 136.5, 134.7, 132.5, 130.8, 129.7, 129.1, 129.0, 128.6, 127.4, 125.5, 124.5, 118.0, 116.6, 116.2, 21.3. ESI-MS m/z: [M + H]+(found) = 355.35, [M + H]+(calcd) = 355.37; Elemental analysis calculated for C22H14N2O3 (%): C, 74.57; H, 3.98; N, 7.91 (found: C, 74.62; H, 3.96; N, 7.97).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 4-methylbenzoate (7e): Yield: 83%; pale yellow powder; m.p. 227.9–228.5 °C; IR, υmax (KBr) cm−1: 1732 (C=O), 1636 (ester), 1421 (C=N). UV λmax (lg ε) (DCM) 364 (4.22) nm, 382 (4.20) nm. 1H-NMR (500 MHz, CDCl3) δ 8.83 (d, J = 5.0 Hz, 1H), 8.71 (d, J = 8.8 Hz, 1H), 8.14 (d, J = 8.1 Hz, 2H), 8.03 (d, J = 9.8 Hz, 1H), 8.01 (d, J = 2.2 Hz, 1H), 7.93 (d, J = 5.0 Hz, 1H), 7.55 (dd, J = 8.8, 2.3 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.00 (d, J = 9.8 Hz, 1H), 2.48 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 165.3, 159.4, 148.8, 146.0, 144.9, 139.7, 137.0, 136.5, 132.6, 130.3, 129.8, 129.5, 129.0, 126.4, 125.5, 124.6, 118.0, 116.6, 116.3, 21.8. ESI-MS m/z: [M + H]+(found) = 355.35, [M + H]+(calcd) = 355.37; Elemental analysis calculated for C22H14N2O3 (%): C, 74.57; H, 3.98; N, 7.91 (found: C, 74.43; H, 3.99; N, 7.86).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 2-chlorobenzoate (7f): Yield: 72%; pale yellow powder; m.p. 208.9–210.3 °C; IR, υmax (KBr) cm−1: 1744 (C=O), 1676 (ester), 1416 (C=N). UV λmax (lg ε) (DCM) 364 (4.31) nm, 382 (4.32) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.72 (d, J = 8.8 Hz, 1H), 8.11 (dd, J = 7.8, 1.2 Hz, 1H), 8.06–8.02 (m, 2H), 7.94 (d, J = 5.0 Hz, 1H), 7.59–7.52 (m, 3H), 7.46–7.41 (m, 1H), 6.99 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 164.1, 159.4, 148.3, 146.0, 139.7, 137.1, 136.5, 134.6, 133.6, 132.6, 132.0, 131.5, 129.7, 129.0, 128.9, 126.9, 125.6, 124.4, 118.1, 116.7, 116.1. ESI-MS m/z: [M + H]+(found) = 375.33, [M + H]+(calcd) = 375.78; Elemental analysis calculated for C21H11ClN2O3 (%): C, 67.30; H, 2.96; N, 7.47 (found: C, 67.22; H, 2.98; N, 7.53).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 3-chlorobenzoate (7g): Yield: 75%; pale yellow powder; m.p. 240.2–241.1 °C; IR, υmax (KBr) cm−1: 1742 (C=O), 1677 (ester), 1422 (C=N). UV λmax (lg ε) (DCM) 364 (4.33) nm, 382 (4.34) nm. 1H-NMR (500 MHz, CDCl3) δ 8.85 (d, J = 5.0 Hz, 1H), 8.75 (d, J = 8.8 Hz, 1H), 8.24 (t, J = 1.8 Hz, 1H), 8.14 (dt, J = 7.8, 1.2 Hz, 1H), 8.05 (d, J = 9.8 Hz, 1H), 8.02 (d, J = 2.2 Hz, 1H), 7.95 (d, J = 5.0 Hz, 1H), 7.69–7.63 (m, 1H), 7.56 (dd, J = 8.8, 2.3 Hz, 1H), 7.51 (t, J = 7.9 Hz, 1H), 7.01 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 164.1, 159.4, 148.4, 146.0, 139.8, 137.2, 136.6, 135.0, 134.0, 132.6, 131.0, 130.3, 130.1, 129.7, 129.0, 128.4, 125.6, 124.4, 118.1, 116.6, 116.1. ESI-MS m/z: [M + H]+(found) = 375.33, [M + H]+(calcd) = 375.78; Elemental analysis calculated for C21H11ClN2O3 (%): C, 67.30; H, 2.96; N, 7.47 (found: C, 67.33; H, 3.04; N, 7.32).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 4-chlorobenzoate (7h): Yield: 69%; pale yellow powder; m.p. 239.8–240.7 °C; IR, υmax (KBr) cm−1: 1734 (C=O), 1684 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 364 (4.53) nm, 382(4.52) nm. 1H-NMR (500 MHz, CDCl3) δ 8.82 (d, J = 5.0 Hz, 1H), 8.71 (d, J = 8.8 Hz, 1H), 8.18 (d, J = 8.6 Hz, 2H), 8.03 (d, J = 9.8 Hz, 1H), 7.99 (d, J = 2.2 Hz, 1H), 7.91 (d, J = 5.0 Hz, 1H), 7.57–7.49 (m, 3H), 6.99 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 164.4, 159.3, 148.4, 146.0, 140.6, 139.7, 137.1, 136.5, 132.6, 131.6, 129.7, 129.1, 129.0, 127.6, 125.6, 124.4, 118.0, 116.6, 116.1. ESI-MS m/z: [M + H]+(found) = 375.35, [M + H]+(calcd) = 375.78; Elemental analysis calculated for C21H11ClN2O3 (%): C, 67.30; H, 2.96; N, 7.47 (found: C, 67.36; H, 2.95; N, 7.53).