

Abstract
A library of thirty eight novel thiazolo[5,4-f]quinazolin-9(8H)-one derivatives (series 8, 10, 14 and 17) was prepared via the Hügershoff reaction and a Cu catalyzed intramolecular C-S bond formation, helped by microwave-assisted technology when required. The efficient multistep synthesis of the key 6-amino-3-cyclopropylquinazolin-4(3H)-one (3) has been reinvestigated and performed on a multigram scale from the starting 5-nitroanthranilic acid. The inhibitory potency of the final products was evaluated against five kinases involved in Alzheimer’s disease and showed that some molecules of the 17 series described in this paper are particularly promising for the development of novel multi-target inhibitors of kinases.
Keywords: Hügershoff reaction; thiazolo[5,4-f]quinazolin-9(8H)-ones; microwave-assisted synthesis; protein kinases; CDK5; GSK-3; CLK1; CK1; DYRK1A
1. Introduction
The occurrence and properties of the thiazole ring in various natural and synthetic products have been the interest of many research groups on account of its useful biological properties [1,2,3,4,5]. Since the 2000s our laboratory has been involved in a research program dealing with the preparation and pharmacological evaluation of original heterocyclic derivatives bearing a thiazole ring, mostly inspired by marine alkaloids [6,7,8,9,10]. In this context, our research group is mainly invested in the synthesis of C,N,S-containing bioactive molecules able to modulate the activity of deregulated kinases (CDK5, GSK-3, CLK1, CK1 and the dual-specificity kinase DYRK1A) involved to some extent in Alzheimer’s disease (AD) [11,12,13,14,15,16,17,18]. Consequently, the reactivity of 4,5-dichloro-1,2,3-dithiazolium chloride (Appel’s salt) [19] has been extensively studied for the synthesis of sulfur-nitrogen heteroarenes bearing a thiazole ring, substituted by a carbonitrile group at C-2. The chemistry of this versatile function was explored allowing access to novel imidate derivatives [11,12,13,14,15,16,17,18]. Among them some thiazolo[5,4-f]quinazolin-9(8H)-ones (Figure 1) have been revealed of particular interest in the design of multi-target-directed ligands (MTDLs), a new strategy for the development of powerful tools against neurodegenerative diseases [20,21,22,23,24].
Figure 1.

General formula of lead kinases inhibitors previously described [25] and new targeted molecules.
The result of preliminary docking studies is schematized in Figure 2 and concerns the ATP-binding site of GSK-3β [11,12]. The nitrogen in position 6 of the thiazolo[5,4-f]quinazolin-9-one core seems to form a hydrogen bond with a NH-residue in the hinge segment, whilst the benzyl-carbimidate function linked to the thiazole moiety may form a polar interaction with the amino group of Lys85. In this hypothesis the bulky substituent may not fit into the hydrophobic backpocket of GSK-3β.
Figure 2.

(a) Schematic representation of binding mode of one of the lead compounds in the ATP-binding site of GSK-3; (b) Description of the aminoalkyl and aryl substituents envisioned in position 2 of the thiazole with the aim of a better affinity and selectivity for target kinases (e.g., GSK-3).
Considering that this region of the ATP-binding site of kinases can be used to gain affinity as well as selectivity, the synthetic route to novel C-2 substituted thiazolo[5,4-f]quinazolin-9-ones (Figure 1) bearing aminoalkyl and aminoaryl substituents at position C-2 of the thiazole moiety was investigated. In this study, the substituent present on position N-8 of the tricyclic structure was restrained to cyclopropyl, which has proved its efficiency in previous studies concerning this kinase group (see IC50 values in Figure 1) [25].
This paper describes the development of various methods allowing the preparation of a library of novel 2-aminoalkyl and 2-aminoarylthiazolo[5,4-f]quinazolin-9(8H)-ones for which interesting multi-target kinase inhibitory activities were expected. Following our previous strategies, the synthetic routes for this work were envisioned starting from commercially available anthranilic acids. A part of the chemistry described in this paper was achieved under microwave irradiation as a continuation of our global strategy consisting in the design of appropriate reagents and techniques offering operational, economic, and environmental benefits over conventional methods [26,27,28].
2. Results and Discussion
2.1. Chemistry
The target molecules were thiazolo[5,4-f]quinazolin-9(8H)-ones substituted in position N-8 by a cyclopropyl chain (Figure 1). The envisioned synthetic route is depicted in Scheme 1. The key intermediate 6-amino-3-cyclopropylquinazolin-4(3H)-one (3) was obtained from the corresponding nitro derivative 2, itself obtained from 5-nitroanthranilic acid (1). The second part of the synthesis consisted in transforming the amine function into its thiourea analogue which can be cyclized into a thiazole ring by various methods. The first planned strategy uses the Hügershoff reaction, a bromine-mediated cyclization process involving electrophilic addition to the thiocarbonyl of the thiourea to afford a transient intermediate (e.g., by the seminal Br2 [29,30] or its recent equivalent such as benzyltrimethylammonium bromide [31]), which is then attacked by the π system of the aromatic ring, followed by rapid formation of thiazole ring. The second route imagined concerns a metal catalyzed intramolecular C-S bond formation [32,33,34,35,36] previously described for the synthesis of variously 2-substituted benzothiazoles from thiobenzanilides (Scheme 1).
Scheme 1.

Envisioned retrosynthetic pathway for the synthesis of the target products, via Hügershoff reaction [29,30,31] or transition metal-catalyzed intramolecular C-S bond formation [32,33,34,35,36].
2.1.1. Synthesis of the Key 6-Amino-3-cyclopropylquinazolin-4(3H)-one (3)
To obtain an efficient route to the key 6-amino-3-cyclopropylquinazolin-4(3H)-one (3), the multistep strategy described in our previous works [11,12] was transformed into a convenient one-pot sequential process which was perfectly controlled using microwave heating at atmospheric pressure. Experimentally, the sequential MCR procedure consisted in irradiating of the starting 5-nitroanthranilic acid (1) and DMFDMA (2.5 equiv) in DMF for 15 min at 100 °C. After evaporation of the basic solvent (DMF), cyclopropylamine (1.1 equiv) and acetic acid were immediately added in the reaction flask and heated under microwaves at 100 °C for a further 15 min.
As depicted in Scheme 2 condensation of cyclopropylamine with the intermediate carbimidate gave 3-cyclopropyl-6-nitroquinazolin-4(3H)-one (2, 85% yield) which was reduced by treatment with ammonium formate in the presence of a catalytic amount of 10% palladium charcoal to afford the strategic 6-amino-3-cyclopropylquinazolin-4(3H)-one (3) in very good yield (85%). Some comments concerning the microwave procedure as well as the technical and practical aspects are warranted. In the case of the microwave-assisted synthesis, DMF and acetic acid present the advantage of having good dielectric properties, thus facilitating an efficient heating of the reaction mixture [37,38,39]. A reactor able to work at atmospheric pressure had some advantages, such as the possibility of easier scale-up and the use of common laboratory glassware. In the synthetic pathway steps described in this paper, an irradiation power at 900 W was enough to efficiently reach the programmed temperature with a short time ramp (2 min, not added to the reaction time indicated in schemes).
Scheme 2.

Sequential MCR procedure for convenient synthesis of 6-amino-3-cyclopropylquinazolin-4(3H)-one (3) from 1. Reagents and conditions: (a) Step 1: DMFDMA (2.5 equiv), DMF, 100 °C (µw), 15 min; Step 2: Cyclopropylamine (1.1 eq), AcOH, 100 °C (µw), 15 min; 85%; (b) HCO2NH4 (5.0 equiv), Pd/C (10%), EtOH, 85 °C (μw), 15 min; 85%.
2.1.2. Synthesis of 2-Aminoarylthiazolo[5,4-f]quinazolin-9-ones via Hügershoff Reaction
The seminal work of Hügershoff described the cyclization of thioureas with liquid bromine in chloroform to form the corresponding 2-aminobenzothiazoles [29,30]. According to these facts, the key isothiocyanate intermediate was needed as further precursor of substituted thioureas and their corresponding thiazoles. As described in Scheme 3, isothiocyanate 4 was obtained in nearly quantitative yield (98%) by treatment of amine 3 in the presence of thiophosgene in a mixture of tetrahydrofuran (THF) and a saturated aqueous solution of NaHCO3. The less toxic 1,1′-thiocarbonyl-diimidazole (TCDI) was found to be less effective.
Scheme 3.

Synthetic routes for access to the key isothiocyanate 4.
According to previous works, the chemistry of 4,5-dichloro-1,2,3-dithiazolium chloride (Appel’s salt) allowed the safer synthesis of the attempted isothiocyanate precursor 4 [40,41]. Treatment of amine 3 with Appel’s salt in dichloromethane (DCM), in the presence of pyridine gave the corresponding imino-1,2,3-dithiazole derivative 5 which was stirred in DCM in the presence of 4 equiv of DBU [40], at −78 °C and then at room temperature, to afford 4 in a quite good yield (65%). Despite a low yield compared to the procedure using thiophosgene, this alternative synthesis has the advantage of being less toxic and safer than the former one. It can be noticed that treating 3 with 3 equiv of DBU led to the unique formation of the intermediate cyanothioformamide 6 (74%) [40]. The addition of 1 equiv. of DBU to 6 and stirring at room temperature for 1 h produced isothiocyanate 4 in a 88% yield (Scheme 3).
Two methods were tested for the synthesis of the expected thioureas 7, as depicted in Scheme 4 and Table 1. The first one depended of the availability of some isothiocyanates from commercial sources. It involved condensation of amine 3 with various aryl isothiocyanates to give compounds 7 (method A). In other cases, isothiocyanate 4 was treated with aniline derivatives to give 4 (method B). It should be noticed that in the case of less nucleophilic aromatic amines (e.g., 4-trifluoro-methylaniline and 3-aminopyridine), heating at 40 °C under microwave irradiation was required for access to the corresponding thioureas 7d and 7g.
Scheme 4.

Synthesis of isothiocyanates 7a–l from amines 3 or from isothiocyanates 4.
Table 1.
Chemical structures and yields obtained for the synthesis of series 7a–l.
| -R a | Compound | Method | Time (h) | Yield b (%) |
|---|---|---|---|---|
| Ph | 7a | A/B | 1/2 | 84/80 |
| 4-Cl-C6H4 | 7b | A | 1 | 91 |
| 4-F-C6H4 | 7c | B | 2 | 90 |
| 4-CF3-C6H4 | 7d | Bc | 1 | 84 |
| 4-NO2-C6H4 | 7e | A | 0.5 | 74 |
| 2,4-diCl-C6H3 | 7f | B | 1 | 89 |
| 3-Py | 7g | Bc | 2 | 89 |
| 4-Me-C6H4 | 7h | A | 2 | 89 |
| 4-OMe-C6H4 | 7i | A | 2 | 87 |
| 4-NMe2-C6H4 | 7j | B | 10 min | 87 |
| 2,4-diOMe-C6H3 | 7k | B | 5 min | 97 |
| 3,4-diOMe-C6H3 | 7l | B | 5 min | 88 |
a This list of anilines was established, based on the Topliss schemes [42], in order to get the most potent compounds as early as possible; b Isolated yield; c heated for 1 h at 40 °C under microwaves.
With thioureas 7a–7l in hand, the conditions of the Hügershoff reaction were examined using the N-phenylthiourea 7a, with various oxidizing agents and solvents. The best results were obtained when 7a was treated with 1.0 equiv of Br2 in acetic acid at room temperature (Scheme 5). However, although cyclization seemed effective, it was not regioselective. In the case of the N-phenyl derivative, the process resulted in a mixture of three isomeric compounds. NMR analysis demonstrated that the minor derivatives were the two tricyclic isomers 8a and 9a, whilst the major product was the benzothiazole derivative 10a resulting from the attack of the benzene ring (A in Scheme 5) on the electrophilic sulfur atom, after oxidation of the thiourea with bromide. This result demonstrated that the benzene group (A) of the starting diarylthiourea 7a possessed the most electron-rich ortho-carbon, compared to the quinazolin-4-one ring (B in Scheme 5).
Scheme 5.

Hügershoff cyclization mechanism applied to thiourea 7a and compounds 8a, 9a and 10a identified.
Furthermore, analysis of isomers 8a and 9a confirmed that the most abundant was the angular derivative 8a which was purified in poor yield (10%) whilst traces of 9a did not allow its complete purification. Although the compound 10a was isolated in a low yield (30%), the quantities obtained were sufficient for the biological experiments. Taking all these facts into account, the experimental conditions described above were extended to series 7b–7g in which electron-deficient phenyl substituents are present in ring A of the thiourea derivatives (Scheme 6). Results are given in Table 2 where it was observed that isomers 9b–e were apparently formed (per NMR analysis of crude reaction mixtures) without being purified. It was noted that compound 8b was not separated from 9b, whatever chromatographic methods were used.
Scheme 6.

Synthesis of 8-cyclopropyl-2-(arylamino)thiazolo[5,4-f]quinazolin-9(8H)-ones 8b–f and their pyrido analog 8g.
Table 2.
Chemical structures and yields obtained for the synthesis of series 8a–g via the Hügershoff reaction.
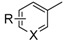 |
Compound | Time (h) | Yield a (%) |
|---|---|---|---|
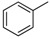 |
8a | 0.5 | 10 b,c |
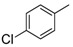 |
8b | 1 | - d |
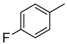 |
8c | 1 | 45 b |
 |
8d | 1 | 79 b |
 |
8e | 4 | 86 b |
 |
8f | 1.5 | 90 |
 |
8g | 4 | 80 |
a Isolated yield; b Traces of compounds of the 9 series were detected but not purified; c In this case only compound 10a was isolated in a 30% yield; d Not isolated.
The Hügershoff conditions were also applied to a thiourea possessing highly electron-rich phenyl substituents (e.g., 3,4-diOMe). Compound 7l was stirred for 30 min at room temperature to give unambiguously the corresponding benzothiazole 10l (90% yield) via the regioselective cyclization of the 3,4-dimethoxyphenylthiourea into the thiazole ring (Scheme 7). At this stage of the study, a small library of 2-(arylamino)-8-cyclopropylthiazolo[5,4-f]quinazolin-9(8H)-ones 8a, 8c–g was prepared accompanied by the two 2-(benzo[d]thiazol-2-ylamino)-8-cyclopropylthiazolo[5,4-f]quinazolin-9(8H)-one derivatives 10a and 10l.
Scheme 7.

Hügershoff conditions applied to 7l giving the corresponding 2-substituted benzothiazole 10l selectively.
