

Abstract
Dimethylarginine dimethylaminohydrolase (DDAH) is a highly conserved hydrolytic enzyme found in numerous species, including bacteria, rodents, and humans. In humans, the DDAH-1 isoform is known to metabolize endogenous asymmetric dimethylarginine (ADMA) and monomethyl arginine (l-NMMA), with ADMA proposed to be a putative marker of cardiovascular disease. Current literature reports identify the DDAH family of enzymes as a potential therapeutic target in the regulation of nitric oxide (NO) production, mediated via its biochemical interaction with the nitric oxide synthase (NOS) family of enzymes. Increased DDAH expression and NO production have been linked to multiple pathological conditions, specifically, cancer, neurodegenerative disorders, and septic shock. As such, the discovery, chemical synthesis, and development of DDAH inhibitors as potential drug candidates represent a growing field of interest. This review article summarizes the current knowledge on DDAH inhibition and the derived pharmacokinetic parameters of the main DDAH inhibitors reported in the literature. Furthermore, current methods of development and chemical synthetic pathways are discussed.
Keywords: dimethylarginine dimethylaminohydrolase, arginine, nitric oxide, asymmetric dimethylarginine, monomethyl arginine, enzyme inhibitors, organic synthesis
1. Introduction
Dimethylarginine dimethylaminohydrolase (DDAH) is a modulator of the nitric oxide (NO) pathway and is responsible for the metabolism of methylated arginines [1]. DDAH is conserved throughout many species and has been demonstrated to pre-date evolution of the Nitric Oxide Synthase (NOS) family [2], suggesting an alternative function for methylated arginines independent to their role as NOS regulatory molecules. Two different isoforms of human DDAH have been identified, with DDAH-1 and DDAH-2 sharing 62% homology. DDAH-1 is primarily expressed in different areas of the brain [3], the dorsal root ganglia and the spinal dorsal horn [4]. DDAH-2 appears prevalent in pancreatic islet cells [5] and infected tissues [6]. In addition, Altman et al. reported a study showing high expression of DDAH-2 in porcine immune tissues [7]. The two isoforms are both expressed in kidney [8,9,10], heart [11,12], adipose tissue [13], placenta and trophoblasts [14], and the liver [15,16,17,18]. Phylogenetic analyses indicate DDAH-1 was generated by duplication of the genomic segment located on chromosome 6 comprising DDAH-2 [2,19]. Although hydrolysis of asymmetrically methylated arginines appears to be the primary function of DDAH-1, alternate roles for both DDAH isoforms are evident when they independently interact with other enzymes [20,21]. For example, DDAH-1 is reported to bind the tumour suppressor protein neurofibromin, resulting in increased phosphorylation of neurofibromin by protein kinase A (PKA) [20] and is additionally reported to regulate the levels of phosphorylated protein kinase B (p-Akt) by increasing Ras activity via a mechanism independent to the NOS pathway [22]. Similarly, DDAH-2 is known to interact with PKA causing the induction of vascular endothelial growth factor (VEGF) via phosphorylation of the Sp1 transcription factor [21].
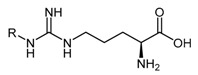
Asymmetric dimethylarginine (ADMA, 1), monomethylarginine (l-NMMA, 2), and symmetric dimethylarginine (SDMA, 3) are endogenous methylated arginines (Figure 1). They are formed by two biochemical processes: (1) the initial post-translational methylation of protein arginine residues which is catalyzed by the protein arginine methyltransferases (PRMT’s); and (2) the subsequent release from their respective methylated protein(s) by proteolysis [23]. Once released into the cytosol, transport and efflux of the methylated arginines is mediated by the y+ cationic amino acid (CAT-1) transporter [24]. ADMA and l-NMMA inhibit all members of the NOS family of enzymes [25,26,27,28,29,30], while SDMA competes with l-arginine, a NOS substrate, for transport by the y+ cation transporter [31]. Different enzymes play a role in the metabolism of methylated arginines, particularly alanine-glyoxylate aminotransferase 2 (AGXT2) [32,33,34]. However, ADMA and l-NMMA, but not SDMA, are primarily converted by DDAH-1 to l-citrulline (4) and dimethylamine (5) or monomethylamine (6), respectively [35] (Scheme 1).
Figure 1.

The endogenous methylated arginines, asymmetric dimethylarginine (ADMA 1), monomethylarginine (l-NMMA 2), and symmetric dimethylarginine (SDMA 3) act as direct or indirect inhibitors of the NOS family of enzymes. ADMA (1) and l-NMMA (2) are substrates for human DDAH-1.
Scheme 1.

ADMA and l-NMMA are substrates for DDAH-1 and are converted to l-citrulline (4) and an amine species 5 or 6.
The role of ADMA accumulation and impaired DDAH expression and/or activity in the pathophysiology of several diseases is well documented [36,37,38,39,40,41,42,43,44]. Increased plasma or serum ADMA concentrations have been associated with coronary events [45,46,47], renal disease [29,37], atherosclerosis [48], and cerebrovascular disease [49,50]. Up-regulation of DDAH expression and activity might, therefore, represent a good strategy in the treatment of the aforementioned pathologies to increase ADMA metabolism and lower its plasma concentrations.
In contrast, reduced ADMA concentrations have been demonstrated in other disease states, such as amyotrophic lateral sclerosis [51], multiple sclerosis [43], Alzheimer’s disease and dementia [52,53]. ADMA has also been shown to have neuroprotective effects in a model of Parkinson’s disease [54]. Furthermore, DDAH overexpression in tumours enhances their metastatic potential [42,55,56]. Several reports also show that inhibition of NO synthesis results in significant anti-angiogenic effects both in vitro and in vivo, with potential beneficial effects in cancer [57,58,59,60,61]. Moreover, excessive NO concentrations play a key role in the pathophysiology of septic shock [62,63,64,65] (an early therapeutic approach for the treatment of septic shock involved the administration of 546C88 (l-NMMA) to inhibit NOS and reduce NO synthesis, however the increase in mortality in patients receiving l-NMMA caused the termination of the clinical trial in phase III [66]. Further analysis of the data revealed that mortality was associated with elevated l-NMMA concentrations; conversely, lower l-NMMA concentrations were associated with improved survival [36]. In this context a more controlled, indirect modulation of methylated arginine concentrations through pharmacological DDAH-1 inhibition (DDAH-2 knockout affects outcome in models of polymicrobial sepsis [44]) might represent an alternative strategy, and appears particularly promising in a rat model of septic shock [67]). In these circumstances, DDAH inhibition could exert a controlled inhibition of the NO synthesis by increasing ADMA concentrations and offering a new approach in the treatment of these diseases.
Therefore, alterations in DDAH and NOS activity might exert either beneficial or toxic effects, according to specific experimental models and disease conditions. The potential of DDAH as a molecular target in the treatment of these pathological conditions has led several groups to study the endogenous mechanisms involved in the modulation of the expression and activity of human DDAH-1 and DDAH-2 [68]. The elucidation of these mechanisms may identify new strategies for the pharmacological activation or inhibition of these enzymes to restore physiological homeostasis after pathological imbalance. Since the discovery of the DDAH family of enzymes in 1987 [35], a number of research groups have identified inhibitors with the aim of developing such therapeutics. While no ‘selective’ DDAH inhibitor has entered clinical trials, numerous compounds have been synthesized that act as prototypic in vitro inhibitors. Herein, we discuss all published DDAH inhibitors according to their chemical structure (‘substrate-like’ and ‘non-substrate-like’), and summarize the details of their synthetic pathways as well as how they alter the pharmacokinetic parameters for l-citrulline formation by DDAH.
2. Endogenous Modulation of DDAH Activity
The production of specific gene products, both in terms of RNA and proteins, is physiologically regulated by a plethora of mechanisms and can be triggered or silenced by various stimuli and factors. Of particular note, DDAH activity is reported to be inhibited by divalent transition metals [69,70] and it is known that elevated zinc(II) concentrations are associated with oxidative stress in numerous pathological conditions [71,72,73].
S-Nitrosylation of the enzyme’s cysteine residues is a further important post-translational regulatory mechanism [74] and is generally associated with increased expression of inducible NOS (iNOS). The induction of iNOS generates excessive NO that can bind covalently (but reversibly) to the thiol (SH) moiety of each DDAH cysteine [74]. The apparent S-nitrosylation of the catalytic DDAH cysteine residue results in DDAH inhibition. This in turn causes the subsequent accumulation of DDAH substrates, ADMA and l-NMMA, which eventually reach concentrations that reversibly inhibit the NOS enzymes [75].
Oxidative stress additionally modulates DDAH activity. Factors associated with oxidative stress, including shear stress [76], tumour necrosis factor α (TNF-α), oxidized low density lipoprotein (LDL) [76], and homocysteine [77] each suppresses DDAH expression and activity. In contrast, antioxidant mediators such as probucol [78], estradiol [79], and interleukin-1β (IL-1β) [80] up-regulate DDAH expression. Moreover, nicotine and cigarette smoking have been shown to decrease DDAH expression and activity in endothelial cells, with consequential accumulation of ADMA, NOS inhibition, and endothelial damage [81,82]. Furthermore, it is known that selective down-regulation of DDAH-1 and up-regulation of DDAH-2 occurs when angiotensin II binds to the type 1 angiotensin receptor (AT1-R), linking DDAH transcription to angiotensin-induced inflammation [8]. In addition, epigenetic modifications are known to alter DDAH-2 regulation at the promoter. For example, DNA hypermethylation has the ability to suppress DDAH-2 transcription, while histone acetylation has the ability to up-regulate DDAH-2 transcription [83].
3. Pharmacological Induction of DDAH
As a result of diminished DDAH activity and/or accumulation of ADMA and l-NMMA in various disease states, several studies have investigated the effects of restoring or increasing DDAH expression. Statin treatment is associated with a significant reduction in ADMA concentrations [84,85,86], and an increase in both DDAH-1 and DDAH-2 expression and activity [87,88,89]. Another class of compounds enhancing ADMA metabolism via DDAH up-regulation is represented by the farnesoid X receptor (FXR) agonists. GW4064 upregulates both DDAH-1 and the cationic transporter CAT-1, leading to increased ADMA cellular uptake and metabolism, and decreased serum ADMA concentrations [15,90]. Another FXR agonist, INT-747, can increase DDAH-1 expression [91,92], however this is not associated with significant changes in ADMA and NO concentrations [91]. The lipid lowering drug probucol also lowers ADMA concentrations [93] via up-regulation of both DDAH expression and activity, and down-regulation of PRMT-1 expression [78,94]. Another lipid lowering drug, fenofibrate, decreases ADMA concentrations by restoring DDAH activity in conditions of oxidative stress [95]. Additionally, the oral antidiabetic drug pioglitazone and the β1 receptor blocker nebivolol both decrease serum ADMA concentrations by increasing DDAH-2 expression [96,97,98,99].
4. Pharmacological Inhibition of DDAH
Contrary to the comments in Section 3 above, there is a growing body of evidence to suggest that the modulation of DDAH enzyme activity may be utilized in conditions characterized by excessive NO production [36,67]. The remainder of this review will focus on highlighting the current knowledge associated with the main DDAH inhibitors reported in the literature to date.
4.1. Substrate-Like Inhibitors
4.1.1. S-Nitroso-l-Homocysteine (HcyNO)
Since the identification of S-nitrosylation as an important mechanism of DDAH regulation, the discovery of l-homocysteine’s ability to inhibit DDAH-1 led to the development of S-nitroso-l-homocysteine (HcyNO, 9), an endogenous S-nitrosothiol and a source of endogenous NO [100,101,102]. Synthesis and characterisation of HcyNO as a bovine DDAH inhibitor was reported by Knipp et al. (2005) [102]. HcyNO was synthesized in near quantitative yield in a two-step reaction (Scheme 2) via alkaline hydrolysis of commercially available l-homocysteine thiolactone hydrochloride (7) to l-homocysteine (8) [103], followed by S-nitrosylation of l-homocysteine with sodium nitrite (NaNO2) and acidification to afford HcyNO (9) [104]. Alternatively, S-nitrosylation can be undertaken directly on commercially available l-homocysteine (8). Mild reaction conditions are required, and excellent yields make HcyNO simple to prepare on a large scale.
Scheme 2.

Synthetic scheme for generating HcyNO (9). Reagents and Conditions: (i) 5 M NaOH, 37 °C, 5 min, 96%; (ii) NaNO2, 2 M HCl, 37 °C, 15 min, >99%.
HcyNO irreversibly inhibits DDAH by covalently binding to the putative catalytic cysteine with an inhibitory constant (Ki) of 690 μM [105]. Mass spectrometry analyses using different isotopic forms of S-nitroso-l-homocysteine (Hcy15NO and HcyN18O) and oxygen-18 labelled water (H218O) demonstrated that HcyNO forms a N-thiosulfoximide adduct with cysteine 273 (Cys273) of bovine DDAH-1. The authors’ proposed mechanism of inhibition requires the lone pair of electrons of the Cys273 SH to attack the S-nitroso group in HcyNO to eliminate a hydroxyl anion (Scheme 3). This mechanism is likely stabilized by neighbouring amino acids, namely His172. A further hydroxyl anion originating from water attacks the cationic sulfur of the conjugated intermediate, and subsequently rearranges to give the covalent thiosulfoximide adduct [102].
Scheme 3.

Schematic of the reaction mechanism proposed by Knipp et al. [102] for the covalent inhibition of DDAH-1 by HcyNO (9) at Cys273. Adapted with permission from [102]. Copyright 2005 American Chemical Society.
4.1.2. 2-Chloroacetamidine and N-But-3-ynyl-2-chloroacetamidine
Stone et al. [106] identified 2-chloroacetamidine (11) as a nonspecific inhibitor of two enzymes of the amidinotransferases superfamily, Pseudomonas aeruginosa DDAH (PaDDAH) and human peptydilarginine deaminase (PAD). 2-Chloroacetamidine (11, Scheme 4) is a commercially available compound bearing a 2-chloromethylene chain and an amidinium group, which partially resembles arginine. It can be synthesized via reaction of chloroacetonitrile (10) with sodium methoxide followed by treatment with ammonium chloride [107,108]. 2-Chloroacetamidine acts as a weak irreversible amidinotransferase inhibitor with greater affinity for PaDDAH, relative to PAD4 (Table 1). Data acquired by electrospray ionization mass spectrometry revealed the formation of an irreversible thiouronium-enzyme adduct whereby the loss of chlorine from the inhibitor is observed [106].
Scheme 4.

Synthetic scheme for generating 2-chloroacetamidine (11) (the conditions described afford the hydrochloride salt). Reagents and Conditions: (i) NaOMe, dry MeOH, overnight; (ii) AcOH, NH4Cl, reflux, 6 h, 89%.
Table 1.
Reported inhibitory constants for 2-chloroacetamidine (11) [106].
| Enzyme | Ki (mM) | Kinact (min−1) |
|---|---|---|
| PaDDAH | 3.1 ± 0.8 | 1.2 ± 0.1 |
| PAD4 | 20 ± 5 | 0.7 ± 0.1 |
The addition of an N-but-3-ynyl substituent to the acetamidine group of 2-chloroacetamidine led to formation of the human DDAH-1 probe, N-but-3-ynyl-2-chloro-acetamidine (18, Scheme 5) [109]. N-But-3-ynyl-2-chloro-acetamidine binds within the putative active-site, while the N-alkynyl substituent is reacted with biotin-PEO3-azide under copper catalysis via click chemistry. Successful conjugation can be easily detected in immunoblots using an anti-biotin antibody [109]. Synthesis of the N-but-3-ynyl-2-chloro-acetamidine probe was achieved in five steps using two modules (Scheme 5). Amine module 15 was prepared in three steps: activation of the terminal hydroxyl group of 3-butyn-1-ol (12) through mesylation to afford 13 in 62% yield, followed by conversion to amine 15 via azide intermediate 14 in 49% yield across two steps. The imidic ester module 17 was obtained in moderate yields (approximately 48%) by reacting chloroacetonitrile (10) with anhydrous ethanol (16) in the presence of dry hydrochloric acid gas. Final coupling between amine 15 and the imidic ester 17 proceeds under basic aqueous conditions to afford probe N-but-3-ynyl-2-chloro-acetamidine (18) in reasonable yields (approximately 63%). To date, no data are available regarding the selectivity of N-but-3-ynyl-2-chloro-acetamidine for human DDAH-1 and the potential cross reactivity of this probe with PaDDAH.
Scheme 5.