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 2, 4-dichlorobenzoate (7i): Yield: 78%; pale yellow powder; m.p. 246.2–247.1 °C; IR, υmax (KBr) cm−1: 1738 (C=O), 1676 (ester), 1423 (C=N). UV λmax (lg ε) (DCM) 364 (4.23) nm, 383 (4.25) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.73 (d, J = 8.8 Hz, 1H), 8.09 (d, J = 8.5, 1H), 8.07–8.01 (m, 2H), 7.95 (d, J = 5.0 Hz, 1H), 7.59 (d, J = 2.0 Hz, 1H), 7.56 (dd, J = 8.8, 2.3 Hz, 1H), 7.43 (dd, J = 8.4, 2.0 Hz, 1H), 7.00 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 163.2, 159.4, 148.2, 146.0, 139.8, 139.6, 137.2, 136.6, 135.9, 133.1, 132.6, 131.5, 129.7, 129.0, 127.4, 127.0, 125.6, 124.3, 118.1, 116.7, 116.1. ESI-MS m/z: [M + H]+(found) = 410.34, [M + H]+(calcd) = 410.22; Elemental analysis calculated for C21H10Cl2N2O3 (%): C, 61.64; H, 2.46; N, 6.85 (found: C, 61.61; H, 2.38; N, 6.77).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 4-methoxybenzoate (7j): Yield: 72%; yellow powder; m.p. > 250 °C; IR, υmax (KBr) cm−1: 1726 (C=O), 1673 (ester), 1419 (C=N). UV λmax (lg ε) (DCM) 364 (3.98) nm, 382 (3.96) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.73 (d, J = 8.8 Hz, 1H), 8.24–8.18 (m, 2H), 8.05 (d, J = 9.8 Hz, 1H), 8.02 (d, J = 2.2 Hz, 1H), 7.94 (d, J = 5.0 Hz, 1H), 7.55 (dd, J = 8.8, 2.3 Hz, 1H), 7.05–6.98 (m, 3H), 3.92 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 165.0, 164.2, 159.4, 148.8, 146.0, 139.7, 137.0, 136.5, 132.6, 132.4, 129.9, 129.0, 129.5, 124.6, 121.4, 118.0, 116.6, 116.3, 114.0, 55.6. ESI-MS m/z: [M + H]+(found) = 371.34, [M + H]+(calcd) = 371.36; Elemental analysis calculated for C22H14N2O4 (%): C, 71.35; H, 3.81; N, 7.56 (found: C, 71.29; H, 3.92; N, 7.49).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 2-fluorobenzoate (7k): Yield: 65%; pale yellow powder; m.p. 245.3–246.2 °C; IR, υmax (KBr) cm−1: 1744 (C=O), 1675 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 364 (4.36) nm, 382 (4.34) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.72 (d, J = 8.8 Hz, 1H), 8.16 (td, J = 7.6, 1.9 Hz, 1H), 8.08–8.00 (m, 2H), 7.94 (d, J = 5.0 Hz, 1H), 7.68–7.62 (m, 1H), 7.57 (dd, J = 8.8, 2.3 Hz, 1H), 7.32 (td, J = 7.6, 1.1 Hz, 1H), 7.29–7.22 (m, 1H), 7.00 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 162.9 (d, J = 4.0 Hz), 162.4 (d, J = 262.7 Hz), 159.4, 148.3, 146.0, 139.7, 137.1, 136.5, 135.6 (d, J = 262.7 Hz), 132.6, 129.8, 129.0, 125.5, 124.5, 124.3, 124.3, 118.0, 117.7 (d, J = 9.7 Hz), 117.4 (d, J = 22.4 Hz), 116.6, 116.2. ESI-MS m/z: [M + H]+(found) = 359.34, [M + H]+(calcd) = 359.33; Elemental analysis calculated for C21H11FN2O3 (%): C, 70.39; H, 3.09; N, 7.82 (found: C, 70.41; H, 3.06; N, 7.81).