The results of this oxidative cyclization of thioureas depend of the substituents present on the aryl groups with a low tolerance. To overcome this drawback a metal catalyzed C-H functionalization/intramolecular C-S bond formation was envisioned, inspired by the work of Doi et al. on the synthesis of variously 2-substituted benzothiazoles from thiobenzanilides [32,33].
2.1.3. Synthesis of 2-Aminoaryl- and 2-Aminoalkylthiazolo[5,4-f]quinazolin-9-ones via Metal Catalyzed C-S Bond Formation
According to the conditions described by Doi et al. [32,33], thiourea 7a was stirred in the presence of palladium dichloride (PdCl2), copper iodide (CuI) and tetrabutylmammonim bromide (TBAB) in a DMSO/NMP (1:1, v/v) mixture as solvent. After 2 h of heating at 120 °C, all the starting material 7a disappeared and workup revealed a mixture of three products. Analyses allowed us to identify 8a, 9a and 10a (see Scheme 5), demonstrating a lack of selectivity under such conditions.
Needing regioselectivity in the cyclization process, we focused our attention on the work described ten years ago by Batey and Evindar on the conversion of ortho-bromoanilides or ortho-bromothioanilides into the corresponding benzoxazoles or benzothiazoles via intramolecular C-O or C-S bond formation [34,35,36]. Optimal conditions for cyclization used a catalyst combination of CuI (5 mol %) and 1.10-phenanthroline (10 mol %) in the presence of Cs2CO3 (2.0 equiv) in dimethoxyethane (DME).
In a first approach, it was decided to prepare the phenylthiourea 12 for testing the metal-catalyzed cyclization conditions. Our study started from the ortho-brominated derivative of 3 which was easily obtained by stirring the starting amine 3 with N-bromosuccinimide (NBS, 1.0 equiv) in DMF at room temperature for 2 h. 6-Amino-5-bromo-3-cyclopropylquinazolin-4(3H)-one (11) was obtained in a high yield of 93% (Scheme 8).
Scheme 8.

Synthesis of thiourea 12 via condensation of aniline with isothiocyanate 13.
The key ortho-brominated thiourea 12 was synthesized according to the aforementioned strategy used for the synthesis of 7a. Unfortunately condensation of amine 11 with phenyl isothiocyanate was not fructuous, certainly due to the steric hindrance of the bromide associated with the low nucleophilicity of the aromatic amino group. To overcome this drawback the 5-bromo-6-isothiocyanatoquinazolin-4(3H)-one (13) was readily prepared (82% yield) by treatment of 11 with thiophosgene in a mixture of THF/NaHCO3 sat. aq. (1:1, v/v), as previously described. The phenyl-thiourea 12 was obtained by condensation of aniline with isothiocyanate 13 in acetonitrile at room temperature for 1 h (82% yield) (Scheme 8).
Experimental conditions for metal-catalyzed cyclization of thiourea 12 were adapted according to Batey et al. [34,35,36]: CuI 10 mol %, 1,10-phenantroline (20 mol %) and Cs2 CO3 (2.0 equiv) as a base. Instead of 24 h of conventional heating conditions, only 1 h of microwave-assisted heating (80 °C) allowed complete conversion of the starting material 12 (according to NMR analysis) into the cyclized product 8a (Table 3, entry 1).
Table 3.
Experimental condition optimization for the intramolecular metal-catalyzed cyclization of 12.
| Entry | CuI (mol %) | Ligand a | Cs2CO3 (Equiv) | Conversion b (%) (Isolated Yield) |
|---|---|---|---|---|
| 1 | 10 | 20 | 2 | 100 |
| 2 | 10 | - | 2 | 100 (80) |
| 3 | 5 | - | 2 | 80 b |
| 4 | 10 | - | - | 65 b |
| 5 | - | - | 2 | - c |
a 1,10-phenanthroline; b Reaction conversion was determined by 1H NMR analysis; c Traces.
Encouraged by this good result, a short optimization study was engaged in order to extend the best experimental conditions to a larger scope of products (Scheme 9, Table 3). Various conditions were screened, changing the quantities of copper catalyst, ligand and base. It was quickly confirmed that 10 mol % of CuI was necessary for a successful cyclization, under ligand-free conditions and in the presence of 2.0 equiv of Cs2 CO3 as a base (Table 3, entry 2).
Scheme 9.

General description of the intramolecular metal-catalyzed cyclization of 12.
Once cyclization conditions were optimized, a sequential one-pot version of the transformation of isothiocyanate 13 into the tricyclic arene 8 series was considered. This approach required the complete conversion of the starting material 13 into the corresponding thiourea intermediate (in square brackets in Scheme 10) before addition of other reagents (CuI and Cs2CO3) and microwave-assisted cyclization by heating for 1 h. Depending on the nature of aniline, the conversion time can vary from 30 min (electron-rich compounds e.g., 4-OMe, 3,4-diOMe or 4-NMe2 anilines) to an overnight stirring (12 h) as observed for the deactivated aniline substituted by a sulfonamide function (see compound 8m in Scheme 10 and Table 4). Because electron-poor anilines required several hours for completion of the first part of the reaction, the Hügershoff reaction was found to be more efficient in these cases (products 8d–f).
Scheme 10.

One-pot sequential CuI catalyzed intramolecular cyclization of intermediates thioureas (from 13) for the synthesis of series 8 and 14.
Table 4.
Chemical structures and yields obtained for the sequential MCR-synthesis of 8a, 8c and 8h–m via Cu catalysed intramolecular cyclization.
| -R1 | -R2 | Compound | Time a (h) | Yield b (%) |
|---|---|---|---|---|
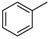 |
H | 8a | 1 | 80 |
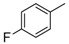 |
H | 8c | 2 | 96 |
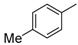 |
H | 8h | 1 | 89 |
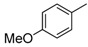 |
H | 8i | 0.5 | 88 |
 |
H | 8j | 0.5 | 87 |
 |
H | 8k | 0.5 | 86 |
 |
H | 8l | 0.5 | 90 |
 |
H | 8m | 12 | 77 |
a Time of the conversion of 13 into the corresponding thioureas (step 1); b Isolated yield.
The methodology described for the anilines 8h–m was then extended to various alkylamines chosen for their frequency in various bioactive molecules. All the thiourea intermediates were formed in situ after 30 min of stirring. Microwave-assisted heating at 80 °C for 1 h complete the sequence to afford the expected cyclized compounds (series 14a–f) in very good yields (80%–96%, see Table 5).
Table 5.
Chemical structures and yields obtained via the sequential MCR synthesis of 14a–14f.
| -R1 | -R2 | Compound | Time a (h) | Yield b (%) |
|---|---|---|---|---|
 |
H | 14a | 0.5 | 80 |
 |
H | 14b | 0.5 | 96 |
 |
H | 14c | 0.5 | 89 |
 |
H | 14d | 0.5 | 88 |
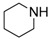 |
14e | 0.5 | 87 | |
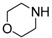 |
14f | 0.5 | 86 |
a Time of the conversion of 13 into the corresponding thioureas (step 1); b Isolated yield.
With the intent to compare the biological results of the aforementioned products 14a–f with their imidate analogues, the synthesis of a small library of 9-oxo-(8H)thiazolo[5,4-f]quinazoline-2-carboximidamides was undertaken via the synthesis of the key 8-cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carbonitrile (16 in Scheme 11). Compound 16 was obtained by reaction of 11 with 4,5-dichloro-1,2,3-dithiazolium chloride (Appel’s salt) following by a copper-mediated cyclization of the intermediate imine 15 (Scheme 11). As described in previous works [16,17] the versatile carbonitrile function in position 2 of the thiazole ring may allow the synthesis of various amidine or imidate derivatives. A new set of six novel carboximidamides 17a–f was prepared by stirring 16 with the appropriate amines (10 equiv) under microwave-assisted heating at 100 °C. The chemical structures and yields obtained for the synthesis of the series of compounds 17a–f prepared are described in Table 6.
Scheme 11.

Synthesis of carboximidamides 17a–f from brominated amine 11.
Table 6.
Chemical structures and yields obtained for the synthesis of 17a–f.
| -R1 | -R2 | Compound | Time a (min) | Yield b (%) |
|---|---|---|---|---|
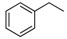 |
H | 17a | 45 | 82 |
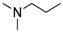 |
H | 17b | 45 | 99 |
 |
H | 17c | 45 | 87 |
 |
H | 17d | 45 | 57 |
 |
17e | 90 | 60 | |
 |
17f | 180 | 54 |
a Time of the conversion of 16 into the corresponding carboximidamides; b Isolated yield.
These studies showed that the synthetic access to fourteen novel 2-arylamino-substituted thiazolo[5,4-f]quinazolin-9(8H)-ones can be performed from ortho-brominated thioureas via a regio-selective intramolecular C-S coupling reaction, catalyzed by copper iodide. Optimization of experimental conditions served to develop a one-pot sequential process helped by microwave assisted heating. This allowed the convenient synthesis of new thiazoloquinazolin-9(8H)-ones substituted in position 2 by various aromatic and aliphatic amine groups for which biological activity was expected.
2.2. Biological Studies
Products of series 7a–l, 8a–m, 10a and 10l, 14a–f and 17a–f were tested in five different in vitro kinase assays (CDK5/p25 (cyclin-dependent kinase), CK1δ/ε (casein kinase 1), GSK-3α/β (glycogen synthase kinase 3), DYRK1A (dual-specificity tyrosine phosphorylation regulated kinase) and CLK1 (cdc2-like kinase 1) to evaluate their inhibition potency [43,44,45,46,47,48]. All compounds were first tested at a final concentration of 10 µM. Compounds showing less than 50% inhibition were considered as inactive (IC50 > 10 µM). Compounds displaying more than 50% inhibition at 10 µM were next tested over a wide range of concentrations (usually 0.01 to 10 µM), and IC50 values were determined from the dose-response curves (Sigma-Plot). Harmine (Table 7) is a β-carboline alkaloid known to be a potent inhibitor of DYRK1A [49]. It was also tested as positive control and its IC50 values were compared to those obtained for the compounds under study.
Table 7.
Kinase inhibitory activity a,b,c of the thiazolo[5,4-f]quinazoline series (7a–l, 8a–m, 10a and 10l, 14a–f and 17a–f).
| Compounds | CDK5/p25 | CK1δ/ε | CLK1 | DYRK1A | GSK-3α/β |
|---|---|---|---|---|---|
| 7a–l | >10 | >10 | >10 | >10 | ≥10 |
| 8a | >10 | >10 | 3.4 | >10 | ≥10 |
| 8c–f | >10 | >10 | >10 | >10 | ≥10 |
| 8g | >10 | >10 | 1.7 | >10 | ≥10 |
| 8h–i | >10 | >10 | >10 | >10 | ≥10 |
| 8j | >10 | >10 | 1.3 | 2.0 | 7.3 |
| 8k–m | >10 | >10 | >10 | >10 | ≥10 |
| 10a | >10 | >10 | 5.2 | 4.8 | ≥10 |
| 10l | >10 | >10 | >10 | >10 | ≥10 |
| 14a | >10 | >10 | 3.4 | >10 | >10 |
| 14b | >10 | >10 | >10 | >10 | ≥10 |
| 14c | >10 | >10 | >10 | >10 | ≥10 |
| 14d | >10 | >10 | 8.1 | 8.2 | ≥10 |
| 14e | >10 | >10 | 5.3 | >10 | ≥10 |
| 14f | >10 | >10 | >10 | >10 | ≥10 |
| 17a | >10 | >10 | 1.0 | 0.67 | 0.4 |
| 17b | >10 | >10 | 0.29 | 4.4 | 1.1 |
| 17c | >10 | >10 | 2.3 | 3.2 | 2.8 |
| 17d | >10 | >10 | 2.1 | 2.9 | 3.9 |
| 17e | >10 | 1.9 | 0.38 | 0.14 | 0.23 |
| 17f | >10 | >10 | 0.61 | 0.82 | 0.49 |
| Harmine | >10 | 1.5 | 0.026 | 0.029 | >10 |
a IC50 values are reported in μM. The most significant results are presented in bold; b Kinases activities were assayed in triplicate; c Typically, the standard deviation of single data points was below 10%.
Results given in Table 7 demonstrate that none of the tricyclic derivatives prepared in series 7, 8, 10 and 14 showed significant inhibitory activity against the set of five kinases tested. Other derivatives substituted in position 2 of the thiazole by an aminoaryl or aminoalkyl group were completely inactive against all kinases.
On a general aspect, most interesting biological activity of the tested compounds was oriented towards one kinase of the initial panel: CLK1. This latter is one of the four isoforms (CLK1-4) of the cdc2-like kinase family. In humans, the highest levels of CLK1 expression were found in the brain. It was described that inhibitors CLK inhibitors may alter the splicing of microtubule-associated protein tau implicated in Alzheimer’s disease and Parkinson’s disease [48]. Three molecules of series 8 (8a, 8g and 8j), compound 10a and products 14a, 14d and 14e, have shown micromolar activity (1.3 μM < IC50 < 8.1 μM) against the CLK1 kinase.
In series 8a–m and in all the compounds prepared in series 7, 8, 10 and 14, only one compound inhibits in the micromolar range (1.3 μM < IC50 < 7.3 μM) CLK1, DYRK1A and GSK3. This compound is the 8-cyclopropyl-2-[(4-(dimethylamino)phenyl]amino-thiazolo [5,4-f]quinazolin-9(8H)-one (8j) which possesses a tertiary amine in the ortho-position of the aminoarene substituent.
Undoubtedly, the most active molecules prepared in this study were members of the 17a–f seriesin which the final carboximidamide functions were obtained after attack of various amines on the initial carbonitrile function. Most of these compounds showed micromolar or submicromolar activities against CLK1 (0.38 μM < IC50 < 0.61 μM), DYRK1A (0.14 μM < IC50 < 0.82 μM) and GSK-3α/β (0.23 μM < IC50 < 0.43 μM).
The two derivatives 17e and 17f are the most active products of this study, their interesting IC50 values being slightly better for 17e than for 17f. These two isosters are also bioisosters, considering the results given in Table 7.
Comparing the results of this study with our previous works, it seems rather obvious that having such molecular scaffolds with submicromolar affinities for various kinases is related to the presence of carboximidamide or carboximidate functions that result from the substitution of a carbonitrile group itself present in position 2 of the thiazole.
Because drugs focusing their activity against a single target may generate low benefits, recent studies are encouraged in the direction of multi-targeting strategies. In the present case, the novel structures studied are not the most valuable for the further discovery of multi-kinases inhibitors. Developing molecules showing submicromolar affinities on a panel of two or three kinases needs to leave a substituted carbon on position 2 of the thiazole part of these scaffolds.
