

Abstract
Flow chemistry-based syntheses of deuterium-labeled analogs of important antidiabetic chalcones were achieved via highly controlled partial C≡C bond deuteration of the corresponding 1,3-diphenylalkynones. The benefits of a scalable continuous process in combination with on-demand electrolytic D2 gas generation were exploited to suppress undesired over-reactions and to maximize reaction rates simultaneously. The novel deuterium-containing chalcone derivatives may have interesting biological effects and improved metabolic properties as compared with the parent compounds.
Keywords: chalcones, continuous-flow reactor, deuterium labeling, heterogeneous catalysis, triple bond reduction
1. Introduction
Chalcones are 1,3-diaryl-2-propen-1-ones which belong to the flavonoid family [1,2,3]. Their naturally occurring representatives and synthetic analogs exert a wide array of pharmacological activities, including anticancer [4], antiviral [5], antibacterial [6], anti-inflammatory [7], antiplatelet [8] and antitubercular [9] effects (Figure 1), making the chalcone skeleton an important scaffold for drug discovery [10,11,12,13,14].
Figure 1.

Examples of chalcone derivatives with various biological effects: (a) antiplatelet activity [8]; (b) antibacterial activity [6]; (c) antitubercular activity [9]; (d) anti-inflammatory effect [7]; (e) antiviral activity [5]; (f) anticancer activity [4].
In recent decades, the metabolic syndrome and diabetes have become ever-increasing health issues worldwide [15]. The associated complications of these disorders, such as stroke, cardiovascular diseases, peripheral vascular diseases, diabetic neuropathy, renal failure, blindness and amputations, lead to reduced life expectancy, the enhancement of disability, and enormous medical costs [16,17,18]. Despite extensive biological studies on chalcones [10,11,12,13,14], their antidiabetic activities have scarcely been investigated [19,20,21]. Our research group has previously reported that certain synthetic chalcones (1‒4) containing various halogen atoms at position 2 of ring A (Figure 2) effectively promote glucose consumption in adipocytes [22]. Structure–activity relationship analyses have established that, besides the halogen substituents on ring A, the presence of a methoxy group on ring B contributes to the potent antidiabetic activities observed.
Figure 2.

Antidiabetic chalcone derivatives (1–4) developed by our research group.
There has recently been an upsurge of interest in isotopic labeling studies not merely in chemistry and biochemistry [23,24,25,26], but also in environmental sciences and pharmacological research [27,28]. Among the stable isotopes used for labeling studies, deuterium is of marked significance [24,25,26,27], thanks to the fact that it has the highest relative mass difference compared to its light isotope (1H), which allows a considerably large isotope effect [29]. Deuterium not only serves as a nonradioactive, stable isotopic tracer in the evaluation of the metabolic pathways [30] or in tracer studies to investigate pharmacokinetics and reaction mechanisms [31,32]; as a result of the large isotope effect, H‒D exchange may also potentiate active pharmaceutical ingredients [27]. As an example, the metabolic rates of nonlabeled derivatives and deuterated compounds are very different, and the use of deuterium-labeled analogs may therefore help in the elimination of unwanted metabolites, which can lead in practice to the diminution of adverse effects [33,34,35].
Inspired by the above findings, we aimed for the synthesis of deuterium-labeled derivatives of antidiabetic chalcones 1‒4 by exploring the partial deuteration of the C≡C bond of the corresponding 1,3-diphenylalkynone analogs (Scheme 1). We presumed that the incorporation of deuterium onto the α,β-unsaturated carbonyl system may not only help potentiate the pharmacological activities, but also exert positive effects on the metabolic pathway of the bioactive chalcones [27]. Selective deuteration of the alkynone core may be expected to constitute a significant synthetic challenge in view of the easy over-reduction of the C=C bond and the carbonyl group [36]; we therefore set out to exploit the benefits of continuous-flow processing [37,38,39,40,41,42,43,44,45,46,47].
Scheme 1.

The proposed synthesis of deuterium-labeled chalcones.
2. Results and Discussion
Most techniques for the synthesis of deuterium-labeled compounds rely on expensive D2 gas as a deuterium source. Similarly to hydrogenations, the handling of D2 gas demands extreme precautions and costly special apparatus to maintain sufficient operational safety. The conventional methods for the production of D2 gas are fractional distillation of liquid hydrogen, pyrolysis of UD3, and reactions of D2O with reducing agents (Na, Fe or Mg) [48]. Unfortunately, these methods involve several drawbacks, such as the production of radioactive waste, the need for cryogenic conditions, and the large quantity of metal sludge produced [48]. Catalytic H–D exchange reactions between H2 and D2O appear more feasible, but often do not yield sufficiently pure D2 and require long reaction times [49,50,51,52,53,54]. Alternatively, the application of deuterated reagents and deuterated solvents as isotopic sources eliminates the hazards of gas handling, but the costs of these special conditions severely limit the practical applicability [55,56,57,58,59].
To overcome the difficulties of conventional deuterium labeling techniques, we have developed a unique flow chemistry-based method for deuteration reactions by changing the hydrogen source to deuterated water in an H-Cube® system (Figure 3) [36,60,61]. This approach is safe, as D2 gas is continuously generated in small aliquots through the electrolytic decomposition of D2O, the consumption of which is very low, implying high deuterium efficiency. The gas generated in situ is combined with the substrate solution via a gas–liquid mixer, and the resulting mixture is transported to a filled catalyst bed, where the triphasic reaction takes place [62,63]. The purity of the D2 gas produced can be as high as 99.9%, which ensures high deuterium incorporation ratios [36,60,61]; moreover, the precise control over the reaction conditions may enhance the efficacy of the deuteration reactions [36,39,47,64,65].
Figure 3.