Synthetic scheme for generating the DDAH-1 biochemical probe N-but-3-ynyl-2-chloro-acetamidine (18). Reagents and Conditions: (i) DCM, TEA, DMAP, MsCl, 0 °C for 2 h then 25 °C for 3 h, 62%; (ii) anhydrous DMF, NaN3, 70 °C, 3.5 h; (iii) Et2O, Ph3P, 0 °C for 2 h, addition of H2O, then 25 °C for 20 h, 49% (two steps); (iv) dry HCl(g) at 0 °C for 1 h, stop HCl(g), then 0 °C for 4 h and RT overnight 48%–76%; (v) H2O, pH = 10, 0 °C for 3 h, 25 °C for 2 h, neutralized, RT for 1.5 days, 62%. Adapted with permission from [109]. Copyright 2009 American Chemical Society.
4.1.3. (S)-2-Amino-4-(3-Methylguanidino)butanoic Acid Analogues
Screening experiments with a library comprising 26 analogues of ADMA (1) and l-NMMA (2) identified (S)-2-amino-4-(3-methylguanidino)butanoic acid (4124W, 21), as a mammalian DDAH inhibitor [110]. The original synthesis of 4124W (21), first described by Ueda et al. in 2003 [80] is shown in Scheme 6, and was achieved by coupling N,S-dimethylthiopseudouronium iodide (19) with the copper complex of 2,4-diamino-n-butyric acid dihydrochloride (20) [32,111]. 4124W (21) was finally crystallized as the hydrochloride salt.
Scheme 6.

Synthetic scheme for generating (S)-2-amino-4-(3-methylguanidino)butanoic acid (4124W), as reported by Ueda et al. [80] using the methods of Ogawa et al. [32] and Corbin et al. [111]. Reagents and Conditions: (i) (1) 2,4-diamino-n-butyric acid dihydrochloride (20), copper acetate, 1:1 NH4OH/H2O; (2) N,S-dimethylthiopseudouronium iodide (19) RT, 24 h; (3) Dowex 50WX4 (NH4+ form), 4124W (21) was crystallized as the hydrochloride salt.
High chemical and structural similarity between 4124W and the DDAH substrate l-NMMA exists (compound 21, Scheme 6, and compound 2, Figure 1), and although exhibiting weak DDAH inhibition (IC50 = 416 μM) [110], this prompted Rossiter et al. [112] to synthesize a further 34 compounds based on the 4124W structure. Furthermore, Rossiter et al. [112] reported an alternative synthetic approach, which although requiring three steps, is synthetically versatile and was applied to develop their library of 4124W analogues. Rossiter’s strategy utilized protected intermediates to avoid complications associated with the purification of polar amino acids. Intermediate 24 (Scheme 7) is generated from commercially available N,N′-bis-tert-butoxycarbonylpyrazole-1H-carbox-amidine (22) by Mitsunobu reaction with alcohol 23 containing the desired R1 group, followed by reaction with (S)-tert-butyl 4-amino-2-((tert-butoxycarbonyl)amino)butanoate (25) to afford the protected amino acid analogue 26 (Scheme 7). Cleavage of the Boc protecting groups is achieved with a 4 M solution of HCl in 1,4-dioxane and produced 4124W (21) in moderate yields (approximately 44%) or analogues 27 in moderate to good yields (12%–67%).
Scheme 7.

Synthetic scheme for generating (S)-2-amino-4-(3-methylguanidino)butanoic acid analogues. Reagents and Conditions: (i) DEAD, PPh3, THF, 0 °C-RT, 3–16 h; (ii) DIPEA, CH3CN, RT, 24 h; (iii) 4M HCl/1,4-dioxane, RT, 24–72 h, 12%–67% overall; (iv) SOCl2, 0 °C for 30 min, reflux for 1 h, RT overnight, 44%–84%. Adapted with permission from [112]. Copyright 2005 American Chemical Society.
Additionally, Rossiter et al. developed a synthetic method for N,N-disubstituted alkylguanidinobutanoic acids and N-arylguanidines (Scheme 8). This involved reacting N,N’-bis-tert-butoxycarbonylpyrazole-1H-carboxamidine (22) with Boc-homoserine-tert-butyl ester (30), whereby the pyrazole group in the intermediate 31 is displaced by the reaction with amine 32 containing the desired R1 and R2 groups.
Scheme 8.

Synthetic scheme for generating N,N-disubstituted alkylguanidinobutanoic acid and N-arylguanidine analogues. Reagents and Conditions: (i) DEAD, Ph3P, THF, 0 °C-RT, 3–16 h; (ii) DIPEA, CH3CN, RT, 24 h; (iii) 4 M HCl/1,4-dioxane, 24–72 h, 0.29%–41% overall. Adapted with permission from [112]. Copyright 2005 American Chemical Society.
Deprotection of the amino acid analogues 33 is carried out using the same acidic conditions described in the synthesis of Rossiter’s 4124W analogues in Scheme 7 and provides analogues 34 in low to moderate yields (0.29%–41%). Each analogue’s inhibitory potential was assessed based on the reduced conversion of [14C]-NMMA to [14C]-citrulline by rat DDAH. Screening experiments revealed N-monosubstitution to be more effective than disubstitution, and that a R1 substituent comprising a N-2-methoxyethyl group increased inhibitor binding affinity (viz. compound 27a; IC50 = 189 μM; Scheme 7). Furthermore, a series of N-2-methoxyethyl guanidinobutanoate esters [112] was synthesized via thionyl chloride promoted esterification with alcohol containing the desired R3 group (Scheme 7). The generated benzyl ester 29a resulted in substantial improvement in DDAH-1 inhibition (IC50 = 27 μM). A series of amide analogues based on structures 29 (structures omitted) exhibited poor DDAH-1 inhibitory potentials.
4.1.4. NG-Substituted-l-Arginine Analogues
Rossiter et al. [112] also synthesized a library of N-substituted arginine analogues. A significant increase in inhibitor binding affinity was obtained for NG-(2-methoxyethyl)-l-arginine (commonly known as L-257 (compound 39; IC50 = 22 μM; Scheme 9) and its corresponding methyl ester (also known as L-291 (compound 40; IC50 = 20 μM; Scheme 9), relative to compound 27a (Scheme 7) that contains one less carbon within the main-chain. Interestingly, the benzyl ester of L-257 did not increase DDAH-1 inhibition as was observed with compound 29a. These data suggest that arginine analogues may bind differently within the putative DDAH-1 active-site. Moreover, neither the corresponding methyl amide of L-257, nor the (R)-isomer of L-257 (structures omitted) resulted in DDAH-1 inhibition.
Scheme 9.

Synthetic scheme for generating L-257 (39) and L-291 (40). Reagents and Conditions: (i) DEAD, Ph3P, THF, 0 °C-RT, 3–16 h; (ii) DIPEA, CH3CN, RT, 24 h; (iii) 4 M HCl/1,4-dioxane, 24–72 h, 44% overall; (iv) SOCl2, 0 °C for 30 min, reflux for 1 h, RT overnight, 80%. Adapted with permission from [112]. Copyright 2005 American Chemical Society.
The synthesis of L-257 (39) is represented in Scheme 9. The reaction sequence follows a similar approach to that described in the synthesis of the (S)-2-amino-4-(3-methylguanidino)butanoic acid analogues, using Boc protected amino acid analogues. Firstly, 2-methoxyethyl pyrazole carboxamidine (36) is prepared under Mitsunobu conditions [113] from 2-methoxyethanol (35) and N,N′-bis-tert-butoxycarbonylpyrazole-1H-carboxamidine (22). This intermediate 36 is reacted with Boc-Orn-OBut (37) to introduce the N-alkyl-guanidino group to compound 38. Final deprotection of 38 with 4 M HCl in 1,4-dioxane afforded L-257 (39) in 44% overall yield. L-257 can be subsequently esterified to afford L-291 (40) in yields of approximately 80%.
Both L-257 (39) and L-291 (40) inhibit DDAH-1 in vivo [112]. They are non-cytotoxic and selective for DDAH-1 against the three main isoforms of NOS [112,114] and arginase [115], an enzyme responsible for the metabolism of l-arginine into urea and ornithine [112,114,115]. L-257 (39) was utilized by Kotthaus et al. [114] as a validated active ‘parent’ compound for determining viable lead compounds, and to obtain initial structure-activity relationships (SARs) for those leads. In particular, the role of the N-substituent in DDAH inhibition was investigated as the guanidino moiety is considered to be important in the proposed catalytic mechanism and binding to the enzyme [116]. In the work undertaken by Kotthaus et al., the NG-(2-methoxyethyl) side-chain was varied along with the degree of guanidine substitution. Similarly, Tommasi et al. synthesized further L-257 derivatives with differing side-chains [117]. In both studies, the NG-(2-methoxyethyl) side-chain conferred inhibitors with activity toward DDAH-1. N-monosubstituted examples are represented in Table 2.
Table 2.
| Entry | R– | Inhibition at 1 mM (%) | IC50 (μM) | Ki (μM) |
|---|---|---|---|---|
| 1 1 | * CH3OCH2CH2– | 83 | 31 | 13 |
| 2 1 | CH3NHCH2CH2– | 29 | - | - |
| 3 1 | (CH3)2NHCH2CH2– | 14 | - | - |
| 4 1 | CH3SHCH2CH2– | 80 | 408 | - |
| 5 1 | FCH2CH2– | 67 | 379 | - |
| 6 1 | BnCH2– | 55 | 866 | - |
| 7 2 | CH3CH2CH2– | 60 | 283 | 90 |
| 8 2 | CH2CHCH2– | 70 | 207 | 58 |
| 9 2 | CH2CHCH2CH2– | 71 | 189 | 57 |
| 10 2 | CHCCH2– | 83 | 55 | 17 |
| 11 2 | F3CCH2– | 41 | - | - |
| 12 2 | O2N– | 6 | - | - |
| 13 2 | NH2COCH2CH2– | 43 | - | - |
* Denotes L-257 (39) (Scheme 9); 1 Measured by Ultra Performance Liquid Chromatography-Mass Spectrometry; 2 Measured by colorimetric assay.
4.1.5. NG-(2-Methoxyethyl)-l-Arginine Carboxylate Bioisosteres
The use of bioisosteric replacements is a well-known strategy adopted by medicinal chemists to improve the pharmacokinetic and pharmacodynamic properties of certain compounds, including but not limited to inhibition potency, lipophilicity, pKa, and metabolic stability [118]. Tommasi et al. [117] reported the synthesis and pharmacological evaluation of five carboxylate bioisosteres of L-257 (compounds 44a–c; Scheme 10, 49a–b; Scheme 11). In compounds 44a–c (Scheme 10), the carboxylic acid moiety is modified by introducing an acyl methyl sulfonamide 44a (2-amino-5-(3-(2-methoxyethyl)guanidino)-N-(methylsulfonyl)-pentanamide; ZST316), an O-methyl-hydroxamate 44b and a hydroxamate 44c. The replacement is introduced on Z-ornithine(Boc)-OH (41) in moderate to good yields (51%–86%) to afford 42, followed by the quantitative cleavage of Boc protected 42 with 4 M HCl in 1,4-dioxane. Guanidination of 42 with 2-methoxyethylpyrazole carboxamidine (36) is undertaken as described by Rossiter et al. [112] (see Section 4.1.3 and Section 4.1.4, Scheme 7 and Scheme 9). Deprotection of the Boc- and Z-appended arginine derivatives 43a–c is achieved in a single step using TFMSA and TFA [117,119] and produces compounds 44a–c.
Scheme 10.

Synthetic scheme for generating NG-(2-methoxyethyl)-l-arginine carboxylate bioisosteres. Reagents and Conditions: (i) CDI, DBU, methanesulfonamide, THF, RT, 7 h, 51%; (ii) HATU, DIPEA, O-methylhydroxylamine hydrochloride, THF, 0 °C-RT, 16 h, 86%; (iii) HATU, DIPEA, O-benzylhydroxylamine, THF, 0 °C-RT, 16 h, 57%; (iv) 4 M HCl/1,4-dioxane, DCM, RT, 30 min, 99%; (v) DIPEA, DCM, RT, 24 h, 43a 42%, 43b 22%, 43c 56%; (vi) TFMSA/TFA, RT, 1 h, 99%. Reproduced in part from [117] with permission of the Royal Society of Chemistry.
Scheme 11.

Synthetic scheme for generating NG-(2-methoxyethyl)-l-arginine carboxylate bioisosteres. Reagents and Conditions: (i) i-BuCOCl, N-methylmorpholine, NH4OH (30%), THF, −10 °C-RT, 3 h, 99%; (ii) TFAA, TEA, THF, 0 °C-RT, 16 h, 47%; (iii) 4 M HCl/1,4-dioxane, DCM, RT, 30 min, 99%; (iv) DIPEA, DCM, RT, 24 h, 40%; (v) NaN3, ZnBr2, H2O/2-propanol, reflux, 16 h, 30%; (vi) (1) NH2OH·HCl, NaHCO3, DMSO, reflux, 16 h; (2) CDI, DBU, THF, reflux, 30 min, 73%; (vii) TFMSA/TFA, DCM, RT, 30 min, 99%. Reproduced in part from [117] with permission of the Royal Society of Chemistry.
Bioisosteric replacement of the carboxylic acid moiety in L-257 with O-methyl hydroxamate 44b and hydroxamate 44c resulted in a decrease in DDAH-1 inhibition (IC50 = 230 and 131 μM, respectively). By contrast, introduction of an acyl methyl sulfonamidic moiety in ZST316 (44a) led to significant improvements in pharmacokinetic parameters (98% inhibition at 1 mM, IC50 = 3 μM and Ki = 1 μM). Compound ZST316 (44a) is the most potent human DDAH-1 inhibitor with ‘substrate-like’ structure reported in the literature to date.
Other noteworthy inhibitors in this series include the tetrazole 1-[4-amino-4-(1H-tetrazol-5-yl)butyl]-3-(2-methoxyethyl)guanidine (ZST086, 49a, Scheme 11) and the 5-oxo-1,2,4-oxadiazole 1-[4-amino-4-(5-oxo-4,5-dihydro-1,2,4-oxadiazol-3-yl)butyl]-3-(2-methoxyethyl)guanidine (ZST152, 49b, Scheme 11) [117]. The synthesis of these inhibitors with azole bioisosteric groups is achieved via conversion of the terminal carboxylic acid in 41 to primary amide 45 in quantitative yields using isobutylchloroformate, N-methylmorpholine, and aqueous ammonia. Primary amide 45 is then converted to nitrile 46 using trifluoroacetic anhydride and triethylamine in 47% yield. Compound 46 is guanylated [112] (see Section 4.1.3 and Section 4.1.4, Scheme 7 and Scheme 9) to afford compound 47, from which the heterocycle is constructed from the nitrile. The tetrazole is introduced by treating nitrile 47 with sodium azide in the presence of zinc(II) bromide to afford compound 48a, using the click chemistry procedure reported by Demko and Sharpless [120]. Although the use of zinc(II) bromide in the synthesis of Boc-amino tetrazoles derived from α-amino acids has been reported [121], the use of a Lewis acid may result in partial deprotection of compound 48a during the reaction. Ammonium chloride represents an alternative reagent [122]. The 5-oxo-1,2,4-oxadiazole 48b is synthesized from nitrile 47 using hydroxylamine hydrochloride and sodium bicarbonate in DMSO, followed by treatment with CDI and DBU [123]. Acidic deprotection of 48a–b is achieved as described previously [117,119]. Despite obtaining lower yields for heterocyclic adducts 49a–b (7%–13%) relative to bioisosteres 44a–c (19%–32%), the inhibitory potential of 5-oxo-1,2,4-oxadiazole ZST152 (49b) was significant (95% at 1 mM, IC50 = 18 μM, Ki = 7 μM), while tetrazole ZST086 (49a) displayed 91% inhibition at 1 mM, with IC50 = 34 μM and Ki = 14 μM [117].
4.1.6. N5-(1-Iminoalk(en)yl)-l-Ornithine Derivatives
Amidines are commonly known bioisosteres of the guanidine moiety [124]. N5-(1-iminoalk(en)yl)-l-ornithines such as N5-(1-imino-3-butenyl)-l-ornithine (L-VNIO, 54a, Figure 2), are reported to inhibit NOS by competing with l-arginine for binding at the enzyme catalytic-site [125,126,127]. Therefore, derivatives of this class of compounds have been extensively studied as DDAH inhibitors. The synthetic approach to afford N5-(1-iminoalk(en)yl)-l-ornithine derivatives 54 (Scheme 12) [127,128], utilized a similar approach to the N-but-3-ynyl-2-chloro-acetamidine probe (18) (see Section 4.1.2, Scheme 5) [109]. The R1 furnished nitrile 50 is converted to its corresponding ethyl imidic ester 51 by bubbling dry hydrochloric acid gas through a solution of anhydrous ethanol (16). The ethyl imidic ester 51 is then reacted with Nα-Boc-l-ornithine (52) to afford the protected adduct 53. Deprotection is obtained by treatment with 6 M HCl to achieve the N5-(1-iminoalk(en)yl)-l-ornithine 54 in excellent overall yields (84%–94%).
Figure 2.

