

Abstract
A FRET-based random screening assay was used to generate hit compounds as sortase A inhibitors that allowed us to identify ethyl 3-oxo-2-(2-phenylhydrazinylidene)butanoate as an example of a new class of sortase A inhibitors. Other analogues were generated by changing the ethoxycarbonyl function for a carboxy, cyano or amide group, or introducing substituents in the phenyl ring of the ester and acid derivatives. The most active derivative found was 3-oxo-2-(2-(3,4dichlorophenyl)hydrazinylidene)butanoic acid (2b), showing an IC50 value of 50 µM. For a preliminary assessment of their antivirulence properties the new derivatives were tested for their antibiofilm activity. The most active compound resulted 2a, which showed inhibition of about 60% against S. aureus ATCC 29213, S. aureus ATCC 25923, S. aureus ATCC 6538 and S. epidermidis RP62A at a screening concentration of 100 µM.
Keywords: sortase A, biofilms, 2-(2-phenylhydrazinylidene)alkanoic acid derivatives, FRET
1. Introduction
Antibiotic resistance is a very important challenge and in 2015 the World Health Organization (WHO) considered it as one of the most important global health problems [1]. The overuse of antibiotics, both in human and animal populations, plays an important part in the appearance of drug-resistant strains.
The drug resistance of Gram-positive pathogens is currently of great significance and in this context Staphylococcus aureus that is responsible of both acute and chronic infectious diseases has an extraordinary ability to develop antibiotic-resistance [2]. Its great versatility as a pathogen is due to a huge number of virulence factors [3]. Among the most important virulence factors that it displays during the pathogenesis, the cell-wall associated proteins called microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) can promote the adherence to host tissue by interacting with fibronectin. Other aspects of pathogenesis such as invasion, escape from host defences and the formation of biofilms, that cause chronic infectious diseases or biomaterial associated infections, are also due to the MSCRAMMs [4,5].
Sortase A (SrtA) is the enzyme that incorporates the MSCRAMMs to the peptidoglycan through the following mechanism: the enzyme first cleaves the bond in the sorting signal between the threonine (T) and the glycine (G) residues of a LPxTG motif of cellular proteins; then it causes the formation of a thioester acyl-enzyme intermediate; the last step is a transpeptidation of an amide bond of the carboxyl terminal of threonine and the amine terminus of a pentaglycine cross bridge in peptidoglycan precursors [6].
S. aureus strains lacking the SrtA gene do not display surface proteins at the cell wall. Therefore, SrtA mutant strains are less virulent than wild strains and they are defective during their pathogenic action [7]. At least twenty different S. aureus surface proteins that carry a C-terminal LPxTG motif have been described. These virulence factors include protein A (Spa), two fibronectin binding proteins (FnbpA and FnbpB) and two clumping factors (ClfA and ClfB). Some of these proteins play key roles in biofilm formation [7,8].
An anti-virulence strategy based on agents that target virulence determinants could be effective in preventing the biofilm formation of Gram positive bacteria that are naturally resistant to current antibiotics. Considering that the first crucial step in staphylococcal pathogenesis and biofilm formation is bacterial adhesion, promoted by the surface exposed proteins at the cell wall, we presume that the new inhibitory agents targeting the sortase enzyme that links surface proteins to the cell wall are potentially more useful rather than any single MSCRAMM involved in the pathogenesis [9]. Consequently, sortase A is a good target to develop novel anti-virulence agents and new classes of SrtA inhibitors could tackle the first stage of infectious disease process and biofilm formation [10].
A number of promising small synthetic organic compounds that work as effective SrtA inhibitors and could be developed as anti-virulence drugs, were recently reviewed [11]. Most of classes of described inhibitors (diarylacrylonitriles [12], rhodanines [13], pyridazinones [13], pyrazolethiones [13], 3,6-disubstituted triazolothiadiazol [14], aryl(β-amino)ethyl ketones [15] and benzo-[d]isothiazol-3(2H)-one adamantanes [16], were identified by high-throughput screening (HTS) or virtual screening. SrtA is an ideal target not associated with bacterial growth and cell death, but rather related to virulence [17]. Moreover, as opposed to conventional antibiotics, in response to which the evolution of resistance by the pathogens is advantageous and nearly unavoidable, in the case of antivirulence agents, the bacterial resistance is potentially costly and therefore less probable.
Starting from these considerations and in continuation of our research work on antibacterial and antibiofilm agents [18,19,20,21,22], we thought it would be of interest to obtain novel SrtA inhibitors, showing more molecular diversity than the known ones, which could interfere with Gram positive virulence mechanisms as well as the biofilm formation. In order to achieve this goal, we randomly screened 200 compounds synthesized in our laboratory hoping to obtain positive hits. A high-throughput FRET-assay that monitors the SrtA-driven hydrolysis of an internally quenched fluorescent substrate (dabcyl-QALPETGEE-edans) was used to test the compounds and select sortase- specific inhibitors [16]. Among the 200 screened compounds, the most active were the phenylhydrazinylidene derivatives 1a and 2, where 1a, with an IC50 value of 192 µM, was selected as a hit (Figure 1).
Figure 1.
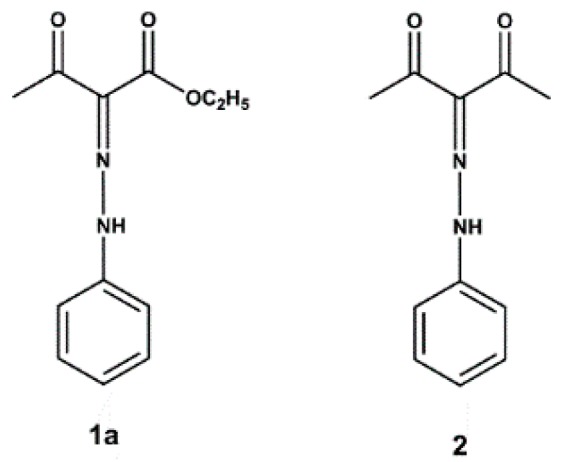
Structure of phenylhydrazinylidene derivatives 1a and 2.
2. Results and Discussion
2.1. Chemistry
A new set of compounds 1b–f, analogs of compound 1a, were prepared. The phenyl ring of 1b–f bears different substituents in order to obtain a good discrimination as regards hydrophobic, electronic and steric effects. We additionally synthesized compounds 5a–f, in which the carboxyethyl group of 1a–f is replaced with a carboxy group and 6a and 7a, which bear a carboxyamido or cyano group replacing the carboxyethyl group of 1a. Finally, analogues 1g,h and 5g,h were prepared, in which the acetyl group is replaced by a benzoyl group. All the compounds were obtained as shown in Scheme 1.
Scheme 1.
Synthetic route for compounds 1a–h, 5a–h, 6a and 7a.