Schematic outline of the continuous-flow deuteration reactor (H-Cube®).
Our synthetic strategy toward the anticipated deuterium-labeled compounds involved continuous-flow deuteration of the corresponding ynone analogs of antiabetic chalcones 1–4 (Scheme 1). Ynones 5–9 were prepared by literature procedures through the coupling of acid chlorides with terminal acetylenes under Sonogashira conditions: PdCl2(PPh3)2/CuI as a catalyst and Et3N as a base in THF as solvent (Scheme 2) [66,67,68]. The mixtures were stirred for 1 h at room temperature (RT) under a N2 atmosphere, and ynones 5–9 were achieved in excellent yields after simple extractive work-up and chromatographic purification (see Experimental Section).
Scheme 2.

Coupling of acid chlorides with terminal acetylenes under Sonogashira conditions to yield the corresponding ynones as starting materials for the continuous-flow deuterations.
We first investigated the deuteration of 1,3-diphenylprop-2-yn-1-one (5) as a nonsubstituted model compound (Table 1). The reactions were carried out in EtOAc as aprotic solvent so as to prevent D–H exchange and to maximize deuterium incorporation. We had previously established that the steric hindrance generated by the halogen atom at position 2 of ring A exerts a significant influence on the rate of deuterium incorporation, as larger substituents may block the central core of the molecule, necessitating harsher conditions for the reaction to take place [36]. The deuteration of 5 was therefore investigated by using Lindlar catalyst (5% Pd on CaCO3, poisoned with lead) at ambient temperature (25 °C) and at pressures in the range 10‒80-bar. The utilization of high pressures in gas–liquid–solid reactions is favorable, as the elevation of pressure increases the solubility of gases, thereby enhancing the reaction rate. Accordingly, it was found that pressurization pushed the reaction to completion, resulting in higher total conversions, though pressures ≥40 bar contributed significantly to over-reaction of the desired dideuteroenone (5a) to give the corresponding tetradeuteroketone (5b) and pentadeuteroalcohol (5c) as side products (Table 1). At 80 bar, for example, a total conversion of 82% was found, but the crude product mixture contained 23% of 5b, and even 5c was formed to an extent of 4% (entry 4). If it is taken into account that 5a can be isolated from the alkynone starting material (5) more easily than from the over-reaction products (5b and 5c), 20 bar can be selected as the pressure of choice, with an acceptable total conversion of 57% and an excellent dideuteroenone/tetradeuteroketone ratio of 86:14, and without detectable pentadeuteroalcohol formation (entry 2). During the reactions, the (Z) deuterated chalcone isomer is formed, as a consequence of the mechanism of the heterogeneous catalytic process [69]. However, the (Z) chalcone readily undergoes thermal or photochemical isomerization to the more stable (E) form [70,71,72], which was detected exclusively after the purification. It must also be noted that an O–D bond on the pentadeuterochalcone side-product (5c) was not observed, as it is converted instantly to O–H due to exchange with moisture when it is exposed to air.
Table 1.
Continuous-flow deuteration of 5 a.
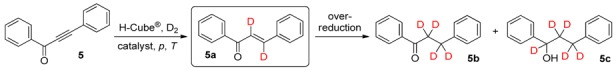
| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 5a c | 5b | 5c | |||||
| 1 | Lindlar catalyst | 10 | 25 | 47 | 92 | 8 | 0 |
| 2 | Lindlar catalyst | 20 | 25 | 57 | 86 | 14 | 0 |
| 3 | Lindlar catalyst | 40 | 25 | 63 | 77 | 23 | 0 |
| 4 | Lindlar catalyst | 80 | 25 | 82 | 73 | 23 | 4 |
a: Conditions: c = 1 mg·mL‒1 in ethyl acetate, 1 mL·min–1 flow rate; b: Determined by GC-MS analysis of the crude material; c: During the reactions, the (Z) deuterated chalcone isomer is formed, but readily isomerizes to the more stable (E) form.
In continuous-flow deuteration of 1-(2-fluorophenyl)-3-(4-methoxyphenyl)prop-2-yn-1-one (6) with Lindlar catalyst, the gradual increase of the pressure from 10 to 80 bar at ambient temperature had a pronounced influence on the rate of the reaction (Table 2). Although lower overall conversions were achieved than in deuterations of the nonsubstituted alkynone (5), compound 6 exhibited a lower tendency to over-reaction, even at pressures ≥40 bar, which could possibly be explained by the steric hindrance generated by the fluorine atom at position 2 of ring A. Thus, 80 bar, selected as optimum pressure, resulted in a total conversion of 63% and a dideuteroenone/tetradeuteroketone ratio of 80:20 without over-reaction to 6c (entry 4).
Table 2.
Continuous-flow deuteration of 6 a.
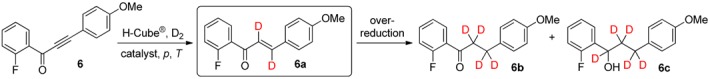
| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 6a c | 6b | 6c | |||||
| 1 | Lindlar catalyst | 10 | 25 | 23 | 90 | 10 | 0 |
| 2 | Lindlar catalyst | 20 | 25 | 40 | 86 | 14 | 0 |
| 3 | Lindlar catalyst | 40 | 25 | 57 | 81 | 19 | 0 |
| 4 | Lindlar catalyst | 80 | 25 | 63 | 80 | 20 | 0 |
a: Conditions: c = 1 mg·mL‒1 in ethyl acetate, 1 mL·min–1 flow rate; b: Determined by GC-MS analysis of the crude material; c: During the reactions, the (Z) deuterated chalcone isomer is formed, but readily isomerizes to the more stable (E) form.