3. Materials and Methods
3.1. General Information
Materials were obtained commercially and used without further purification. All reactions were monitored by thin-layer chromatography with silica gel 60 F254 precoated aluminium plates (0.25 mm). Visualization was performed with UV light at wavelengths of 254 and 312 nm. Purifications were conducted with a flash column chromatography system equipped with a dual UV/Vis spectrophotometer (200–600 nm), a fraction collector (176 tubes), a dual piston pump (1 to 200 mL/min, Pmax = 15 bar), which allowed quaternary gradients, and an additional inlet for air purge. The melting points of solid compounds were measured with a STUART Melting Point SMP3 instrument (Bibby Scientific Ltd., Roissy, France) with a precision of 1.5 °C. IR spectra were recorded with a Spectrum 100 Series FTIR spectrometer (PerkinElmer, Villebon S/Yvette, France). Liquids and solids were investigated with a single-reflection attenuated total reflectance (ATR) accessory; the absorption bands are given in cm−1. The NMR spectra (1H and 13C) were acquired at 295 K using a WP 300 spectrometer AVANCE 300 MHz spectrometer (Bruker, Wissembourg, France) at 300 and 75.4 MHz, using TMS as an internal standard. Coupling constants J are in Hz, and chemical shifts are given in ppm. Signals in 13C spectra were assigned based on the result of 13C DEPT135 experiments (see Supplementary Materials). Mass spectrometry was performed by the Mass Spectrometry Laboratory of the University of Rouen. The mass spectra [ESI, EI, and field desorption (FD)] were recorded with a LCP 1er XR spectrometer (WATERS, Guyancourt, France). Microwave experiments were conducted in a commercial microwave reactor especially designed for synthetic chemistry. Start S™ (Milestone S.r.l., Bergamo, Italy) is a multi-mode cavity with a microwave power delivery system ranging from 0 to 1200 W. The temperatures of the reactions were mainly monitored via contact-less infrared pyrometer which was calibrated in control experiments with a fibre-optic contact thermometer protected in a Teflon coated ceramic well inserted directly in the reaction mixture. Open vessel experiments were carried out in a 50–250 mL round bottom flask fitted with a reflux condenser. The vessel contents were stirred by means of an adjustable rotating magnetic plate located below the floor of the microwave cavity and a Teflon-coated magnetic stir bar inside the vessel. Temperature and power profiles were monitored in both cases through the EASY-Control software provided by the manufacturer (Milestone S.r.l., Bergamo, Italy). The times indicated in the various protocols are the times measured when the mixtures reached the programmed temperature after a ramp period of 2 min.
3.2. Chemistry
3-Cyclopropyl-6-nitroquinazolin-4(3H)-one (2). A stirred suspension of 5-nitroanthranilic acid 1 (1.5 mmol, 1 equiv.) in DMFDMA (500 µL, 2.5 equiv.) and DMF (1.5 mL) was heated at 100 °C for 15 min under microwave (after a ramp period of 2 min at 900 W). Solvents were removed in vacuo and cyclopropylamine (1.65 mmol, 1.1 equiv.) was added followed with AcOH (1.5 mL). The mixture was irradiated at 100 °C (900 W) for 15 min (ramp: 2 min). Evaporation of the solvent gave a crude product which was purified by column chromatography using petroleum ether/methylene chloride (100:0 to 0:100, v/v) as eluent. Compound 2 was isolated as a light yellow solid (295 mg, 85%), mp. 174–176 °C; 1H-NMR (DMSO-d6) δ 8.82 (d, J = 2.7 Hz, 1H, H5), 8.54 (dd, J = 9.0, 2.7 Hz, 1H, H7), 8.45 (s, 1H, H2), 7.86 (d, J = 9.0 Hz, 1H, H8), 3.34–3.25 (m, 1H, NCH), 1.08–0.99 (m, 4H, CH);13C-NMR (DMSO-d6) δ 160.6, 151.6, 151.3, 145.1, 128.9, 128.1, 121.9, 121.4, 29.5, 5.9; νmax 3356, 3102, 1675, 1600, 1512, 1300, 1275, 936, 855, 752 cm−1; HRMS calcd for C11H10N3O3 [M + H]+ 232.0722 found 232.0719.
6-Amino-3-cyclopropylquinazolin-4(3H)-one (3). A stirred mixture of 6-nitro-3-cyclopropyl-quinazolin-4(3H)-one 2 (15 mmol, 1.0 equiv.), ammonium formate (5.0 equiv.) and a catalytic (10 mol %) amount of 10% palladium charcoal in ethanol (0.2 M solution) was heated under microwaves for 20 min (300 W, 85 °C). The reaction mixture was filtered through Celite® and washed with hot ethanol. The solvent was removed in vacuo to give the crude product which was dissolved in ethyl acetate, washed with water, dried over MgSO4 and concentrated under reduced pressure to give the reduced compound as a pale yellow solid (2.6 g, 85%), mp. 172–174 °C; 1H-NMR (DMSO-d6) δ 7.94 (s, 1H, H2), 7.37 (d, J = 8.7 Hz, 1H, H8), 7.22 (d, J = 2.7 Hz, 1H, H5), 7.07 (dd, J = 8.7, 2.7 Hz, 1H, H7), 5.66 (br s, 2H, NH2), 3.20–3.16 (m, 1H, NCH), 1.03–1.01 (m, 2H, CH), 0.90 (m, 2H, CH); 13C-NMR (DMSO-d6) δ 161.3, 148.1, 142.7, 138.3, 127.9, 122.3, 122.0, 106.1, 28.9, 6.0; νmax 3420, 3336, 3222, 1653, 1627, 1600, 1491, 1325, 830, 791 cm−1; HRMS calcd for C11H12N3O [M + H]+ 202.0980 found 202.0974.
3-Cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (4). Method A: A solution of DBU (542 mg, 3.57 mmol, 3.0 equiv.) in methylene chloride (1.0 mL) was added drop wise to a stirred solution of 6-aminoquinazolin-4(3H)-one 5 (400 mg, 1.19 mmol) in methylene chloride (7 mL) maintained at −78 °C. The resulting mixture was warmed to room temperature and stirred for 2 h. One equivalent of DBU (181 mg, 1.19 mmol) was added and the black solution was stirred for 1 h. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (0/100 to 50/50, v/v) as eluent to furnish 4 as a white solid (188 mg, 65%). Method B: Thiophosgene (0.57 mL, 7.47 mmol, 1.5 equiv.) was added drop wise to a stirred solution of 6-aminoquinazolin-4(3H)-one 3 (1.00 g, 4.97 mmol) in a mixture of THF (5.0 mL) and a saturated solution of NaHCO3 (5.0 mL) maintained at 0 °C. The resulting mixture was warmed to room temperature and stirred for 30 min. On completion, the reaction mixture was diluted with water (25 mL) and ethyl acetate (25 mL). The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed with water and brine. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (0/100 to 50/50, v/v) as eluent to furnish 4 as a colorless solid (1.18 g, 98%); mp. 146–148 °C; 1H-NMR (DMSO-d6) δ 8.31 (s, 1H, H2), 8.04 (d, J = 2.2 Hz, 1H, H5), 7.82 (dd, J = 8.6, 2.2 Hz, 1H, H7), 7.70 (d, J = 8.6 Hz, 1H, H8), 3.28–3.21 (m, 1H, NCH), 1.08–0.97 (m, 4H, CH); 13C-NMR (CDCl3) δ 161.2, 147.4, 146.2, 137.8, 131.7, 130.6, 129.2, 123.3, 122.9, 29.6, 6.7; νmax 2118, 1669, 1602, 833, 535 cm−1; HRMS calcd for C12H10N3OS [M + H]+ 244.0545 found 244.0537.
6-[(4-Chloro-5H-1,2,3-dithiazol-5-yl)amino]-3-cyclopropylquinazolin-4(3H)-one (5). A suspension of 6-amino-3-cyclopropylquinazolin-4(3H)-one (3, 300 mg, 1.49 mmol) and 4,5-dichloro-1,2,3-dithiazolium chloride (1.2 equiv.) in DCM (0.1 M solution) was stirred for 1 h at room temperature under an argon atmosphere. Pyridine (2.0 equiv.) was added and the mixture was stirred again for 2 h at room temperature. The resulting solution was concentrated under vacuo to give a crude residue which was purified by chromatography on silica gel with EtOAc/DCM (5/95 then 50/50, v/v) to give the expected iminodithiazole as a yellow solid (382 mg, 76%), mp. 190–192 °C; 1H-NMR (DMSO-d6) δ 8.29 (s, 1H, H2), 7.97 (d, J = 2.4 Hz, 1H, H5), 7.78 (d, J = 8.7 Hz, 1H, H8), 7.65 (dd, J = 8.7, 2.4 Hz, 1H, H7), 3.20–3.16 (m, 1H, NCH), 1.09–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 161.0, 160.4, 149.0, 147.7, 147.0, 145.4, 129.1, 128.2, 122.3, 114.2, 29.3, 5.9; νmax 3073, 1659, 1592, 1530, 1454, 1305, 951, 842, 581 cm−1; HRMS calcd for C13H10N4OS2Cl2 [M + H]+ 336.9985 found 336.9974.
(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)carbamothioyl cyanide (6). A solution of DBU (542 mg, 3.57 mmol, 3.0 equiv.) in methylene chloride (1.0 mL) was added drop wise to a stirred solution of iminodithiazole 5 (400 mg, 1.19 mmol) in methylene chloride (7.0 mL) maintained at −78 °C. The resulting mixture was warmed to room temperature and stirred for 2 h. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate/methylene chloride (0/100 to 50/50, v/v) as eluent to furnish 6 as an orange solid (250 mg, 74%), mp. 196–198 °C; 1H-NMR (DMSO-d6) δ 13.72 (br s, 1H, NH), 8.91 (d, J = 2.4 Hz, 1H, H5), 8.32 (s, 1H, H2), 8.16 (dd, J = 9.0, 2.7 Hz, 1H, H7), 7.76 (d, J = 9.0 Hz, H8), 3.20–3.16 (m, 1H, NCH), 1.09–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 161.1, 160.8, 148.4, 146.0, 136.9, 128.7, 128.0, 121.4, 118.7, 114.0, 29.3, 5.9; νmax 3519, 3430, 2944, 2055, 1641, 1601, 1487, 1389, 1256, 1193, 1021, 847, 739 cm−1; HRMS calcd for C13H11N4OS [M + H]+ 271.0654 found 271.0655.
6-Amino-5-bromo-3-cyclopropylquinazolin-4(3H)-one (11). To a stirred solution of 6-amino-3-cyclo-propylquinazolinone 3 (5.5 g, 27.3 mmol) was added NBS (4.9 g, 27.3 mmol, 1.0 equiv.) at room temperature. After 2 h of stirring at room temperature, the solvent was removed under vacuum and the crude residue was purified by chromatography on silica gel using EtOAc/DCM (20/80 to 50/50, v/v) as eluent to give the compound 11 as a colorless solid (7.1 g, 93%), mp. 172–174 °C; 1H-NMR (DMSO-d6) δ 8.03 (s, 1H, H2), 7.40 (d, J = 8.7 Hz, 1H, H8), 7.28 (d, J = 8.7 Hz, 1H, H7), 5.86 (br s, 2H, NH2), 3.18–3.13 (m, 1H, NCH), 1.03–0.99 (m, 2H, CH), 0.91–0.85 (m, 2H, CH); 13C-NMR (DMSO-d6) δ 159.6, 146.1, 144.0, 140.4, 127.4, 121.7, 119.5, 100.5, 29.4, 6.0; νmax 3088, 3017, 2926, 2848, 1675, 1508, 1335, 1141, 855, 752, 699 cm−1; HRMS calcd for C11H11N3O79Br [M + H]+ 280.0085 found 280.0095 (100%), calcd for C11H11N3O81Br [M + H]+ 282.0065 found 282.0072 (93%).
1-(5-Bromo-3-cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-3-phenylthiourea (12). A solution of isothiocyanate 13 (0.200 g, 0.62 mmol) and aniline (61 µL, 0.65 mmol, 1.05 equiv.) in acetonitrile (2.0 mL) was stirred at room temperature overnight. the solvent was removed under vacuum und the crude residue was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (0/1 to 5/5, v/v) to furnish the expected thiourea 12 as a colorless solid (0.211 g, 82%), mp. 212–214 °C; 1H-NMR (DMSO-d6) δ 10.11 (s, 1H, NH), 9.47 (s, 1H, NH), 8.31 (s, 1H, H2), 7.92 (d, J = 8.7 Hz, 1H, H8), 7.62 (d, J = 8.7 Hz, 1H, H7), 7.57–7.55 (m, 2H, Ph), 7.39–7.34 (m, 2H, Ph), 7.19–7.14 (m, 1H, Ph), 3.22–3.17 (m, 1H, NCH), 1.04–0.92 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.2, 159.6, 148.2, 147.8, 139.0, 137.9, 135.1, 128.6, 126.4, 124.8, 123.7, 119.6, 119.1, 29.2, 6.0; νmax 3315, 2930, 1677, 1619, 1515, 1324, 1297, 897, 696, 501 cm−1; HRMS calcd for C18H16N4OS79Br [M + H]+ 415.0228 found 415.0233 (93%), for C18H16N4OS81Br [M + H]+ 417.0208 found 417.0197 (100%).
5-Bromo-3-cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (13). According to the previous procedure described for compound 4, 6-amino-5-bromoquinazolin-4(3H)-one (11, 1.00 g, 3.57 mmol) was stirred with thiophosgene (0.410 g, 5.35 mmol, 1.5 equiv.) in a mixture of THF (3.5 mL) and a saturated solution of NaHCO3 (3.5 mL) to furnish 13 as a colorless solid (0.940 g, 82%), mp. 210–212 °C; 1H-NMR (DMSO-d6) δ 8.36 (s, 1H, H2), 7.90 (d, J = 8.7 Hz, 1H, H8), 7.67 (d, J = 8.7 Hz, 1H, H7), 3.23–3.16 (m, 1H, NCH), 1.05–0.93 (m, 4H, CH); 13C-NMR (CDCl3) δ 159.9, 148.0, 147.6, 138.7, 132.1, 131.3, 128.3, 121.1, 120.0, 30.1, 6.7 (2C); νmax 3061, 2921, 2122, 1688, 1588, 1454, 1314, 1259, 838 cm−1; HRMS calcd for C12H9N3OS79Br [M + H]+ 321.9650 found 321.9647, for C12H9N3OS81Br [M + H]+ 323.9629 found 323.9630.