| 3,4 | a | b | c | d | e | f |
|---|---|---|---|---|---|---|
| R | H | 3-4,Cl2 | 3-Cl | 4-NO2 | 3,4,5-OCH3 | 4-CH3 |
| 1,5,6,7 | a | b | c | d | e | f | g | h |
|---|---|---|---|---|---|---|---|---|
| R | H | 3-4,Cl2 | 3-Cl | 4-NO2 | 3,4,5-OCH3 | 4-CH3 | H | 3-4,Cl2 |
| R1 | CH3 | CH3 | CH3 | CH3 | CH3 | CH3 | C6H5 | C6H5 |
The diazonium salts 4a–f obtained from the anilines 3a–f were reacted with ethyl acetoacetate or ethyl benzoylacetate to give the ester derivatives 1a–h, which, in turn, were transformed into the corresponding acids 5a–h (Scheme 1). Similarly, the analogue 6a was obtained by reacting 4a with 3-aminocrotononitrile. Finally, the carboxyamido derivative 7a was obtained by reacting 1a with ammonia.
Esters 1a–d,f,g, the cyano and carboxyamido derivatives 6a and 7a were already described. The reported synthetic method was further modified, and the abovementioned esters 1a–d,f,g, and compound 7a were obtained.
It is well established that the above class of derivatives exist in the arylhydrazinylidene (arylhydrazone) form [23,24], however one needs to assign the geometrical structure of the substituted C=N double bond. The structure of compounds 1a–d,f,g is missing in some previously reported articles or a mixture of the E and Z forms is reported [25,26,27,28]. Moreover, opposite geometries were proposed for the same phenylhydrazinylidene derivative [29,30]. However, the crystallographically determined geometrical structure for compounds 1a,f (E isomers) [31,32] is in agreement with that obtained by IR and 1H-NMR spectra [29,32]. At this point it was thought of interest to establish the geometrical structure of all the remaining compounds as this class of derivatives is not sufficiently investigated. The reported 1H-NMR assignment of the geometrical structures of compounds 1a,f is based on the NH and CH3CO chemical shifts. For the compounds that bear the E structure, in which the NH and acetyl groups are intramolecularly bonded (see Figure 2), the NH and methyl signals are located to lower field as compared to the Z isomer: NH(E) about δ 14.85, NH(Z) about δ 12.80; CH3CO(E) about δ 2.6, CH3CO(Z) about δ 2.5. Considering these data, the geometrical structure of compounds 1b–e was assigned on the basis of their 1H-NMR spectra. The esters 1b,c,e, show in the 1H-NMR spectra the signal for NH in the range δ 14.61–14.90 and that for the methyl moiety of the acetyl group in the range δ 2.59–2.61, therefore the same structure of 1a,f (E form) was assigned. As regards compound 1d, its 1H-NMR spectrum shows the NH signal at δ 12.70 and the methyl one at δ 2.53, values which are compatible with the Z form. The geometrical structures of ethyl benzoylacetate derivatives 1g,h, were assigned on the basis of the comparison between the 1H-NMR spectra of these compounds and that of ethyl 2-(2-phenyl-hydrazinyilidene)mesoxalate (8, see Figure 2) [23]. The 1H-NMR spectrum of compound 8 shows a singlet at δ 12.76 for the NH group intramolecularly bonded to the carboxylate one. The 1H-NMR spectra of 1g,h show the NH signal at chemical shift values very near to δ 12.76, that is δ 12.74 and 12.60, respectively, therefore the Z structure was assigned to these compounds. The cyano derivative 6a shows a signal in the 1H-NMR spectrum at δ 9.49 which excludes the H-bond of the NH with the carbonyl group. This view is supported by the 1H-NMR spectrum of 2-(2-phenylhydrazinylidene)propanodinitrile (9) [33] (see Figure 2), which shows the NH chemical shift at δ 9.57, a value very near to δ 9.49. Compound 6 might presumably be arranged to a dimeric form of type 10 where each single molecule show the E structure (see Figure 3). Finally, the acetoacetamide derivative 7a exists in the Z form, as indicated by 13C- and 15N-NMR studies reported in the literature (see Figure 3) [34].
Figure 2.

Reported NH chemical shift values of the geometrical structures of compounds 1a,f, ethyl (2-phenylhydrazinylidene)mesoxalate (8) and 2-(2-phenylhydrazinylidene)propanedinitrile (9).
Figure 3.

Dimeric structure 10 of (2E)-3-oxo-2-(2-phenylhydrazinylidene)butanenitrile (6a) and structures of (2Z)-3-oxo-2-(2-phenylazohydrazinylidene)butanamide 7a and (2Z)-acids 5a–h.
All the spectroscopic data of 5a,d,f that were only partially and inaccurately reported in a previously published article [35], were de novo recorded and are reported herein. The 1H-NMR spectra of all compounds 5 showed the same pattern, i.e., two distinct signals in the range δ 13.70–14.35, attributable to the NH and carboxy groups. The IR spectra showed absorptions in the range of 1697–1704 cm−1 for bonded carbonyl groups, and a very broad band in the 3200–2500 cm−1 range for the carboxy group. These values are compatible with a Z structure in which two strong intramolecular hydrogen bonds exist (see Figure 3). This type of structural arrangement has been confirmed by X-ray diffraction studies on compound 5a [36].
2.2. Biology
The esters 1b–h, the acids 5a–h and compounds 6a and 7a were tested for their inhibitory activity, utilizing the assay monitoring enzymatic hydrolysis of sortase A FRET substrate analogue dabcyl-QALPETGEE-edans (Table 1). In accordance with preliminary data the esters 1b–h were poorly active or inactive. The acids 5a–h showed IC50 values in the range of 50–100 µM. The phenyl substitution in the phenylhydrazinylidene moiety does not play a determinant role for the activity. However, it seems that strong electron withdrawing groups in the phenyl ring, such as two chlorine atoms or a nitro group, can afford a moderate increase in the inhibitory activity in comparison to unsubstituted phenyl, possibly due to a more acidic NH group. On the contrary, the presence of electron releasing groups, such as a methyl or three methoxyl groups, produce a slight decrease of the activity. Replacement of the acetyl group with the benzoyl one in compounds 5 afforded a slight decrease of activity. Finally, the cyano derivative 6a showed the same activity as compared to 5a, whereas the amide 7a, was less active than all the acids.
Table 1.
Activity as sortase A inhibitors of compounds 1a–h, 5a–h, 6a, 7a.
| Entry | Compounds | IC50 µM |
|---|---|---|
| 1 | 1a | 192 |
| 2 | 1b | ns |
| 3 | 1c | ns |
| 4 | 1d | 100 |
| 5 | 1e | 110 |
| 6 | 1f | ns |
| 7 | 1g | ns |
| 8 | 1h | ns |
| 9 | 5a | 80 |
| 10 | 5b | 50 |
| 11 | 5c | 87 |
| 12 | 5d | 57 |
| 13 | 5e | 92 |
| 14 | 5f | 100 |
| 15 | 5g | 98 |
| 16 | 5h | 100 |
| 17 | 6a | 80 |
| 18 | 7a | 120 |
| 19 | PVS 1 | 736 |
| 20 | BC 2 | 120 |
| 21 | DPDAP 3 | 10 |
1 Phenyl vinyl sulfone; 2 Berberine chloride; 3 1-(3,4-dichlorophenyl)-3-dimethylamino-1-propanone; ns = not significant; IC50 ≥ 500 µM.