N5-(1-Iminoalk(en)yl)-l-ornithine derivatives, L-VNIO (54a), L-IPO (54b), and Cl-NIO (54c), as synthesized by Kotthaus et al. [114] and Wang et al. [128,129].
Scheme 12.

Synthetic scheme for generating N5-(1-iminoalk(en)yl)-l-ornithine derivatives. Reagents and Conditions: (i) dry HCl(g) at 0 °C for 1 h, stop HCl(g), then 0 °C for 4 h and RT overnight; (ii) H2O, 5 °C, pH = 10, 1.5 h, adjust pH = 7, overnight, Dowex 50WX8-400 (H2O then 10% aqueous pyridine); (iii) 6 M HCl, EtOAc, 84%–94% from Nα-Boc-l-ornithine (52). Adapted with permission from [127]. Copyright 1999 American Chemical Society.
The significant structural similarity of the N5-(1-iminoalk(en)yl)-l-ornithine derivatives with DDAH substrates, ADMA (1) and L-NMMA (2) led different groups to study this class of compounds as potential DDAH inhibitors [114,128]. Kotthaus et al. [114] examined five N5-(1-iminoalk(en)yl)-l-ornithines with varying side-chain lengths and bond saturation for their ability to inhibit l-citrulline formation by human DDAH-1 using both high performance liquid chromatography and colorimetry. L-VNIO (54a, Figure 2) emerged as the best DDAH-1 inhibitor of this series of derivatives (97% inhibition at 1 mM, IC50 = 13 μM, Ki = 2 μM). However, increasing the main-chain length by one carbon atom from ornithine to lysine reduced its DDAH inhibitory potential.
Wang et al. [128] investigated this class of compounds as dual DDAH-1 and NOS inhibitors, reporting the synthesis and kinetic parameters for four N5-(1-iminoalkyl)-l-ornithines. N5-(1-Iminopropyl)-l-ornithine (L-IPO 54b, Figure 2) was identified as the best compound exhibiting inhibition of both human DDAH-1 (Ki = 52 μM) and rat nNOS (Ki = 3 μM). Wang et al. [129] additionally developed an irreversible human DDAH-1 inhibitor, N5-(1-imino-2-chloroethyl)-l-ornithine (Cl-NIO, 54c, Figure 2), whose chemical structure reflects a combination of both L-IPO (54b) and 2-chloroacetamidine (11). The synthesis of Cl-NIO (54c) is similar to that described for the N5-(1-iminoalk(en)yl)-l-ornithine derivatives (Scheme 12). Here, Nα-Boc-l-ornithine is reacted with 2-chloro-1-ethoxyethanimine, and the intermediate obtained is deprotected with 4.6 M HCl in dioxane to generate Cl-NIO (54c, Figure 2) in low yields (10%). The inhibition potency of Cl-NIO (54c) was assessed via colorimetric assay and exhibited Ki = 1.3 μM. In summary, several of the N5-(1-iminoalk(en)yl)-l-ornithine derivatives (54a–c, Figure 2) are potent human DDAH-1 inhibitors. Off-target effects on the broader NO pathway (NOSs, arginases, AGTX-2) may either represent an advantage (strong NO suppression), or limit their application (loss of NO physiological functions) as potential therapeutics.
4.2. Non-Substrate-Like Inhibitors
4.2.1. Pentafluorophenyl (PFP) Sulfonate Esters
Vallance et al. [130] identified pentafluorophenyl (PFP) sulfonate esters 61 as nonspecific PaDDAH and bacterial arginine deaminase (ADI) inhibitors, after highlighting their structural similarity with the cysteine protease family of enzymes. This class of inhibitor can be obtained in moderate to good yields (44%–75%) via the 1,3-dipolar cycloaddition between nitrone 60 and the electron deficient vinyl-appended PFP sulfonate 57 (Scheme 13) [131]. The shelf-stable PFP vinylsulfonate 57 is synthesized in moderate yield (59%) from pentafluorophenol (55) and 2-chloroethane-1-sulfonyl chloride (56) [132], while various nitrones 60 can be generated from their corresponding aldehydes 58 by reaction with N-methyl hydroxylamine (59) under mild conditions and in good yield (for examples, please see [133,134]).
Scheme 13.

Synthetic scheme for generating PFP sulfonate esters 61. Reagents and Conditions: (i) DCM, TEA, 0 °C, 59%; (ii) Method A: NaHCO3, anhydrous MgSO4, DCM, reflux, 24 h, 73%–90% [133]; Method B: NaOAc, EtOH, RT, 90% [134]; (iii) dry toluene, reflux, 1–24 h, 44%–75%. Reproduced in part from [130,131,132,133] with permission of the Royal Society of Chemistry and the American Chemical Society.
The electron deficiency of the vinyl PFP sulfonate dipolarophile 57 affords the reversed regioselective 4C-substituted adduct of the isoxazolidine in compound 61, contrary to the 5C-substituted adduct typically observed with olefins. Although this was previously observed for electron deficient dipolarophiles [135,136,137,138], Caddick et al. [131] reported the 4C-substituted isoxazolidine 61 is increasingly favoured at higher temperatures. Each of the nitrones 60 reported by Caddick et al. [131] participates in cycloaddition with the vinyl PFP sulfonate 57, including C-aryl- and C-alkyl-N-methyl species with both electron-donating and electron-withdrawing substituents in addition to poly- and hetero-aromatic substituents. This robust synthetic methodology provides the ability to generate a structurally diverse library of PFP sulfonate esters 61 with moderate ease and high yields [130]. Several PFP sulfonate esters 61 have been screened for their activity as PaDDAH and ADI inhibitors at two concentrations (500 μM and 50 μM), with five compounds emerging as relatively potent PaDDAH inhibitors (IC50 = 16–58 μM, see [130] for structures). Vallance et al. revealed the mode of inhibition was competitive utilising time-dependence and reversibility experiments [130]. Interestingly, the PFP sulfonate esters 61 have been proposed as potential antibacterial agents, however no data has been reported identifying if PaDDAH inhibition modulates bacterial growth or viability. Likewise, no studies utilizing human DDAH have been reported identifying whether the PFP sulfonate esters exhibit specificity for bacterial PaDDAH. Non-specific PFP sulfonate ester binding to both human DDAH and PaDDAH would be counterproductive for their proposed application as antibiotics.
4.2.2. Indolylthiobarbituric Acid Derivatives
The indolylthiobarbituric acid derivatives 68 [139] were identified as PaDDAH inhibitors by a combination of virtual screening using a compound library comprising 308,000 structures and subsequent biological screening. Among the 109 highest scoring compounds, 90 were commercially available and used in colorimetric enzyme activity assays. Three compounds consisting of two chemical scaffolds were identified as potential PaDDAH inhibitors. One of these scaffolds, indolylthiobarbituric acid (68a), together with additional compounds 68b–c identified from a similarity search, emerged as promising inhibitors of PaDDAH (IC50 = 2–16.9 μM). The most potent inhibitor of this series was SR445 (68c, IC50 = 2 μM), and no member of this class of compounds showed any human DDAH inhibition [114].
This class of inhibitors is comprised of thiobarbituric acid module 64 and indolyl module 67 (Scheme 14). The thiobarbituric acid module 64 is synthesized using a modified version of the method described by Fisher and Mering in 1903, by reaction of N-substituted 2-thiourea 63 with diethyl malonate (62) [140,141]. The indolyl module 67 is alkylated by deprotonation of indole-3-carbaldehyde (65) and reaction with 2-chloro-N-furan-2-ylmethyl-acetimide (66) using a method previously reported (yield 83%) [142]. The aldehyde substituent of the indolyl derivative 67 is subsequently reacted with the thiobarbituric acid derivative 64 under acidic conditions to afford 68 (yields not reported) [139,143]. This produced a mixture of (E)- and (Z)-isomers about the linking double bond.
Scheme 14.

Synthetic scheme for generating indolylthiobarbituric acid derivatives 68. Reagents and Conditions: (i) NaOMe, MeOH, 60 °C, 6 h, yields not reported; (ii) NaH, DMF, RT, overnight, 83%; (iii) 6 M HCl, EtOH, RT, 2–3 h, yields not reported.
4.2.3. Ebselen
Ebselen (2-phenyl-1,2-benzisoselenazol-3(2H)-one, 73), a selenazole heterocyclic compound with well-known antioxidant properties [144], was recently identified as a potent DDAH-1 inhibitor. This was achieved by utilizing a high throughput screening (HTS) assay of two commercial libraries comprising 2446 compounds [145]. The HTS identified each compound’s ability to inhibit the conversion of the synthetic substrate, S-methyl-l-thiocitrulline (SMTC) to citrulline and methanethiol by PaDDAH. Compounds exhibiting PaDDAH inhibitory potentials were validated for their ability to inhibit human DDAH-1 using ADMA as the substrate. The quantification of inhibition was measured using derivatized citrulline formation and colorimetric detection. Characterisation of PaDDAH and human DDAH-1 inhibition by ebselen was subsequently determined by dose-response, reversibility, and ESI-MS experiments.
While ebselen (73) is commercially available, it can be prepared via two main methods [146,147,148]. The method described in Scheme 15 [147,148] is preferred due to its shorter reaction times and elementary design. This method involves the treatment of benzoic acid (69) with thionyl chloride and aniline to afford benzanilide (70) followed by ortho-lithiation to dianion 71 using n-butyl lithium. Selenium is then inserted into the carbon-lithium bond of 71, forming dianion intermediate 72, whose cyclization to ebselen (73) is promoted by copper(II) bromide oxidant.
Scheme 15.

Synthetic scheme for generating ebselen (73). Reagents and Conditions: (i) (1) DCM, pyridine, SOCl2, reflux, 2 h; (2) toluene, aniline, reflux, 3 h, 80%; (ii) dry THF, n-BuLi, nitrogen atmosphere, 0 °C, 30 min, 76% (determined via methylation of 72); (iii) same solution as step (ii), Se, 0 °C, 30 min; (iv) same solution as step (iii), CuBr2, 30 min at −78 °C, then 2 h at RT, 63%. Adapted with permission from [147], copyright 1989 American Chemical Society, and [148], copyright 2003 The Pharmaceutical Society of Japan.
Ebselen is reported to irreversibly inhibit both PaDDAH (IC50 = 96 ± 2 nM) and human DDAH-1 (IC50 = 330 ± 30 nM), forming a covalent selenosulfide bond with each enzyme’s respective catalytic cysteine. In silico docking experiments suggest inhibitor binding is supported by π-stacking interactions with Phe76 in addition to other polar interactions involving His162. In vitro experiments demonstrated ebselen exhibits no effect on human DDAH-1 expression [145].
Ebselen is the most potent DDAH inhibitor with ‘non-substrate-like’ structure reported in the literature to date and demonstrates an anti-inflammatory response [149]. However, its application as a DDAH inhibitor is limited due to its poor DDAH selectivity. A wide range of molecular targets are modulated by ebselen including: NADPH oxidase 2 [150,151], H+/K+ ATPase transporter [152], Ca2+- and phospholipid-dependent protein kinase C [151], lipoxygenase [153], thioredoxin reductase [154], and metallothionein [155]). Moreover, cellular toxicity in leukocytes and hepatocytes has been reported, further reducing the likelihood of ebselen’s use in clinical practice [156,157].
4.2.4. 4-Hydroxy-2-Nonenal
4-Hydroxy-2-nonenal (4-HNE, 76) is a cytotoxic aldehyde generated from lipid peroxidation during inflammation [158,159] and is abundant in the body. 4-HNE is highly reactive toward proteinaceous nucleophilic groups, such as thiol and imidazole moieties. Nucleophilic attack affords Michael adducts, whilst Schiff bases are formed with primary amines (e.g., lysine residues) [160,161,162,163]. Different methods have been reported for the synthesis of 4-HNE (76) and its derivatives [164,165,166]. The method reported by Soulér et al. [166] is described here due to its simplicity, speed, and versatility (Scheme 16). The synthesis is completed in a single cross-metathesis reaction between octen-3-ol (75) and 2-propenal (74) in the presence of Hoveyda-Grubbs 2nd generation ruthenium catalyst under a nitrogen atmosphere and in dry oxygen-free solvent (Scheme 16). The reaction proceeds at room temperature for 25 h affording the (E)-isomer of 4-HNE (76) in 75% yield.
Scheme 16.