(Z)-5-Bromo-6-[(4-chloro-5H-1,2,3-dithiazol-5-ylidene)amino]-3-cyclopropylquinazolin-4(3H)-one (15). A suspension of 6-amino-5-bromo-3-cyclopropylquinazolin-4(3H)-one (11, 2.0 g, 1.0 equiv.) and 4,5-dichloro-1,2,3-dithiazolium chloride (1.2 equiv.) in DCM (0.1 M solution) was stirred for 1 h at room temperature under an argon atmosphere. Pyridine (2.0 equiv.) was added and the mixture was stirred again for 2 h at room temperature. The resulting solution was concentrated under vacuo to give a crude residue which was purified by chromatography on silica gel with EtOAc/DCM (5/95 then 50/50, v/v) to give the desired iminodithiazole as an orange solid (2.3 g, 76%), mp. 204–206 °C; 1H-NMR (DMSO-d6) δ 8.33 (s, 1H, H2), 7.75 (d, J = 8.7 Hz, 1H, H7), 7.58 (d, J = 8.7 Hz, 1H, H8), 3.24–3.17 (m, 1H, NCH), 1.06–1.00 (m, 2H, CH), 0.97–0.94 (m, 2H, CH); 13C-NMR (DMSO-d6) δ 163.2, 159.6, 150.5, 147.9, 146.8, 145.8, 129.0, 124.6, 120.3, 111.3, 29.7, 6.0 (2C); νmax 3003, 1862, 1675, 1593, 1452, 1325, 1293, 1148, 826, 759 cm−1; HRMS calcd for C13H9N4OS279BrCl [M + H]+ 414.9090 found 414.9088.
8-Cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carbonitrile (16). A suspension of imine 15 (2.00 g, 1.0 equiv.), copper iodide (CuI, 2.0 equiv.) in pyridine (0.33 M solution) was irradiated under microwaves at 115 °C (power input: 300 W) for 20 min. After cooling, the mixture was diluted in EtOAc, washed with a saturated aqueous solution of sodium thiosulfate. The organic layer was dried over MgSO4 and the solvent was removed in vacuo. The crude residue was purified by column chromatography on silica gel with EtOAc/methylene chloride (0/100 to 20/80, v/v) as eluent to give the expected compound as a colorless solid (1.05 g, 81%), mp. 248–250 °C; 1H-NMR (DMSO-d6) δ 8.63 (d, J = 8.7 Hz, 1H, H4), 8.61 (s, 1H, H2), 7.99 (d, J = 9.0 Hz, 1H, H5), 3.42–3.36 (m, 1H, NCH), 1.13–1.09 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 160.2, 150.3, 149.5, 148.2, 139.2, 131.6, 129.9, 127.8, 115.4, 113.5, 29.7, 5.9 (2C); νmax 3067, 2233 (C≡N), 1664, 1579, 1441, 1353, 1303, 1222, 1038, 839, 692 cm−1; HRMS calcd for C13H9N4OS [M + H]+ 269.0497 found 269.0487.
3.2.1. General Procedures for the Synthesis of N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-aryl or Alkylthioureas 7a–l from 3-Cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (4) or 6-Amino-3-cyclopropylquinazolin-4(3H)-one (3)
Method A: To a stirred solution of isothiocyanate 4 (250 mg, 1.03 mmol) in acetonitrile (3 mL) was added the appropriate amine (1.08 mmol, 1.05 equiv.) at room temperature. The resulting mixture was stirred at to room temperature for nucleophilic amines for 15 min to 2 h or under microwave at 40 °C for 1 h. On completion, the solvent was evaporated under vacuum. Trituration of crude mixture in diethyl ether (6 mL) followed by filtration furnished the expected thiourea derivatives 7a, c–d, g, j–l.
Method B: To a stirred solution of 6-amino-3-cyclopropylquinazolinone (3, 250 mg, 1.24 mmol) in DMF (4 mL) was added the appropriate arylisothiocyanate (1.05 equiv.) at room temperature. The resulting mixture was stirred at room temperature for 15 min to 4 h. On completion, the solvent was evaporated under vacuum. Trituration of crude mixture in diethyl ether (8 mL) followed by filtration furnished the expected thiourea derivatives 7a–b; e–f; h, i.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-phenylthiourea (7a): According to the method A (40 °C, 1 h) or the method B (r.t., 2 h), product 7a was obtained as a colorless solid (291 mg, 84%) or (333 mg, 80%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.09 (s, 1H, NH), 10.00 (s, 1H, NH), 8.25 (d, J = 1.8 Hz, 1H, H5), 8.21 (s, 1H, H2), 7.92 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.60 (d, J = 8.7 Hz, 1H, H8), 7.46 (d, J = 8.0 Hz, 2H, Ph), 7.34 (t, J = 8.0 Hz, 2H, Ph), 7.14 (t, J = 8.0 Hz, 1H, Ph), 3.26–3.19 (m, 1H, NCH), 1.03–0.92 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.0, 146.8, 144.1, 139.1, 138.3, 130.1, 128.5, 127.0, 124.5, 123.6, 121.1, 118.8, 29.1, 5.9; νmax 3292, 3069, 2990, 1649, 1592, 1511, 1492, 1303, 1246, 1163, 1037, 821, 610, 580 cm−1; HRMS calcd for C18H17N4OS [M + H]+ 337.1123 found 337.1109.
N-(4-Chlorophenyl)-N′-(3-cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)thiourea (7b): According to the method B (r.t., 1 h), compound 7b was obtained as a colorless solid (418 mg, 91%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.17 (s, 1H, NH), 10.06 (s, 1H, NH), 8.26 (d, J = 1.8 Hz, 1H, H5), 8.23 (s, 1H, H2), 7.93 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.63 (d, J = 8.7 Hz, 1H, H8), 7.53 (d, J = 8.7 Hz, 2H, ArH), 7.42 (d, J = 8.7 Hz, 2H, ArH), 3.28–3.18 (m, 1H, NCH), 1.08–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.8, 161.1, 147.0, 144.2, 138.2, 130.2, 128.5, 128.4, 127.2, 125.3, 121.3, 119.0, 29.2, 6.0; νmax 3290, 1645, 1506, 1488, 1305, 1245, 1087, 787, 654, 525 cm−1; HRMS calcd for C18H16N4OS Cl [M + H]+ 371.0733 found 371.0721.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-fluorophenyl)thiourea (7c): According to the method A (r.t., 2 h), compound 7c was isolated as a colorless solid (331 mg, 90%), mp. 194–196 °C; 1H-NMR (DMSO-d6) δ 10.07 (s, 1H, NH), 9.94 (s, 1H, NH), 8.26 (d, J = 2.4 Hz, 1H, H5), 8.22 (s, 1H, H2), 7.93 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.62 (d, J = 8.7 Hz, 1H, H8), 7.50–7.46 (m, 2H, ArH), 7.22–7.16 (m, 2H, ArH), 3.25–3.20 (m, 1H, NCH), 1.08–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.1, 161.1, 159.3 (d, J = 240 Hz, C4′-F), 146.9, 144.1, 138.3, 135.5, 130.2, 127.1, 126.2 (d, J = 8.25 Hz, 2C, C2′-F), 121.2, 119.0, 115.2 (d, J = 22.5 Hz, 2C, C3′-F), 29.2, 6.0; νmax 3324, 3195, 3055, 1651, 1633, 1606, 1510, 1300, 1221, 836, 717, 545 cm−1; HRMS calcd for C18H16N4OFS [M + H]+ 355.1029 found 355.1014.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-[4-(trifluoromethyl)phenyl)]-thiourea (7d): According to the method A (40 °C, 1 h), compound 7d was isolated as a light yellow solid (351 mg, 84%), mp. 198–200 °C; 1H-NMR (DMSO-d6) δ 10.33 (s, 1H, NH), 10.29 (s, 1H, NH), 8.28 (d, J = 2.7 Hz, 1H, H5), 8.23 (s, 1H, H2), 7.94 (dd, J = 8.7, 2.7 Hz, 1H, H7), 7.76 (d, J = 8.7 Hz, 2H, ArH), 7.67 (d, J = 8.7 Hz,2H, ArH), 7.64 (d, J = 8.7 Hz, 1H, H8), 3.25–3.21 (m, 1H, NCH), 1.08–0.94 (m, 4H, CH); 19F NMR (282 MHz, DMSO-d6) δ −60.4 (3F, CF3); 13C-NMR (DMSO-d6) δ 179.8, 161.1, 147.1, 144.3, 143.1, 138.0, 130.2, 127.2, 125.7 (q, J = 37.5 Hz, 2C, C3′-F), 124.1 (q, J = 32.0 Hz, C4′-F), 122.9, 121.2, 119.0, 29.2, 6.0; νmax 3167, 2988, 1684, 1604, 1487, 1323, 1289, 1190, 1068, 835, 613 cm−1; HRMS calcd for C19H16N4OF3S [M + H]+ 405.0997 found 405.0996.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-nitrophenyl)thiourea (7e): According to the method B (r.t., 30 min.), compound 7e was isolated as a yellow solid (351 mg, 74%), mp. 198–200 °C; 1H-NMR (DMSO-d6) δ 10.56 (s, 1H, NH), 10.55 (s, 1H, NH), 8.31 (d, J = 2.3 Hz, 1H, H5), 8.24 (s, 1H, H2), 8.22 (d, J = 9.1 Hz, 2H, ArH), 7.95 (dd, J = 8.7, 2.3 Hz, 1H, H7), 7.85 (d, J = 8.7 Hz, 2H, ArH), 7.65 (d, J = 8.7 Hz, 1H, H8), 3.28–3.22 (m, 1H, NCH), 1.06–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.5, 161.0, 147.2, 146.0, 144.4, 142.4, 137.8, 130.1, 127.3, 122.4, 121.3, 119.1, 29.2, 6.0; νmax 3331, 3215, 3094, 3067, 1650, 1626, 1490, 1322, 1246, 1106, 847, 705, 527 cm−1; HRMS calcd for C18H16N5O3S [M + H]+ 382.0974 found 382.0959.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(2,4-dichlorophenyl)thiourea (7f): According to the method B (r.t., 1 h), compound 7f was isolated as a colorless solid (446 mg, 89%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.28 (s, 1H, NH), 9.70 (s, 1H, NH), 8.32 (d, J = 2.4 Hz, 1H, H5), 8.24 (s, 1H, H2), 7.95 (dd, J = 2.4, 8.7 Hz, 1H, H7), 7.72 (d, J = 2.4 Hz, 1H, H3′); 7.64 (d, J = 8.7 Hz, 1H, H8), 7.61 (d, J = 8.4 Hz, 1H, H6′), 7.46 (dd, J = 2.4, 8.4 Hz, 1H, H5′), 3.28–3.20 (m, 1H, NCH), 1.08–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.5, 161.1, 147.1, 144.3, 138.0, 135.4, 131.4, 131.2, 131.0, 130.2, 129.0, 127.5, 127.2, 121.3, 119.3, 29.2, 6.0; νmax 3234, 1650, 1518, 1445, 1346, 1209, 840, 808, 665, 499 cm−1; HRMS calcd for C18H15N4OSCl2 [M + H]+ 405.0344 found 405.0344.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(pyridin-3-yl)thiourea (7g): According to the method A (40 °C (µW), 1 h), compound 7g was isolated as a colorless solid (309 mg, 89%), mp. 188–190 °C; 1H-NMR (DMSO-d6) δ 10.29 (s, 1H, NH), 10.05 (s, 1H, NH), 8.62 (d, J = 2.4 Hz, 1H, H2′), 8.35 (d, J = 4.5 Hz, 1H, H6′), 8.27 (d, J = 2.1 Hz, 1H, H5), 8.24 (s, 1H, H2), 7.96–7.92 (m, 2H, H7 + H4′), 7.64 (d, J = 8.7 Hz, 1H, H8), 7.39 (dd, J = 2.4, 4.5 Hz, 1H, H5′), 3.27–3.22 (m, 1H, NCH), 1.06–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.4, 161.1, 147.0, 145.44, 145.37, 144.3, 138.0, 136.1, 131.4, 130.3, 127.2, 123.2, 121.3, 119.1, 29.2, 6.0; νmax 3217, 3011, 1656, 1539, 1485, 1363, 1257, 839, 707, 558 cm−1; HRMS calcd for C17H16N5OS [M + H]+ 338.1076 found 338.1071.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(p-tolyl)thiourea (7h): According to the method B (r.t., 2 h), compound 7h was obtained as a colorless solid (386 mg, 89%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 9.99 (s, 1H, NH), 9.90 (s, 1H, NH), 8.27 (d, J = 1.8 Hz, 1H, H5), 8.22 (s, 1H, H2), 7.94 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.61 (d, J = 8.7 Hz, 1H, H8), 7.35 (d, J = 8.1 Hz, 2H, ArH), 7.16 (d, J = 8.1 Hz, 2H, ArH), 3.26–3.19 (m, 1H, NCH), 2.30 (s, 3H, CH3), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.1, 146.8, 144.0, 138.5, 136.5, 133.9, 130.2, 129.0, 127.0, 123.9, 121.2, 118.8, 29.2, 20.5, 6.0; νmax 3290, 3072, 2993, 1648, 1596, 1513, 1492, 1303, 1246, 1163, 1037, 827, 654, 608, 580 cm−1; HRMS calcd for C19H19N4OS [M + H]+ 351.1280 found 351.1276.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-methoxyphenyl)thiourea (7i): According to the method B (r.t., 2 h), compound 7i was isolated as a colorless solid (395 g, 87%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 9.91 (s, 1H, NH), 9.81 (s, 1H, NH), 8.27 (d, J = 1.8 Hz, 1H, H5), 8.22 (s, 1H, H2), 7.94 (dd, J = 8.7, 1.8 Hz, 1H, H7), 7.61 (d, J = 8.7 Hz, 1H, H8), 7.34 (d, J = 8.1 Hz, 2H, ArH), 6.93 (d, J = 8.1 Hz, 2H, ArH), 3.76 (s, 3H, OCH3), 3.26–3.19 (m, 1H, NCH), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.0, 161.1, 156.7, 146.8 144.0, 138.54 131.8, 130.2, 127.0, 126.0, 121.2, 118.9, 113.8, 55.2, 29.2, 6.0; νmax 3296, 1648, 1565, 1514, 1306, 817, 719, 574 cm−1; HRMS calcd for C19H19N4O2S [M + H]+ 367.1229 found 367.1222.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(4-methoxyphenyl)thiourea (7j): According to the method A (r.t., 10 min.), compound 7j was isolated as a grey solid (340 mg, 87%), mp. 210–212 °C; 1H-NMR (DMSO-d6) δ 9.77 (s, 1H, NH), 9.70 (s, 1H, NH), 8.27 (d, J = 2.4 Hz, 1H, H5), 8.20 (s, 1H, H2), 7.93 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.59 (d, J = 8.7 Hz, 1H, H8), 7.22 (d, J = 9.0 Hz, 2H, ArH), 6.72 (d, J = 9.0 Hz, 2H, ArH), 2.89 (s, 6H, NCH3), 3.33–3.21 (m, 1H, NCH), 1.05–0.93 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.1, 148.3, 146.7, 143.9, 138.7, 130.2, 127.9, 126.9, 125.7, 121.1, 118.8, 112.3, 40.3, 29.1, 6.0; νmax 3290, 3245, 1649, 1524, 1311, 1243, 704 cm−1; HRMS calcd for C20H22N5OS [M + H]+ 380.1545 found 380.1530.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(2,4-dimethoxyphenyl)thiourea (7k): According to the method A (r.t., 30 min.), product 7k was isolated as a black solid (396 mg, 97%), mp. 166–168 °C; 1H-NMR (DMSO-d6) δ 9.90 (s, 1H, NH), 9.22 (s, 1H, NH), 8.33 (d, J = 2.4 Hz, 1H, H5), 8.21 (s, 1H, H2), 7.95 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.60 (d, J = 8.7 Hz, 1H, H8), 7.47 (d, J = 8.4 Hz, 1H, H6′), 6.64 (d, J = 2.7 Hz, 1H, H3′), 6.53 (dd, J = 8.4, 2.7 Hz, 1H, H5′), 3.83 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.25–3.20 (m, 1H, NCH), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 180.1, 161.1, 159.5, 154.2, 146.8, 143.9, 138.6, 130.1, 128.2, 126.9, 121.1, 120.1, 118.8, 104.3, 99.0, 55.7, 55.3, 29.1, 6.0; νmax 3156, 2969, 1674, 1601, 1510, 1487, 1282, 1236, 1206, 1031, 834, 686, 540 cm−1; HRMS calcd for C20H21N4O3S [M + H]+ 397.1334 found 397.1319.