Tests for the antibiofilm activity of the most active SrtA inhibitors 5a–h, 6a and 7a were performed to assess their preliminary antivirulence properties. The compounds were tested for their ability to interfere with biofilm formation of S. aureus ATCC 29213, S. aureus ATCC 25923, S. aureus ATCC 6538 and S. epidermidis RP62A at a screening concentration of 100 µM (see Table 2), a concentration at which planktonic strains were not susceptible (MIC > 270 µM). We found that all the above phenylhydrazinylidene derivatives interfered with biofilm formation. With the exception of S. aureus ATCC 2913, the presence of substituents in the phenyl ring of derivatives 5b–f, independently of their hydrophobic and electronic properties, does not offer any advantage for the biofilm inhibitory activity. In fact, the most active compound was 5a, showing inhibition of biofilm formation of about 60% at 100 µM against all the tested strains. A slight positive effect on the inhibition of biofilm formation of S. aureus 29213 strains was observed for compounds 5b–d, which bear an electron-withdrawing substituent in the phenyl ring.
Table 2.
Inhibition of biofilm formation of compounds 5a–h, 6a and 7a at 100 μM concentration.
| Comp. | Percentages of Inhibition of Biofilm Formation | |||
|---|---|---|---|---|
| S. aureus 25923 | S. aureus 29213 | S. aureus 6538 | S. epidermidis RP62A | |
| 5a | 68.3 ± 2.0 | 62.3 ± 1.5 | 60.1 ± 1.7 | 61.5 ± 1.2 |
| 5b | 39.2 ± 1.4 | 71.3 ± 6.4 | 22.0 ± 0.9 | 29.1 ± 1.4 |
| 5c | 41.1 ± 0.7 | 69 ± 4.6 | 30.3 ± 0.4 | 22.9 ± 1.3 |
| 5d | 45.3 ± 1.7 | 73.7 ± 0.8 | 53.6 ± 1.8 | 48.6 ± 1.6 |
| 5e | 28.8 ± 2.9 | 34.8 ± 1.3 | 48.2 ± 0.9 | 27.1 ± 1.2 |
| 5f | 33.8 ± 2.7 | 40.8 ± 2.1 | 40.3 ± 1.9 | 24.4 ± 0.9 |
| 5g | 36.8 ± 2.1 | 36.9 ± 1.2 | 41.9 ± 1.7 | 23.7 ± 0.7 |
| 5h | 55.9 ± 2.6 | 42.7 ± 3.7 | 70.1 ± 2.1 | 45.4 ± 1.6 |
| 6a | 45.1 ± 2.9 | 46.2 ± 2.3 | 51 ± 0.9 | 34.8 ± 1.8 |
| 7a | 42.1 ± 1.8 | 48.2 ± 2.9 | 42.8 ± 1.1 | 45.9 ± 2.1 |
Replacement of the acetyl group with the benzoyl one (5g) in compound 5a led to a decrease of activity for all tested strains. For the analogue compound 5h, bearing two chlorine atoms linked to the phenyl ring, the antibiofilm activity was slightly increased only against the S. aureus 6538 strain. Finally, replacement of the carboxy group of 5a with the cyano and carboxamide groups that resulted respectively in derivatives 6a and 7a, allowed only to obtain the analogues less active than 5a.
3. Experimental Section
3.1. Chemistry
3.1.1. General
All commercial chemicals were purchased from Aldrich (Sigma-Aldrich, St. Louis, MO, USA). Reaction progress was monitored by TLC on silica gel plates (Merck 60, F254, 0.2 mm, Merck spa, Vimodrone, Italy) and visualization on TLC was achieved by UV light. Organic solutions were dried over Na2SO4. Evaporation refers to the removal of solvent on a rotary evaporator under reduced pressure. All melting points were determined on a Büchi 530 capillary melting point apparatus (Buchi Italia srl, Cornaredo, Italy) and are uncorrected. IR spectra were recorded with a Spectrum RXI FT-IR System spectrophotometer (Perkin Elmer Italia spa, Milano, Italy) as solids in KBr discs. UV spectra were recorded with a Cary 50 Scan UV-Visible Spectrophotometer (Varian Medical System Italia, Cernusco sul Naviglio, Italy). 1H-NMR and 13C-NMR spectra were recorded in CDCl3 at 300.13 and 75.47 MHz respectively, using an AC series 300 MHz spectrometer (Bruker, Milano, Italia; tetramethylsilane as the internal standard): chemical shifts are expressed in δ values (ppm). Microanalyses data (C, H, N) were obtained by an Elemental Vario EL III apparatus (Elemetal Analysensysteme, Hanau, Germany) and are within ±0.4% of the theoretical values. Yields refer to products after crystallization. The name of the compounds was obtained using the ACD/Chem Sketch FREEWARE ver. 14.00 (ACD/Lab, Toronto, ON, Canada).
Physicochemical and spectroscopic data are also reported for the previously reported derivatives 1b–d,f,g. Spectroscopic data allow for the correct assignment of the geometry. The synthetic procedure and a more detailed physicochemical and structural characterization of compound 7a are also reported as only 13C- and 15N-NMR data of this compound were previously reported [34].
Some 1H-NMR and 13C-NMR spectras were presented in Supplementary Materials.
3.1.2. General Procedure for the Synthesis of Compounds 1a–h
The appropriate aniline (0.012 mol) in 5 N aqueous HCl solution (6 mL) was diazotized under stirring at 0–5 °C by addition of a solution of NaNO2 (0.88 g in 3 mL of water). After 10 min a saturated NaOAc aqueous solution was added until pH 5, followed dropwise by that of ethyl acetoacetate (1.53 mL) and NaOAc (1.44 g in 2.4 mL water) in ethanol (9 mL), maintaining the temperature under 10 °C. The reaction mixture was stirred at 5–10 °C for 30 min then at room temperature for 90 min. The precipitate of the arylhydrazinylidene derivative was filtered off, washed with water and crystallized from the appropriate solvent or purified by preparative TLC (silica gel plate, layer thickness 2 mm).
Ethyl (2E)-3-Oxo-2-(2-phenylhydrazinylidene)butanoate (1a). Yield 91%; m.p.: 78–80 °C (MeOH), lit. [29] 80 °C (MeOH), spectroscopic data are in accord with those reported in the literature [25,29].