When the fluorine atom on phenyl ring A was replaced by chlorine (1-(2-chlorophenyl)-3-(4-methoxyphenyl)prop-2-yn-1-one, 7), deuteration with Lindlar catalyst at 20‒80 bar resulted in conversions of only 33%‒47% (Table 3, entries 1‒3). 5% Pd/BaSO4 was therefore investigated next as a more active heterogeneous catalyst (keeping in mind that activated charcoal as a catalyst support may contain protic contamination that can negatively influence deuterium incorporation) [73]. The Pd/BaSO4-mediated reactions at RT gave excellent total conversions (75% and 86% at 20 and 40 bar, respectively), but formation of the desired dideuteroenone 7a was significantly suppressed by undesired over-reduction to 7b, and even to 7c, affording dideuteroenone/tetradeuteroketone/pentadeuteroalcohol ratios of 62:38:0 and 60:38:2, at 20 and 40 bar, respectively (entries 4 and 5).
Table 3.
Continuous-flow deuteration of 7 a.
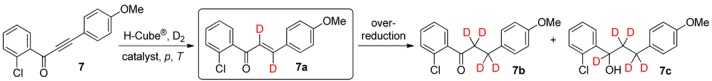
| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 7a c | 7b | 7c | |||||
| 1 | Lindlar catalyst | 20 | 25 | 33 | 89 | 11 | 0 |
| 2 | Lindlar catalyst | 40 | 25 | 43 | 88 | 12 | 0 |
| 3 | Lindlar catalyst | 80 | 25 | 47 | 79 | 21 | 0 |
| 4 | 5% Pd/BaSO4 | 20 | 25 | 75 | 62 | 38 | 0 |
| 5 | 5% Pd/BaSO4 | 40 | 25 | 86 | 60 | 38 | 2 |
a: Conditions: c = 1 mg·mL‒1 in ethyl acetate, 1 mL·min–1 flow rate; b: Determined by GC-MS analysis of the crude material; c: During the reactions, the (Z) deuterated chalcone isomer is formed, but readily isomerizes to the more stable (E) form.
In the case of bromine-substituted ynone 8 (1-(2-bromophenyl)-3-(4-methoxyphenyl)prop-2-yn-1-one), the efficacy of 5% Pd/BaSO4 was tested first at 40 bar employing temperatures of 50 and 70 °C, to compensate the larger steric hindrance generated by the halogen atom at position 2 of ring A (Table 4). Despite the achievement of acceptable conversions of 50% and 53%, significant over-reduction occurred at both temperatures, and dideuteroenone 8a was formed to extents of only 69% and 56% at 50 and 70 °C, respectively (entries 1 and 2). We were delighted to find that elevation of the pressure to 60 bar at ambient temperature resulted in a slight increase in conversion to 66% and a significant improvement in chemoselectivity, with an acceptable dideuteroenone (8a)/tetradeuteroketone (8b) ratio of 77:23, without detectable pentadeuteroalcohol (8c) present (entry 3). An effort to further enhance the amount of 8a by raising the temperature to 50 °C was unsuccessful because of the intensified 8a‒8b over-reduction (entry 4).
Table 4.
Continuous-flow deuteration of 8 a.
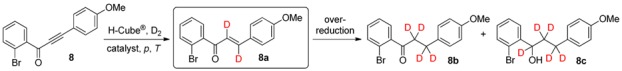
| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 8a c | 8b | 8c | |||||
| 1 | 5% Pd/BaSO4 | 40 | 50 | 50 | 69 | 31 | 0 |
| 2 | 5% Pd/BaSO4 | 40 | 70 | 53 | 56 | 44 | 0 |
| 3 | 5% Pd/BaSO4 | 60 | 25 | 66 | 77 | 23 | 0 |
| 4 | 5% Pd/BaSO4 | 60 | 50 | 82 | 61 | 39 | 0 |
a: Conditions: c = 1 mg·mL‒1 in ethyl acetate, 1 mL·min–1 flow rate; b: Determined by GC-MS analysis of the crude material; c: During the reactions, the (Z) deuterated chalcone isomer is formed, but readily isomerizes to the more stable (E) form.
Finally, the continuous-flow deuteration of 1-(2-iodophenyl)-3-(4-methoxyphenyl)prop-2-yn-1-one (9) was attempted by using 5% Pd/BaSO4 as catalyst in combination with harsher reaction conditions, with regard to the significant steric hindrance generated by iodine (Table 5) [36]. Although the total conversion at 80 bar and 50 °C was only 17%, formation of the corresponding dideuteroenone (9a) was exclusive under these conditions (entry 1). We were pleased to find that when the temperature was elevated to 100 °C, the conversion rose to an acceptable level of 56%, still without the formation of over-reaction products (entry 2). In attempts to improve the rate of the reaction, the use of 5% Pt/Al2O3 as a more active heterogeneous catalyst led to higher total conversions at RT and 20‒80 bar, but it had a pronounced negative effect on the chemoselectivity, with the unwanted tetradeuteroketone (9b) becoming the main product, together with significant amounts of pentadeuteroalcohol 9c (entries 3‒5).
Table 5.
Continuous-flow deuteration of 9 a.
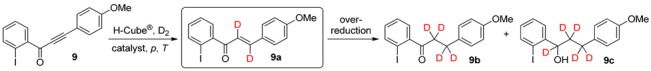
| Entry | Catalyst | p (bar) | T (°C) | Total Conversion b (%) | Product Ratio b (%) | ||
|---|---|---|---|---|---|---|---|
| 9a c | 9b | 9c | |||||
| 1 | 5% Pd/BaSO4 | 80 | 50 | 17 | 100 | 0 | 0 |
| 2 | 5% Pd/BaSO4 | 80 | 100 | 56 | 100 | 0 | 0 |
| 3 | 5% Pt/Al2O3 | 20 | 25 | 89 | 28 | 67 | 5 |
| 4 | 5% Pt/Al2O3 | 40 | 25 | 95 | 18 | 68 | 14 |
| 5 | 5% Pt/Al2O3 | 80 | 25 | 99 | 7 | 59 | 34 |
a: Conditions: c = 1 mg·mL‒1 in ethyl acetate, 1 mL·min–1 flow rate; b: Determined by GC-MS analysis of the crude material; c: During the reactions, the (Z) deuterated chalcone isomer is formed, but readily isomerizes to the more stable (E) form.