Synthetic scheme for generating 4-hydroxy-2-nonenal (4-HNE, 76). Reagents and Conditions: (i) dry oxygen-free DCM, RT, 25 h, 75%, Hoveyda-Grubbs 2nd generation (0.025 eq.).
4-HNE is reported to react with cysteine and histidine residues and it was this reactivity that led Forbes et al. to investigate its use as a human DDAH-1 inhibitor [167]. In Forbes’ experiments, DDAH activity was measured via both colorimetric and radioisotope activity assays, with 4-HNE showing dose-dependent inhibition (IC50 = 50 μM) and near complete inhibition at 500 μM. Mass spectrometry experiments elucidated the mechanism of inhibition, whereby the formation of a Michael adduct between 4-HNE and His173 of the DDAH-1 active-site occurs.
4.2.5. PD 404182
6H-6-Imino-(2,3,4,5-tetrahydropyrimido)[1,2-c]-[1,3]benzothiazine (PD 404182, 79), is a commercially available heterocyclic pyrimidobenzothiazine and a suitable lead compound for SAR studies due to its simple synthetic methodology [168]. PD 404182 (79) is a compound listed in the Library of Pharmacologically Active Compounds (LOPAC) and is reported to have antibacterial [168,169] and antiviral [170,171] activity. Ghebremariam et al. [61] utilized a colorimetric assay to screen a library of over 1200 compounds for their ability to inhibit the conversion of ADMA to l-citrulline and dimethylamine by recombinant human DDAH-1. Compounds with reasonable inhibitory potentials were subjected to kinetic characterisation via fluorimetric assay using the synthetic substrate, S-methylthiocitrulline. Reversibility and ESI-MS studies were additionally undertaken. PD 404182 was subsequently evaluated for its ability to reduce angiogenesis and to protect against lipopolysaccharide-induced NO production. PD 404182 (79) was found to be a potent irreversible competitive human DDAH-1 inhibitor (IC50 = 9 μM) and showed promise as an anti-inflammatory and antiangiogenic agent.
The synthesis of PD404182 can be achieved via two routes [172,173]. Both approaches generate reasonable to high yields (61%–88%) over several synthetic steps. The first approach involves the reaction of 2-(2′-haloaryl)-tetrahydropyrimidine 77 with carbon disulfide in the presence of sodium hydride, followed by hydrolysis of the carbamodithioate derivative 78 and subsequent treatment with cyanogen bromide to afford 79 (Scheme 17; Route A). Alternatively, the 2-(2′-haloaryl)-tetrahydropyrimidine 77 can be reacted with tert-butylisothiocyanate (80) in the presence of sodium hydride to afford intermediate 81, followed by treatment with trifluoroacetic acid to afford 79 (Scheme 17; Route B).
Scheme 17.

Synthetic scheme for generating PD 404182 (79). Reagents and Conditions: Route A: (i) DMF, NaH, CS2, argon atmosphere, 80 °C, 12 h, 88%; (ii) (1) 0.1 M NaOH, MeOH/H2O (9:1), reflux, 12 h; (2) anhydrous EtOH, BrCN, argon atmosphere, reflux, 2 h, 61% (two steps). Route B: (iii) DMF, NaH, argon atmosphere, 80 °C, 2 h, 62%; (iv) TFA, CHCl3, molecular sieves 4 Å, reflux, 1 h, 85%. Adapted with permission from [172,173]. Copyright 1975, 2010 American Chemical Society.
4.2.6. Proton Pump Inhibitors
Proton H+/K+ ATPase pump inhibitors (PPIs) are widely prescribed to inhibit gastric acid secretion and are characterized by a 2-pyridylmethylsulfinylbenzimidazole scaffold. The chemical structures of the commonly prescribed PPIs 82a–f are shown in Figure 3.
Figure 3.

Chemical structures of commonly prescribed proton pump inhibitors (PPIs) 82a–f.
HTS experiments utilising a library of 130,000 compounds and recombinant enzyme identified PPIs as human DDAH-1 inhibitors [174,175]. Initial screening was undertaken using a colorimetric assay with ADMA as the substrate. Further characterization of compounds of interest was achieved via a fluorimetric assay using S-methylthiocitrulline [174,175]. Surface plasmon resonance (SPR) was additionally utilized to detect the binding of PPIs to amine-coupled DDAH-1 (CM5 sensor chip) [175]. All commercially available PPIs were found to reversibly inhibit DDAH-1 (IC50 = 51–63 μM). Interestingly, in vitro experiments using human endothelial cells and in vivo experiments in mice demonstrated that PPIs increase ADMA concentrations [175] further supporting the evidence that prolonged use of PPIs is associated with an increased risk of cardiovascular disease [176,177].
PPIs are comprised of pyridine and benzimidazole moieties with different substituents. Their syntheses are protected by current patents (with the exception of omeprazole (82a)). The original patent for omeprazole (82a) [178] details preparation of the 2-(chloromethyl)pyridine derivative 84. Condensation [179] of compound 84 with 2-mercaptobenzimidazole derivative 83 affords the thioether precursor 85, which is subsequently oxidized to the sulfoxide in omeprazole (82a) using conditions such as hydrogen peroxide and a vanadium catalyst [180] (Scheme 18; Route A).
Scheme 18.

Synthetic scheme for generating omeprazole (82a). Reagents and Conditions: Route A: (i) 1 M NaOH (2 eq.), EtOH, RT, 8 h; (ii) catalyst: V2O5, NaVO3, NH4VO3, [(CH3COCH2COCH3)2VO] (0.0001–0.1 eq.), H2O2: 10%–70% aqueous or organic solution (1–3 eq.), solvent: halogenated hydrocarbons, ethers, alcohols, ketones, nitriles, or H2O, temperature: 0 °C to boiling point of the solvent, 0.5–24 h, 89%–93%. Route B: (iii) condensation, conditions not specified in patent; (iv) [(CH3COCH2COCH3)2VO] (0.006 eq.), 30% H2O2, acetone, 0–5 °C for 1 h then 20–22 °C for 1 h, 90%; (v) 10% NaOH, nitrogen atmosphere, 50 °C, 3 h; (vi) CO2, extraction into 1:1 v/v isopropanol/toluene, reflux, 20–30 min, 42% (across steps (v) and (vi)).
Further development of the synthetic method to yield omeprazole (82a) [180] identified that the coupling between the pyridine and benzimidazole moieties can be modified to simplify purification. Principally, this method avoids the formation of the thioether intermediate 85. Furthermore, the crude product containing omeprazole (82a) produced from the original synthesis is prone to discolouration during the oxidation step [180]. The revised synthesis (Scheme 18; Route B) proceeds via condensation of 2-chloromercaptobenzimidazole derivative 86 with a primary amide pyridine derivative 87 to afford the acetamide thioether intermediate 88, which is oxidized to the acetamide sulfoxide 89. Compound 89 is subsequently hydrolysed to the carboxylic acid salt 90 under alkaline conditions, with decarboxylation yielding omeprazole (82a) or lansoprazole (82c), depending on the benzimidazole 86 and the pyridine 87 derivatives used as the starting materials.
4.2.7. 1,2,3-Triazoles
The synthesis of 1,2,3-triazoles 95 (Scheme 19) comprised of 4-halopyridine and benzimidazole units have been explored as ‘non-substrate-like’ DDAH-1 inhibitors [117]. The synthesis of these derivatives was undertaken based on reports that identified binding of both the 4-halopyridine and benzimidazole fragments to human DDAH-1 [181]. A triazole linker was selected to connect these fragments via copper(I)-catalysed ‘click’ cycloaddition between the terminal alkyne 92 and azide 94 (Scheme 19). The alkyne 92 is synthesized by Sonogashira coupling of the alkyne derivative with the required halogenated species 91 [182,183]. The azide 94 is synthesized by reaction of diphenylphosphoryl azide with the required primary alcohol 93 [184].
Scheme 19.