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 3-fluorobenzoate (7l): Yield: 62%; pale yellow powder; m.p. 233.4–234.2 °C; IR, υmax (KBr) cm−1: 1747 (C=O), 1675 (ester), 1422 (C=N). UV λmax (lg ε) (DCM) 364 (4.16) nm, 382 (4.15) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.73 (d, J = 8.8 Hz, 1H), 8.07–8.02 (m, 2H), 8.01 (d, J = 2.3 Hz, 1H), 7.95–7.90 (m, 2H), 7.57–7.51 (m, 2H), 7.41–7.36 (m, 1H), 7.00 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 164.2 (d, J = 3.2 Hz), 163.7 (d, J = 247.0 Hz), 159.4, 148.4, 146.0, 139.7, 137.1, 136.6, 132.6, 131.3 (d, J = 7.7 Hz), 130.4 (d, J = 7.8 Hz), 129.7, 129.0, 126.0 (d, J = 3.1 Hz), 125.6, 124.4, 121.1 (d, J = 21.6 Hz), 118.1, 117.2 (d, J = 22.2 Hz), 116.6, 116.1. ESI-MS m/z: [M + H]+(found) = 359.33, [M + H]+(calcd) = 359.33; Elemental analysis calculated for C21H11FN2O3 (%): C, 70.39; H, 3.09; N, 7.82 (found: C, 70.44; H, 3.12; N, 7.79).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 4-fluorobenzoate (7m): Yield: 66%; pale yellow powder; m.p. 226.2–227.4 °C; IR, υmax (KBr) cm−1: 1736 (C=O), 1687 (ester), 1417 (C=N). UV λmax (lg ε) (DCM) 364 (4.15) nm, 382 (4.12) nm. 1H-NMR (500 MHz, CDCl3) δ 8.83 (d, J = 5.0 Hz, 1H), 8.72 (d, J = 8.8 Hz, 1H), 8.30–8.24 (m, 2H), 8.03 (d, J = 9.8 Hz, 1H), 8.00 (d, J = 2.3 Hz, 1H), 7.92 (d, J = 5.0 Hz, 1H), 7.54 (dd, J = 8.8, 2.3 Hz, 1H), 7.25–7.20 (m, 2H), 6.99 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 167.4 (d, J = 257.0 Hz), 164.3, 159.4, 148.5, 146.0, 139.7, 137.1, 136.5, 132.6, 132.4 (d, J = 9.6), 129.7, 129.0, 125.6, 125.4 (d, J = 3.0 Hz), 124.4, 118.0, 116.6, 116.2, 116.0 (d, J = 23.1 Hz). ESI-MS m/z: [M + H]+(found) = 359.34, [M + H]+(calcd) = 359.33; Elemental analysis calculated for C21H11FN2O3 (%): C, 70.39; H, 3.09; N, 7.82 (found: C, 70.26; H, 2.98; N, 7.87).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 2-(trifluoromethyl) benzoate (7n): Yield: 61%; pale yellow powder; m.p. 191.3–192.1 °C; IR, υmax (KBr) cm−1: 1754 (C=O), 1681 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 363 (4.26) nm, 382 (4.24) nm. 1H-NMR (500 MHz, CDCl3) δ 8.85 (d, J = 5.0 Hz, 1H), 8.72 (d, J = 8.8 Hz, 1H), 8.08–8.00 (m, 3H), 7.97 (d, J = 5.0 Hz, 1H), 7.90–7.86 (m, 1H), 7.76–7.71 (m, 2H), 7.57 (dd, J = 8.8, 2.3 Hz, 1H), 6.99 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 165.2, 159.4, 148.2, 146.0, 139.8, 137.2, 136.6, 132.6, 132.1, 132.0, 130.8, 130.0, 129.7, 129.2 (d, J = 42.5 Hz), 127.1 (d, J = 5.4 Hz), 127.1 (d, J = 5.5 Hz), 125.6, 124.1, 123.5 (d, J = 273.8 Hz), 118.1, 116.7, 115.8. ESI-MS m/z: [M + H]+(found) = 409.38, [M + H]+(calcd) = 409.34; Elemental analysis calculated for C22H11F3N2O3 (%): C, 64.71; H, 2.72; N, 6.86 (found: C, 64.66; H, 2.73; N, 6.89).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 3-(trifluoromethyl) benzoate (7o): Yield: 62%; pale yellow powder; m.p. 199.9–200.8 °C; IR, υmax (KBr) cm−1: 1742 (C=O), 1687 (ester), 1422 (C=N). UV λmax (lg ε) (DCM) 364 (4.31) nm, 382 (4.33) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.74 (d, J = 8.8 Hz, 1H), 8.52 (s, 1H), 8.44 (d, J = 7.8 Hz, 1H), 8.07–8.00 (m, 2H), 7.96–7.91 (m, 2H), 7.71 (t, J = 7.8 Hz, 1H), 7.57 (dd, J = 8.8, 2.3 Hz, 1H), 7.00 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 164.0, 159.4, 148.3, 146.0, 139.8, 137.2, 136.6, 133.4, 132.6, 131.6 (d, J = 33.4 Hz), 130.4 (d, J = 3.8 Hz), 130.1, 129.6, 129.5, 129.0, 127.2 (d, J = 3.9 Hz), 125.6, 124.6 (d, J = 272.7 Hz), 124.3, 118.1, 116.6, 116.0. ESI-MS m/z: [M + H]+(found) = 409.40, [M + H]+(calcd) = 409.34; Elemental analysis calculated for C22H11F3N2O3 (%): C, 64.71; H, 2.72; N, 6.86 (found: C, 64.80; H, 2.74; N, 6.90).