Ethyl (2E)-3-Oxo-2-(2-(3,4-dichlorophenyl)hydrazinylidene)butanoate (1b). Yield 62%; m.p.: 100–102 °C (cycloexane), lit. [27] 71 °C (ethanol, E/Z mixture); IR ν[cm−1]: 3157 (NH), 1708 (CO); 1H-NMR (CDCl3) δ [ppm]: 1.40 (t, 3H, CH3, J = 7.2 Hz), 2.59 (s, 3H, CH3), 4.35 (q, 2H, CH2, J = 7.2 Hz); 7.19–7.55 (m, 3H, aromatic protons); 14.60 (s, 1H, exchangeable with D2O, NH); 13C-NMR (CDCl3) δ [ppm]: 14.31 (CH3); 30.82 (CH3); 61.26 (CH2); 115.52 (CH); 117.84 (CH); 127.06 (C); 128.76(C); 131.14 (CH); 133.77 (C); 141.21 (C); 164.53 (COOC2H5); 197.43 (CH3CO).
Ethyl (2E)-3-Oxo-2(2-(3-chlorophenyl)hydrazinylidene)butanoate (1c). Yield: 45%; m.p.: 70–72 °C (cyclohexane), lit. [26] 83–85 °C, (crude E/Z mixture); IR ν[cm−1]: 3100 (NH), 1703 (CO); UV (ethanol) λmax [nm], Log ε: 234, 4.14; 350, 4.25; 1H-NMR (CDCl3) δ [ppm]: 1.42 (t, 3H, CH3, J = 7.2 Hz), 2.59 (s, 3H, CH3), 4.34 (q, 2H, CH2, J = 7.2 Hz), 7.11–7.47 (m, 4H, aromatic protons), 14.63 (s, 1H, exchangeable with D2O, NH). 13C-NMR (CDCl3) δ [ppm]: 14.31 (CH3); 30.81 (CH3); 61.13 (CH2); 114.53 (CH); 116.28 (CH); 125.45 (CH); 126.64 (C); 130.49 (CH); 135.50 (C); 142.85 (C); 164.67 (COOC2H5); 197.26 (CH3CO).
Ethyl (2Z)-3-Oxo-2(2-(4-nitrophenyl)hydrazinylidene)butanoate (1d). Yield: 70%; m.p.: 120–122 °C (ethanol), lit. [26] 127.5–128 °C, (crude E/Z mixture); IR ν[cm−1]: 3156 (NH); 1684 (CO). 1H-NMR (CDCl3) δ [ppm]: 1.42 (t, 3H, CH3, J = 7.2 Hz), 2.53 (s, 3H, CH3), 4.40 (q, 2H, CH2, J = 7.2 Hz), 7.41 (d, 2H, aromatic protons, J = 8.7 Hz), 8.28 (d, 2H, aromatic protons, J = 8.7 Hz), 12.70 (s, 1H, exchangeable with D2O, NH); 13C-NMR (CDCl3) δ [ppm]: 14.03 (CH3); 26.91 (CH3); 62.09 (CH2); 115.10 (2 × CH); 126.04 (2 CH); 130.35 (C); 143.88(C); 146.50 (C); 163.10 (COOC2H5); 194.15 (CH3CO).
Ethyl (2E)-3-Oxo-2-(2-(3,4,5-trimethoxyphenyl)hydrazinylidene)butanoate (1e). Yield: 20%; m.p. 78–80 °C (cyclohexane); IR ν[cm−1]: 3142 (NH), 1701 (CO); 1H-NMR (CDCl3) δ [ppm]: 1.40 (t, 3H, CH3, J = 7.2 Hz), 2.61 (s, 3H, CH3), 3.86 (s, 3H, CH3), 3.91 (s, 6H, 2 CH3), 4.34 (q, 2H, CH2, J = 7.2 Hz), 6.92 e 6.70 (s, 2H, aromatic protons), 14.84 (s, 1H, exchangeable with D2O, NH). 13C-NMR (CDCl3) δ [ppm]: 14.26 (CH3); 30.77 (CH3); 56.10 (2 OCH3); 60.87 (CH2); 61.07 (OCH3); 93.75 (2 CH); 125.53 (C); 135.89 (C); 137.73 (C); 154.08 (2 C); 164.92 (COOC2H5); 196.94 (CH3CO). Anal. Calc. for C15H20N2O6: C, 55.55%; H, 6.22%; N, 8.64%. Found: C, 55.25%; H, 5.92%; N, 8.39%.
Ethyl (2E)-3-Oxo-2-(2-(4-methylphenyl)hydrazinylidene)butanoate (1f). Yield: 63%; m.p.: 70–72 °C (cyclohexane), lit. [32] 80–81 °C, (E/Z mixture); IR ν[cm−1]: 3447 (NH); 1700 (CO); 1H-NMR (CDCl3) δ [ppm]: 1.40 (t, 3H, CH3, J = 7.2 Hz), 2.34 (s, 3H, CH3), 2.58 (s, 3H, CH3), 4.33 (q, 2H, CH2, J = 7.2 Hz), 7.18 (d, 2H, aromatic protons, J = 8.4 Hz), 7.32 (d, 2H, aromatic protons, J = 8.7 Hz), 14.90 (s, 1H, exchangeable with D2O, NH). 13C-NMR (CDCl3) δ [ppm]: 14.35 (CH3); 21.03 (CH3); 30.77 (CH3); 60.87 (CH2); 116.40 (2 CH); 125.48 (C); 130.09 (2 CH); 135.77 (C); 139.56 (C); 165.14 (COOC2H5); 197.07 (CH3CO).
Ethyl (2Z)-3-Oxo-3-phenyl-2-(2-phenylhydrazinylidene)propanoate (1g). Yield 60%; m.p. 63–65 °C (MeOH), lit. [23] 65 °C (MeOH); IR ν[cm−1]: 3229, 3196 (NH), 1675 (CO); 1H-NMR (CDCl3) δ [ppm]: 1.37 (t, 3H, CH3, J = 7.2 Hz), 4.37 (q, 2H, CH2, J = 7.2 Hz), 7.10–7.95 (a set of signal, 10H, 2 C6H5), 12.74 (s, 1H, exchangeable with D2O, NH ).
Ethyl (2Z)-3-oxo-3-phenyl-2-(2-(3,4-dichlorophenyl)hydrazinylidene)-propanoate (1h). Yield: 62%; m.p. 96–98 °C (cycloexane); IR ν[cm−1]: 3168, 3200 (NH), 1673 (CO); 1H-NMR (CDCl3) δ [ppm]: 1.34 (t, 3H, CH3, J = 7.2 Hz), 4.36 (q, 2H, CH2, J = 7.2 Hz), 6.98–7.94 (a set of signal, 8H, C6H5 and C6H3), 12.60 (s, 1H, NH). Anal. Calc. for C17H14Cl2N2O3: C, 55.91%; H, 3.86%; N, 7.67%. Found: C, 56.14%; H, 3.58%; N, 7.50%.