N-(3-Cyclopropyl-4-oxo-3,4-dihydroquinazolin-6-yl)-N′-(3,4-dimethoxyphenyl)thiourea (7l): According to the method A (r.t., 5 min.), compound 7l was isolated as a black solid (359 mg, 88%), mp. 196–198 °C; 1H-NMR (DMSO-d6) δ 9.87 (s, 1H, NH), 9.83 (s, 1H, NH), 8.24 (d, J = 2.4 Hz, 1H, H5), 8.21 (s, 1H, H2), 7.93 (dd, J = 8.7, 2.4 Hz, 1H, H7), 7.60 (d, J = 8.7 Hz, 1H, H8), 7.10 (s, 1H, ArH), 6.93 (s, 2H, ArH), 3.75 (s. 3H, OCH3), 3.74 (s, 3H, OCH3), 3.26–3.16 (m, 1H, NCH), 1.05–0.94 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 179.7, 161.1, 148.4, 146.8, 146.4, 144.1, 138.5, 131.9, 130.4, 126.9, 121.2, 119.1, 116.5, 111.7, 109.3, 55.7, 55.5, 29.1, 6.0; νmax 3271, 1670, 1602, 1511, 1233, 1130, 1024, 835, 566 cm−1; HRMS calcd for C20H21N4O3S [M + H]+ 397.1334 found 397.1342.
3.2.2. General Procedure for the Synthesis of 8-Cyclopropyl-2-arylaminothiazolo[5,4-f]quinazolin-9(8H)-ones 8d–g and 3-Cyclopropyl-6-[(benzo[d]thiazol-2-yl)amino]quinazolin-4(3H)-ones 10a; 10l via the Hügershoff Reaction
To a solution of appropriate thiourea derivative 7 (125 mg, 1 equiv.) in acetic acid (0.2 M solution) maintained at 0 °C was added dropwise bromine (Br2, 1.0 equiv.). The resulting mixture was warmed to room temperature and stirred for 30 min. to 4 h. On completion, the solvent was removed under vacuum. The crude residue was dissolved in dichloromethane (25 mL). The organic layer was washed with a saturated solution of NaHCO3, then with water and brine. Evaporation of solvent gave a crude mixture which was adsorbed on Celite® and purified by flash chromatography using ethyl acetate and methylene chloride (100:0 to 0:100%, v/v) as eluent to furnish the expected fused 2-arylaminothiazole 8 or 10.
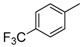
8-Cyclopropyl-2-([4-(trifluoromethyl)phenyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (8d): from 7d (125 mg, 0.31 mmol) in presence of bromine (16.0 µL, 0.31 mmol, 1.0 equiv.), 1 h at room temperature. After purification, 8d was isolated as a colorless solid (98 mg, 79%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 11.06 (s, 1H, NH), 8.31 (s, 1H, H2), 8.09 (d, J = 8.2 Hz, 1H, H4), 8.03 (d, J = 9.0 Hz, 2H, Ph), 7.75 (d, J = 9.0 Hz, 2H, Ph), 7.67 (d, J = 8.2 Hz, 1H, H5), 3.36–3.28 (m, 1H, NCH), 1.10–1.03 (m, 4H, CH); 19F NMR (282 MHz, DMSO-d6) δ −60.4 (3F, CF3); 13C-NMR (DMSO-d6) δ 164.5, 160.5, 150.7, 146.2, 143.8, 144.3, 143.1, 126.3, 125.9, 125.6, 124.8 (q, J = 37.5 Hz, 2C, C3′-F), 124.1 (q, J = 32.0 Hz, C4′-F), 122.9 (2C), 121.2, 117.5, 115.4, 29.3, 5.9; νmax 3263, 3061, 3018, 1661, 1532, 1318, 1066, 1108, 831 cm−1; HRMS calcd for C19H14N4OSF3 [M + H]+ 403.0842 found 403.0840.
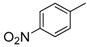
8-Cyclopropyl-2-[(4-nitrophenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8e): from 7e (125 mg, 0.33 mmol) in presence of bromine (16.8 µL, 0.33 mmol, 1.0 equiv.), 4 h at room temperature. After purification, 8e was isolated as a yellow solid (108 mg, 86%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 11.41 (s, 1H, NH), 8.34 (s, 1H, H7), 8.31 (d, J = 9.3 Hz, 2H, Ph), 8.15 (d, J = 8.6 Hz, 1H, H4), 8.07 (d, J = 9.3 Hz, 2H, Ph), 7.72 (d, J = 8.6 Hz, 1H, H5), 3.38–3.24 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 164.0, 160.5, 150.4, 146.4, 146.2, 144.0, 140.8, 125.9, 125.3, 125.2, 117.1, 115.4, 29.3, 5.9; νmax 3291, 3111, 1643, 1560, 1500, 1327, 1307, 1254, 1110, 826 cm−1; HRMS calcd for C18H14N5O3S [M + H]+ 380.0817 found 380.0824.
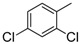
8-Cyclopropyl-2-(2,4-dichlorophenylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8f): from 7f (125 mg, 0.31 mmol) in presence of bromine (15.8 µL, 0.31 mmol, 1.0 equiv.), 1 h 30 at room temperature. After purification, 8f was isolated as a colorless solid (112 mg, 90%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.25 (s, 1H, NH), 8.47 (d, J = 8.9 Hz, 1H, H6′), 8.31 (s, 1H, H7), 8.03 (d, J = 8.6 Hz,1H, H4), 7.71 (d, J = 2.4 Hz, 1H, H3′), 7.65 (d, J = 8.4 Hz, 1H, H5), 7.51 (dd, J = 2.4, 8.6 Hz, 1H, H5′), 3.33–3.28 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 165.9, 160.5, 150.4, 146.1, 143.6, 136.0, 129.1, 127.8, 127.5, 125.4, 124.9, 124.7, 124.0, 115.4, 29.3, 6.0; νmax 3217, 3094, 1662, 1584, 1521, 1456, 1298, 901, 831, 622, 536 cm−1; HRMS calcd for C18H13N4OSCl2 [M + H]+ 403.0187 found 403.0180.
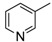
8-Cyclopropyl-2-(pyridin-3-ylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8g): from 7g (125 mg, 0.37 mmol) and bromine (19.0 µL, 0.37 mmol, 1.0 equiv.), 2 h at room temperature. After purification, 8g was isolated as a colorless solid (100 mg, 80%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.89 (s, 1H, NH), 8.94 (d, J = 2.4 Hz, 1H, H2′), 8.39 (dd, J = 2.4, 8.2 Hz, 1H, H6′), 8.31 (s, 1H, H7), 8.26 (dd, J = 2.4, 4.5 Hz, 2H, H4′), 8.08 (d, J = 8.6 Hz, 1H, H4), 7.67 (d, J = 8.6 Hz, 1H, H5), 7.43 (dd, J = 4.5, 8.2 Hz, 1H, H5′), 3.38–3.24 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 164.8, 160.5, 150.8, 146.1, 143.6, 142.9, 139.5, 137.2, 125.6, 125.5, 125.0, 124.3, 123.8, 115.4, 29.3, 5.9; νmax 3659, 3256, 2973, 1662, 1521, 1426, 1280, 826, 799 cm−1; HRMS calcd for C17H14N5OS [M + H]+ 336.0919 found 336.0910.
3-Cyclopropyl-6-[(benzo[d]thiazol-2-yl)amino]quinazolin-4(3H)-one (10a): from 7a (125 mg, 0.37 mmol) in presence of bromine (18.9 µL, 0.37 mmol, 1.0 equiv.), after 1 h at room temperature. After purification, 10a was isolated as a colorless solid (37 mg, 30%), mp. 258–260 °C; 1H-NMR (DMSO-d6) δ 10.93 (s, 1H, NH), 8.72 (d, J = 2.3 Hz, 1H, H5), 8.18 (s, 1H, H2), 8.10 (dd, J = 2.3, 8.7 Hz, 1H, H7), 7.85 (d, J = 8.7 Hz, 1H, H8), 7.69–7.64 (m, 2H, H5′+H6′), 7.40–7.35 (m, 1H, H4′), 7.22–7.17 (m, 1H, H7′), 3.31–3.21 (m, 1H, NCH), 1.09–0.95 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 161.3, 161.2, 151.9, 146.0, 142.5, 139.4, 130.1, 128.0, 126.1, 124.7, 122.7, 122.0, 121.3, 119.5, 112.4, 29.2, 6.0; νmax 3401, 1657, 1623, 1599, 1537, 1490, 1315 cm−1; HRMS calcd for C18H15N4OS [M + H]+ 335.0967 found 335.0966.
3-Cyclopropyl-6-((5,6-dimethoxybenzo[d]thiazol-2-yl)amino)quinazolin-4(3H)-one (10l): from 7l (125 mg, 0.32 mmol) and bromine (16.4 µL, 0.32 mmol, 1.0 equiv.), 30 min at room temperature. After purification, 10l was isolated as a colorless solid (113 mg, 90%), mp. 200 °C; 1H-NMR (DMSO-d6) δ 10.73 (s, 1H, NH), 8.71 (d, J = 2.5 Hz, 1H, H5), 8.35 (s, 1H, H2), 8.07 (dd, J = 2.5, 8.8 Hz, 1H, H7), 7.68 (d, J = 8.8 Hz, 1H, H8), 7.48 (s, 1H, H4′), 7.28 (s, 1H, H7′), 3.85 (s, 3H, OCH3), 3.79 (s, 3H, OCH3), 3.33–3.28 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 160.3, 160.1, 150.4, 148.6, 147.3, 146.1, 145.3, 140.3, 137.5, 125.0, 121.4, 120.7, 104.3, 103.3, 56.0, 55.7, 30.0, 6.0; νmax 3380, 3240, 2999, 1693, 1552, 1482, 1219, 625, 527 cm−1; HRMS calcd for C20H19N4O3S [M + H]+ 395.1178 found 395.1173.
3.2.3. General Procedure for the Synthesis of 8-Cyclopropyl-2-arylaminothiazolo[5,4-f]quinazolin-9(8H)-ones 8a; c; h–n and 8-Cyclopropyl-2-alkylaminothiazolo[5,4-f]quinazolin-9(8H)-ones 14a–f from 5-Bromo-3-cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (13) via a One-Pot Regio-Controlled Copper(I) Mediated C-S Coupling of Intermediates 7
To a solution of 5-bromo-3-cyclopropyl-6-isothiocyanatoquinazolin-4(3H)-one (13, 100 mg, 0.31 mmol, 1.0 equiv.) in DME (3 mL) was added the appropriate amine (0.33 mmol, 1.05 equiv.) at room temperature for 30 min. to 12 h. After completion, copper(I) iodide (5.7 mg, 0.03 mmol, 10 mol %) and cesium carbonate (208 mg, 0.64 mmol, 2.0 equiv.) were added and the resulting suspension was heated under microwave at 80 °C for 1 h. The resulting mixture was adsorbed on Celite® and purified by flash chromatography using ethyl acetate/methylene chloride (0/100 to 50/50, v/v) as eluent to furnish the expected fused 2-arylaminothiazoloquinazolinones 8a; c; h–n and 14a; f or methanol/methylene chloride (0:100 to 5:95, v/v) as eluent to furnish the 2-alkylaminothiazoloquinazolinones 14b–e.
8-Cyclopropyl-2-(phenylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8a): According to the general procedure, thiourea intermediate was obtained from 13 and aniline (30.7 mg) after 12 h at room temperature. After purification, 8a was isolated as a colorless solid (82.9 mg, 80%), mp. 204–206 °C; 1H-NMR (DMSO-d6) δ 10.69 (s, 1H, NH), 8.30 (s, 1H, H7), 8.04 (d, J = 8.7 Hz, 1H, H4), 7.85–7.83 (m, 2H, H2 + H6′), 7.64 (d, J = 8.7 Hz, 1H, H5), 7.41–7.37 (m, 2H, Ph), 7.07–7.03 (m, 1H, Ph), 3.38–3.24 (m, 1H, NCH), 1.09–1.02 (m, 4H, CH); 13C-NMR (75 MHz, -d6) δ 165.1, 160.6, 151.2, 145.9, 143.3, 140.5, 129.0 (2C), 125.5, 125.2, 124.8, 122.2, 117.8, 115.4, 20.3, 6.0; νmax 3257, 3078, 1674, 1516, 1317, 1243, 828, 751, 696 cm−1; HRMS calcd for C18H15N4OS [M + H]+ 335.0967 found 335.0951.