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 4-(trifluoromethyl) benzoate (7p): Yield: 62%; pale yellow powder; m.p. 223.8–224.9 °C; IR, υmax (KBr) cm−1: 1743 (C=O), 1677 (ester), 1415 (C=N). UV λmax (lg ε) (DCM) 364 (4.21) nm, 382 (4.22) nm. 1H-NMR (500 MHz, CDCl3) δ 8.85 (d, J = 5.0 Hz, 1H), 8.74 (d, J = 8.8 Hz, 1H), 8.37 (d, J = 8.2 Hz, 2H), 8.07–7.99 (m, 2H), 7.94 (d, J = 5.0 Hz, 1H), 7.83 (d, J = 8.3 Hz, 2H), 7.56 (dd, J = 8.8, 2.3 Hz, 1H), 7.00 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 164.1, 159.4, 148.3, 146.0, 139.8, 137.2, 136.6, 135.4 (d, J = 33.0 Hz), 132.6, 132.4, 130.7, 129.6, 129.0, 125.8 (d, J = 4.0 Hz), 125.6, 124.5 (d, J = 273.6 Hz), 124.3, 118.1, 116.6, 116.0. ESI-MS m/z: [M + H]+(found) = 409.37, [M + H]+(calcd) = 409.34; Elemental analysis calculated for C22H11F3N2O3 (%): C, 64.71; H, 2.72; N, 6.86 (found: C, 64.79; H, 2.77; N, 6.81).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 1-naphthoate (7q): Yield: 83%; white powder; m.p. 221.3–222.4 °C; IR, υmax (KBr) cm−1: 1732 (C=O), 1678 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 364 (4.57) nm, 382 (4.55) nm. 1H-NMR (500 MHz, CDCl3) δ 9.06 (dd, J = 8.6, 1.1 Hz, 1H), 8.83 (d, J = 5.0 Hz, 1H), 8.74 (d, J = 8.7 Hz, 1H), 8.55 (dd, J = 7.3, 1.3 Hz, 1H), 8.14 (dt, J = 8.2, 1.1 Hz, 1H), 8.07 (d, J = 2.3 Hz, 1H), 8.03 (d, J = 9.8 Hz, 1H), 7.97–7.92 (m, 2H), 7.68–7.55 (m, 4H), 7.00 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 165.8, 159.4, 148.7, 146.0, 139.7, 137.1, 136.5, 134.8, 134.0, 132.6, 131.8, 131.5, 129.8, 129.0, 128.8, 128.4, 126.6, 125.6, 125.6, 125.3, 124.7, 124.6, 118.1, 116.6, 116.4. ESI-MS m/z: [M + H]+(found) = 391.39, [M + H]+(calcd) = 391.40; Elemental analysis calculated for C25H14N2O3 (%): C, 76.92; H, 3.61; N, 7.18 (found: C, 76.99; H, 3.57; N, 7.24).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl 2-naphthoate (7r): Yield: 80%; white powder; m.p. 234.0–235.1 °C; IR, υmax (KBr) cm−1: 1735 (C=O), 1685 (ester), 1419 (C=N). UV λmax (lg ε) (DCM) 364 (4.59) nm, 382 (4.58) nm. 1H-NMR (500 MHz, CDCl3) δ 8.86–8.79 (m, 2H), 8.73 (d, J = 8.8 Hz, 1H), 8.22 (dd, J = 8.6, 1.7 Hz, 1H), 8.06 (d, J = 2.2 Hz, 1H), 8.03 (d, J = 9.8 Hz, 1H), 8.01 (d, J = 8.2, 1H), 7.96 (d, J = 8.6, 1H), 7.94–7.89 (m, 2H), 7.67–7.57 (m, 3H), 6.99 (d, J = 9.8 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ 165.4, 159.4, 148.8, 146.0, 139.7, 137.0, 136.5, 136.0, 132.6, 132.5, 132.2, 129.8, 129.5, 129.0, 128.9, 128.6, 127.9, 127.0, 126.3, 125.6, 125.4, 124.6, 118.0, 116.6, 116.3. ESI-MS m/z: [M + H]+(found) = 391.38, [M + H]+(calcd) = 391.40; Elemental analysis calculated for C25H14N2O3 (%): C, 76.92; H, 3.61; N, 7.18 (found: C, 76.85; H, 3.57; N, 7.12).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl acetate (7s): Yield: 52%; yellow solid; m.p. 198.5–199.3 °C; IR, υmax (KBr) cm−1: 1758 (C=O), 1675 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 364 (3.87) nm, 382 (3.85) nm. 1H-NMR (500 MHz, CDCl3) δ 8.80 (d, J = 5.0 Hz, 1H), 8.63 (d, J = 8.8 Hz, 1H), 8.00 (d, J = 9.8 Hz, 1H), 7.88 (d, J = 5.0 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.40 (dd, J = 8.8, 2.3 Hz, 1H), 6.96 (d, J = 9.8 Hz, 1H), 2.38 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ 169.5, 159.3, 148.4, 145.9, 139.7, 136.9, 136.5, 132.5, 129.7, 129.0, 125.4, 124.3, 117.9, 116.6, 116.0, 21.2. ESI-MS m/z: [M + H]+(found) = 279.26, [M + H]+(calcd) = 279.27; Elemental analysis calculated for C16H10N2O3 (%): C, 69.06; H, 3.62; N, 10.07 (found: C, 69.01; H, 3.58; N, 10.13).