Synthetic scheme for generating 1,2,3-triazoles 95. Reagents and Conditions: (i) 92a (Ph3P)2PdCl2, CuI, TEA, TIPS-acetylene, DMF, 80 °C, 72 h, 30%, 92b (Ph3P)2PdCl2, CuI, TEA, ethynyl-TMS, 80 °C, 20 h, 48%; (ii) DPPA, DBU, toluene, RT, 16 h, 94a 55%, 94b 83%; (iii) CuI, sodium ascorbate, TEA, DCM, RT, 24 h, 95a 65%, 95b 30%. Reproduced in part from [117] with permission of the Royal Society of Chemistry.
Human DDAH-1 inhibition, albeit low (IC50 = 422 μM, 57%, inhibition at 1 mM), was reported with triazole 95a. In general, 95a–b failed to show significant inhibitory potential and may be due to the triazole linker not facilitating a productive inhibitory binding conformation of the fragments in the enzyme active-site.
5. Conclusions
Current evidence suggest that inhibiting the DDAH family of enzymes may be used as a therapeutic tool to indirectly inhibit the NOS enzymes via increasing concentrations of endogenous NOS inhibitors, such as ADMA and l-NMMA. Therefore the development and synthesis of DDAH inhibitors is emerging as a promising field of clinical research. Furthermore, engineering compounds that target bacterial DDAH may represent a suitable strategy for the development of new antimicrobial agents; however further investigations are required. This has led to different classes of DDAH inhibitors being reported within the last decade characterized by alternative features in terms of chemical structure, potency, enzyme selectivity, and mechanism of action. The importance placed on each of these features depends on the end application of the inhibitor. Table 3 summarizes the most potent DDAH inhibitors documented in the literature to date. Some compounds, namely PFP sulfonate esters 61 or indolylthiobarbituric acid derivatives 68, exhibit significant PaDDAH inhibition. In particular, selectivity experiments using indolylthiobarbiturates failed to exhibit inhibitory effects on the human isoenzyme, therefore derivatives of this structural class are well positioned for further investigation in antibiotic applications. Among classes of molecules which are active on the human isoenzyme (amino acid derivatives 27, 29, 39, 40, 44, 49, 54, ebselen (73), 4-HNE (76), PD 404182 (79), PPIs 82, 1,2,3-triazoles 95), L-257 (39) and L-291 (40) are highly selective for DDAH-1, while ornithine derivatives such as L-VNIO (54a) or L-IPO (54b) are known to inhibit not only DDAH, but also arginase and the NOS family of enzymes. Furthermore, compounds such as ebselen (73) and 4-HNE (76), although very potent DDAH inhibitors, are well known to exhibit a range of off-target effects, thus are more suitable for experiments in vitro rather than in vivo.
Table 3.
Summary of the main DDAH inhibitors and their kinetic parameters.
| Chemical Number | Chemical Name | Chemical Structure | Enzyme | IC50 (µM) | Ki (µM) | Kinact (min−1) | Reference |
|---|---|---|---|---|---|---|---|
| (9) | S-Nitroso-l-homocysteine (HcyNO) | 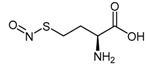 |
Bovine DDAH-1 | 75 | 690 | 0.38 | [105] |
| (11) | 2-Chloroacetamidine | 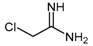 |
PaDDAH 1 | - | 3100 | 1.2 | [106] |
| (18) | N-But-3-ynyl-2-chloro-acetamidine | 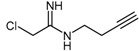 |
Human DDAH-1 | 350 | - | - | [109] |
| (21) | (S)-2-Amino-4-(3-methylguanidino)butanoic acid (4124W) | 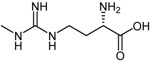 |
Rat DDAH-1 | 416 | - | - | [110] |
| (39) | NG-(2-Methoxyethyl)-l-arginine (L-257) | 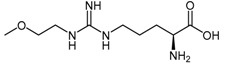 |
Human DDAH-1 | 31 2 | 13 2 | - | [112] |
| (40) | NG-(2-Methoxyethyl)-l-arginine methyl ester (L-291) | 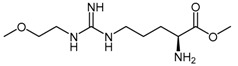 |
Rat DDAH-1 | 20 | - | - | [112] |
| (44a) | 2-Amino-5-(3-(2-methoxyethyl)guanidino)-N-(methylsulfonyl)-pentanamide (ZST316) | 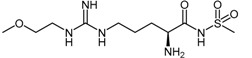 |
Human DDAH-1 | 3 | 1 | - | [117] |
| (54a) | N5-(1-Imino-3-butenyl)-l-ornithine (L-VNIO) | 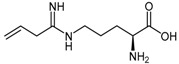 |
Human DDAH-1 | 13 | 2 | - | [114] |
| (54b) | N5-(1-Iminopropyl)-l-ornithine (L-IPO) | 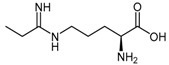 |
Human DDAH-1 | - | 52 | - | [128] |
| (54c) | N5-(1-Imino-2-chloroethyl)-l-ornithine (Cl-NIO) | 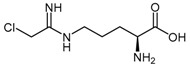 |
Human DDAH-1 | - | 1.3 | 0.34 | [129] |
| 61 | Pentafluorophenyl (PFP) sulfonate esters | 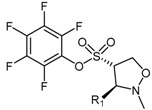 |
PaDDAH 1 | 16–58 | - | - | [130] |
| 68a–c | Indolylthiobarbituric acid derivatives | 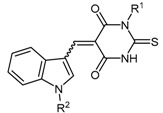 |
PaDDAH 1 | 2–17 | - | - | [139] |
| (73) | Ebselen | 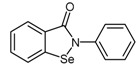 |
Human DDAH-1 | 0.33 | - | - | [145] |
| (76) | 4-Hydroxy-2-nonenal (4-HNE) | 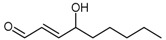 |
Human DDAH-1 | 50 | - | - | [167] |
| (79) | 6H-6-Imino-(2,3,4,5-tetrahydropyrimido)[1,2-c]-[1,3]benzothiazine (PD 404182) | 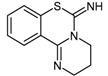 |
Human DDAH-1 | 9 | - | - | [61] |
| 82a–f | Proton H+/K+ ATPase pump inhibitors (PPIs) | 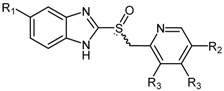 |
Human DDAH-1 | 51–63 | - | - | [175] |
1 Pa: Pseudomonas aeruginosa; 2 as determined by Tommasi et al. [117].
In terms of the varied approaches to chemical synthesis of the DDAH inhibitors discussed here, many can be synthesized in moderate to excellent overall yields and use reaction sequences that can be easily modified to enable the design of a large number of analogues. The commercial availability of several of the compounds determined to be DDAH inhibitors (2-chloroacetamidine (11), ebselen (73) 4-HNE (76) and the PPIs 82) demonstrates the versatility of some of the synthetic routes for large scale production.
Based on the in vitro pharmacokinetic and/or enzyme selectivity data compiled in this review, there appear to be three promising human DDAH-1 inhibitors that have the potential to progress to clinical trial: ZST316 (Ki = 1 μM), Cl-NIO (Ki = 1.3 μM), and L-257 (Ki = 13 μM) (Table 3). Despite exhibiting the most potent inhibitory response in vitro, a paucity of data exists with respect to the in vivo pharmacokinetic and pharmacodynamic profiling of ZST316. Literature reports involving the enzyme selectivity of Cl-NIO shows considerable promise, with no measurable cross-reactivity in experiments specifically involving eNOS [129]. However, data are not available regarding this inhibitor‘s interactions with iNOS, nNOS, or the arginase family of enzymes. Contrary to this, data reported for L-257 by Kotthaus et al. [115] demonstrates that L-257 is highly selective for DDAH-1. The administration of L-257 in a rodent model of septic shock not only improved survival, but additionally improved haemodynamic and organ function [67], indicating a potential clinical role for L-257.
Interestingly, little is understood about the regulation and pharmacological modulation of human DDAH-2. As such, the development of improved metabolomic approaches for the sensitive measurement of DDAH-2 activity in vitro is an important step forward in profiling the endogenous substrates and biochemical role(s) of this important enzyme in humans.
The continued exploration of DDAH inhibition and the synthesis of new chemical entities with improved pharmacokinetic parameters are therefore at an exciting junction in the development of novel medicines.
Acknowledgments
The authors thank Flinders Medical Centre Foundation and Flinders Faculty of Health Sciences, the Scottish Universities Life Sciences Alliance (SULSA), and the NHS Grampian Endowment Fund for providing financial support which assisted with reading available evidence and conducting experimental work.
Abbreviations
The following abbreviations are used in this manuscript:
| DDAH | Dimethylarginine dimethylamino hydrolase |
| ADMA | Asymmetric dimethylarginine |
| L-NMMA | Monomethyl arginine |
| NOS | Nitric oxide synthase |
| NO | Nitric oxide |
| SDMA | Symmetric dimethylarginine |
| PRMTs | Protein arginine methyl transferases |
| CAT | Cationic amino acid transporter |
| AGXT2 | Alanine-glyoxylate amino transferase 2 |
| PKA | Protein kinase A |
| Akt | Protein kinase B |
| VEGF | Vascular endothelial growth factor |
| iNOS | Inducible nitric oxide synthase |
| TNF-α | Tumour necrosis factor α |
| LDL | Low density lipoprotein |
| IL-1β | Interleukin-1β |
| AT1-R | Type 1 angiotensin receptor |
| DNA | Deoxyribonucleic acid |
| FXR | Farnesoid X receptor |
| HcyNO | S-Nitroso-l-homocysteine |
| NaNO2 | Sodium nitrite |
| NaOH | Sodium hydroxide |
| HCl | Hydrochloric acid |
| Min | Minutes |
| Ki | Inhibition constant |
| µM | Micromolar |
| H2O | Water |
| Cys | Cysteine |
| His | Histidine |
| PaDDAH | Pseudomonas aeruginosa dimethylarginine dimethylamino hydrolase |
| PAD | Peptydilarginine deaminase |
| NaOMe | Sodium methoxide |
| MeOH | Methanol |
| AcOH | Acetic acid |
| NH4Cl | Ammonium chloride |
| PEO | Poly(ethylene oxide) |
| DCM | Dichloromethane |
| TEA | Triethylamine |
| DMAP | 4-Dimethylaminopyridine |
| MsCl | Methanesulfonyl chloride |
| Et2O | Diethyl ether |
| RT | Room temperature |
| NaN3 | Sodium azide |
| DMF | N,N-Dimethylformamide |
| Ph3P | Triphenylphosphine |
| 4124W | (S)-2-Amino-4-(3-methylguanidino)butanoic acid |
| Boc | tert-Butyloxycarbonyl |
| M | Molar |
| DEAD | Diethyl azodicarboxylate |
| THF | Tetrahydrofuran |
| DIPEA | N,N-Diisopropylethylamine |
| CH3CN | Acetonitrile |
| SOCl2 | Thionyl chloride |
| SAR | Structure activity relationship |
| IC50 | Inhibition constant |
| L-257 | NG-(2-Methoxyethyl)-l-arginine |
| L-291 | NG-(2-Methoxyethyl)-l-arginine methyl ester |
| Orn | Ornithine |
| Bu | Butyl |
| Z | Carboxybenzyl |
| TFMSA | Trifluoromethanesulfonic acid |
| TFA | Trifluoroacetic acid |
| CDI | 1,1′-Carbonyldiimidazole |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| h | Hours |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| mM | Millimolar |
| DMSO | Dimethyl sulfoxide |
| i-BuCOCl | Isobutyl chloroformate |
| NH4OH | Ammonium hydroxide |
| TFAA | Trifluoroacetic anhydride |
| ZnBr2 | Zinc bromide |
| NH2OH.HCl | Hydroxylamine hydrochloride |
| NaHCO3 | Sodium bicarbonate |
| L-VNIO | N5-(1-Imino-3-butenyl)-l-ornithine |
| EtOH | Ethanol |
| EtOAc | Ethyl acetate |
| L-IPO | N5-(1-Iminopropyl)-l-ornithine |
| Cl-NIO | N5-(1-Imino-2-chloroethyl)-l-ornithine |
| PFP | Pentafluorophenyl |
| NaOAc | Sodium acetate |
| ADI | Arginine deaminase |
| HTS | High throughput screening |
| SMTC | S-methyl-l-thiocitrulline |
| ESI-MS | Electrospray ionization mass spectrometry |
| BuLi | Butyl lithium |
| Se | Selenium |
| CuBr2 | Copper(II) bromide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| H+/K+ ATPase | Proton/potassium ATPase |
| Ca2+ | Calcium 2+ cation |
| 4-HNE | 4-Hydroxy-2-nonenal |
| LOPAC | Library of Pharmacologically Active Compounds |
| NaH | Sodium hydride |
| CS2 | Carbon disulfide |
| BrCN | Cyanogen bromide |
| Å | Angstrom |
| PPIs | Proton pump inhibitors |
| SPR | Surface plasmon resonance |
| CM | Carboxymethylated |
| (Ph3P)2PdCl2 | Bis(triphenylphosphine)palladium chloride |
| CuI | Copper(I) iodide |
| TIPS | Triisopropylsilyl |
| TMS | Trimethylsilane |
| DPPA | Diphenylphosphoryl azide |
Author Contributions
The manuscript was conceived by A.A.M., R.B.M. and S.T. collected the primary data and compiled draft manuscripts. B.C.L. and A.A.M. supervised development of the manuscript, and assisted in data interpretation, manuscript evaluation and editing.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Vallance P., Leiper J. Blocking no synthesis: How, where and why? Nat. Rev. Drug Discov. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- 2.Tran C.T., Fox M.F., Vallance P., Leiper J.M. Chromosomal localization, gene structure, and expression pattern of DDAH1: Comparison with DDAH2 and implications for evolutionary origins. Genomics. 2000;68:101–105. doi: 10.1006/geno.2000.6262. [DOI] [PubMed] [Google Scholar]
- 3.Greco R., Ferrigno A., Demartini C., Zanaboni A., Mangione A.S., Blandini F., Nappi G., Vairetti M., Tassorelli C. Evaluation of ADMA-DDAH-NOS axis in specific brain areas following nitroglycerin administration: Study in an animal model of migraine. J. Headache Pain. 2015;16, 74:1–8. doi: 10.1186/s10194-015-0560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D’Mello R., Sand C.A., Pezet S., Leiper J.M., Gaurilcikaite E., McMahon S.B., Dickenson A.H., Nandi M. Dimethylarginine dimethylaminohydrolase 1 is involved in spinal nociceptive plasticity. Pain. 2015;156:2052–2060. doi: 10.1097/j.pain.0000000000000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasegawa K., Wakino S., Kimoto M., Minakuchi H., Fujimura K., Hosoya K., Komatsu M., Kaneko Y., Kanda T., Tokuyama H., et al. The hydrolase DDAH2 enhances pancreatic insulin secretion by transcriptional regulation of secretagogin through a sirt1-dependent mechanism in mice. FASEB J. 2013;27:2301–2315. doi: 10.1096/fj.12-226092. [DOI] [PubMed] [Google Scholar]
- 6.Amrouni D., Meiller A., Gautier-Sauvigné S., Piraud M., Bouteille B., Vincendeau P., Buguet A., Cespuglio R. Cerebral changes occurring in arginase and dimethylarginine dimethylaminohydrolase (DDAH) in a rat model of sleeping sickness. PLoS ONE. 2011;6:e16891. doi: 10.1371/journal.pone.0016891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altmann K.S., Havemeyer A., Beitz E., Clement B. Dimethylarginine-dimethylaminohydrolase-2 (DDAH-2) does not metabolize methylarginines. Chembiochem. 2012;13:2599–2604. doi: 10.1002/cbic.201200499. [DOI] [PubMed] [Google Scholar]
- 8.Onozato M.L., Tojo A., Leiper J., Fujita T., Palm F., Wilcox C.S. Expression of ng,ng-dimethylarginine dimethylaminohydrolase and protein arginine n-methyltransferase isoforms in diabetic rat kidney: Effects of angiotensin ii receptor blockers. Diabetes. 2008;57:172–180. doi: 10.2337/db06-1772. [DOI] [PubMed] [Google Scholar]
- 9.Tomlinson J.A.P., Caplin B., Boruc O., Bruce-Cobbold C., Cutillas P., Dormann D., Faull P., Grossman R.C., Khadayate S., Mas V.R., et al. Reduced renal methylarginine metabolism protects against progressive kidney damage. J. Am. Soc. Nephrol. 2015;26:3045–3059. doi: 10.1681/ASN.2014030280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo Z., Aslam S., Welch W.J., Wilcox C.S. Activation of NRF2 coordinates DDAH/PPAR-γ/ENOS pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension. 2015;65:896–902. doi: 10.1161/HYPERTENSIONAHA.114.04760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu H., Qu X., Liang Z., Chen W., Xia W., Song Y. Variance of DDAH/PRMT/ADMA pathway in atrial fibrillation dogs. Biochem. Biophys. Res. Commun. 2008;377:884–888. doi: 10.1016/j.bbrc.2008.10.080. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y., Park S., Li Y., Missov E., Hou M., Han X., Hall J.L., Miller L.W., Bache R.J. Alterations of gene expression in failing myocardium following left ventricular assist device support. Physiol. Genom. 2003;14:251–260. doi: 10.1152/physiolgenomics.00022.2003. [DOI] [PubMed] [Google Scholar]
- 13.Zheng J., Wang K., Jin P., Dong C., Yuan Q., Li Y., Yang Z. The association of adipose-derived dimethylarginine dimethylaminohydrolase-2 with insulin sensitivity in experimental type 2 diabetes mellitus. Acta Biochim. Biophys. Sin. 2013;45:641–648. doi: 10.1093/abbs/gmt058. [DOI] [PubMed] [Google Scholar]