8-Cyclopropyl-2-[(4-fluorophenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8c): According to the general procedure, thiourea intermediate was obtained from 13 and 4-fluoroaniline (56.8 mg) after 2 h at room temperature. After purification, 8c was isolated as a white solid (104.9 mg, 96%), mp > 265 °C; 1H-NMR (300 MHz,DMSO-d6) δ 10.69 (s, 1H, NH), 8.29 (s, 1H, H7), 8.05 (d, J = 8.7 Hz, 1H, H4), 7.88–7.86 (m, 1H, Ph), 7.66 (d, J = 8.7 Hz, 1H, H5), 7.24 (m, 2H, Ph), 3.38–3.24 (m, 1H, NCH), 1.09–1.02 (m, 4H, CH); 19F NMR (282 MHz, DMSO-d6) δ −120.8; 13C-NMR (DMSO-d6) δ 165.1, 160.6, 157.4 (d, J = 237 Hz, C4′-F), 151.1, 145.9, 143.4, 137.0, 125.4, 125.2, 124.9, 119.5 (d, J = 7.50 Hz, 2C, C2′-F), 115.7, 115.4 (d, J = 22.5 Hz, 2C, C3′-F), 29.2, 5.9 (2C); νmax 3291, 3111, 1643, 1560, 1500, 1327, 1307, 1254, 1110, 826 cm−1; HRMS calcd for C18H14N4OSF [M + H]+ 353.0872 found 353.0861.
8-Cyclopropyl-2-(p-tolylamino)thiazolo[5,4-f]quinazolin-9(8H)-one (8h): According to the general procedure, thiourea intermediate was obtained from 13 and p-toluidine (35.4 mg,) after 2 h at room temperature. After purification, 8h was isolated as a colorless solid (96 mg, 89%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.56 (s, 1H, NH), 8.28 (s, 1H, H7), 8.02 (d, J = 8.7 Hz, 1H, H4), 7.71 (d, J = 8.1 Hz, 2H, Ph), 7.63 (d, J = 8.7 Hz, 1H, H5), 7.20 (d, J = 8.1 Hz, 2H, Ph), 3.38–3.24 (m, 1H, NCH), 2.29 (s, 3H, CH3), 1.10–1.01 (m, 4H, CH); 13C-NMR (75 MHz,DMSO-d6) δ 165.2, 160.6, 151.3, 145.8, 143.3, 138.0, 131.1, 129.4, 125.4, 125.1, 124.8, 118.0, 115.4, 29.3, 20.4, 5.9; νmax 3191, 3041, 1640, 1564, 1501, 1327, 1317, 1254, 1140, 840 cm−1; HRMS calcd for C19H17N4OS [M + H]+ 349.1118 found 349.1123.
8-Cyclopropyl-2-[(4-methoxyphenyl]amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8i): According to the general procedure, thiourea intermediate was obtained from 13 and p-anisidine (40.6 mg) after 2 h at room temperature. After purification, 8i was isolated as a colorless solid (99.4 mg, 88%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.50 (s, 1H, NH), 8.26 (s, 1H, H7), 7.78 (d, J = 8.7 Hz, 1H, H4), 7.72 (d, J = 9.0 Hz, 2H, Ph), 7.62 (d, J = 8.7 Hz, 1H, H5), 6.98 (d, J = 9.0 Hz, 2H, Ph), 3.78 (s, 3H, OCH3), 3.38–3.26 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 165.6, 160.6, 154.8, 151.4, 145.7, 143.1, 133.8, 126.5, 125.3, 124.9, 124.8, 119.8, 115.4, 114.2, 115.4, 55.2, 29.1, 5.9; νmax 3267, 3195, 2994, 1679, 1565, 1505, 1295, 1228, 1029, 826, 522 cm−1; HRMS calcd for C19H17N4O2S [M + H]+ 365.1072 found 365.1066.
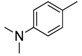
8-Cyclopropyl-2-([4-(dimethylamino)phenyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (8j): According to the general procedure, thiourea intermediate was obtained from 13 and 4-(dimethylamino)aniline (44.9 mg) after 1 h at room temperature. After purification, 8j was isolated as a pale yellow solid (101.7 mg, 87%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.27 (s, 1H, NH), 8.24 (s, 1H, H7), 7.94 (d, J = 8.7 Hz, 1H, H4), 7.61–7.56 (m, 3H, Ph + H5), 7.78 (d, J = 9.1 Hz, 1H, Ph), 3.38–3.24 (m, 1H, NCH), 2.88 (s, 6H, NCH3), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 166.2, 160.6, 151.7, 146.9, 145.5, 142.9, 130.4, 125.3, 124.7, 120.3, 115.4, 113.1, 40.3, 29.2, 5.9; νmax 3184, 2992, 1644, 1574, 1501, 1427, 1347, 1244, 840 cm−1; HRMS calcd for C20H20N5OS [M + H]+ 378.1389 found 378.1376.
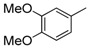
8-Cyclopropyl-2-[(2,4-dimethoxyphenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8k): According to the general procedure, thiourea intermediate was obtained from 13 and 2,4-dimethoxyaniline (50.5 mg) after 30 min. at room temperature. After purification, 8k was isolated as a brown solid (105.1 mg, 86%), mp. 240–242 °C; 1H-NMR (DMSO-d6) δ 9.75 (s, 1H, NH), 8.24 (s, 1H, H7), 8.03 (d, J = 8.7 Hz, 1H, H6′), 7.92 (d, J = 8.6 Hz, 1H, H4), 7.59 (d, 1H, J = 8.6 Hz, H5), 6.69 (d, J = 2.4 Hz, 1H, H3′), 6.60 (dd, J = 2.4, 8.7 Hz, 1H, H5′), 3.85 (s, 3H, OCH3), 3.80 (s, 3H, OCH3), 3.38–3.24 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 167.9, 160.6, 151.8, 151.4, 145.5, 142.9, 125.9, 124.6, 124.5, 123.4, 122.3, 115.4, 104.4, 99.1, 55.7, 55.3, 29.2, 5.9; νmax 3402, 3061, 2932, 2826, 1669, 1563, 1532, 1451, 1178, 1023, 822, 514 cm−1; HRMS calcd for C20H19N4O3S [M + H]+ 395.1178 found 395.1178.
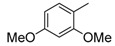
8-Cyclopropyl-2-[(3,4-dimethoxyphenyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (8l): According to the general procedure, thiourea intermediate was obtained from 13 and 4-aminoveratrole (50.5 mg) after 30 min. at room temperature. After purification, 8l was isolated as a colorless solid (110 mg, 90%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 10.47 (s, 1H, NH), 8.27 (s, 1H, H7), 7.99 (d, J = 8.6 Hz, 1H, H4), 7.63 (d, J = 8.6 Hz, 1H, H5), 7.46 (d, J = 2.4 Hz, 1H, H2′), 7.34 (d, J = 2.4, 8.8 Hz, 1H, H6′), 6.98 (d, J = 8.8 Hz, 1H, H5′), 3.81 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 3.38–3.24 (m, 1H, NCH), 1.08–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 165.5, 160.6, 151.4, 148.9, 145.7, 144.4, 143.2, 134.2, 125.3, 125.0, 124.8, 115.4, 112.5, 110.1, 103.8, 55.8, 55.4, 29.2, 5.9; νmax 3273, 3201, 2937, 2832, 1659, 1564, 1516, 1455, 1226, 1025, 826, 537 cm−1; HRMS calcd for C20H19N4O3S [M + H]+ 395.1178 found 395.1166.
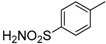
4-[(8-Cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazolin-2-yl)amino]benzenesulfonamide (8m): According to the general procedure, thiourea intermediate was obtained from 13 and 4-aminobenzenesulfonamide (56.8 mg) after 12 h at room temperature. After purification, 8l was isolated as a beige solid (128.1 mg, 77%), mp > 265 °C; 1H-NMR (DMSO-d6) δ 11.10 (s, 1H, NH), 8.30 (s, 1H, H7), 8.10 (d, J = 8.6 Hz, 1H, H4), 8.00 (d, J = 8.8, 2H, Ph), 7.83 (d, J = 8.8 Hz, Ph), 7.67 (d, J = 8.6 Hz, 1H, H5), 7.28 (br, 2H, NH2), 3.39–3.28 (m, 1H, NCH), 1.10–1.01 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 164.5, 160.5, 150.7, 146.2, 143.7, 143.2, 136.9, 127.0, 125.6, 125.0, 117.2, 115.4, 29.3, 6.0; νmax 3486, 3296, 3067, 1655, 1591, 1556, 1518, 1153, 832, 534 cm−1; HRMS calcd for C18H16N5O3S2 [M + H]+ 414.0695 found 414.0692.
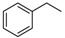
2-(Benzylamino)-8-cyclopropylthiazolo[5,4-f]quinazolin-9(8H)-one (14a): According to the general procedure, thiourea intermediate was obtained from 13 and benzylamine (35.4 mg) after 30 min. at room temperature. After purification, 14a was isolated as a colorless solid (91.8 mg, 85%), mp. 262–264 °C; 1H-NMR (DMSO-d6) δ 8.73 (t, J =5.7 Hz, 1H, NH), 8.21 (s, 1H, H7), 7.84 (d, J = 8.7 Hz, 1H, H4), 7.55 (d, J = 8.7 Hz, 1H, H5), 7.40–7.27 (m, 5H Ph), 4.64 (d, J =5.7 Hz, 2H, CH2), 3.33–3.28 (m, 1H, NCH), 1.07–0.99 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 169.8, 160.5, 151.6, 145.2, 142.4, 138.7, 128.4 (2C), 127.4 (2C), 127.1, 125.7, 124.5, 124.2, 115.4, 47.2, 29.1, 5.9; νmax 3223, 3067, 2907, 1658, 1589, 1455, 1311, 826, 703, 686 cm−1; HRMS calcd for C19H17N4OS [M + H]+ 349.1117 found 349.1123.
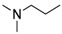
8-Cyclopropyl-2-([2-(dimethylamino)ethyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (14b): According to the general procedure, thiourea intermediate was obtained from 13 and N,N-dimethylethylenediamine (29.1 mg) after 30 min. at room temperature. After purification, 14b was isolated as a colorless solid (98.0 mg, 96%), mp. 210–212 °C; 1H-NMR (DMSO-d6/CDCl3) δ 8.12 (s, 1H, H7), 7.90 (d, J = 8.6 Hz, 1H, H4), 7.63 (d, 1H, J = 8.6 Hz, H5), 7.37 (s, 1H, NH), 3.70 (m, 2H, CH2NH), 3.35–3.30 (m, 1H, NCH), 2.89–2.85 (m, 2H, NCH2), 2.50 (s, 6H, NCH3), 1.28 (m, 2H, CH), 1.04 (m, 2H, CH); 13C-NMR (DMSO-d6/CDCl3) δ 170.1, 160.8, 151.6, 143.9, 142.2, 126.0, 124.2, 124.1, 115.3, 56.9, 44.3, 41.1, 25.7, 6.0; νmax 3201, 2942, 2776, 1659, 1592, 1571, 1456, 1340, 1314, 1164, 825 cm−1; HRMS calcd for C16H20N5OS [M + H]+ 330.1389 found 330.1392.
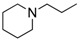
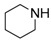
8-Cyclopropyl-2-([2-(piperidin-1-yl)ethyl]amino)thiazolo[5,4-f]quinazolin-9(8H)-one (14c): According to the general procedure, thiourea intermediate was obtained from 13 and 2-piperiridinylethylamine (43.0 mg) after 30 min. at room temperature. After purification, 14c was isolated as a colorless solid (104 mg, 91%), mp. 218–220 °C; 1H-NMR (DMSO-d6) δ 8.25–8.20 (m, 2H, H7 + NH), 7.82 (d, J = 8.4 Hz, 1H, H4), 7.54 (d, 1H, J = 8.4 Hz, H5), 3.56–3.54 (m, 2H, NCH2), 3.41–3.31 (m, 2H, CH2NH), 3.30–3.24 (m, 1H, NCH), 2.65–2.59 (m, 2H, CH2N), 1.55–1.54 (m, 4H, CH), 1.41–1.39 (m, 4H, CH), 1.10–1.01 (m, 4H,CH); 13C-NMR (DMSO-d6) δ 169.6, 160.5, 151.7, 145.2, 142.3, 125.6, 124.4, 124.0, 115.4, 64.8, 54.1, 53.8, 40.1, 29.1, 15.1, 5.9; νmax 3279, 2960, 2860, 1660, 1566, 1529, 1453, 1297, 1113, 833 cm−1; HRMS calcd for C18H22N5O2S [M + H]+ 372.1494 found 372.1487.
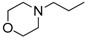
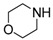
8-Cyclopropyl-2-[(2-morpholinoethyl)amino]thiazolo[5,4-f]quinazolin-9(8H)-one (14d): According to the general procedure, thiourea intermediate was obtained from 13 and 2-morpholinoethylamine (43.0 mg) after 30 min. at room temperature. After purification, 14d was isolated as a colorless solid (102 mg, 89%), mp. 222–224 °C; 1H-NMR (DMSO-d6) δ 8.27 (s, 1H, H7), 8.25 (br, 1H, NH), 7.89 (d, J = 8.4 Hz, 1H, H4), 7.61 (d, 1H, J = 8.4 Hz, H5), 3.67–3.63 (m, 4H, OCH2), 3.60–3.57 (m, 2H, CH2NH), 3.30–3.24 (m, 1H, NCH), 2.57–2.53 (m, 2H, CH2N), 2.44–2.42 (m, 4H, NCH2), 1.09–1.00 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 169.6, 160.6, 151.8, 145.1, 142.3, 125.6, 124.4, 124.0, 115.4, 66.1, 57.0, 53.3, 41.1, 29.1, 5.9; νmax 3279, 2960, 1665, 1566, 1529, 1454, 1111, 835 cm−1.
8-Cyclopropyl-2-(piperidin-1-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (14e): According to the general procedure, thiourea intermediate was obtained from 13 and piperidine (26.3 mg) after 30 min. at room temperature. After purification, 14e was isolated as a colorless solid (90.1 mg, 89%), mp. 244–246 °C; 1H-NMR (CDCl3) δ 8.05 (s, 1H, H7), 7.88 (d, J = 8.4 Hz, 1H, H4), 7.63 (d, 1H, J = 8.4 Hz, H5), 3.68 (m, 4H, NCH2), 3.30–3.25 (m, 1H, NCH), 1.71 (m, 6H, CH2), 1.21 (m, 2H, CH), 0.97 (m, 2H, CH); 13C-NMR (CDCl3) δ 172.3, 161.6, 152.8, 144.0, 142.0, 126.1, 125.1, 124.9, 49.5, 29.3, 25.3, 24.2, 6.5; νmax 2949, 2860, 1659, 1522, 1432, 1254, 1120, 824 cm−1; HRMS calcd for C17H19N4OS [M + H]+ 327.1280 found 327.1283.