The above optimization reactions (Table 1, Table 2, Table 3, Table 4 and Table 5) were typically carried out on a 0.1 mmol scale. For preparative purposes, a 10-fold scale-up was made under the selected conditions (see Table 1, entry 2; Table 2, entry 4; Table 3, entry 4; Table 4, entry 3; and Table 5, entry 2) simply by pumping in a larger amount of starting material. Even gram-scale deuterations can readily be performed in flow as a function of the starting material quantity and the operation time, without any change in the reaction conditions and without the need for re-optimization.
As a consequence of the high purity of the D2 gas employed, the deuterated target compounds were obtained with deuterium contents (which reflect the deuterium incorporation ratio over incidental hydrogen addition) of ≥98%. It should also be noted that neither over-reaction products containing reduced or partially reduced aromatic rings, nor halogen–deuterium exchange on ring A were detected, even when Pt/Al2O3 was used as a catalyst.
3. Materials and Methods
3.1. Materials
The reagents and materials were of the highest commercially available grade and were used without further purification. Cartridges containing 5% Pt/Al2O3, 5% Pd/BaSO4 and Lindlar catalyst were purchased from ThalesNano Inc. (Budapest, Hungary).
3.2. Synthesis of the Starting Materials
The corresponding acid chloride (1.0 mmol, 1 equiv.), terminal acetylene (1.0 equiv.), PdCl2(PPh3)2 (0.03 equiv.), CuI (0.06 equiv.) and Et3N (1.5 equiv.) were combined in THF (6 mL), and the mixture was stirred for 1 h at ambient temperature under a N2 atmosphere. It was next diluted with water to 12 mL, and the resulting solution was extracted with CH2Cl2 (2 × 15 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue obtained was purified by column chromatography on silica gel, with a mixture of n-hexane/EtOAc as eluent; ynones 5‒9 were achieved in yields of 83%‒92% [66,67,68].
3.3. Continuous-Flow Deuterations
Deuterations were carried out in an H-Cube® system (ThalesNano Inc.) with replacement of the H2O hydrogen source to D2O (VWR, 99.96%) [36,60,61]. The catalyst cartridges, with internal dimensions of 30 × 4 mm, contained approximately 100 mg of 5% Pt/Al2O3, 5% Pd/BaSO4 or Lindlar catalyst. The selected cartridge was placed into a heating unit controlled by a built-in Peltier system (maximum temperature: 100 °C), and the system also contained a backpressure regulator that ensured constant pressures up to a maximum of 100 bar. The continuous stream of the reaction solution was provided by an HPLC pump (Knauer WellChrom K-120). For each reaction, a 1-mg·mL−1 solution of the corresponding ynone (5‒9) was prepared in ethyl acetate (HPLC grade). The mixture was homogenized by sonication for 2 min and then pumped through the H-Cube® under the appropriate conditions. Between two reactions, the catalyst bed was washed for 10 min with ethyl acetate at 1 mL min‒1. The crude products were purified by column chromatography on silica gel with mixtures of n-hexane/EtOAc as eluent.
3.4. Product Analysis
Ynones 5‒9 and deuterium-labeled chalcones 5a‒9a were characterized by NMR and GC-MS. 1H-NMR and 13C-NMR spectra were recorded in CDCl3 as an applied solvent on Bruker Avance DRX 400 and JEOL ECS 400 instruments with TMS as the internal standard, at 400.1 and 100.6 MHz, respectively. GC-MS analyses were carried out with a Thermofisher Scientific DSQ II Single Quadrupole GC/MS, on a 30 m × 0.25 mm × 0.25 μm HP-5MS capillary column (Agilent J & W Scientific). The measurement parameters: column oven temperature: from 50 to 300 °C at 10 °C·min−1 (0‒25 min), and 300 °C (25‒30 min); injection temperature: 250 °C; ion source temperature: 200 °C; EI: 70 eV; carrier gas: He, at 1 mL·min‒1; injection volume: 5 μL; split ratio: 1:50; and mass range: 45–800 m/z. Conversions and product ratios were determined from the GC-MS spectra of the crude materials. Deuterium contents were calculated from the relative intensities of the 1H-NMR indicator signals. Product characterization data can be found in the Supporting Information.
4. Conclusions
Scalable continuous-flow syntheses of the deuterium-labeled derivatives of antidiabetic chalcones were achieved in the present study. Our synthetic strategy relied on the selective C≡C bond deuteration of the corresponding alkynone analogs in an H-Cube® system following change of the hydrogen source to high-purity deuterated water. The methodology applied was simple and safe as it eliminated potentially dangerous D2 gas-handling. The effects of the most important reaction conditions (catalyst, pressure and temperature) were investigated in order to determine the optimum deuteration conditions. In spite of the possibility of facile over-reactions, the desired deuterium-labeled compounds were attained with high selectivities.
A patent application has been filed on deuterium-labeled chalcones 6a‒9a. Their biological effects and kinetic profiles are currently under investigation in our laboratories; these results will be reported in due course.