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl propionate (7t): Yield: 51%; yellow solid; m.p. 166.7–168.1 °C; IR, υmax (KBr) cm−1: 1753 (C=O), 1681 (ester), 1414 (C=N). UV λmax (lg ε) (DCM) 364 (3.89) nm, 382 (3.85) nm. 1H-NMR (500 MHz, CDCl3) δ 8.81 (d, J = 5.0 Hz, 1H), 8.65 (d, J = 8.8 Hz, 1H), 8.01 (d, J = 9.8 Hz, 1H), 7.89 (d, J = 5.0 Hz, 1H), 7.86 (d, J = 2.2 Hz, 1H), 7.40 (dd, J = 8.8, 2.3 Hz, 1H), 6.97 (d, J = 9.8 Hz, 1H), 2.68 (q, J = 7.6 Hz, 2H), 1.32 (t, J = 7.6 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 173.0, 159.3, 148.5, 145.9, 139.7, 136.9, 136.5, 132.5, 129.8, 129.0, 125.4, 124.4, 117.9, 116.6, 116.0, 27.8, 9.1. ESI-MS m/z: [M + H]+(found) = 293.27, [M + H]+(calcd) = 293.29; Elemental analysis calculated for C17H12N2O3 (%): C, 69.86; H, 4.14; N, 9.58 (found: C, 69.79; H, 4.17; N, 9.63).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl butyrate (7u): Yield: 55%; yellow solid; m.p. 138.2–139.5 °C; IR, υmax (KBr) cm−1: 1753 (C=O), 1674 (ester), 1418 (C=N). UV λmax (lg ε) (DCM) 364 (3.87) nm, 382 (3.86) nm. 1H-NMR (500 MHz, CDCl3) δ 8.83 (d, J = 5.0 Hz, 1H), 8.67 (d, J = 8.8 Hz, 1H), 8.02 (d, J = 9.8 Hz, 1H), 7.92 (d, J = 5.0 Hz, 1H), 7.87 (d, J = 2.2 Hz, 1H), 7.41 (dd, J = 8.8, 2.3 Hz, 1H), 6.98 (d, J = 9.8 Hz, 1H), 2.63 (t, J = 7.4 Hz, 2H), 1.89–1.81 (m, 2H), 1.10 (t, J = 7.4 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 172.2, 159.4, 148.5, 145.9, 139.7, 136.9, 136.5, 132.6, 129.8, 129.0, 125.5, 124.4, 117.9, 116.6, 116.0, 36.3, 18.5, 13.7. ESI-MS m/z: [M + H]+(found) = 307.28, [M + H]+(calcd) = 307.32; Elemental analysis calculated for C18H14N2O3 (%): C, 70.58; H, 4.61; N, 9.15 (found: C, 70.55; H, 4.66; N, 9.18).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl pentanoate (7v): Yield: 56%; yellow solid; m.p. 141.6–142.7 °C; IR, υmax (KBr) cm−1: 1755 (C=O), 1675 (ester), 1417 (C=N). UV λmax (lg ε) (DCM) 364 (3.87) nm, 382 (3.85) nm. 1H-NMR (500 MHz, CDCl3) δ 8.81 (d, J = 5.0 Hz, 1H), 8.64 (d, J = 8.8 Hz, 1H), 8.00 (d, J = 9.8 Hz, 1H), 7.90 (d, J = 5.0 Hz, 1H), 7.84 (d, J = 2.3 Hz, 1H), 7.39 (dd, J = 8.8, 2.3 Hz, 1H), 6.96 (d, J = 9.8 Hz, 1H), 2.64 (t, J = 7.5 Hz, 2H), 1.83–1.76 (m, 2H), 1.54–1.45 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3) 172.4, 159.3, 148.5, 145.9, 139.6, 136.9, 136.5, 132.5, 129.7, 129.0, 125.4, 124.4, 117.9, 116.6, 116.0, 34.1, 27.0, 22.3, 13.8. ESI-MS m/z: [M + H]+(found) = 321.29, [M + H]+(calcd) = 321.35; Elemental analysis calculated for C19H16N2O3 (%): C, 71.24; H, 5.03; N, 8.74 (found: C, 71.16; H, 5.11; N, 8.69).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl hexanoate (7w): Yield: 53%; yellow solid; m.p. 112.8–113.7 °C; IR, υmax (KBr) cm−1: 1761 (C=O), 1675 (ester), 1417 (C=N). UV λmax (lg ε) (DCM) 364 (3.85) nm, 382 (3.86) nm. 1H-NMR (500 MHz, CDCl3) δ 8.81 (d, J = 5.0 Hz, 1H), 8.64 (d, J = 8.8 Hz, 1H), 8.01 (d, J = 9.8 Hz, 1H), 7.90 (d, J = 5.0 Hz, 1H), 7.85 (d, J = 2.3 Hz, 1H), 7.39 (dd, J = 8.8, 2.3 Hz, 1H), 6.97 (d, J = 9.8 Hz, 1H), 2.63 (t, J = 7.5 Hz, 2H), 1.85–1.78 (m, 2H), 1.48–1.38 (m, 4H), 0.96 (t, J = 7.1 Hz, 3H). 13C-NMR (126 MHz, CDCl3) δ 172.4, 159.3, 148.5, 145.9, 139.7, 136.9, 136.5, 132.5, 129.8, 129.0, 125.4, 124.4, 117.9, 116.6, 116.0, 34.4, 31.3, 24.6, 22.4, 13.9. ESI-MS m/z: [M + H]+(found) = 335.35, [M + H]+(calcd) = 335.38; Elemental analysis calculated for C20H18N2O3 (%): C, 71.84; H, 5.43; N, 8.38 (found: C, 71.85; H, 5.46; N, 8.43).