8-Cyclopropyl-2-(piperidin-1-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (14f): According to the general procedure, thiourea intermediate was obtained from 13 and morpholine (28.7 mg) after 30 min. at room temperature. After purification, 14f was isolated as a colorless solid (93.6 mg, 92%), mp. 230–232 °C; 1H-NMR (DMSO-d6) δ 8.26 (s, 1H, H7), 7.92 (d, J = 8.7 Hz, 1H, H4), 7.62 (d, J = 8.7 Hz, 1H, H5), 3.79–3.75 (m, 4H, OCH2), 3.65–3.60 (m, 4H, NCH2), 3.33–3.28 (m, 1H, NCH), 1.08–1.00 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 171.7, 160.5, 151.4, 145.6, 142.7, 125.4, 125.0, 124.8, 115.5, 65.5, 47.9, 29.2, 6.0; νmax 3061, 2971, 2860, 1655, 1561, 1456, 1432, 1340, 1112, 823 cm−1; HRMS calcd for C16H17N4O2S [M + H]+ 329.1072 found 329.1060.
3.2.4. General Procedure for the Synthesis of N-Alkyl-8-cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamides 17a–f from 16
In a sealed tube, a solution of carbonitrile derivative 16 (75 mg, 0.28 mmol) and the appropriate amine (2.8 mmol, 10 equiv.) in THF (1 mL) was heated under microwaves at 100 °C for 45 min under argon atmosphere. The solvent was removed under vacuum and the crude residue was adsorbed on Celite® and purified by flash chromatography on silica gel with methanol/methylene chloride (0/100 to 2/98, v/v) as eluent to furnish the desired amidine compound.
N-Benzyl-8-cyclopropyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17a): beige powder (105 mg, 93%), mp. 148–150 °C; 1H-NMR (DMSO-d6) δ 8.48 (s, 1H, H7), 8.43 (d, J = 9.0 Hz, 1H, H4), 7.84 (d, J = 9.0 Hz, 1H, H5), 7.49–7.23 (m, 5H, Ph), 6.99 (br s, 2H, NH2), 4.48 (s, 2H, CH2Ph), 3.39–3.33 (m, 1H, NCH), 1.14–1.01 (m, 4H, CH); 13C-NMR (CDCl3) δ 161.0, 152.1, 146.9, 146.8, 129.4, 129.0, 128.6, 127.5, 127.1, 126.1, 29.7, 6.6; νmax 3456, 3328, 3187, 1699, 1653, 1604, 1586, 1448, 1342, 1311, 1091, 826, 738 cm−1; HRMS calcd for C20H18N5OS [M + H]+ 376.1232 found 376.1240.
(Z)-8-Cyclopropyl-N′-[2-(dimethylamino)ethyl]-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17b): colorless solid (99 mg, 99%), mp. 168–170 °C; 1H-NMR (DMSO-d6) δ 8.46 (s, 1H, H7), 8.40 (d, J = 8.7 Hz, 1H, H4), 7.82 (d, J = 8.7 Hz, 1H, H5), 6.75 (br s, 2H, NH2), 3.38–3.33 (m, 1H, NCH), 3.31 (m, 2H, NCH2), 2.56 (m, 2H, NCH2), 2.23 (s, 6H, NCH3), 1.11–1.04 (m, 4H, CH); νmax 3398, 3329, 3203, 2829, 1653, 1589, 1507, 1453, 1350, 1314, 829 cm−1; HRMS calcd for C17H21N6OS [M + H]+ 357.1498 found 357.1500.
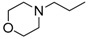
(Z)-8-Cyclopropyl-N′-(2-morpholinoethyl)-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17c): colorless solid (63 mg, 57%), mp. 154–156 °C; 1H-NMR (CDCl3) δ 8.32 (d, J = 8. 7 Hz, 1H, H4), 8.22 (s, 1H, H7), 7.80 (d, J = 8.7 Hz, 1H, H5), 3.76–3.74 (m, 4H, OCH2), 3.48 (t, J = 6.0 Hz, H1′), 3.37–3.32 (m, 1H, NCH), 2.76 (t, J = 6.0 Hz, H2′), 2.63–2.60 (m, 4H, NCH2), 1.30–1.23 (m, 2H, CH), 1.22–1.03 (m, 2H, CH), 13C-NMR (CDCl3) δ 169.2, 161.0, 153.0, 152.2, 146.9, 133.4, 129.5, 126.3, 116.4, 67.0, 58.3, 53.9, 42.6, 29.7, 6.7; νmax 3480, 3374, 2966, 1642, 1316, 1110, 832, 521 cm−1; HRMS calcd for C19H23N6O2S [M + H]+ 399.1603 found 399.1604.
(Z)-8-Cyclopropyl-9-oxo-N′-[2-(piperidin-1-yl)ethyl]-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carboximidamide (17d): colorless solid (96 mg, 87%), mp. 180–182 °C; 1H-NMR (CDCl3) δ 8.33 (d, J = 8. 7 Hz, 1H, H4), 8.22 (s, 1H, H7), 7.80 (d, J = 8.7 Hz, 1H, H5), 3.52–3.48 (m, 2H, H1′), 3.37–3.33 (m, 1H, NCH), 2.70–2.66 (m, 1H, H2′), 2.52–2.50 (m, 4H, NCH), 1.65–1.59 (m, 4H, CH), 1.49–1.30 (m, 4H, CH), 1.30–1.26 (m, 2H, CH), 1.22–1.03 (m, 2H, CH), 13C-NMR (CDCl3) δ 169.8, 161.0, 152.3, 146.8, 133.5, 129.4, 126.1, 116.5, 54.9, 29.7, 26.1, 24.4, 6.7; νmax 3463, 3325, 2933, 1650, 1588, 1345, 1040, 825, 568 cm−1; HRMS calcd for C20H25N6OS [M + H]+ 397.1811 found 397.1811.
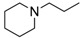
8-Cyclopropyl-2-[imino(morpholino)methyl]thiazolo[5,4-f]quinazolin-9(8H)-one (17e): colorless solid (53.7 mg, 54%), mp. 184–186 °C ; 1H-NMR (DMSO-d6) δ 8.48 (s, 1H, H7), 8.45 (d, J = 9.0 Hz, 1H, H4), 8.83 (d, J = 9.0 Hz, 1H, H5), 3.71–3.68 (m, 4H, OCH2), 3.48–3.45 (m, 4H, NCH2), 3.39–3.33 (m, 1H, NCH), 1.12–1.07 (m, 4H, CH); 13C-NMR (DMSO-d6) δ 162.0, 160.8, 159.5, 151.6, 148.7, 147.1, 131.4, 129.7, 126.7, 116.0, 66.3, 46.9, 30.0, 6.4; νmax 3279, 2955, 2854, 1660, 1586, 1496, 1436, 1347, 1109, 830 cm−1; HRMS calcd for C17H25N5O2S. [M + H]+ 356.1188 found 356.1190.
8-Cyclopropyl-2-[imino(piperidin-1-yl)methyl]thiazolo[5,4-f]quinazolin-9(8H)-one (17f): colorless solid (54.0 mg, 59%), mp. 204–206 °C ; 1H-NMR (DMSO-d6) δ 8.50 (s, 1H, H7), 8.48 (d, J = 8.7 Hz, 1H, H4), 7.18 (br s, 1H, NH), 7.87 (d, J = 8.7 Hz, 1H, H5), 3.43 (m, 4H, NCH2), 3.39–3.33 (m, 1H, NCH), 1.70–1.56 (m, 6H, CH2), 1.11–1.05 (m, 4H, CH); νmax 3162, 2936, 2848, 1678, 1587, 1345, 829 cm−1; HRMS calcd for C17H25N5O2S. [M + H]+ 354.1389 found 354.1389.
3.3. In Vitro Kinase Preparation and Assays
3.3.1. Buffers
Buffer A: MgCl2 (10 mM), 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 1 mM dithiothreitol (DTT), 25 mM Tris-HCl pH 7.5, 50 μg heparin/mL.
Buffer B: β-Glycerophosphate (60 mM), 30 mM p-nitrophenylphosphate, 25 mM 3-(N-morpholino)propanesulfonic acid (Mops) (pH 7.2), 5 mM EGTA, 15 mM MgCl2, 1 mM DTT, 0.1mM sodium vanadate.
3.3.2. Kinase Preparations and Assays
Kinase activities were assayed in triplicates in buffer A or B, for 30 min. at 30 °C, at a final adenosine triphosphate (ATP) concentration of 15 μM. Blank values were substracted and activities expressed in % of the maximal activity, i.e., in the absence of inhibitors. Controls were performed with appropriate dilutions of dimethylsulfoxide (DMSO). IC50 values were calculated from dose-response curves established by Sigma-Plots. The GSK-3, CK1, DYRK1A and CLK1 peptide substrates were obtained from Proteogenix (Oberhausbergen, France) [43,44].
CDK5/p25. (Human, recombinant) was prepared as previously described [45]. Its kinase activity was assayed in buffer A, with 1 mg of histone H1/mL, in the presence of 15 μM [γ-33P] ATP (3000 Ci/mmol; 10 mCi/mL) in a final volume of 30 μL. After 30 min incubation at 30 °C, 25 μL aliquots of supernatant were spotted onto sheets of Whatman P81 phosphocellulose paper, and 20 s later, the filters were washed eight times (for at least 5 min each time) in a solution of 10 mL phosphoric acid/L of water. The wet filters were counted in the presence of 1 mL ACS (Amersham) scintillation fluid.
GSK-3α/β. (Porcine brain, native) was assayed, as described for CDK5/p25 but in buffer A and using a GSK-3 specific substrate (GS-1: YRRAAVPPSPSLSRHSSPHQpSEDEEE) (pS stands for phosphorylated serine) [46].
CK1δ/ε. (Porcine brain, native) was assayed as described for CDK5/p25 but using the CK1-specific peptide substrate RRKHAAIGpSAYSITA [47].
DYRK1A. (Rat, recombinant, expressed in E. coli as a glutathione transferase (GST) fusion protein) was purified by affinity chromatography on glutathione-agarose and assayed, as described for CDK5/p25 using Woodtide (KKISGRLSPIMTEQ) (1.5 µg/assay) as a substrate.
CLK1. (Human, recombinant, expressed in E. coli as GST fusion protein) was assayed in buffer A (+0.15 mg BSA/mL) with RS peptide (GRSRSRSRSRSR) (1 μg/assay).
4. Conclusions
A library of thirty eight novel thiazolo[5,4-f]quinazoline derivatives (series 8, 10, 14 and 17) has been prepared, using microwave-assisted technology. An efficient multistep synthesis of 6-amino-3-cyclopropylquinazolin-4(3H)-one (3) was developed and optimized to the multigram scale, affording the synthesis of large quantities of a versatile and efficient precursor for the various target molecules in this study. The synthetic routes to these variously 8-aminoaryl thiazolo[5,4-f]quinazolin-9(8H)-ones, incited us to re- explore some aspects of the Hügershoff reaction involving a first bromination on thiocarbonyl group of a thiourea, followed by intramolecular electrophilic aromatic substitution. Whilst the second route was envisioned via a CuI catalyzed ligand-free intramolecular C-S bond formation allowing the target 2-substituted benzothiazoles from thiourea precursors. The inhibitory potency of the final products against five kinases involved in AD was evaluated. Our study demonstrates that molecules of the 8, 10 and 14 series described in this paper are not pertinent for the development of kinases inhibitors. The most active compounds are carboximidamides analogues of the target compounds of this study. They showed submicromolar IC50 values for CLK1, DYRK1A and GSK-3α/β over the other tested enzymes, suggesting that only chemical functions derived from a carbonitrile or a carbonyl carbon may generate affinity and specificity for a set of specific kinases.
Acknowledgments
Financial support from the MESR (French Ministère de l’Enseignement Supérieur & de la Recherche) is gratefully acknowledged for the doctoral fellowships to D.H. We thank the LABEX SynOrg (ANR-11-LABX-0029) for financial support (J.G.). We also acknowledge Milestone S.r.l. (Italy) for provision of multi-mode microwave reactor (Start S™) and for technical support. This research was partly supported by grants from the “Fonds Unique Interministériel” (FUI) TRIAD (LM) projects, the “Fondation Jérôme Lejeune” (LM), and an FP7-KBBE-2012 grant (BlueGenics) to LM.
Abbreviations
The following abbreviations are used in this manuscript:
| ATP | Adenosine triphosphate |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| CMGC group | Group of kinases including Cyclin-dependent kinases (CDKs), Mitogen-activated protein kinases (MAP kinases), Glycogen synthase kinases (GSK) and Cyclin dependent kinases (CDK-like kinases) |
| DMF | N,N-Dimethylformamide |
| DMFDMA | N,N-Dimethylformamide dimethyl acetal |
| MCR | Multi-component reaction |
| MTDL | Multi-target-directed ligand |
| NBS | N-Bromosuccinimide |
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/6/794/s1.
Author Contributions
T.B. conceived the project, helped by C.F., T.B. and D.H. designed the experiments and D.H. executed the chemical synthesis accompanied by C.D., N.L. and L.M. designed and performed the biological experiments. T.B. and C.F. wrote the paper. All authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 8, 10, 14 and 17 are not available from the authors.