Acknowledgments
This study was supported by a bilateral mobility grant from the Ministry of Science and Technology of Taiwan and the Hungarian Academy of Sciences (MOST 104-2911-1-037-051 and SNK-79/2013). The projects of Ministry of Science and Technology of Taiwan (MOST 104-2811-B-037-018) and Kaohsiung Medical University, Taiwan (KMU-TP104E39), awarded to F.-R.C., are greatly appreciated. We are grateful to the Hungarian Research Foundation (OTKA K115731) for financial support. S.B.Ö. acknowledges the award of a János Bolyai Research Scholarship and a postdoctoral fellowship (Postdoctoral Research Program) from the Hungarian Academy of Sciences.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/3/318/s1.
Author Contributions
S.B.Ö., F.F. and F.-R.C. designed the experiments, interpreted the results and wrote the manuscript. S.B.Ö. and C.-T.H. carried out the flow and batch reactions. Y.-C.W. and J.-H.L. carried out the analytical investigations. C.-T.H. and J.-H.L. purified the crude products. All authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of compounds 5‒9 are available from the authors in mg quantities.
References
- 1.Bukhari S.N.A., Jasamai M., Jantan I. Synthesis and biological evaluation of chalcone derivatives (mini review) Mini-Rev. Med. Chem. 2012;12:1394–1403. doi: 10.2174/13895575112091394. [DOI] [PubMed] [Google Scholar]
- 2.Moore B.S., Hertweck C., Hopke J.N., Izumikawa M., Kalaitzis J.A., Nilsen G., O’Hare T., Piel J., Shipley P.R., Xiang L., et al. Plant-like biosynthetic pathways in bacteria: From benzoic acid to chalcone1. J. Nat. Prod. 2002;65:1956–1962. doi: 10.1021/np020230m. [DOI] [PubMed] [Google Scholar]
- 3.Tomás-Barberán F.A., Clifford M.N. Flavanones, chalcones and dihydrochalcones–nature, occurrence and dietary burden. J. Sci. Food Agric. 2000;80:1073–1080. doi: 10.1002/(SICI)1097-0010(20000515)80:7<1073::AID-JSFA568>3.0.CO;2-B. [DOI] [Google Scholar]
- 4.Nishimura R., Tabata K., Arakawa M., Ito Y., Kimura Y., Akihisa T., Nagai H., Sakuma A., Kohno H., Suzuki T. Isobavachalcone, a chalcone constituent of angelica keiskei, induces apoptosis in neuroblastoma. Biol. Pharm. Bull. 2007;30:1878–1883. doi: 10.1248/bpb.30.1878. [DOI] [PubMed] [Google Scholar]
- 5.Deng J., Kelley J.A., Barchi J.J., Sanchez T., Dayam R., Pommier Y., Neamati N. Mining the NCI antiviral compounds for HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2006;14:3785–3792. doi: 10.1016/j.bmc.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 6.Haraguchi H., Tanimoto K., Tamura Y., Mizutani K., Kinoshita T. Mode of antibacterial action of retrochalcones from glycyrrhiza inflata. Phytochemistry. 1998;48:125–129. doi: 10.1016/S0031-9422(97)01105-9. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura C., Kawasaki N., Miyataka H., Jayachandran E., Kim I.H., Kirk K.L., Taguchi T., Takeuchi Y., Hori H., Satoh T. Synthesis and biological activities of fluorinated chalcone derivatives. Bioorg. Med. Chem. 2002;10:699–706. doi: 10.1016/S0968-0896(01)00319-4. [DOI] [PubMed] [Google Scholar]
- 8.Fontenele J.B., Leal L.K.A.M., Ferreira M.A.D., Silveira E.R., Viana G.S.B. Antiplatelet effect of lonchocarpin and derricin isolated from lonchocarpus sericeus. Pharm. Biol. 2005;43:726–731. doi: 10.1080/13880200500387406. [DOI] [Google Scholar]
- 9.Sharma M., Chaturvedi V., Manju Y.K., Bhatnagar S., Srivastava K., Puri S.K., Chauhan P.M.S. Substituted quinolinyl chalcones and quinolinyl pyrimidines as a new class of anti-infective agents. Eur. J. Med. Chem. 2009;44:2081–2091. doi: 10.1016/j.ejmech.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Sahu N.K., Balbhadra S.S., Choudhary J., Kohli D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012;19:209–225. doi: 10.2174/092986712803414132. [DOI] [PubMed] [Google Scholar]
- 11.Yadav V.R., Prasad S., Sung B., Aggarwal B.B. The role of chalcones in suppression of NF-κB-mediated inflammation and cancer. Int. Immunopharmacol. 2011;11:295–309. doi: 10.1016/j.intimp.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batovska D.I., Todorova I.T. Trends in utilization of the pharmacological potential of chalcones. Curr. Clin. Pharmacol. 2010;5:1–29. doi: 10.2174/157488410790410579. [DOI] [PubMed] [Google Scholar]
- 13.Nowakowska Z. A review of anti-infective and anti-inflammatory chalcones. Eur. J. Med. Chem. 2007;42:125–137. doi: 10.1016/j.ejmech.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 14.Ni L., Meng C.Q., Sikorski J.A. Recent advances in therapeutic chalcones. Expert. Opin. Ther. Pat. 2004;14:1669–1691. doi: 10.1517/13543776.14.12.1669. [DOI] [Google Scholar]