3.1.3. General Procedure for the Preparation of Compounds 5a–h
To a solution of arylhydrazinylidene derivative 1a–h (5 g) in ethanol (50 mL) an aqueous solution of 5 N NaOH (12 mL) was added. The mixture was stirred at room temperature for 20 h. The collected sodium salt was washed with ethanol and solubilized in cold water. The water solution was acidified with 37% aqueous hydrochloric acid and the solid was filtered off, washed with water and recrystallized from a suitable solvent.
(2Z)-3-Oxo-2-(2-phenylhydrazinylidene)butanoic acid (5a). Yield: 68%; m.p. 155–157 °C (ethanol), lit. [35] 160–161 °C (aqueous ethanol); IR ν[cm−1]: 3200–2500 (NH, and OH), 1698 (CO); 1H-NMR (CDCl3) δ [ppm]: 2.60 (s, 3H, CH3), 7.24–7.47 (m, 3H, aromatic protons), 13.93 (s, 1H, exchangeable with D2O, NH), 14.09 (s, 1H, exchangeable with D2O, COOH). Anal. Calc. for C10H10N2O3: C, 58.25%; H, 4.89%; N, 13.59%. Found: C, 58.51%; H, 4.58%; N, 13.76%.
(2Z)-3-Oxo-2-(2-(3,4-dichlorophenyl)hydrazinylidene)butanoic acid (5b). Yield: 56%; m.p.: 218–220 °C (ethanol); IR ν[cm−1]: 3200–2500 (NH, and OH), 1697 (CO); 1H-NMR (CDCl3) δ [ppm]: 2.61 (s, 3H, CH3), 7.24–7.58 (m, 3H, aromatic protons), 13.81 (s, 1H, exchangeable with D2O, NH), 14.01 (s, 1H, exchangeable with D2O, COOH). Anal. Calc. for C10H8Cl2N2O3: C, 43.66%; H, 2.93%; N, 10.18%. Found: C, 43.99%; H, 2.61%; N, 10.30%.
(2Z)-3-Oxo-2-(2-(3-chlorophenyl)hydrazinylidene)butanoic acid (5c). Yield: 50%; m.p.: 148–150 °C (ethanol); IR ν[cm−1]: 3200–2500 (NH and OH), 1698 (CO); UV (ethanol) λmax [nm], Log ε: 255, 3.91; 360, 4.01; 1H-NMR (CDCl3) δ [ppm]: 2.61 (s, 3H, CH3), 7.21–7.49 (m, 4H, aromatic protons), 13.83 (s, 1H, exchangeable with D2O, NH), 14.00 (s, 1H, exchangeable with D2O, OH); 13C-NMR (CDCl3) δ [ppm]: 24.57 (CH3), 115.03 (CH), 116.47 (CH), 124.10 (C), 126.70 (CH); 130.86 (CH), 136.23 (C), 141.87(C); 165.56 (COOH); 202.23 (CH3CO). Anal. Calc. for C10H9ClN2O3: C, 49.91%; H, 3.77%; N, 11.64%. Found: C, 50.25%; H, 3.82%; N, 11.89%.
(2Z)-3-Oxo-2-(2-(4-nitrophenyl)hydrazinylidene)butanoic acid (5d). Yield: 41%; m.p.: 202–204 °C (1,4-dioxane), lit. [35] 194–195 °C (aqueous ethanol); IR ν[cm−1]: 3200–2400 (NH and OH), 1700 (CO); 1H-NMR (CDCl3) δ [ppm]: 2.65 (s, 3H, CH3); 7.56 (d, 2H, aromatic protons, J = 9.0 Hz), 8.34 (d, 2H, aromatic protons, J = 9.0 Hz), 13.70 (s, 1H, exchangeable with D2O, NH), 14.11 (s, 1H, exchangeable with D2O, OH); 13C-NMR (CDCl3) δ [ppm]: 24.74 (CH3), 116.45 (2×CH), 125.65 (C), 125.87 (2 × CH), 145.33 (C), 145.47 (C), 164.85 (COOH), 202.27 (CH3CO). Anal. Calc. for C10H9N3O5: C, 47.81%; H, 3.61%; N, 16.73%. Found: C, 48.15%; H, 3.32%; N, 16.64%.
(2Z)-3-Oxo-2-(2-(3,4,5-trimethoxyphenyl)hydrazinylidene)butanoic acid (5e). Yield: 32%; m.p.: 150–152 °C (ethanol); IR ν[cm−1]: 3200–2500 (NH and OH); 1693 (CO). 1H-NMR (CDCl3) δ [ppm]: 2.60 (s, 3H, CH3); 3.87 (s, 3H, CH3); 3.92 (s, 6H, 2 × CH3); 6.70 (s, 2H, aromatic protons); 14.00 (s, 1H, exchangeable with D2O, NH); 14.10 (s, 1H, exchangeable with D2O, OH); 13C-NMR (CDCl3) (δ): 23.82 (CH3); 55.76 (2 × CH3); 60.63 (CH3); 93.63 (2 CH); 122.73 (C); 136.24 (C); 136.58 (C); 153.78 (2 C); 165.31 (COOH); 201.10 (CH3CO). Anal. Calc. for C13H16N2O6: C, 52.70%; H, 5.44%; N, 9.46%. Found: C, 52.55%; H, 5.30%; N, 9.58%.
(2Z)-3-Oxo-2-(2-(4-methylphenyl)hydrazinylidene)butanoic acid (5f). Yield: 75%; m.p.: 182–184 °C (ethanol/NNDMF), lit. [35] 198–199 °C (aqueous ethanol); IR: ν[cm−1]: 3200–2655 (NH and OH); 1695 (CO); 1H-NMR (CDCl3) δ [ppm]: 2.38 (s, 3H, CH3); 2.58 (s, 3H, CH3); 7.22–7.36 (dd, 4H, aromatic protons); 13.98 (s, 1H, exchangeable with D2O, NH); 14.08 (s, 1H, exchangeable with D2O, OH). 13C-NMR (CDCl3) δ [ppm]: 21.12 (CH3); 21.03 (CH3); 24.37 (CH3); 116.64 (2 × CH); 123.13 (C); 130.08 (2 × CH); 137.18 (C); 138.51 (C); 165.81 (COOH); 201.80 (CH3CO). Anal. Calc. for C11H12N2O3: C, 59.99%; H, 5.49%; N, 12.72%. Found: C, 59.97%; H, 5.35%; N, 12.66%.