- 14.Ayling L.J., Whitley G.S.J., Aplin J.D., Cartwright J.E. Dimethylarginine dimethylaminohydrolase (DDAH) regulates trophoblast invasion and motility through effects on nitric oxide. Hum. Reprod. 2006;21:2530–2537. doi: 10.1093/humrep/del111. [DOI] [PubMed] [Google Scholar]
- 15.Hu T., Chouinard M., Cox A.L., Sipes P., Marcelo M., Ficorilli J., Li S., Gao H., Ryan T.P., Michael M.D., et al. Farnesoid x receptor agonist reduces serum asymmetric dimethylarginine levels through hepatic dimethylarginine dimethylaminohydrolase-1 gene regulation. J. Biol. Chem. 2006;281:39831–39838. doi: 10.1074/jbc.M606779200. [DOI] [PubMed] [Google Scholar]
- 16.Wang J., Sim A.S., Wang X.L., Wilcken D.E. l-Arginine regulates asymmetric dimethylarginine metabolism by inhibiting dimethylarginine dimethylaminohydrolase activity in hepatic (HEPG2) cells. Cell. Mol. Life Sci. 2006;63:2838–2846. doi: 10.1007/s00018-006-6271-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishima T., Hamada T., Ui-Tei K., Takahashi F., Miyata Y., Imaki J., Suzuki H., Yamashita K. Expression of DDAH1 in chick and rat embryos. Dev. Brain Res. 2004;148:223–232. doi: 10.1016/j.devbrainres.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 18.Matsuguma K., Ueda S., Yamagishi S.-I., Matsumoto Y., Kaneyuki U., Shibata R., Fujimura T., Matsuoka H., Kimoto M., Kato S., et al. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J. Am. Soc. Nephrol. 2006;17:2176–2183. doi: 10.1681/ASN.2005121379. [DOI] [PubMed] [Google Scholar]
- 19.Leiper J.M., Santa Maria J., Chubb A., MacAllister R.J., Charles I.G., Whitley G.S., Vallance P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J. 1999;343:209–214. doi: 10.1042/bj3430209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokuo H., Yunoue S., Feng L., Kimoto M., Tsuji H., Ono T., Saya H., Araki N. Phosphorylation of neurofibromin by camp-dependent protein kinase is regulated via a cellular association of ng,ng-dimethylarginine dimethylaminohydrolase. FEBS Lett. 2001;494:48–53. doi: 10.1016/S0014-5793(01)02309-2. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa K., Wakino S., Tanaka T., Kimoto M., Tatematsu S., Kanda T., Yoshioka K., Homma K., Sugano N., Kurabayashi M., et al. Dimethylarginine dimethylaminohydrolase 2 increases vascular endothelial growth factor expression through sp1 transcription factor in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006;26:1488–1494. doi: 10.1161/01.ATV.0000219615.88323.b4. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P., Hu X., Xu X., Chen Y., Bache R.J. Dimethylarginine dimethylaminohydrolase 1 modulates endothelial cell growth through nitric oxide and akt. Arterioscler. Thromb. Vasc. Biol. 2011;31:890–897. doi: 10.1161/ATVBAHA.110.215640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyake M., Kakimoto Y. Synthesis and degradation of methylated proteins of mouse organs: Correlation with protein synthesis and degradation. Metab. Clin. Exp. 1976;25:885–896. doi: 10.1016/0026-0495(76)90121-9. [DOI] [PubMed] [Google Scholar]
- 24.Teerlink T. Adma metabolism and clearance. Vasc. Med. 2005;10(Suppl. S1):S73–S81. doi: 10.1177/1358836X0501000111. [DOI] [PubMed] [Google Scholar]
- 25.Olken N.M., Rusche K.M., Richards M.K., Marletta M.A. Inactivation of macrophage nitric oxide synthase activity by NG-Methyl-l-arginine. Biochem. Biophys. Res. Commun. 1991;177:828–833. doi: 10.1016/0006-291X(91)91864-9. [DOI] [PubMed] [Google Scholar]
- 26.Palmer R.M.J., Rees D.D., Ashton D.S., Moncada S. l-Arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 1988;153:1251–1256. doi: 10.1016/S0006-291X(88)81362-7. [DOI] [PubMed] [Google Scholar]
- 27.Hibbs J.B., Jr., Taintor R.R., Vavrin Z. Macrophage cytotoxicity: Role for l-arginine deiminase and imino nitrogen oxidation to nitrite. Science. 1987;235:473–476. doi: 10.1126/science.2432665. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H.Q., Fast W., Marletta M.A., Martasek P., Silverman R.B. Potent and selective inhibition of neuronal nitric oxide synthase by N omega-propyl-l-arginine. J. Med. Chem. 1997;40:3869–3870. doi: 10.1021/jm970550g. [DOI] [PubMed] [Google Scholar]
- 29.Vallance P., Leone A., Calver A., Collier J., Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 30.Cardounel A.J., Zweier J.L. Endogenous methylarginines regulate neuronal nitric-oxide synthase and prevent excitotoxic injury. J. Biol. Chem. 2002;277:33995–34002. doi: 10.1074/jbc.M108983200. [DOI] [PubMed] [Google Scholar]
- 31.Closs E.I., Basha F.Z., Habermeier A., Förstermann U. Interference of l-Arginine analogues with l-Arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide. 1997;1:65–73. doi: 10.1006/niox.1996.0106. [DOI] [PubMed] [Google Scholar]
- 32.Ogawa T., Kimoto M., Watanabe H., Sasaoka K. Metabolism of ng,ng- and ng,ng-dimethylarginine in rats. Arch. Biochem. Biophys. 1987;252:526–537. doi: 10.1016/0003-9861(87)90060-9. [DOI] [PubMed] [Google Scholar]
- 33.Rodionov R.N., Murry D.J., Vaulman S.F., Stevens J.W., Lentz S.R. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J. Biol. Chem. 2010;285:5385–5391. doi: 10.1074/jbc.M109.091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caplin B., Wang Z., Slaviero A., Tomlinson J., Dowsett L., Delahaye M., Salama A., Wheeler D.C., Leiper J. Alanine-glyoxylate aminotransferase-2 metabolizes endogenous methylarginines, regulates no, and controls blood pressure. Arterioscler. Thromb. Vasc. Biol. 2012;32:2892–2900. doi: 10.1161/ATVBAHA.112.254078. [DOI] [PubMed] [Google Scholar]
- 35.Ogawa T., Kimoto M., Sasaoka K. Occurrence of a new enzyme catalyzing the direct conversion of ng,ng-dimethyl-l-arginine to l-citrulline in rats. Biochem. Biophys. Res. Commun. 1987;148:671–677. doi: 10.1016/0006-291X(87)90929-6. [DOI] [PubMed] [Google Scholar]
- 36.Leiper J., Nandi M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nat. Rev. Drug Discov. 2011;10:277–291. doi: 10.1038/nrd3358. [DOI] [PubMed] [Google Scholar]
- 37.Zoccali C., Bode-Böger S.M., Mallamaci F., Benedetto F.A., Tripepi G., Malatino L.S., Cataliotti A., Bellanuova I., Fermo I., Frölich J.C., et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: A prospective study. Lancet. 2001;358:2113–2117. doi: 10.1016/S0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- 38.Arrigoni F.I. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation. 2003;107:1195–1201. doi: 10.1161/01.CIR.0000051466.00227.13. [DOI] [PubMed] [Google Scholar]
- 39.Alpoim P.N., Godoi L.C., Freitas L.G., Gomes K.B., Dusse L.M. Assessment of l-arginine asymmetric 1 dimethyl (ADMA) in early-onset and late-onset (severe) preeclampsia. Nitric Oxide. 2013;33:81–82. doi: 10.1016/j.niox.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Anderssohn M., Maass L.M., Diemert A., Luneburg N., Atzler D., Hecher K., Boger R.H. Severely decreased activity of placental dimethylarginine dimethylaminohydrolase in pre-eclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2012;161:152–156. doi: 10.1016/j.ejogrb.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 41.Sydow K., Mondon C.E., Schrader J., Konishi H., Cooke J.P. Dimethylarginine dimethylaminohydrolase overexpression enhances insulin sensitivity. Arterioscler. Thromb. Vasc. Biol. 2008;28:692–697. doi: 10.1161/ATVBAHA.108.162073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacobi J., Sydow K., von Degenfeld G., Zhang Y., Dayoub H., Wang B., Patterson A.J., Kimoto M., Blau H.M., Cooke J.P. Overexpression of dimethylarginine dimethylaminohydrolase reduces tissue asymmetric dimethylarginine levels and enhances angiogenesis. Circulation. 2005;111:1431–1438. doi: 10.1161/01.CIR.0000158487.80483.09. [DOI] [PubMed] [Google Scholar]
- 43.Ljubisavljevic S., Stojanovic I., Pavlovic R., Sokolovic D., Pavlovic D., Cvetkovic T., Stevanovic I. Modulation of nitric oxide synthase by arginase and methylated arginines during the acute phase of experimental multiple sclerosis. J. Neurol. Sci. 2012;318:106–111. doi: 10.1016/j.jns.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 44.Lambden S., Kelly P., Ahmetaj-Shala B., Wang Z., Lee B., Nandi M., Torondel B., Delahaye M., Dowsett L., Piper S., et al. Dimethylarginine dimethylaminohydrolase 2 regulates nitric oxide synthesis and hemodynamics and determines outcome in polymicrobial sepsis. Arterioscler. Thromb. Vasc. Biol. 2015;35:1382–1392. doi: 10.1161/ATVBAHA.115.305278. [DOI] [PubMed] [Google Scholar]
- 45.Schulze F., Lenzen H., Hanefeld C., Bartling A., Osterziel K.J., Goudeva L., Schmidt-Lucke C., Kusus M., Maas R., Schwedhelm E., et al. Asymmetric dimethylarginine is an independent risk factor for coronary heart disease: Results from the multicenter coronary artery risk determination investigating the influence of adma concentration (cardiac) study. Am. Heart J. 2006;152:493.e491–493.e498. doi: 10.1016/j.ahj.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Maas R., Schulze F., Baumert J., Lowel H., Hamraz K., Schwedhelm E., Koenig W., Boger R.H. Asymmetric dimethylarginine, smoking, and risk of coronary heart disease in apparently healthy men: Prospective analysis from the population-based monitoring of trends and determinants in cardiovascular disease/kooperative gesundheitsforschung in der region augsburg study and experimental data. Clin. Chem. 2007;53:693–701. doi: 10.1373/clinchem.2006.081893. [DOI] [PubMed] [Google Scholar]
- 47.Duckelmann C., Mittermayer F., Haider D.G., Altenberger J., Eichinger J., Wolzt M. Asymmetric dimethylarginine enhances cardiovascular risk prediction in patients with chronic heart failure. Arterioscler. Thromb. Vasc. Biol. 2007;27:2037–2042. doi: 10.1161/ATVBAHA.107.147595. [DOI] [PubMed] [Google Scholar]
- 48.Miyazaki H., Matsuoka H., Cooke J.P., Usui M., Ueda S., Okuda S., Imaizumi T. Endogenous nitric oxide synthase inhibitor : A novel marker of atherosclerosis. Circulation. 1999;99:1141–1146. doi: 10.1161/01.CIR.99.9.1141. [DOI] [PubMed] [Google Scholar]
- 49.Wanby P., Teerlink T., Brudin L., Brattström L., Nilsson I., Palmqvist P., Carlsson M. Asymmetric dimethylarginine (ADMA) as a risk marker for stroke and tia in a swedish population. Atherosclerosis. 2006;185:271–277. doi: 10.1016/j.atherosclerosis.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 50.Chen S., Li N., Deb-Chatterji M., Dong Q., Kielstein J.T., Weissenborn K., Worthmann H. Asymmetric dimethyarginine as marker and mediator in ischemic stroke. Int. J. Mol. Sci. 2012;13:15983–16004. doi: 10.3390/ijms131215983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Isobe C., Abe T., Terayama Y. Decrease in asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in cerebrospinal fluid during elderly aging and in patients with sporadic form of amyotrophic lateral sclerosis. Neuro Signals. 2010;18:43–48. doi: 10.1159/000312527. [DOI] [PubMed] [Google Scholar]
- 52.Arlt S., Schulze F., Eichenlaub M., Maas R., Lehmbeck J.T., Schwedhelm E., Jahn H., Boger R.H. Asymmetrical dimethylarginine is increased in plasma and decreased in cerebrospinal fluid of patients with Alzheimer’s disease. Dement. Geriatr. Cognit. Disord. 2008;26:58–64. doi: 10.1159/000144026. [DOI] [PubMed] [Google Scholar]
- 53.Abe T., Tohgi H., Murata T., Isobe C., Sato C. Reduction in asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in the cerebrospinal fluid during aging and in patients with Alzheimer’s disease. Neurosci. Lett. 2001;312:177–179. doi: 10.1016/S0304-3940(01)02214-5. [DOI] [PubMed] [Google Scholar]
- 54.Tang X.Q., Fang H.R., Li Y.J., Zhou C.F., Ren Y.K., Chen R.Q., Wang C.Y., Hu B. Endogenous hydrogen sulfide is involved in asymmetric dimethylarginine-induced protection against neurotoxicity of 1-methyl-4-phenyl-pyridinium ion. Neurochem. Res. 2011;36:2176–2185. doi: 10.1007/s11064-011-0542-y. [DOI] [PubMed] [Google Scholar]
- 55.Kostourou V., Robinson S.P., Whitley G.S., Griffiths J.R. Effects of overexpression of dimethylarginine dimethylaminohydrolase on tumor angiogenesis assessed by susceptibility magnetic resonance imaging. Cancer Res. 2003;63:4960–4966. [PubMed] [Google Scholar]
- 56.Kostourou V., Robinson S.P., Cartwright J.E., Whitley G.S.J. Dimethylarginine dimethylaminohydrolase i enhances tumour growth and angiogenesis. Br. J. Cancer. 2002;87:673–680. doi: 10.1038/sj.bjc.6600518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu L.B., Dong X.S., Sun W.Z., Zhao D.L., Yang Y. Effect of a nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester on invasion of human colorectal cancer cell line SL-174T. World J. Gastroenterol. 2005;11:6385–6388. doi: 10.3748/wjg.v11.i40.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Punathil T., Katiyar S.K. Inhibition of non-small cell lung cancer cell migration by grape seed proanthocyanidins is mediated through the inhibition of nitric oxide, guanylate cyclase, and ERK1/2. Mol. Carcinog. 2009;48:232–242. doi: 10.1002/mc.20473. [DOI] [PubMed] [Google Scholar]
- 59.Vanella L., di Giacomo C., Acquaviva R., Santangelo R., Cardile V., Barbagallo I., Abraham N.G., Sorrenti V. The DDAH/NOS pathway in human prostatic cancer cell lines: Antiangiogenic effect of L-name. Int. J. Oncol. 2011;39:1303–1310. doi: 10.3892/ijo.2011.1107. [DOI] [PubMed] [Google Scholar]
- 60.Fiedler L.R., Bachetti T., Leiper J., Zachary I., Chen L., Renne T., Wojciak-Stothard B. The ADMA/DDAH pathway regulates vegf-mediated angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009;29:2117–2124. doi: 10.1161/ATVBAHA.109.194035. [DOI] [PubMed] [Google Scholar]
- 61.Ghebremariam Y.T., Erlanson D.A., Cooke J.P. A novel and potent inhibitor of dimethylarginine dimethylaminohydrolase: A modulator of cardiovascular nitric oxide. J. Pharmacol. Exp. Ther. 2014;348:69–76. doi: 10.1124/jpet.113.206847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schumer W. Pathophysiology and treatment of septic shock. Am. J. Emerg. Med. 1984;2:74–77. doi: 10.1016/0735-6757(84)90112-8. [DOI] [PubMed] [Google Scholar]
- 63.Thiemermann C. Nitric oxide and septic shock. Gen. Pharmacol. Vasc. Syst. 1997;29:159–166. doi: 10.1016/S0306-3623(96)00410-7. [DOI] [PubMed] [Google Scholar]
- 64.Kilbourn R.G., Traber D.L., Szabó C. Nitric oxide and shock. Dis. Mon. 1997;43:279–348. doi: 10.1016/S0011-5029(97)90028-6. [DOI] [PubMed] [Google Scholar]
- 65.Titheradge M.A. Nitric oxide in septic shock. Biochim. Biophys. Acta Bioenerg. 1999;1411:437–455. doi: 10.1016/S0005-2728(99)00031-6. [DOI] [PubMed] [Google Scholar]
- 66.Lopez A., Lorente J.A., Steingrub J., Bakker J., McLuckie A., Willatts S., Brockway M., Anzueto A., Holzapfel L., Breen D., et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: Effect on survival in patients with septic shock. Crit. Care Med. 2004;32:21–30. doi: 10.1097/01.CCM.0000105581.01815.C6. [DOI] [PubMed] [Google Scholar]
- 67.Wang Z., Lambden S., Taylor V., Sujkovic E., Nandi M., Tomlinson J., Dyson A., McDonald N., Caddick S., Singer M., et al. Pharmacological inhibition of ddah1 improves survival, haemodynamics and organ function in experimental septic shock. Biochem. J. 2014;460:309–316. doi: 10.1042/BJ20131666. [DOI] [PubMed] [Google Scholar]
- 68.Wadham C., Mangoni A.A. Dimethylarginine dimethylaminohydrolase regulation: A novel therapeutic target in cardiovascular disease. Expert Opin. Drug Metab. Toxicol. 2009;5:303–319. doi: 10.1517/17425250902785172. [DOI] [PubMed] [Google Scholar]