- 15.Simmons D. Prevention of gestational diabetes mellitus: Where are we now? Diabetes Obes. Metab. 2015;17:824–834. doi: 10.1111/dom.12495. [DOI] [PubMed] [Google Scholar]
- 16.Bailey C.J. The current drug treatment landscape for diabetes and perspectives for the future. Clin. Pharmacol. Ther. 2015;98:170–184. doi: 10.1002/cpt.144. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasan S., Florez J.C. Therapeutic challenges in diabetes prevention: We have not found the “exercise pill”. Clin. Pharmacol. Ther. 2015;98:162–169. doi: 10.1002/cpt.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seuring T., Archangelidi O., Suhrcke M. The economic costs of type 2 diabetes: A global systematic review. Pharmacoeconomics. 2015;33:811–831. doi: 10.1007/s40273-015-0268-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shukla P., Srivastava S.P., Srivastava R., Rawat A.K., Srivastava A.K., Pratap R. Synthesis and antidyslipidemic activity of chalcone fibrates. Bioorg. Med. Chem. Lett. 2011;21:3475–3478. doi: 10.1016/j.bmcl.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 20.Li X.-H., Zou H.-J., Wu A.-H., Ye Y.-L., Shen J.-H. Structure-based drug design of a novel family of chalcones as PPARα agonists: Virtual screening, synthesis, and biological activities in vitro. Acta Pharmacol. Sin. 2007;28:2040–2052. doi: 10.1111/j.1745-7254.2007.00670.x. [DOI] [PubMed] [Google Scholar]
- 21.Enoki T., Ohnogi H., Nagamine K., Kudo Y., Sugiyama K., Tanabe M., Kobayashi E., Sagawa H., Kato I. Antidiabetic activities of chalcones isolated from a japanese herb, angelica keiskei. J. Agric. Food Chem. 2007;55:6013–6017. doi: 10.1021/jf070720q. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh C.-T., Hsieh T.-J., El-Shazly M., Chuang D.-W., Tsai Y.-H., Yen C.-T., Wu S.-F., Wu Y.-C., Chang F.-R. Synthesis of chalcone derivatives as potential anti-diabetic agents. Bioorg. Med. Chem. Lett. 2012;22:3912–3915. doi: 10.1016/j.bmcl.2012.04.108. [DOI] [PubMed] [Google Scholar]
- 23.Hiroaki H. Recent applications of isotopic labeling for protein nmr in drug discovery. Expert. Opin. Drug Discov. 2013;8:523–536. doi: 10.1517/17460441.2013.779665. [DOI] [PubMed] [Google Scholar]
- 24.Percy A.J., Rey M., Burns K.M., Schriemer D.C. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry—A review. Anal. Chim. Acta. 2012;721:7–21. doi: 10.1016/j.aca.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 25.Gevaert K., Impens F., Ghesquiere B., Van Damme P., Lambrechts A., Vandekerckhove J. Stable isotopic labeling in proteomics. Proteomics. 2008;8:4873–4885. doi: 10.1002/pmic.200800421. [DOI] [PubMed] [Google Scholar]
- 26.Lloyd-Jones G.C., Munoz M.P. Isotopic labelling in the study of organic and organometallic mechanism and structure: An account. J. Label. Compd. Radiopharm. 2007;50:1072–1087. doi: 10.1002/jlcr.1382. [DOI] [Google Scholar]
- 27.Gant T.G. Using deuterium in drug discovery: Leaving the label in the drug. J. Med. Chem. 2014;57:3595–3611. doi: 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- 28.Iglesias J., Sleno L., A Volmer D. Isotopic labeling of metabolites in drug discovery applications. Curr. Drug Metab. 2012;13:1213–1225. doi: 10.2174/138920012803341357. [DOI] [PubMed] [Google Scholar]
- 29.Krumbiegel P. Large deuterium isotope effects and their use: A historical review. Isot. Environ. Health Stud. 2011;47:1–17. doi: 10.1080/10256016.2011.556725. [DOI] [PubMed] [Google Scholar]
- 30.Robins R., Billault I., Duan J.-R., Guiet S., Pionnier S., Zhang B.-L. Measurement of 2H distribution in natural products by quantitative 2H-NMR: An approach to understanding metabolism and enzyme mechanism? Phytochem. Rev. 2003;2:87–102. doi: 10.1023/B:PHYT.0000004301.52646.a8. [DOI] [Google Scholar]
- 31.Kharasch E.D., Bedynek P.S., Park S., Whittington D., Walker A., Hoffer C. Mechanism of ritonavir changes in methadone pharmacokinetics and pharmacodynamics: I. Evidence against CYP3A mediation of methadone clearance. Clin. Pharmacol. Ther. 2008;84:497–505. doi: 10.1038/clpt.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baldwin J.E., Raghavan A.S., Hess B.A., Jr., Smentek L. Thermal 1,5 hydrogen sigmatropic shifts in cis,cis-1,3-cyclononadienes probed by gas-phase kinetic studies and density functional theory calculations. J. Am. Chem. Soc. 2006;128:14854–14862. doi: 10.1021/ja065656s. [DOI] [PubMed] [Google Scholar]
- 33.Stringer R.A., Williams G., Picard F., Sohal B., Kretz O., McKenna J., Krauser J.A. Application of a deuterium replacement strategy to modulate the pharmacokinetics of 7-(3,5-dimethyl-1H-1,2,4-triazol-1-yl)-3-(4-methoxy-2-methylphenyl)-2,6-dimethylpyrazolo[5,1-b]oxazole, a novel CRF1 antagonist. Drug Metab. Dispos. 2014;42:954–962. doi: 10.1124/dmd.114.057265. [DOI] [PubMed] [Google Scholar]