(2Z)-3-Oxo-3-phenyl-2-(2-phenylhydrazinylidene)propanoic acid (5g). Yield: 35%; m.p. 136–138°C (ethanol); IR: ν[cm−1]: 3200–2500, multiple bands (NH and COOH); 1H-NMR (CDCl3) δ [ppm]: 7.24–7.90 (a set of signal, 10 H, 2 × C6H5), 14,28 (s, 1H, NH or COOH), 14.35 (s, 1H, COOH or NH). 13C-NMR (CDCl3) (δ): 116.88 (CH), 122.89 (C), 126.87 (CH), 127.94 (CH), 129.85 (CH), 130.70 (CH), 132.64 (CH), 136.50 (C), 140.90 (C), 166.41 (COOH), 195.65 (C6H5CO). Anal. Calc. for C15H12N2O3: C, 67.16%; H, 4.51%; N, 10.44%. Found: C, 67.15%; H, 4.88%; N, 10.37%.
(2Z)-3-Oxo-2-(2-(3,4-dichlorophenyl)hydrazinylidene)-3-phenylpropanoic acid (5h). Yield: 60% m.p. 202–204 °C (ethyl acetate); IR: ν[cm−1]: 3200–2400, multiple bands, NH and COOH; 1H-NMR (CDCl3) δ [ppm]: 7.11–7.88 (a set of signal, 8H, C6H5 and C6H3), 14.15 (s, 2H, NH and COOH). 13C-NMR (DMSO) (δ): 115.85 (CH), 117.28 (CH), 127.94 (CH), 125.42 (C), 128.66 (CH), 130.42 (CH), 130.86 (C), 131.67 (CH), 132.24 (C), 133.24 (CH), 137.30 (C), 143.06 (C), 164.13 (COOH), 190.79 (C6H5CO). Anal. Calc. for C15H10Cl2N2O3: C, 53.44%; H, 2.99%; N, 8.31%. Found: C, 53.45%; H, 3.19%; N, 8.36%.
3.1.4. (2E)-3-Oxo-2-(2-phenylhydrazinylidene)butanenitrile (6a)
See reference [37].
3.1.5. (2Z)-3-Oxo-2-(2-phenylhydrazinylidene)butanamide (7a)
A solution of ethyl (2E)-3-oxo-2-(2-(phenylhydrazinylidene)butanoate (290 mg, 1.5 mmol) in ethanol (3 mL) was treated with 30% (w/v) aqueous ammonia (9 mL) under reflux for 8 h. The mixture was allowed to stand at r.t. for 12 h. The resulting solid product was filtered off and recrystallized from cyclohexane. Yield: 30%; m.p.: 134–136 °C; IR: ν[cm−1]: 3181, 3220s, 3355; 1H-NMR (CDCl3) δ [ppm]: 2.54 (s, 3H, CH3), 5.73 (s, 1H, NH), 7.18–7.39 (a set of signal, 5H, C6H5), 9.09 (s, 1H, NH), 14.60 (s, 1H, NH). 13C-NMR resonance values are according with literature data [34].
3.2. Biology
3.2.1. Enzyme Activity Assay
3.2.1.1 Expression of Recombinant Sortase A
Recombinant and catalytically active sortase A with the N-terminal deletion of residues 1–59 and possessing C-terminal hexahistidine sequence (SrtAΔN59-6His) was used for the enzyme activity assay. SrtAΔN59-6His was prepared according to a slightly modified previously published method [16]. Briefly, after the IPTG induction of the E. coli BL21 (DE3) transformed strain, the cell pellet was collected by centrifugation and resuspended in lysis buffer, and the recombinant protein was purified by affinity chromatography on a Ni-NTA column (Qiagen spa, Milano, Italy). The enzyme was eluted with an imidazole gradient and the fractions containing the protein were further purified by the gel filtration. A Superdex 75 (GE Healthcare srl, Milano, Italy) column, which was equilibrated with 10 mM sodium phosphate (pH 7.0) buffer containing 100 mM NaCl and 1 mM DTT, was used for the final purification. The fractions containing the protein were collected and concentrated to 10 mg/mL. The purified protein was analyzed using matrix-assisted laser desorption-ionization time-of-flight (MALDI-TOF) mass spectroscopy and SDS-PAGE Coomassie Blue staining.
3.2.1.2. HTS Screening of Candidate Compounds and Secondary Assays of the Selected Hits and Synthesized Analogues 1a–h, 2a–h, 6a and 7a
All of the compounds were prepared as 10 mM stock solutions in dimethyl sulfoxide (DMSO) and used for the IC50 determination. The compounds were screened at a single dose of 100 µM (1% DMSO) in black 384-well plates (Greiner Bio-One, Kremsmunster, Australia). Two known sortase inhibitors, phenyl vinyl sulfone [38] and 1-(3,4-dichlorophenyl)-3-(dimethylamino)- propan-1-one [15], were used as the positive controls. The inhibitory activity of all of the compounds was assessed by quantifying the increase in fluorescence intensity upon cleavage of the FRET-peptide dabcyl-QALPETGEE-edans, which was used as the sortase substrate. A previously published method [39] was used with slight modifications. Briefly, the reactions were performed in a volume of 100 µL containing 50 mM Tris-HCl, 5 mM CaCl2, 150 mM NaCl, pH 7.5, 10 µM S. aureus SrtA, 20 µM fluorescent peptide substrate dabcyl-QALPETGEE-edans, and the prescribed concentrations of the test compounds or positive controls.
The peptide substrate without the recombinant SrtA was incubated in the same manner and used as a negative control. The reactions were conducted for 24 h at 37 °C, and the fluorescence emitted with an excitation wavelength of 350 nm and an emission wavelength of 495 nm after substrate cleavage was recorded.
End-point determination of product formation was used as a criterion for the primary screening. This determination was made by measuring the total product fluorescence 24 h after the initiation of the reaction. The relative inhibition activity was determined as %I = 100% − (Fsample/Fcontrol*100%), where Fsample is the fluorescence intensity of the well containing the corresponding test compound and Fcontrol is the fluorescence of the positive control reaction without inhibition.
For the IC50 determination, 10 μM S. aureus sortase A was preincubated in the reaction buffer with increasing concentrations of the inhibitory compounds (x–y μM) for 1 h at 37 °C prior to the addition of the dabcyl-QALPETGEE-edans substrate to a final concentration of 50 µM. The total fluorescence was recorded at 1-min intervals for 1 h, and the progress curves were constructed. The initial velocities of the biphasic reactions were obtained through nonlinear regression, as previously described [40,41]. The IC50 values were determined by fitting the obtained data to the following default four-parameter variable slope sigmoidal function in SigmaPlot 12.5 using a nonlinear least squares algorithm:
| (1) |
3.3. Antibacterial and Antibiofilm Evaluation
3.3.1. Microbial Strains
The reference strains S. aureus ATCC 29213, S. aureus ATCC 25923, S. aureus ATCC 6538 and S. epidermidis RP62A were used in the assays.
3.3.2. Minimum Inhibitory Concentrations (MIC)
MICs of phenylhydrazinylidene-derivatives were determined by a micro-method as previously described [42]. Tryptic Soy Broth (TSB, Sigma-Aldrich srl, Milano, Italy) containing 2% glucose was used as medium.