- 69.Knipp M., Charnock J.M., Garner C.D., Vasak M. Structural and functional characterization of the zn(ii) site in dimethylargininase-1 (DDAH-1) from bovine brain. Zn(ii) release activates ddah-1. J. Biol. Chem. 2001;276:40449–40456. doi: 10.1074/jbc.M104056200. [DOI] [PubMed] [Google Scholar]
- 70.Murray-Rust J., Leiper J., McAlister M., Phelan J., Tilley S., Santa Maria J., Vallance P., McDonald N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat. Struct. Biol. 2001;8:679–683. doi: 10.1038/90387. [DOI] [PubMed] [Google Scholar]
- 71.Bush A.I., Multhaup G., Moir R.D., Williamson T.G., Small D.H., Rumble B., Pollwein P., Beyreuther K., Masters C.L. A novel zinc (ii) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer’s disease. J. Biol. Chem. 1993;268:16109–16112. [PubMed] [Google Scholar]
- 72.Aizenman E., Stout A.K., Hartnett K.A., Dineley K.E., McLaughlin B., Reynolds I.J. Induction of neuronal apoptosis by thiol oxidation: Putative role of intracellular zinc release. J. Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- 73.Bush A.I., Pettingell W.H., Multhaup G., Paradis M.D., Vonsattel J.P., Gusella J.F., Beyreuther K., Masters C.L., Tanzi R.E. Rapid induction of Alzheimer a beta amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 74.Palm F., Onozato M.L., Luo Z., Wilcox C.S. Dimethylarginine dimethylaminohydrolase (DDAH): Expression, regulation, and function in the cardiovascular and renal systems. Am. J. Physiol. Heart Circ. Physiol. 2007;293:H3227–H3245. doi: 10.1152/ajpheart.00998.2007. [DOI] [PubMed] [Google Scholar]
- 75.Leiper J., Murray-Rust J., McDonald N., Vallance P. S-Nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: Further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc. Natl. Acad. Sci. USA. 2002;99:13527–13532. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Osanai T., Saitoh M., Sasaki S., Tomita H., Matsunaga T., Okumura K. Effect of shear stress on asymmetric dimethylarginine release from vascular endothelial cells. Hypertension. 2003;42:985–990. doi: 10.1161/01.HYP.0000097805.05108.16. [DOI] [PubMed] [Google Scholar]
- 77.Stuhlinger M.C., Tsao P.S., Her J.H., Kimoto M., Balint R.F., Cooke J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation. 2001;104:2569–2575. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- 78.Jiang J.L., Zhang X.H., Li N.S., Rang W.Q., Feng Y., Hu C.P., Li Y.J., Deng H.W. Probucol decreases asymmetrical dimethylarginine level by alternation of protein arginine methyltransferase I and dimethylarginine dimethylaminohydrolase activity. Cardiovasc. Drugs Ther. 2006;20:281–294. doi: 10.1007/s10557-006-9065-1. [DOI] [PubMed] [Google Scholar]
- 79.Holden D.P., Cartwright J.E., Nussey S.S., Whitley G.S.J. Estrogen stimulates dimethylarginine dimethylaminohydrolase activity and the metabolism of asymmetric dimethylarginine. Circulation. 2003;108:1575–1580. doi: 10.1161/01.CIR.0000091083.61609.DF. [DOI] [PubMed] [Google Scholar]
- 80.Ueda S., Kato S., Matsuoka H., Kimoto M., Okuda S., Morimatsu M., Imaizumi T. Regulation of cytokine-induced nitric oxide synthesis by asymmetric dimethylarginine: Role of dimethylarginine dimethylaminohydrolase. Circ. Res. 2003;92:226–233. doi: 10.1161/01.RES.0000052990.68216.EF. [DOI] [PubMed] [Google Scholar]
- 81.Jiang D.J., Jia S.J., Yan J., Zhou Z., Yuan Q., Li Y.J. Involvement of DDAH/ADMA/NOS pathway in nicotine-induced endothelial dysfunction. Biochem. Biophys. Res. Commun. 2006;349:683–693. doi: 10.1016/j.bbrc.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 82.Staab E.B., Weigel J., Xiao F., Madayiputhiya N., Wyatt T.A., Wells S.M. Asymmetric dimethyl-arginine metabolism in a murine model of cigarette smoke-mediated lung inflammation. J. Immunotoxicol. 2015;12:273–282. doi: 10.3109/1547691X.2014.961619. [DOI] [PubMed] [Google Scholar]
- 83.Tomikawa J., Fukatsu K., Tanaka S., Shiota K. DNA methylation-dependent epigenetic regulation of dimethylarginine dimethylaminohydrolase 2 gene in trophoblast cell lineage. J. Biol. Chem. 2006;281:12163–12169. doi: 10.1074/jbc.M513782200. [DOI] [PubMed] [Google Scholar]
- 84.Nishiyama Y., Ueda M., Otsuka T., Katsura K.-I., Abe A., Nagayama H., Katayama Y. Statin treatment decreased serum asymmetric dimethylarginine (ADMA) levels in ischemic stroke patients. J. Atheroscler. Thromb. 2011;18:131–137. doi: 10.5551/jat.5553. [DOI] [PubMed] [Google Scholar]
- 85.Serban C., Sahebkar A., Ursoniu S., Mikhailidis D.P., Rizzo M., Lip G.Y.H., Kees Hovingh G., Kastelein J.J.P., Kalinowski L., Rysz J., et al. A systematic review and meta-analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Sci. Rep. 2015;5 doi: 10.1038/srep09902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu T.M., Ding Y.A., Leu H.B., Yin W.H., Sheu W.H., Chu K.M. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am. J. Cardiol. 2004;94:157–161. doi: 10.1016/j.amjcard.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 87.Ivashchenko C.Y., Bradley B.T., Ao Z., Leiper J., Vallance P., Johns D.G. Regulation of the ADMA-DDAH system in endothelial cells: A novel mechanism for the sterol response element binding proteins, SREBP1c and -2. Am. J. Physiol. Heart Circ. Physiol. 2010;298:H251–H258. doi: 10.1152/ajpheart.00195.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen P., Xia K., Zhao Z., Deng X., Yang T. Atorvastatin modulates the DDAH1/ADMA system in high-fat diet-induced insulin-resistant rats with endothelial dysfunction. Vasc. Med. 2012;17:416–423. doi: 10.1177/1358863X12467492. [DOI] [PubMed] [Google Scholar]
- 89.Yin Q.-F., Xiong Y. Pravastatin restores ddah activity and endothelium-dependent relaxation of rat aorta after exposure to glycated protein. J. Cardiovasc. Pharmacol. 2005;45:525–532. doi: 10.1097/01.fjc.0000159642.44523.7f. [DOI] [PubMed] [Google Scholar]
- 90.Li J., Wilson A., Gao X., Kuruba R., Liu Y., Poloyac S., Pitt B., Xie W., Li S. Coordinated regulation of dimethylarginine dimethylaminohydrolase-1 and cationic amino acid transporter-1 by farnesoid x receptor in mouse liver and kidney and its implication in the control of blood levels of asymmetric dimethylarginine. J. Pharmacol. Exp. Ther. 2009;331:234–243. doi: 10.1124/jpet.109.153510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ghebremariam Y.T., Yamada K., Lee J.C., Johnson C.L.C., Atzler D., Anderssohn M., Agrawal R., Higgins J.P., Patterson A.J., Böger R.H., et al. FXR agonist INT-747 upregulates ddah expression and enhances insulin sensitivity in high-salt fed Dahl rats. PLoS ONE. 2013;8:e60653. doi: 10.1371/journal.pone.0060653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vairappan B., Sharma V., Davies N., Jalan R., Mookherjee R. Activation of farnesoid x receptor (FXR) by its agonist INT-747 restores hepatic endothelial nos activity and lowers portal pressure in cirrhotic rats by modulating the DDAH-ADMA pathway. J. Clin. Exp. Hepatol. 2013;3:S11. doi: 10.1016/j.jceh.2013.03.025. [DOI] [Google Scholar]
- 93.Jiang J.L., Li N.S., Li Y.J., Deng H.W. Probucol preserves endothelial function by reduction of the endogenous nitric oxide synthase inhibitor level. Br. J. Pharmacol. 2002;135:1175–1182. doi: 10.1038/sj.bjp.0704563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang K.-Q., Chen D., Sun D.-Q., Zhang H., Li B., Fu Q. Probucol improves erectile function by restoring endothelial function and preventing cavernous fibrosis in streptozotocin-induced diabetic rats. Urology. 2016;91:241.e9–241.e16. doi: 10.1016/j.urology.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 95.Yang T.-L., Chen M.-F., Luo B.-L., Xie Q.-Y., Jiang J.-L., Li Y.-J. Fenofibrate decreases asymmetric dimethylarginine level in cultured endothelial cells by inhibiting NF-κB activity. Naunyn-Schmiedeberg's Arch. Pharmacol. 2005;371:401–407. doi: 10.1007/s00210-005-1060-8. [DOI] [PubMed] [Google Scholar]
- 96.Wakino S., Hayashi K., Tatematsu S., Hasegawa K., Takamatsu I., Kanda T., Homma K., Yoshioka K., Sugano N., Saruta T. Pioglitazone lowers systemic asymmetric dimethylarginine by inducing dimethylarginine dimethylaminohydrolase in rats. Hypertens. Res. 2005;28:255–262. doi: 10.1291/hypres.28.255. [DOI] [PubMed] [Google Scholar]
- 97.Garbin U., Pasini A.F., Stranieri C., Manfro S., Boccioletti V., Cominacini L. Nebivolol reduces asymmetric dimethylarginine in endothelial cells by increasing dimethylarginine dimethylaminohydrolase 2 (DDAH2) expression and activity. Pharmacol. Res. 2007;56:515–521. doi: 10.1016/j.phrs.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 98.Wang Y., Zhang M., Liu Y., Liu Y., Chen M. The effect of nebivolol on asymmetric dimethylarginine system in spontaneously hypertension rats. Vasc. Pharmacol. 2011;54:36–43. doi: 10.1016/j.vph.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 99.Pasini A.F., Garbin U., Stranieri C., Boccioletti V., Mozzini C., Manfro S., Pasini A., Cominacini M., Cominacini L. Nebivolol treatment reduces serum levels of asymmetric dimethylarginine and improves endothelial dysfunction in essential hypertensive patients. Am. J. Hypertens. 2008;21:1251–1257. doi: 10.1038/ajh.2008.260. [DOI] [PubMed] [Google Scholar]
- 100.Knipp M., Braun O., Gehrig P.M., Sack R., Vasak M. Zn(II)-free dimethylargininase-1 (DDAH-1) is inhibited upon specific Cys-S-nitrosylation. J. Biol. Chem. 2003;278:3410–3416. doi: 10.1074/jbc.M209088200. [DOI] [PubMed] [Google Scholar]
- 101.Tyagi N., Sedoris K.C., Steed M., Ovechkin A.V., Moshal K.S., Tyagi S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H2649–H2656. doi: 10.1152/ajpheart.00548.2005. [DOI] [PubMed] [Google Scholar]
- 102.Knipp M., Braun O., Vasak M. Searching for ddah inhibitors: S-nitroso-l-homocysteine is a chemical lead. J. Am. Chem. Soc. 2005;127:2372–2373. doi: 10.1021/ja0430200. [DOI] [PubMed] [Google Scholar]
- 103.Duerre J.A., Miller C.H. Preparation of l-homocysteine from l-homocysteine thiolactone. Anal. Biochem. 1966;17:310–315. doi: 10.1016/0003-2697(66)90209-0. [DOI] [PubMed] [Google Scholar]
- 104.Feelisch M., Stamler J. Methods in Nitric Oxide Research. 36. Preparation and Detection of S-Nitrosothiols. John Wiley & Sons Ltd; Hoboken, NJ, USA: 1996. pp. 521–539. [Google Scholar]
- 105.Braun O., Knipp M., Chesnov S., Vasak M. Specific reactions of S-nitrosothiols with cysteine hydrolases: A comparative study between dimethylargininase-1 and ctp synthetase. Protein Sci. 2007;16:1522–1534. doi: 10.1110/ps.062718507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stone E.M., Schaller T.H., Bianchi H., Person M.D., Fast W. Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: Dimethylargininase and peptidylarginine deiminase. Biochemistry. 2005;44:13744–13752. doi: 10.1021/bi051341y. [DOI] [PubMed] [Google Scholar]
- 107.Wharton C.J., Wrigglesworth R. Inhibitors of pyrimidine biosynthesis. Part 2. The synthesis of amidine phosphonates as potential inhibitors of carbamoyl phosphate synthase. J. Chem. Soc. Perkin Trans. 1981;1:433–436. doi: 10.1039/p19810000433. [DOI] [Google Scholar]
- 108.Schaefer F.C., Peters G.A. Base-catalyzed reaction of nitriles with alcohols. A convenient route to imidates and amidine salts. J. Org. Chem. 1961;26:412–418. doi: 10.1021/jo01061a034. [DOI] [Google Scholar]
- 109.Wang Y., Hu S., Fast W. A click chemistry mediated in vivo activity probe for dimethylarginine dimethylaminohydrolase. J. Am. Chem. Soc. 2009;131:15096–15097. doi: 10.1021/ja906432e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.MacAllister R.J., Parry H., Kimoto M., Ogawa T., Russell R.J., Hodson H., Whitley G.S., Vallance P. Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br. J. Pharmacol. 1996;119:1533–1540. doi: 10.1111/j.1476-5381.1996.tb16069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Corbin J.L., Reporter M. Ng-methylated arginines; a convenient preparation of ng-methylarginine. Anal. Biochem. 1974;57:310–312. doi: 10.1016/0003-2697(74)90080-3. [DOI] [PubMed] [Google Scholar]
- 112.Rossiter S., Smith C.L., Malaki M., Nandi M., Gill H., Leiper J.M., Vallance P., Selwood D.L. Selective substrate-based inhibitors of mammalian dimethylarginine dimethylaminohydrolase. J. Med. Chem. 2005;48:4670–4678. doi: 10.1021/jm050187a. [DOI] [PubMed] [Google Scholar]
- 113.Kim H.-O., Mathew F., Ogbu C. A convenient synthesis of disubstituted guanidines via the mitsunobu protocol. Synlett. 1999;1999:193–194. doi: 10.1055/s-1999-2584. [DOI] [Google Scholar]
- 114.Kotthaus J., Schade D., Muschick N., Beitz E., Clement B. Structure-activity relationship of novel and known inhibitors of human dimethylarginine dimethylaminohydrolase-1: Alkenyl-amidines as new leads. Bioorg. Med. Chem. 2008;16:10205–10209. doi: 10.1016/j.bmc.2008.10.058. [DOI] [PubMed] [Google Scholar]
- 115.Kotthaus J., Schade D., Kotthaus J., Clement B. Designing modulators of dimethylarginine dimethylaminohydrolase (DDAH): A focus on selectivity over arginase. J. Enzyme Inhib. Med. Chem. 2012;27:24–28. doi: 10.3109/14756366.2011.573480. [DOI] [PubMed] [Google Scholar]
- 116.Stone E.M., Costello A.L., Tierney D.L., Fast W. Substrate-assisted cysteine deprotonation in the mechanism of dimethylargininase (DDAH) from pseudomonas aeruginosa. Biochemistry. 2006;45:5618–5630. doi: 10.1021/bi052595m. [DOI] [PubMed] [Google Scholar]
- 117.Tommasi S., Zanato C., Lewis B.C., Nair P.C., Dall’Angelo S., Zanda M., Mangoni A.A. Arginine analogues incorporating carboxylate bioisosteric functions are micromolar inhibitors of human recombinant DDAH-1. Org. Biomol. Chem. 2015;13:11315–11330. doi: 10.1039/C5OB01843A. [DOI] [PubMed] [Google Scholar]
- 118.Lima L.M., Barreiro E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
- 119.Yusuf A.O., Bhatt B.M., Gitu P.M. Solid-phase peptide synthesis of isotocin with amide of asparagine protected with 1-tetralinyl. Trifluoromethanesulphonic acid (TFMSA) deprotection, cleavage and air oxidation of mercapto groups to disulphide. Bull. Chem. Soc. Ethiop. 2001;15:143–149. [Google Scholar]
- 120.Demko Z.P., Sharpless K.B. Preparation of 5-substituted 1 H-tetrazoles from nitriles in water. J. Org. Chem. 2001;66:7945–7950. doi: 10.1021/jo010635w. [DOI] [PubMed] [Google Scholar]
- 121.Sureshbabu V.V., Naik S.A., Nagendra G. Synthesis of boc-amino tetrazoles derived from α-amino acids. Synth. Commun. 2009;39:395–406. doi: 10.1080/00397910802374133. [DOI] [Google Scholar]
- 122.Anderson D.W., Campbell M.M., Malik M. Tetrazolyl peptides and the tetrazole analogue of 3-aminonocardicinic acid. Tetrahedron Lett. 1990;31:1755–1758. doi: 10.1016/S0040-4039(00)88873-5. [DOI] [Google Scholar]
- 123.Keil S., Urmann M., Bernardelli P., Glien M., Wendler W., Chandross K., Lee L. Phenyl-1,2,4-Oxadiazolone Derivatives, Processes for Their Preparation and Methods for their Use as Pharmaceuticals. 7,709,481 B2. U.S. Patent. 2010 May 4;
- 124.Greenhill J.V., Lue P. Amidines and guanidines in medicinal chemistry. Prog. Med. Chem. 1993;30:203–326. doi: 10.1016/s0079-6468(08)70378-3. [DOI] [PubMed] [Google Scholar]
- 125.Babu B.R., Griffith O.W. N5-(1-imino-3-butenyl)-l-ornithine. A neuronal isoform selective mechanism-based inactivator of nitric oxide synthase. J. Biol. Chem. 1998;273:8882–8889. doi: 10.1074/jbc.273.15.8882. [DOI] [PubMed] [Google Scholar]