6-Oxo-6H-Indolo [3,2,1-de] [1,5] naphthyridin-10-yl dodecanoate (7x): Yield: 49%; yellow solid; m.p. 65.4–66.6 °C; IR, υmax (KBr) cm−1: 1757 (C=O), 1682 (ester), 1420 (C=N). UV λmax (lg ε) (DCM) 364 (3.83) nm, 383 (3.82) nm. 1H-NMR (500 MHz, CDCl3) δ 8.84 (d, J = 5.0 Hz, 1H), 8.67 (d, J = 8.8 Hz, 1H), 8.07 (d, J = 9.8 Hz, 1H),7.94 (d, J = 5.0 Hz, 1H), 7.87 (d, J = 2.3 Hz, 1H), 7.41 (dd, J = 8.8, 2.3 Hz, 1H), 7.00 (d, J = 9.8 Hz, 1H), 2.64 (t, J = 7.5 Hz, 2H), 2.34 (t, J = 7.5 Hz, 4H), 1.85–1.76 (m, 2H), 1.69–1.57 (m, 4H), 1.53–1.41 (m, 2H), 0.90–0.86 (m, 9H). 13C-NMR (125 MHz, CDCl3) δ 172.4, 159.4, 148.5, 145.7, 139.5, 137.0, 136.4, 132.6, 130.0, 129.1, 125.4, 124.5, 118.0, 116.7, 116.1, 34.4, 34.1, 31.9, 29.6, 29.5, 29.3, 29.3, 29.1, 25.0, 24.8, 22.7. ESI-MS m/z: [M + H]+(found) = 419.53, [M + H]+(calcd) = 419.54; Elemental analysis calculated for C26H30N2O3 (%): C, 74.61; H, 7.23; N, 6.69 (found: C, 74.58; H, 7.19; N, 6.67).
3.3. Procedure for Determining Antifungal Activity
The in vitro antifungal activities of compounds 4–6 and 7a–7x against three phytopathogenic fungi (A. solani, F. graminearum, and F. solani) were investigated using the mycelium linear growth rate method [24]. The test compounds and TBZ were completely dissolved in 0.5 mL of DMSO and the solution added to 9.5 mL of sterile water. The resulting solution was added to 90 mL of melted potato dextrose agar (PDA) medium at a temperature below 55 °C. After quickly and completely mixing, the medium containing the compounds at a concentration of 50 μg/mL was poured into a sterilized Petri dish for screening. DMSO without any compounds mixed with PDA served as the negative control. When the medium in the plates was partially solidified, a 4 mm thick, 5 mm diameter disc of fungus cut from previously subcultured Petri dishes was picked up and inoculated in the center of the PDA dishes. The dishes were kept in an incubator at 28 °C for 72 h. Three replicates were performed for each experiment. The inhibitory rate was calculated according to the following formula:
| inhibition rate (%) = [(dc − d0) − (ds − d0)]/(dc − d0) × 100% | (1) |
where d0 is the diameter of the fungus cut, dc is the diameter of the untreated control fungus, and ds is the diameter of the sample-treated fungus.
3.4. Procedure for Determining Antibacterial Activity
The test compounds were dissolved in dichloromethane at a concentration of 2 mg/mL, and then 5 μL of each solution was transferred onto sterile paper discs (6.0 mm diameter), so that the disc contained 10 μg of the compound. Melted LB agar medium (10 mL) was then mixed with 100 μL of bacterial suspension at 45 °C and poured into a sterilized Petri dish. The discs were placed gently on the surface of the media inoculated with the microorganism. Penicillin sodium was used as a positive control and dichloromethane as a negative control. The plates were allowed to stand for 2 h at 4 °C to allow for diffusion and then incubated at 37 °C for 12 h. The diameter of the zone of growth inhibition around the disc was measured. Experiments were repeated in triplicate.
The MICs of some synthesized compounds against four bacterial strains were evaluated using the micro-broth dilution method using Muller-Hinton broth in 96-well plates. The test compounds and penicillin sodium were first dissolved in DMSO at a concentration of 10 mg/mL, and then diluted 20-fold with sterile water to give the stock solution. An aliquot (50 μL) of this solution was pipetted into the first well of each line in a 96-well plate. The stock solution was then serially diluted to give two-fold serial dilutions of the test compounds and positive control in the subsequent wells. Then 50 μL of bacterial suspension (106 cfu/mL) was added to each well and the plate set aside for incubation. One well containing bacterial cells and 2.5% DMSO was used as growth control, and another well containing only growth medium was used as a blank control. After incubation for 12 h at 37 °C, MICs were measured. Three replicates were performed for each experiment.