3.3.3. Biofilm Capability Evaluation (Safranin Method)
The staphylococcal strains were tested for their ability to form biofilms. Briefly, bacteria were grown in TSB containing 2% glucose overnight at 37 °C in a shaking bath and then diluted 1:200 to a suspension with optical density (OD) of about 0.040 at 570 nm [21]. Polystyrene 24-well tissue culture plates were filled with 1 mL of diluted suspension and incubated for 24 h at 37 °C. The wells were then washed three times with 1 mL of sterile phosphate-buffered saline (PBS) and stained with 1 mL of safranin 0.1% v/v for 1 min. The excess stain was removed by placing the plates under running tap water. Plates were dried overnight in an inverted position at 37 °C. Safranin-stained adherent bacteria in each well were re-dissolved to homogeneity in 1 mL of 30% v/v glacial acetic acid, and the OD was read at 492 nm. Each assay was performed in triplicate and repeated at least twice.
3.3.4. Interference with Biofilm Formation Assay
The procedure described above was used to evaluate the activity of phenyl-hydrazinylidene-derivatives in the prevention of biofilm formation. Polystyrene 24-well tissue culture plates were filled with 1 mL of diluted bacterial suspension, obtained and diluted as previously described, and a concentration of 100 µM of each compound was directly added to the bacterial suspension at time zero and incubated at 37 °C for 24 h. The wells were then washed and stained with safranin as per the biofilm forming assay. By comparing the average optical density (O.D.) of the growth control wells with the sample wells, the following formula was used to calculate the percentages of inhibition for each concentration of the sample:
| (2) |
Each assay was performed in triplicate and assays were repeated at least twice.
4. Conclusions
In conclusion, we have synthesized a novel class of small molecules displaying micromolar inhibitory activity against S. aureus SrtA. These derivatives were screened for their activity against the enzyme using a FRET assay. Structure-activity relationship studies on compound 1a (IC50 = 192 µM) have led us to obtain a more active derivative 5b (IC50 = 50 µM) that could be the subject of further studies for the development of new anti-virulence agents.
Acknowledgments
Financial support from “Fondo di Finanziamento della Ricerca di Ateneo (ex 60%) 2007” is gratefully acknowledged.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/2/241/s1.
Author Contributions
Benedetta Maggio, Schillaci Domenico and Giuseppe Daidone conceived and designed the experiments, wrote the paper; Benedetta Maggio, Maria Valeria Raimondi, Fabiana Plescia, Maria Grazia Cusimano, Ainars Leonchiks, Dmitrijs Zhulenkovs and Stella Cascioferro performed experiments; Demetrio Raffa performed experiments and analyzed the data.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of all compounds are available from the authors.
References
- 1.World Health Organization Combat Antimicrobial Resistance. [(accessed on 16 October 2015)]. Available online: http://www.who.int/world-health-day/2011/WHD201_FS_EN.pdf.
- 2.Lowy F.D. Staphylococcus aureus infections. Engl. J. Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 3.Tang Y.-W., Stratton C.W. Staphylococcus aureus: An old pathogen with new weapons. Clin. Lab. Med. 2010;30:179–208. doi: 10.1016/j.cll.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Scott J.R., Barnett T.C. Surface proteins of Gram-positive bacteria and how they get there. Annu. Rev. Microbiol. 2006;60:397–423. doi: 10.1146/annurev.micro.60.080805.142256. [DOI] [PubMed] [Google Scholar]
- 5.Høiby N., Bjarnsholt T., Givskov M., Molin S., Ciofu O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents. 2010;35:322–332. doi: 10.1016/j.ijantimicag.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Fischetti V.A., Pancholi V., Schneewind O. Conservation of a hexapeptide sequence in the anchor region of surface proteins from Gram-positive cocci. Mol. Microbiol. 1990;4:1603–1605. doi: 10.1111/j.1365-2958.1990.tb02072.x. [DOI] [PubMed] [Google Scholar]
- 7.Mazmanian S.K., Liu G., Jensen E.R., Lenoy E., Schneewind O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. USA. 2000;97:5510–5515. doi: 10.1073/pnas.080520697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsompanidou E., Denham E.L., Sibbald M.J.J.B., Yang X.-M., Seinen J., Friedrich A.W., Buist G., van Dijl J.M. The sortase A substrates FnbpA, FnbpB, ClfA and ClfB antagonize colony spreading of Staphylococcus aureus. PLoS ONE. 2012;7:e44646. doi: 10.1371/journal.pone.0044646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L., Wen Y. The role of bacterial biofilm in persistent infections and control strategies. Int. J. Oral Sci. 2011;3:66–73. doi: 10.4248/IJOS11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cascioferro S., Cusimano M.G., Schillaci D. Antiadhesion agents against Gram-positive pathogens. Future Microbiol. 2014;9:1209–1220. doi: 10.2217/fmb.14.56. [DOI] [PubMed] [Google Scholar]
- 11.Cascioferro S., Raffa D., Maggio B., Raimondi M.V., Schillaci D., Daidone G. Sortase A Inhibitors: Recent Advances and Future Perspectives. J. Med. Chem. 2015;58:9108–9123. doi: 10.1021/acs.jmedchem.5b00779. [DOI] [PubMed] [Google Scholar]
- 12.Oh K.-B., Kim S.-H., Lee J., Cho W.-J., Lee T., Kim S. Discovery of Diarylacrylonitriles as a Novel Series of Small Molecule Sortase A Inhibitors. J. Med. Chem. 2004;47:2418–2421. doi: 10.1021/jm0498708. [DOI] [PubMed] [Google Scholar]