- 126.Bretscher L.E., Li H., Poulos T.L., Griffith O.W. Structural characterization and kinetics of nitric-oxide synthase inhibition by novel N5-(iminoalkyl)- and N5-(iminoalkenyl)-ornithines. J. Biol. Chem. 2003;278:46789–46797. doi: 10.1074/jbc.M306787200. [DOI] [PubMed] [Google Scholar]
- 127.Fast W., Nikolic D., van Breemen R.B., Silverman R.B. Mechanistic studies of the inactivation of inducible nitric oxide synthase by N5-(1-iminoethyl)-l-ornithine (L-NIO) J. Am. Chem. Soc. 1999;121:903–916. doi: 10.1021/ja982318l. [DOI] [Google Scholar]
- 128.Wang Y., Monzingo A.F., Hu S., Schaller T.H., Robertus J.D., Fast W. Developing dual and specific inhibitors of dimethylarginine dimethylaminohydrolase-1 and nitric oxide synthase: Toward a targeted polypharmacology to control nitric oxide. Biochemistry. 2009;48:8624–8635. doi: 10.1021/bi9007098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Y., Hu S., Gabisi A.M., Jr., Er J.A., Pope A., Burstein G., Schardon C.L., Cardounel A.J., Ekmekcioglu S., Fast W. Developing an irreversible inhibitor of human DDAH-1, an enzyme upregulated in melanoma. ChemMedChem. 2014;9:792–797. doi: 10.1002/cmdc.201300557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vallance P., Bush H.D., Mok B.J., Hurtado-Guerrero R., Gill H., Rossiter S., Wilden J.D., Caddick S. Inhibition of dimethylarginine dimethylaminohydrolase (DDAH) and arginine deiminase (ADI) by pentafluorophenyl (PFP) sulfonates. Chem. Commun. 2005:5563–5565. doi: 10.1039/b510709a. [DOI] [PubMed] [Google Scholar]
- 131.Caddick S., Bush H.D. Synthesis of functionalized sulfonamides via 1,3-dipolar cycloaddition of pentafluorophenyl vinylsulfonate. Org. Lett. 2003;5:2489–2492. doi: 10.1021/ol0347388. [DOI] [PubMed] [Google Scholar]
- 132.Caddick S., Wilden J.D., Bush H.D., Wadman S.N., Judd D.B. A new route to sulfonamides via intermolecular radical addition to pentafluorophenyl vinylsulfonate and subsequent aminolysis. Org. Lett. 2002;4:2549–2551. doi: 10.1021/ol026181m. [DOI] [PubMed] [Google Scholar]
- 133.Chan K.S., Yeung M.L., Chan W.-K., Wang R.-J., Mak T.C.W. Chromium and tungsten pentacarbonyl groups as reactivity and selectivity auxiliaries in [3 + 2] cycloaddition of alkynyl fischer carbene complexes with n-alkyl nitrones. J. Org. Chem. 1995;60:1741–1747. doi: 10.1021/jo00111a036. [DOI] [Google Scholar]
- 134.Baruah A.K., Prajapati D., Sandhu J. Some novel aspects of regioselectivity in 1,3 dipolar cycloadditions of 4h-1-benzopyran-4-thione. Tetrahedron. 1988;44:6137–6142. doi: 10.1016/S0040-4020(01)89803-3. [DOI] [Google Scholar]
- 135.Sims J., Houk K.N. Reversal of nitrone cycloaddition regioselectivity with electron-deficient dipolarophiles. J. Am. Chem. Soc. 1973;95:5798–5800. doi: 10.1021/ja00798a079. [DOI] [Google Scholar]
- 136.Bimanand A.Z., Houk K.N. The synthesis and cycloadditions of c-cyclopropyl nitrones. Tetrahedron Lett. 1983;24:435–438. doi: 10.1016/S0040-4039(00)81429-X. [DOI] [Google Scholar]
- 137.Padwa A., Fisera L., Koehler K.F., Rodriguez A., Wong G.S.K. Regioselectivity associated with the 1,3-dipolar cycloaddition of nitrones with electron-deficient dipolarophiles. J. Org. Chem. 1984;49:276–281. doi: 10.1021/jo00176a012. [DOI] [Google Scholar]
- 138.Black D.S.C., Crozier R.F., Davis V.C. 1,3-Dipolar cycloaddition reactions of nitrones. Synthesis. 1975;1975:205–221. doi: 10.1055/s-1975-23713. [DOI] [Google Scholar]
- 139.Hartzoulakis B., Rossiter S., Gill H., O'Hara B., Steinke E., Gane P.J., Hurtado-Guerrero R., Leiper J.M., Vallance P., Rust J.M., et al. Discovery of inhibitors of the pentein superfamily protein dimethylarginine dimethylaminohydrolase (DDAH), by virtual screening and hit analysis. Bioorg. Med. Chem. Lett. 2007;17:3953–3956. doi: 10.1016/j.bmcl.2007.04.095. [DOI] [PubMed] [Google Scholar]
- 140.Fischer E., von Mering J. Ueber eine neue klasse von schlafmitteln. Ther. Ggw. 1903;44:97–101. [Google Scholar]
- 141.Nightingale D., Alexander C.H. Some nitrogen substituted barbituric acids and their derivatives. J. Am. Chem. Soc. 1936;58:794–796. doi: 10.1021/ja01296a033. [DOI] [Google Scholar]
- 142.Zhao R., Wang B., Wu H., Hynes J., Jr., Leftheri K., Balasubramanian B., Barrish J.C., Chen B.-C. Improved procedure for the preparation of 7-methoxy-2-methyl-1-(2-morpholinoethyl)-1H-indole-3-carboxylic acid, key intermediate in the synthesis of novel 3-amidoindole and indolopyridone cannabinoid ligands. Arkivoc. 2009;2010:89–95. [Google Scholar]
- 143.Dox A.W., Plaisance G.P. Condensation of thiobarbituric acid with aromatic aldehydes. J. Am. Chem. Soc. 1916;38:2164–2166. doi: 10.1021/ja02267a027. [DOI] [Google Scholar]
- 144.Azad G.K., Tomar R.S. Ebselen, a promising antioxidant drug: Mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 2014;41:4865–4879. doi: 10.1007/s11033-014-3417-x. [DOI] [PubMed] [Google Scholar]
- 145.Linsky T., Wang Y., Fast W. Screening for dimethylarginine dimethylaminohydrolase inhibitors reveals ebselen as a bioavailable inactivator. ACS Med. Chem. Lett. 2011;2:592–596. doi: 10.1021/ml2000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bhabak K.P., Mugesh G. Synthesis, characterization, and antioxidant activity of some ebselen analogues. Chem. Eur. J. 2007;13:4594–4601. doi: 10.1002/chem.200601584. [DOI] [PubMed] [Google Scholar]
- 147.Engman L., Hallberg A. Expedient synthesis of ebselen and related compounds. J. Org. Chem. 1989;54:2964–2966. doi: 10.1021/jo00273a035. [DOI] [Google Scholar]
- 148.Chang T.C., Huang M.L., Hsu W.L., Hwang J.M., Hsu L.Y. Synthesis and biological evaluation of ebselen and its acyclic derivatives. Chem. Pharm. Bull. 2003;51:1413–1416. doi: 10.1248/cpb.51.1413. [DOI] [PubMed] [Google Scholar]
- 149.Schewe T. Molecular actions of ebselen—An antiinflammatory antioxidant. Gen. Pharmacol. Vasc. Syst. 1995;26:1153–1169. doi: 10.1016/0306-3623(95)00003-J. [DOI] [PubMed] [Google Scholar]
- 150.Smith S.M., Min J., Ganesh T., Diebold B., Kawahara T., Zhu Y., McCoy J., Sun A., Snyder J.P., Fu H., et al. Ebselen and congeners inhibit nadph oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem. Biol. 2012;19:752–763. doi: 10.1016/j.chembiol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cotgreave I.A., Duddy S.K., Kass G.E.N., Thompson D., Moldéus P. Studies on the anti-inflammatory activity of Ebselen: Ebselen interferes with granulocyte oxidative burst by dual inhibition of NADPH oxidase and protein kinase C? Biochem. Pharmacol. 1989;38:649–656. doi: 10.1016/0006-2952(89)90211-6. [DOI] [PubMed] [Google Scholar]
- 152.Beil W., Staar U., Sewing K.-F. Interaction of the anti-inflammatory seleno-organic compound ebselen with acid secretion in isolated parietal cells and gastric H+/K+-atpase. Biochem. Pharmacol. 1990;40:1997–2003. doi: 10.1016/0006-2952(90)90229-E. [DOI] [PubMed] [Google Scholar]
- 153.Walther M., Holzhutter H.G., Kuban R.J., Wiesner R., Rathmann J., Kuhn H. The inhibition of mammalian 15-lipoxygenases by the anti-inflammatory drug ebselen: Dual-type mechanism involving covalent linkage and alteration of the iron ligand sphere. Mol. Pharmacol. 1999;56:196–203. doi: 10.1124/mol.56.1.196. [DOI] [PubMed] [Google Scholar]
- 154.Zhao R., Masayasu H., Holmgren A. Ebselen: A substrate for human thioredoxin reductase strongly stimulating its hydroperoxide reductase activity and a superfast thioredoxin oxidant. Proc. Natl. Acad. Sci. USA. 2002;99:8579–8584. doi: 10.1073/pnas.122061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jacob C., Maret W., Vallee B.L. Ebselen, a selenium-containing redox drug, releases zinc from metallothionein. Biochem. Biophys. Res. Commun. 1998;248:569–573. doi: 10.1006/bbrc.1998.9026. [DOI] [PubMed] [Google Scholar]
- 156.Caeran Bueno D., Meinerz D.F., Allebrandt J., Waczuk E.P., dos Santos D.B., Mariano D.O., Rocha J.B. Cytotoxicity and genotoxicity evaluation of organochalcogens in human leucocytes: A comparative study between ebselen, diphenyl diselenide, and diphenyl ditelluride. BioMed Res. Int. 2013;2013 doi: 10.1155/2013/537279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Meotti F.C., Borges V.C., Zeni G., Rocha J.B.T., Nogueira C.W. Potential renal and hepatic toxicity of diphenyl diselenide, diphenyl ditelluride and ebselen for rats and mice. Toxicol. Lett. 2003;143:9–16. doi: 10.1016/S0378-4274(03)00090-0. [DOI] [PubMed] [Google Scholar]
- 158.Usatyuk P.V., Parinandi N.L., Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J. Biol. Chem. 2006;281:35554–35566. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- 159.Benedetti A., Comporti M., Esterbauer H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta Lipids Lipid Metab. 1980;620:281–296. doi: 10.1016/0005-2760(80)90209-X. [DOI] [PubMed] [Google Scholar]
- 160.Uchida K., Stadtman E.R. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc. Natl. Acad. Sci. USA. 1992;89:4544–4548. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Uchida K. Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 2000;28:1685–1696. doi: 10.1016/S0891-5849(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 162.Uchida K., Stadtman E.R. Selective cleavage of thioether linkage in proteins modified with 4-hydroxynonenal. Proc. Natl. Acad. Sci. USA. 1992;89:5611–5615. doi: 10.1073/pnas.89.12.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sayre L.M., Arora P.K., Iyer R.S., Salomon R.G. Pyrrole formation from 4-hydroxynonenal and primary amines. Chem. Res. Toxicol. 1993;6:19–22. doi: 10.1021/tx00031a002. [DOI] [PubMed] [Google Scholar]
- 164.Santaniello E., Repetto A., Chiesa L.M., Biondi P.A. Synthesis and characterization of 4-hydroxy-2-nonenal derivatives for gas chromatographic analysis with electron capture detection (GC-ECD) Redox Rep. Commun. Free Radic. Res. 2007;12:55–58. doi: 10.1179/135100007X162293. [DOI] [PubMed] [Google Scholar]
- 165.Chandra A., Srivastava S.K. A synthesis of 4-hydroxy-2-trans-nonenal and 4-(3H) 4-hydroxy-2-trans-nonenal. Lipids. 1997;32:779–782. doi: 10.1007/s11745-997-0100-6. [DOI] [PubMed] [Google Scholar]
- 166.Soulère L., Queneau Y., Doutheau A. An expeditious synthesis of 4-hydroxy-2(E)-nonenal (4-HNE), its dimethyl acetal and of related compounds. Chem. Phys. Lipids. 2007;150:239–243. doi: 10.1016/j.chemphyslip.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 167.Forbes S.P., Druhan L.J., Guzman J.E., Parinandi N., Zhang L., Green-Church K.B., Cardounel A.J. Mechanism of 4-HNE mediated inhibition of hddah-1: Implications in no regulation. Biochemistry. 2008;47:1819–1826. doi: 10.1021/bi701659n. [DOI] [PubMed] [Google Scholar]
- 168.Birck M.R., Holler T.P., Woodard R.W. Identification of a slow tight-binding inhibitor of 3-deoxy-d-manno-octulosonic acid 8-phosphate synthase. J. Am. Chem. Soc. 2000;122:9334–9335. doi: 10.1021/ja002142z. [DOI] [Google Scholar]
- 169.Kosa N.M., Foley T.L., Burkart M.D. Fluorescent techniques for discovery and characterization of phosphopantetheinyl transferase inhibitors. J. Antibiot. 2014;67:113–120. doi: 10.1038/ja.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chamoun A.M., Chockalingam K., Bobardt M., Simeon R., Chang J., Gallay P., Chen Z. Pd 404,182 is a virocidal small molecule that disrupts hepatitis c virus and human immunodeficiency virus. Antimicrob. Agents Chemother. 2012;56:672–681. doi: 10.1128/AAC.05722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chamoun-Emanuelli A.M., Bobardt M., Moncla B., Mankowski M.K., Ptak R.G., Gallay P., Chen Z. Evaluation of PD 404,182 as an anti-HIV and anti-Herpes simplex virus microbicide. Antimicrob. Agents Chemother. 2014;58:687–697. doi: 10.1128/AAC.02000-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hagen H., Fleig H. Neue synthesen mit elementarem schwefel. Darstellung von 2-(1,3-diaza-2-cycloalken-2-yl)thiophenolen und ihre umwandlung zu 1,4-benzothiazepinen und 1,3,4-benzo-thiadiazepinen. Justus Liebigs Ann. Chem. 1975;1975:1994–2005. doi: 10.1002/jlac.197519751110. [DOI] [Google Scholar]
- 173.Mizuhara T., Oishi S., Fujii N., Ohno H. Efficient synthesis of pyrimido[1,2-c] [1,3]benzothiazin-6-imines and related tricyclic heterocycles by SNAr-type C-S, C-N, or C-O bond formation with heterocumulenes. J. Org. Chem. 2010;75:265–268. doi: 10.1021/jo902327n. [DOI] [PubMed] [Google Scholar]
- 174.Ghebremariam Y.T., Erlanson D.A., Yamada K., Cooke J.P. Development of a dimethylarginine dimethylaminohydrolase (DDAH) assay for high-throughput chemical screening. J. Biomol. Screen. 2012;17:651–661. doi: 10.1177/1087057112441521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ghebremariam Y.T., LePendu P., Lee J.C., Erlanson D.A., Slaviero A., Shah N.H., Leiper J., Cooke J.P. Unexpected effect of proton pump inhibitors: Elevation of the cardiovascular risk factor asymmetric dimethylarginine. Circulation. 2013;128:845–853. doi: 10.1161/CIRCULATIONAHA.113.003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Charlot M., Grove E.L., Hansen P.R., Olesen J.B., Ahlehoff O., Selmer C., Lindhardsen J., Madsen J.K., Kober L., Torp-Pedersen C., et al. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: Nationwide propensity score matched study. Br. Med. J. 2011;342 doi: 10.1136/bmj.d2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Charlot M., Ahlehoff O., Norgaard M.L., Jorgensen C.H., Sorensen R., Abildstrom S.Z., Hansen P.R., Madsen J.K., Kober L., Torp-Pedersen C., et al. Proton-pump inhibitors are associated with increased cardiovascular risk independent of clopidogrel use: A nationwide cohort study. Ann. Intern. Med. 2010;153:378–386. doi: 10.7326/0003-4819-153-6-201009210-00005. [DOI] [PubMed] [Google Scholar]
- 178.Brandstrom A.E., Lamm B.R. Processes for the Preparation of Omeprazole and Intermediates Therefore. 4,620,008 A. U.S. Patent. 1986 Oct 28;
- 179.Crowe A.M., Ife R.J., Mitchell M.B., Saunders D. The preparation of 14C, 35S and 13C labelled forms of omeprazole. J. Label. Compd. Radiopharm. 1986;23:21–33. doi: 10.1002/jlcr.2580230104. [DOI] [Google Scholar]
- 180.Slemon C., Macel B. Preparation of Omeprazole and Lansoprazole, and Intermediates Useful Therein. No. 5,470,983. U.S. Patent. 1995 Nov 28;
- 181.Linsky T.W., Fast W. Discovery of structurally-diverse inhibitor scaffolds by high-throughput screening of a fragment library with dimethylarginine dimethylaminohydrolase. Bioorg. Med. Chem. 2012;20:5550–5558. doi: 10.1016/j.bmc.2012.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zornik D., Meudtner R.M., El Malah T., Thiele C.M., Hecht S. Designing structural motifs for clickamers: Exploiting the 1,2,3-triazole moiety to generate conformationally restricted molecular architectures. Chem. Eur. J. 2011;17:1473–1484. doi: 10.1002/chem.201002491. [DOI] [PubMed] [Google Scholar]
- 183.Gijsen H.J.M., Cleyn M.A.J.D., Surkyn M., Verbist B.M.P. Benzimidazole Cannabinoid Agonists Bearinga Substituted Heterocyclic Group. 8,524,757. U.S. Patent. 2013 Sep 3;
- 184.Thompson A.S., Humphrey G.R., DeMarco A.M., Mathre D.J., Grabowski E.J.J. Direct conversion of activated alcohols to azides using diphenyl phosphorazidate. A practical alternative to mitsunobu conditions. J. Org. Chem. 1993;58:5886–5888. doi: 10.1021/jo00074a008. [DOI] [Google Scholar]