4. Conclusions
In this study, two naturally occurring canthine-6-ones and 24 novel ester derivatives of 10-hydroxycanthin-6-one were synthesized and their in vitro antimicrobial activity was evaluated. Most of the synthesized compounds showed potential antibacterial and antifungal activity. Compound 7s represents a promising lead compound for further study, as it had significant antimicrobial activity against all the fungi and bacterial strains tested. Further research is required into more extensive structural modification, the structure-activity relationship and the mechanism of action of canthin-6-ones.
Acknowledgments
This project was financially supported by the National Natural Science Foundation of China (Grant No. 31270388) and the Fundamental Research Funds for the Central Universities of China (Grant No. QN 2011066).
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/3/390/s1.
Author Contributions
Jun-Ru Wang, Fei Zhao, and Jiang-Kun Dai conceived and designed the experiments; Fei Zhao performed the experiments; Jiang-Kun Dai, Dan Liu, and Shi-Jun Wang analyzed the data; Fei Zhao wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 5, 6, and 7a–7x are available from the authors.
References
- 1.Haynes H.F., Nelson E.R., Price J.R. Alkaloids of the australian rutaceae: Pentaceras australis hook. F. II. Identification of 5-methoxycanthinone. Aust. J. Sci. Res. Ser. B. 1952;A5:563–569. [Google Scholar]
- 2.Showalter H.D.H. Progress in the synthesis of canthine alkaloids and ring-truncated congeners. J. Nat. Prod. 2013;76:455–467. doi: 10.1021/np300753z. [DOI] [PubMed] [Google Scholar]
- 3.Brahmbhatt K.G., Ahmed N., Sabde S., Mitra D., Singh I.P., Bhutani K.K. Synthesis and evaluation of β-carboline derivatives as inhibitors of human immunodeficiency virus. Bioorg. Med. Chem. Lett. 2010;20:4416–4419. doi: 10.1016/j.bmcl.2010.06.052. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y., Liu Y.B., Li Y., Ma S.G., Li L., Qu J., Zhang D., Chen X.G., Jiang J.D., Yu S.S. Sesquiterpenes and alkaloids from the roots of alangium chinense. J. Nat. Prod. 2013;76:1058–1063. doi: 10.1021/np4000747. [DOI] [PubMed] [Google Scholar]
- 5.Xu Z., Chang F.R., Wang H.K., Kashiwada Y., McPhail A.T., Bastow K.F., Tachibana Y., Cosentino M., Lee K.H. Anti-HIV agents 45 and antitumor agents 205. Two new sesquiterpenes, leitneridanins A and B, and the cytotoxic and anti-HIV principles from Leitneria floridana. J. Nat. Prod. 2000;63:1712–1715. doi: 10.1021/np000260u. [DOI] [PubMed] [Google Scholar]
- 6.Bharitkar Y.P., Hazra A., Poduri N.S.A., Ash A., Maulik P.R., Mondal N.B. Isolation, structural elucidation and cytotoxicity evaluation of a new pentahydroxy-pimarane diterpenoid along with other chemical constituents from Aerva lanata. Nat. Prod. Res. 2015;29:253–261. doi: 10.1080/14786419.2014.971794. [DOI] [PubMed] [Google Scholar]
- 7.Dejos C., Voisin P., Bernard M., Regnacq M., Berges T. Canthin-6-one displays antiproliferative activity and causes accumulation of cancer cells in the G2/M phase. J. Nat. Prod. 2014;77:2481–2487. doi: 10.1021/np500516v. [DOI] [PubMed] [Google Scholar]
- 8.Rivero-Cruz J.F., Lezutekong R., Lobo-Echeverri T., Ito A., Mi Q., Chai H.B., Soejarto D.D., Cordell G.A., Pezzuto J.M., Swanson S.M., et al. Cytotoxic constituents of the twigs of Simarouba glauca collected from a plot in Southern Florida. Phytother. Res. 2005;19:136–140. doi: 10.1002/ptr.1642. [DOI] [PubMed] [Google Scholar]
- 9.Hussain H., Al-Harrasi A., Al-Rawahi A., Green I.R., Gibbons S. Fruitful decade for antileishmanial compounds from 2002 to late 2011. Chem. Rev. 2014;114:10369–10428. doi: 10.1021/cr400552x. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira M.E., Nakayama H., Rojas de Arias A., Schinini A., Vera de Bilbao N., Serna E., Lagoutte D., Soriano-Agaton F., Poupon E., Hocquemiller R., et al. Effects of canthin-6-one alkaloids from Zanthoxylum chiloperone on trypanosoma cruzi-infected mice. J. Ethnopharmacol. 2007;109:258–263. doi: 10.1016/j.jep.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 11.Ferreira M.E., de Rojas Arias A., de Torres Ortiz S., Inchausti A., Nakayama H., Thouvenel C., Hocquemiller R., Fournet A. Leishmanicidal activity of two canthin-6-one alkaloids, two major constituents of Zanthoxylum chiloperone var. Angustifolium. J. Ethnopharmacol. 2002;80:199–202. doi: 10.1016/S0378-8741(02)00025-9. [DOI] [PubMed] [Google Scholar]