References
- 1.Wells G., Bradshaw T.D., Diana P., Seaton A., Shi D.-F., Westwell A.D., Stevens M.F.G. Antitumor benzothiazoles. Part 10. The synthesis and antitumor activity of benzothiazole substituted quinol derivatives. Bioorg. Med. Chem. Lett. 2000;10:513–515. doi: 10.1016/s0960-894x(00)00027-5. [DOI] [PubMed] [Google Scholar]
- 2.Bradshaw T.D., Wrigley S., Shi D.-F., Schultz R.J., Paull K.D., Stevens M.F.G. 2-(4-Aminophenyl)benzothiazoles: Novel agents with selective profiles of in vitro anti-tumour activity. Br. J. Cancer. 1998;77:745–752. doi: 10.1038/bjc.1998.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molinski T.F. Marine pyridoacridine alkaloids: structure, synthesis, and biological chemistry. Chem. Rev. 1993;93:1825–1838. doi: 10.1021/cr00021a009. [DOI] [Google Scholar]
- 4.Gunawardana G.P., Kohmoto S., Gunasekara S.P., McConnel O.J., Koehn F.E. Dercitine, a new biologically active acridine alkaloid from a deep water marine sponge, Dercitus sp. J. Am. Chem. Soc. 1988;110:4856–4858. doi: 10.1021/ja00222a071. [DOI] [Google Scholar]
- 5.Gunawardana G.P., Kohmoto S., Burres N.S. New cytotoxic acridine alkaloids from two deep water marine sponges of the family Pachastrellidae. Tetrahedron Lett. 1989;30:4359–4362. doi: 10.1016/S0040-4039(00)99360-2. [DOI] [Google Scholar]
- 6.Bénéteau V., Besson T. Synthesis of novel pentacyclic pyrrolothiazolobenzoquinolinones, analogs of natural marine alkaloids. Tetrahedron Lett. 2001;42:2673–2676. doi: 10.1016/S0040-4039(01)00258-1. [DOI] [Google Scholar]
- 7.Lamazzi C., Chabane H., Thiéry V., Pierre A., Léonce S., Pfeiffer B., Renard P., Guillaumet G., Besson T. Synthesis and cytotoxic evaluation of novel thiazolocarbazoles. J. Enzyme Inhib. Med. Chem. 2002;17:397–401. doi: 10.1080/1475636021000005703. [DOI] [PubMed] [Google Scholar]
- 8.Frère S., Thiéry V., Bailly C., Besson T. Novel 6-substituted benzothiazol-2-yl indolo[1,2-c]quinazolines and benzimidazo[1,2-c]quinazolines. Tetrahedron. 2003;59:773–779. doi: 10.1016/S0040-4020(02)01593-4. [DOI] [Google Scholar]
- 9.Chabane H., Pierre A., Léonce S., Pfeiffer B., Renard P., Thiéry V., Guillaumet G., Besson T. Synthesis and cytotoxic activity of thiazolofluorenone derivatives. J. Enzyme Inhib. Med. Chem. 2004;19:567–575. doi: 10.1080/14756360400004607. [DOI] [PubMed] [Google Scholar]
- 10.Testard A., Picot L., Fruitier-Arnaudin I., Piot J.M., Chabane H., Domon L., Thiéry V., Besson T. Microwave-assisted synthesis of novel thiazolocarbazoles and evaluation as potential anticancer agents. Part III. J. Enzyme Inhib. Med. Chem. 2004;19:467–473. doi: 10.1080/14756360412331280491. [DOI] [PubMed] [Google Scholar]
- 11.Logé C., Testard A., Thiéry V., Lozach O., Blairvacq M., Robert J.-M., Meijer L., Besson T. Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2008;43:1469–1477. doi: 10.1016/j.ejmech.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 12.Testard A., Logé C., Léger B., Robert J.-M., Lozach O., Blairvacq M., Meijer L., Thiéry V., Besson T. Thiazolo[5,4-f]quinazolin-9-ones, inhibitors of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2006;16:3419–3423. doi: 10.1016/j.bmcl.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 13.Loidreau Y., Deau E., Marchand P., Nourrisson M.-R., Logé C., Coadou J.M., Loaëc N., Meijer L., Besson T. Synthesis and molecular modelling studies of 8-arylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-amines as multitarget Ser/Thr kinases inhibitors. Eur. J. Med. Chem. 2015;92:124–134. doi: 10.1016/j.ejmech.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 14.Loidreau Y., Marchand P., Dubouilh-Benard C., Nourrisson M.-R., Duflos M., Loaëc N., Meijer L., Besson T. Synthesis and biological evaluation of N-aryl-7-methoxybenzo[b]furo[3,2-d]pyrimidin-4-amines and their N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amine analogues as dual inhibitors of CLK1 and DYRK1A kinases. Eur. J. Med. Chem. 2013;59:283–295. doi: 10.1016/j.ejmech.2012.11.030. [DOI] [PubMed] [Google Scholar]
- 15.Loidreau Y., Marchand P., Dubouilh-Benard C., Nourrisson M.-R., Duflos M., Lozach O., Loaëc N., Meijer L., Besson T. Synthesis and biological evaluation of N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amines and their pyrido and pyrazino analogues as Ser/Thr kinase inhibitors. Eur. J. Med. Chem. 2012;58:171–183. doi: 10.1016/j.ejmech.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Foucourt A., Hédou D., Dubouilh-Benard C., Désiré L., Casagrande A.-S., Leblond B., Loaëc N., Meijer L., Besson T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part I. Molecules. 2014;19:15546–15571. doi: 10.3390/molecules191015546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foucourt A., Hédou D., Dubouilh-Benard C., Désiré L., Casagrande A.-S., Leblond B., Loaëc N., Meijer L., Besson T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part II. Molecules. 2014;19:15411–15439. doi: 10.3390/molecules191015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leblond B., Casagrande A.-S., Désiré L., Foucourt A., Besson T. DYRK1 inhibitors and uses thereof WO 2013026806. Chem. Abstr. 2013;158:390018. [Google Scholar]
- 19.Appel R., Janssen H., Siray M., Knoch F. Synthese und Reaktionen des 4,5-Dichlor-1,2,3-dithiazolium-chlorids. Chem. Ber. 1985;118:1632–1643. doi: 10.1002/cber.19851180430. [DOI] [Google Scholar]
- 20.Esvan Y.J., Zeinyeh W., Boibessot T., Nauton L., Théry V., Knapp S., Chaikuad A., Loaëc N., Meijer L., Anizon F., et al. Discovery of pyrido[3,4-g]quinazoline derivatives as CMGC family protein kinase inhibitors: Design, synthesis, inhibitory potency and X-ray co–crystal structure. Eur. J. Med. Chem. 2016 doi: 10.1016/j.ejmech.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Schmitt C., Miralinaghi P., Mariano M., Hartmann R.W., Engel M. Hydroxybenzothiophene ketones are efficient pre-mRNA splicing modulators due to dual inhibition of Dyrk1A and Clk1/4. ACS Med. Chem. Lett. 2014;5:963–967. doi: 10.1021/ml500059y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dehbi O., Tikad A., Bourg S., Bonnet P., Lozach O., Meijer L., Aadil M., Akssira M., Guillaumet G., Routier S. Synthesis and optimization of an original V-shaped collection of 4-7-disubstituted pyrido[3,2-d]pyrimidines as CDK5 and DYRK1A Inhibitors. Eur. J. Med. Chem. 2014;80:352–363. doi: 10.1016/j.ejmech.2014.04.055. [DOI] [PubMed] [Google Scholar]
- 23.Bajda M., Guzior N., Ignasik M., Malawska B. Multi-target-directed ligands in Alzheimer’s disease treatment. Curr. Med. Chem. 2011;18:4949–4975. doi: 10.2174/092986711797535245. [DOI] [PubMed] [Google Scholar]
- 24.Cavalli A., Bolognesi M.L., Minarini A., Rosini M., Tumiatti V., Recanatini M., Melchiorre C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008;51:347–372. doi: 10.1021/jm7009364. [DOI] [PubMed] [Google Scholar]
- 25.Hédou D., Godeau J., Loaëc N., Meijer L., Fruit C., Besson T. Synthesis of Thiazolo[5,4-f]quinazolin-9(8H)-ones as Multi-Target Directed Ligands of Ser/Thr Kinases. Molecules. 2016;21:578. doi: 10.3390/molecules21050578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexandre F.R., Domon L., Frère S., Testard A., Thiéry V., Besson T. Microwaves in drug discovery and multi-step synthesis. Mol. Divers. 2003;7:273–280. doi: 10.1023/B:MODI.0000006860.56083.2b. [DOI] [PubMed] [Google Scholar]
- 27.Alexandre F.R., Berecibar A., Wrigglesworth R., Besson T. Efficient synthesis of thiazoloquinazolinone derivatives. Tetrahedron Lett. 2003;44:4455–4458. doi: 10.1016/S0040-4039(03)01026-8. [DOI] [Google Scholar]
- 28.Besson T., Guillard J., Rees C.W. Multistep synthesis of thiazoloquinazolines under microwave irradiation in solution. Tetrahedron Lett. 2000;41:1027–1030. doi: 10.1016/S0040-4039(99)02221-2. [DOI] [Google Scholar]
- 29.Hugershoff A. Einwirkung von Brom auf aromatische Thioharnstoffe. Ber. Dtsch. Chem. Ges. 1903;36:3121–3134. doi: 10.1002/cber.19030360388. [DOI] [Google Scholar]
- 30.Hugershoff A. Einwirkung von Halogenen auf Thioharnstoffe. Ber. Dtsch. Chem. Ges. 1901;34:3130–3135. doi: 10.1002/cber.190103402286. [DOI] [Google Scholar]
- 31.Jordan D.A., Luo C., Reitz A. Efficient conversion of substituted aryl thioureas to 2-aminobenzothiazoles using benzyltrimethylammonium bromide. J. Org. Chem. 2003;68:8693–8696. doi: 10.1021/jo0349431. [DOI] [PubMed] [Google Scholar]
- 32.Inamoto K., Hasegawa C., Kawasaki J., Hiroya K., Doi T. Use of molecular oxygen as a reoxidant in the synthesis of 2-substituted benzothiazoles via palladium-catalyzed C-H functionalization/intramolecular C-S bond formation. Adv. Synth. Catal. 2010;352:2643–2655. doi: 10.1002/adsc.201000604. [DOI] [Google Scholar]
- 33.Inamoto K., Hasegawa C., Hiroya K., Doi T. Palladium-catalyzed synthesis of 2-substituted Benzothiazoles via C-H functionalization/intramolecular C-S bond formation process. Org. Lett. 2008;10:5147–5150. doi: 10.1021/ol802033p. [DOI] [PubMed] [Google Scholar]
- 34.Joyce L.L., Batey R.A. Heterocycle formation via palladium-catalyzed intramolecular oxidative C-H bond functionalization: An Efficient Strategy for the Synthesis of 2-aminobenzothiazoles. Org. Lett. 2009;11:2792–2795. doi: 10.1021/ol900958z. [DOI] [PubMed] [Google Scholar]
- 35.Evindar G., Batey R.A. Parallel Synthesis of a library of benzoxazoles and Benzothiazoles using ligand-accelerated copper-catalyzed cyclizations of ortho-halobenzanilides. J. Org. Chem. 2006;71:1802–1808. doi: 10.1021/jo051927q. [DOI] [PubMed] [Google Scholar]
- 36.Joyce L.L., Evindar G., Batey R.A. Copper- and palladium-catalyzed intramolecular C-S bond formation: A convenient synthesis of 2-aminobenzothiazoles. Chem. Commun. 2004:446–447. doi: 10.1039/B311591G. [DOI] [PubMed] [Google Scholar]
- 37.Hédou D., Harari M., Godeau J., Dubouilh-Benard C., Fruit C., Besson T. Synthesis of polyfunctionalized benzo[d]thiazoles as novel anthranilic acid derivatives. Tetrahedron Lett. 2015;56:4088–4092. doi: 10.1016/j.tetlet.2015.05.018. [DOI] [Google Scholar]
- 38.Hédou D., Deau E., Harari M., Sanselme M., Fruit C., Besson T. Rational multistep synthesis of a novel polyfunctionalized benzo[d]thiazole and its thiazolo[5,4-b]pyridine analogue. Tetrahedron. 2014;70:5541–5549. doi: 10.1016/j.tet.2014.06.103. [DOI] [Google Scholar]
- 39.Hédou D., Guillon R., Lecointe C., Logé C., Chosson E., Besson T. Novel synthesis of angular thiazolo[5,4-f] and [4,5-h]quinazolines, preparation of their linear thiazolo[4,5-g] and [5,4-g]quinazoline analogs. Tetrahedron. 2013;69:3182–3191. doi: 10.1016/j.tet.2013.02.066. [DOI] [Google Scholar]
- 40.Michaelidou S.S., Koutentis P.A. The synthesis of 2-cyano-cyanothioformanilides from 2-(4-chloro-5H-1,2,3-dithiazol-5-ylideneamino)benzonitriles using DBU. Synthesis. 2009:4167–4174. [Google Scholar]
- 41.Besson T., Guillard J., Rees C.W., Thiéry V. New syntheses of aryl isothiocyanates. J. Chem. Soc. Perkin Trans. 1998;1:889–892. doi: 10.1039/a707801c. [DOI] [Google Scholar]
- 42.Topliss J.G. Utilization of operational schemes for analog synthesis in drug design. J. Med. Chem. 1972;15:1006–1011. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 43.Giraud F., Alves G., Debiton E., Nauton L., Thery V., Durieu E., Ferandin Y., Lozach O., Meijer L., Anizon F., et al. Synthesis, protein kinase inhibitory potencies, and in vitro antiproliferative activities of meridianin derivatives. J. Med. Chem. 2011;54:4474–4489. doi: 10.1021/jm200464w. [DOI] [PubMed] [Google Scholar]
- 44.Bach S., Knockaert M., Reinhardt J., Lozach O., Schmitt S., Baratte B., Koken M., Coburn S.P., Tang L., Jiang T., et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem. 2005;280:31208–31219. doi: 10.1074/jbc.M500806200. [DOI] [PubMed] [Google Scholar]
- 45.Leclerc S., Garnier M., Hoessel R., Marko D., Bidd J.A., Snyder G.L., Greengard P., Biernat J., Mandelkow E.-M., Eisenbrand G., et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/P25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s Disease: A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 46.Primot A., Baratte B., Gompel M., Borgne A., Liabeuf S., Romette J.L., Jho E.H., Costantini F., Meijer L. Purification of GSK-3 by affinity chromatography on immobilized axin. Protein Expr. Purif. 2000;20:394–404. doi: 10.1006/prep.2000.1321. [DOI] [PubMed] [Google Scholar]
- 47.Reinhardt J., Ferandin Y., Meijer L. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2007;54:101–109. doi: 10.1016/j.pep.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 48.Jain P., Karthikeyan C., Moorthy N.S.H.N., Waiker D.K., Jain A.K., Trivedi P. Human CDC2-like kinase 1 (CLK1): A novel target for Alzheimer’s disease. Curr. Drug Targets. 2014;15:539–550. doi: 10.2174/1389450115666140226112321. [DOI] [PubMed] [Google Scholar]
- 49.Patel K., Gadewar M., Tripathi R., Prasad S.K., Patel D.K. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta-carboline alkaloid “Harmine”. Asian Pac. J. Trop. Biomed. 2012;2:660–664. doi: 10.1016/S2221-1691(12)60116-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.