- 34.Braman V., Graham P., Cheng C., Turnquist D., Harnett M., Sabounjian L., Shipley J. A randomized phase I evaluation of CTP-499, a novel deuterium-containing drug candidate for diabetic nephropathy. Clin. Pharmacol. Drug Dev. 2013;2:53–66. doi: 10.1002/cpdd.3. [DOI] [PubMed] [Google Scholar]
- 35.Nelson S.D., Trager W.F. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab. Dispos. 2003;31:1481–1497. doi: 10.1124/dmd.31.12.1481. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh C.-T., Ötvös S.B., Wu Y.-C., Mándity I.M., Chang F.-R., Fülöp F. Highly selective continuous-flow synthesis of potentially bioactive deuterated chalcone derivatives. ChemPlusChem. 2015;80:859–864. doi: 10.1002/cplu.201402426. [DOI] [PubMed] [Google Scholar]
- 37.Wirth T., editor. Microreactors in Organic Chemistry and Catalysis. John Wiley & Sons; Hoboken, NJ, USA: 2013. [Google Scholar]
- 38.Ley S.V., Fitzpatrick D.E., Ingham R.J., Myers R.M. Organic synthesis: March of the machines. Angew. Chem. Int. Ed. 2015;54:3449–3464. doi: 10.1002/anie.201410744. [DOI] [PubMed] [Google Scholar]
- 39.Mándity I.M., Ötvös S.B., Fülöp F. Strategic application of residence-time control in continuous-flow reactors. ChemistryOpen. 2015;4:212–223. doi: 10.1002/open.201500018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ötvös S.B., Fülöp F. Flow chemistry as a versatile tool for the synthesis of triazoles. Catal. Sci. Technol. 2015;5:4926–4941. doi: 10.1039/C5CY00523J. [DOI] [Google Scholar]
- 41.Vaccaro L., Lanari D., Marrocchi A., Strappaveccia G. Flow approaches towards sustainability. Green Chem. 2014;16:3680–3704. doi: 10.1039/C4GC00410H. [DOI] [Google Scholar]
- 42.McQuade D.T., Seeberger P.H. Applying flow chemistry: Methods, materials, and multistep synthesis. J. Org. Chem. 2013;78:6384–6389. doi: 10.1021/jo400583m. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida J.-I., Takahashi Y., Nagaki A. Flash chemistry: Flow chemistry that cannot be done in batch. Chem. Commun. 2013;49:9896–9904. doi: 10.1039/C3CC44709J. [DOI] [PubMed] [Google Scholar]
- 44.Hessel V., Kralisch D., Kockmann N., Noël T., Wang Q. Novel process windows for enabling, accelerating, and uplifting flow chemistry. ChemSusChem. 2013;6:746–789. doi: 10.1002/cssc.201200766. [DOI] [PubMed] [Google Scholar]
- 45.Hartman R.L., McMullen J.P., Jensen K.F. Deciding whether to go with the flow: Evaluating the merits of flow reactors for synthesis. Angew. Chem. Int. Ed. 2011;50:7502–7519. doi: 10.1002/anie.201004637. [DOI] [PubMed] [Google Scholar]
- 46.Wegner J., Ceylan S., Kirschning A. Ten key issues in modern flow chemistry. Chem. Commun. 2011;47:4583–4592. doi: 10.1039/c0cc05060a. [DOI] [PubMed] [Google Scholar]
- 47.Irfan M., Glasnov T.N., Kappe C.O. Heterogeneous catalytic hydrogenation reactions in continuous-flow reactors. ChemSusChem. 2011;4:300–316. doi: 10.1002/cssc.201000354. [DOI] [PubMed] [Google Scholar]
- 48.Herber R.H., editor. Inorganic Isotopic Syntheses. Benjamin Inc.; New York, NY, USA: 1962. [Google Scholar]
- 49.Sawama Y., Monguchi Y., Sajiki H. Efficient H–D exchange reactions using heterogeneous platinum-group metal on carbon–H2–D2O system. Synlett. 2012;23:959–972. doi: 10.1002/chin.201225254. [DOI] [Google Scholar]
- 50.Modutlwa N., Maegawa T., Monguchi Y., Sajiki H. Synthesis of deuterium-labelled drugs by hydrogen–deuterium (H–D) exchange using heterogeneous catalysis. J. Label. Compd. Radiopharm. 2010;53:686–692. doi: 10.1002/jlcr.1848. [DOI] [Google Scholar]
- 51.Maegawa T., Fujiwara Y., Inagaki Y., Esaki H., Monguchi Y., Sajiki H. Mild and efficient H/D exchange of alkanes based on C‒H activation catalyzed by rhodium on charcoal. Angew. Chem. Int. Ed. 2008;47:5394–5397. doi: 10.1002/anie.200800941. [DOI] [PubMed] [Google Scholar]
- 52.Kurita T., Aoki F., Mizumoto T., Maejima T., Esaki H., Maegawa T., Monguchi Y., Sajiki H. Facile and convenient method of deuterium gas generation using a Pd/C-catalyzed H2–D2 exchange reaction and its application to synthesis of deuterium-labeled compounds. Chem. Eur. J. 2008;14:3371–3379. doi: 10.1002/chem.200701245. [DOI] [PubMed] [Google Scholar]
- 53.Maegawa T., Fujiwara Y., Inagaki Y., Monguchi Y., Sajiki H. A convenient and effective method for the regioselective deuteration of alcohols. Adv. Synth. Catal. 2008;350:2215–2218. doi: 10.1002/adsc.200800407. [DOI] [Google Scholar]
- 54.Esaki H., Ito N., Sakai S., Maegawa T., Monguchi Y., Sajiki H. General method of obtaining deuterium-labeled heterocyclic compounds using neutral D2O with heterogeneous Pd/C. Tetrahedron. 2006;62:10954–10961. doi: 10.1016/j.tet.2006.08.088. [DOI] [Google Scholar]