- 12.Jiao W.H., Gao H., Zhao F., Lin H.W., Pan Y.M., Zhou G.X., Yao X.S. Anti-inflammatory alkaloids from the stems of Picrasma quassioides bennet. Chem. Pharm. Bull. 2011;59:359–364. doi: 10.1248/cpb.59.359. [DOI] [PubMed] [Google Scholar]
- 13.Siveen K.S., Kuttan G. Modulation of humoral immune responses and inhibition of proinflammatory cytokines and nitric oxide production by 10-methoxycanthin-6-one. Immunopharmacol. Immunotoxicol. 2012;34:116–125. doi: 10.3109/08923973.2011.586703. [DOI] [PubMed] [Google Scholar]
- 14.Dai J.K., Dan W.J., Li N., Du H.T., Zhang J.W., Wang J.R. Synthesis, in vitro antibacterial activities of a series of 3-N-substituted canthin-6-ones. Bioorg. Med. Chem. Lett. 2016;26:580–583. doi: 10.1016/j.bmcl.2015.11.070. [DOI] [PubMed] [Google Scholar]
- 15.Fournet A.R.F.M., Lagoutte D., Poupon E., Soriano-Agaton F. Use of Canthin-6-one and Its Analogs in the Treatment of Mycobacteria-Linked Pathologier. 0059977 A1. U.S. Patent. 2011 Mar 10;
- 16.O’Donnell G., Gibbons S. Antibacterial activity of two canthin-6-one alkaloids from Allium neapolitanum. Phytother. Res. 2007;21:653–657. doi: 10.1002/ptr.2136. [DOI] [PubMed] [Google Scholar]
- 17.Soriano-Agaton F., Lagoutte D., Poupon E., Roblot F. Extraction, hemisynthesis, and synthesis of canthin-6-one analogues. Evaluation of their antifungal activities. J. Nat. Prod. 2005;68:1581–1587. doi: 10.1021/np050250z. [DOI] [PubMed] [Google Scholar]
- 18.Lagoutte D., Nicolas V., Poupon E., Fournet A., Hocquemiller R., Libong D., Chaminade P., Loiseau P.M. Antifungal canthin-6-one series accumulate in lipid droplets and affect fatty acid metabolism in Saccharomyces cerevisiae. Biomed. Pharmacother. 2008;62:99–103. doi: 10.1016/j.biopha.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 19.Wang J.R., Dai J.K., Zhao F., Dan W.J., Yin D.Y., Gao Y., Qin W.J., Zhang J.W. Preparation of Canthin-6-one Quaternary Ammonium Salts as Antibacterial Agents. 104530047A. C.N. Patent. 2014 Dec 16;
- 20.Fisher M.C., Henk D.A., Briggs C.J., Brownstein J.S., Madoff L.C., McCraw S.L., Gurr S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–194. doi: 10.1038/nature10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newman D.J., Cragg G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang R., Gao Z.F., Zhao J.Y., Li W.B., Zhou L., Miao F. New class of 2-aryl-6-chloro-3, 4-dihydroisoquinolinium salts as potential antifungal agents for plant protection: Synthesis, bioactivity and structure-activity relationships. J. Agric. Food Chem. 2015;63:1906–1914. doi: 10.1021/jf505609z. [DOI] [PubMed] [Google Scholar]
- 23.Czerwinski K.M., Zificsak C.A., Stevens J., Oberbeck M. An improved synthesis of canthin-6-one. Synth. Commun. 2003;33:1225–1231. doi: 10.1081/SCC-120017199. [DOI] [Google Scholar]
- 24.Zhang J.W., Li S.K., Wu W.J. The main chemical composition and in vitro antifungal activity of the essential oils of Ocimum basilicum linn. Var. Pilosum (willd.) benth. Molecules. 2009;14:273–278. doi: 10.3390/molecules14010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R., Zhang H., Yuan M., Zhou J., Tu Q., Liu J.J., Wang J. Synthesis and biological evaluation of apigenin derivatives as antibacterial and antiproliferative agents. Molecules. 2013;18:11496–11511. doi: 10.3390/molecules180911496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin D.B.C., Vanderwal C.D. Efficient access to the core of the strychnos, aspidosperma and iboga alkaloids. A short synthesis of norfluorocurarine. J. Am. Chem. Soc. 2009;131:3472–3473. doi: 10.1021/ja900640v. [DOI] [PubMed] [Google Scholar]
- 27.Orbe J., Sanchez-Arias J.A., Rabal O., Rodriguez J.A., Salicio A., Ugarte A., Belzunce M., Xu M., Wu W., Tan H., et al. Design, synthesis, and biological evaluation of novel matrix metalloproteinase inhibitors as potent antihemorrhagic agents: From hit identification to an optimized lead. J. Med. Chem. 2015;58:2465–2488. doi: 10.1021/jm501940y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.