- 55.Mutsumi T., Iwata H., Maruhashi K., Monguchi Y., Sajiki H. Halogen–deuterium exchange reaction mediated by tributyltin hydride using THF-d8 as the deuterium source. Tetrahedron. 2011;67:1158–1165. doi: 10.1016/j.tet.2010.12.007. [DOI] [Google Scholar]
- 56.Di Giuseppe A., Castarlenas R., Pérez-Torrente J.J., Lahoz F.J., Polo V., Oro L.A. Mild and selective H/D exchange at the β-position of aromatic α-olefins by N-heterocyclic carbene–hydride–rhodium catalysts. Angew. Chem. Int. Ed. 2011;50:3938–3942. doi: 10.1002/anie.201007238. [DOI] [PubMed] [Google Scholar]
- 57.Lockley W.J.S., Hesk D. Rhodium- and ruthenium-catalysed hydrogen isotope exchange. J. Label. Compd. Radiopharm. 2010;53:704–715. doi: 10.1002/jlcr.1815. [DOI] [Google Scholar]
- 58.Derdau V., Atzrodt J., Zimmermann J., Kroll C., Brückner F. Hydrogen–deuterium exchange reactions of aromatic compounds and heterocycles by NaBD4-activated rhodium, platinum and palladium catalysts. Chem. Eur. J. 2009;15:10397–10404. doi: 10.1002/chem.200901107. [DOI] [PubMed] [Google Scholar]
- 59.Derdau V. Deuterated ammonium formate as deuterium source in a mild catalytic deuterium transfer reaction of pyridines, pyrazines and isoquinolines. Tetrahedron Lett. 2004;45:8889–8893. doi: 10.1016/j.tetlet.2004.09.165. [DOI] [Google Scholar]
- 60.Ötvös S.B., Mándity I.M., Fülöp F. Highly selective deuteration of pharmaceutically relevant nitrogen-containing heterocycles: A flow chemistry approach. Mol. Divers. 2011;15:605–611. doi: 10.1007/s11030-010-9276-z. [DOI] [PubMed] [Google Scholar]
- 61.Mándity I.M., Martinek T.A., Darvas F., Fülöp F. A simple, efficient, and selective deuteration via a flow chemistry approach. Tetrahedron Lett. 2009;50:4372–4374. doi: 10.1016/j.tetlet.2009.05.050. [DOI] [Google Scholar]
- 62.Jones R.V., Godorhazy L., Varga N., Szalay D., Urge L., Darvas F. Continuous-flow high pressure hydrogenation reactor for optimization and high-throughput synthesis. J. Comb. Chem. 2006;8:110–116. doi: 10.1021/cc050107o. [DOI] [PubMed] [Google Scholar]
- 63.Desai B., Kappe C.O. Heterogeneous hydrogenation reactions using a continuous flow high pressure device. J. Comb. Chem. 2005;7:641–643. doi: 10.1021/cc050076x. [DOI] [PubMed] [Google Scholar]
- 64.Habraken E., Haspeslagh P., Vliegen M., Noël T. Iridium(I)-catalyzed ortho-directed hydrogen isotope exchange in continuous-flow reactors. J. Flow. Chem. 2015;5:2–5. doi: 10.1556/JFC-D-14-00033. [DOI] [Google Scholar]
- 65.Noël T., Su Y., Hessel V. Beyond organometallic flow chemistry: The principles behind the use of continuous-flow reactors for synthesis. Top. Organomet. Chem. 2015 doi: 10.1007/3418_2015_152. [DOI] [Google Scholar]
- 66.Chuang D.-W., El-Shazly M., Balaji D.B., Chung Y.-M., Chang F.-R., Wu Y.-C. Synthesis of flavones and γ-benzopyranones using mild sonogashira coupling and 18-crown-6 ether mediated 6-endo cyclization. Eur. J. Org. Chem. 2012:4533–4540. doi: 10.1002/ejoc.201200529. [DOI] [Google Scholar]
- 67.Cox R.J., Ritson D.J., Dane T.A., Berge J., Charmant J.P.H., Kantacha A. Room temperature palladium catalysed coupling of acyl chlorides with terminal alkynes. Chem. Commun. 2005:1037–1039. doi: 10.1039/b414826f. [DOI] [PubMed] [Google Scholar]
- 68.Karpov A.S., Müller T.J.J. Straightforward novel one-pot enaminone and pyrimidine syntheses by coupling-addition-cyclocondensation sequences. Synthesis. 2003:2815–2826. doi: 10.1002/chin.200421059. [DOI] [Google Scholar]
- 69.Siegel S. Heterogeneous catalytic hydrogenation of C=C and C≡C. In: Trost B.M., Fleming I., editors. Comprehensive Organic Synthesis. Volume 8. Pergamon Press; New York, NY, USA: 1991. pp. 417–442. [Google Scholar]
- 70.Lee Y., Motoyama Y., Tsuji K., Yoon S.-H., Mochida I., Nagashima H. (Z)-selective partial hydrogenation of internal alkynes by using palladium nanoparticles supported on nitrogen-doped carbon nanofiber. ChemCatChem. 2012;4:778–781. doi: 10.1002/cctc.201200058. [DOI] [Google Scholar]
- 71.Yoshizawa K., Shioiri T. Convenient stereoselective synthesis of (Z)-chalcone derivatives from 1,3-diaryl-2-propynyl silyl ethers. Tetrahedron Lett. 2006;47:4943–4945. doi: 10.1016/j.tetlet.2006.05.021. [DOI] [Google Scholar]
- 72.Nicodem D.E., de M.G. Matos J.A. Photoisomerization of chalcone: Wavelength dependence. J. Photochem. 1981;15:193–202. doi: 10.1016/0047-2670(81)87003-7. [DOI] [Google Scholar]
- 73.Oba M., Ohkuma K., Hitokawa H., Shirai A., Nishiyama K. Convenient synthesis of deuterated glutamic acid, proline and leucine via catalytic deuteration of unsaturated pyroglutamate derivatives. J. Label. Compd. Radiopharm. 2006;49:229–235. doi: 10.1002/jlcr.1038. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.