- 13.Suree N., Yi S.W., Thieu W., Marohn M., Damoiseaux R., Chan A., Jung M.E., Clubb R.T. Discovery and structure–activity relationship analysis of Staphylococcus aureus sortase A inhibitors. Bioorg. Med. Chem. 2009;17:7174–7185. doi: 10.1016/j.bmc.2009.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J., Liu H., Zhu K., Gong S., Dramsi S., Wang Y.-T., Li J., Chen F., Zhang R., Zhou L., et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA. 2014;111:13517–13522. doi: 10.1073/pnas.1408601111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maresso A.W., Wu R., Kern J.W., Zhang R., Janik D., Missiakas D.M., Duban M.-E., Joachimiak A., Schneewind O. Activation of Inhibitors by Sortase Triggers Irreversible Modification of the Active Site. J. Biol. Chem. 2007;282:23129–23139. doi: 10.1074/jbc.M701857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhulenkovs D., Rudevica Z., Jaudzems K., Turks M., Leonchiks A. Discovery and structure-activity relationship studies of irreversible benzisothiazolinone-based inhibitors against Staphylococcus aureus sortase A transpeptidase. Bioorg. Med. Chem. 2014;22:5988–6003. doi: 10.1016/j.bmc.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 17.Cascioferro S., Totsika M., Schillaci D. Sortase A: An ideal target for anti-virulence drug development. Microb. Pathog. 2014;77:105–112. doi: 10.1016/j.micpath.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Daidone G., Plescia S., Raffa D., Maggio B., Schillaci D. Synthesis and evaluation of antimicrobial activity of new 4-nitroso and 4-diazopyrazole derivatives. Farmaco (Soc. Chim. Ital. 1989) 1992;47:203–217. [PubMed] [Google Scholar]
- 19.Daidone G., Bajardi M., Plescia S., Raffa D., Schillaci D., Maggio B., Benetollo F., Bombieri G. One-step synthesis, crystallographic studies and antimicrobial activity of new 4-diazopyrazole derivatives. Eur. J. Med. Chem. 1996;31:461–468. doi: 10.1016/0223-5234(96)85166-X. [DOI] [Google Scholar]
- 20.Daidone G., Maggio B., Plescia S., Raffa D., Musiu C., Milia C., Perra G., Marongiu M.E. Antimicrobial and antineoplastic activities of new 4-diazopyrazole derivatives. Eur. J. Med. Chem. 1998;33:375–382. doi: 10.1016/S0223-5234(98)80004-4. [DOI] [Google Scholar]
- 21.Schillaci D., Maggio B., Raffa D., Daidone G., Cascioferro S., Cusimano M.G., Raimondi M.V. 4-Diazopyrazole Derivatives as Potential New Antibiofilm Agents. Chemotherapy. 2008;54:456–462. doi: 10.1159/000159271. [DOI] [PubMed] [Google Scholar]
- 22.Raimondi M.V., Maggio B., Raffa D., Plescia F., Cascioferro S., Cancemi G., Schillaci D., Cusimano M.G., Vitale M., Daidone G. Synthesis and anti-staphylococcal activity of new 4-diazopyrazole derivatives. Eur. J. Med. Chem. 2012;58:64–71. doi: 10.1016/j.ejmech.2012.09.041. [DOI] [PubMed] [Google Scholar]
- 23.Elguero J., Jaequier R., Tarrago J.G. Structure des produits de copulation du chlorurre de phényldiazonium avec les β-dicétones e les β-cétoesters. Bull. Soc. Chim. France. 1966:2981–2989. [Google Scholar]
- 24.Bose A.K., Kugajevsky I. NMR spectral studies—IV: Some 15NH coupling constants. Tetrahedron. 1967;23:1489–1497. doi: 10.1016/0040-4020(67)85102-0. [DOI] [Google Scholar]
- 25.Bandyopadhyay P., Guha L., Seenivasagan T., Sathe M., Sharma P., Parashar B.D., Kaushik M.P. Synthesis and bio-evaluation of aryl hydrazono esters for oviposition responses in Aedes albopictus. Bioorg. Med. Chem. Lett. 2011;21:794–797. doi: 10.1016/j.bmcl.2010.11.101. [DOI] [PubMed] [Google Scholar]
- 26.Ferguson G.N., Valant C., Horne J., Figler H., Flynn B.L., Linden J., Chalmers D.K., Sexton P.M., Christopoulos A., Scammells P.J. 2-Aminothienopyridazines as Novel Adenosine A1 Receptor Allosteric Modulators and Antagonists. J. Med. Chem. 2008;51:6165–6172. doi: 10.1021/jm800557d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pareek A.K., Joseph P.E., Seth D.S. A convenient route for the synthesis and spectral characterization of substituted pyrazolones. Orient. J. Chem. 2009;25:735–738. [Google Scholar]
- 28.Ballatore C., Brunden K., Crowe A., Huryn D., Lee V., Trojanowski J., Smith A., Huang R., Huang W., Johnson R., et al. Aminothienopyridazine Inhibitors of Tau Assembly. Patent WO2011037985 A8. 2011 Mar 31;
- 29.Mitchell A., Nonhebel D.C. Spectroscopic studies of tautomeric systems—III: 2-Arylhydrazones of 1,2,3-triketones. Tetrahedron. 1979;35:2013–2019. doi: 10.1016/S0040-4020(01)88971-7. [DOI] [Google Scholar]
- 30.Jollimore J.V., Vacheresse M., Vaughan K., Hooper D.L. The effect of ortho and para substituents on the formation of the E and Z isomers of the arylhydrazones obtained from diazonium coupling with methyl 3-aminocrotonate and 3-aminocrotononitrile. Can. J. Chem. 1996;74:254–262. doi: 10.1139/v96-029. [DOI] [Google Scholar]
- 31.Gupta S.C., Mandal D.K., Rani A., Sahay A., Prasad S.M. Ethyl 3-oxo-2-(2-phenyl-hydrazinylidene)butanoate: A re-determination. Acta Crystallogr. Sect. E Struct. Rep. Online. 2011;67 doi: 10.1107/S1600536811002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khudina O.G., Burgart Y.V., Shchegol’kov E.V., Saloutin V.I., Kazheva O.N., Chekhlov A.N., D’yachenko O.A. Steric structure of alkyl 2-aryl(hetaryl)hydrazono-3-fluoroalkyl-3-oxopropionates. Russ. J. Org. Chem. 2009;45:801–809. doi: 10.1134/S1070428009060013. [DOI] [Google Scholar]
- 33.Saez R., Otero M.D., Batanero B., Barba F. Microwave reaction of diazonium salts with nitriles. J. Chem. Res. 2008;2008:492–494. doi: 10.3184/030823408X340780. [DOI] [Google Scholar]
- 34.Jirman J., Lyčka A. 13C- and 15N-NMR spectra of phenylazoacetoacetamides and similar compounds. Dyes Pigm. 1987;8:55–62. doi: 10.1016/0143-7208(87)85005-2. [DOI] [Google Scholar]
- 35.Prasad N., Prasad R.M., Sahay A., Srivastava A.K. Prasad, Studied on Arylhydrazones, Part IX: Action of Perchloric Acid Formic Acid on Diethyl Mesoxalate Phenylhydrazones, 2,3-Dioxo-2-(phenylhydrazono)butyrate and Cyano Phenylhydrazones. J. Asian J. Chem. 1994;6:901–910. [Google Scholar]
- 36.Rani A., Saha A.P., Prasad S.M. 3-Oxo-2-(pheylhydrazono)butanoic acid. Acta Crystallogr. Sect. E Struct. Rep. Online. 2002;58:o1001–o1002. doi: 10.1107/S1600536802014757. [DOI] [Google Scholar]
- 37.Al-Mousawi S.M., Moustafa M.S. 2-Arylhydrazononitriles as building blocks in heterocyclic synthesis: A novel route to 2-substituted- 1,2,3-triazoles and 1,2,3-triazolo[4,5-b]pyridines. Beilstein J. Org. Chem. 2007;3 doi: 10.1186/1860-5397-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frankel B.A., Bentley M., Kruger R.G., McCafferty D.G. Vinyl sulfones: inhibitors of SrtA, a transpeptidase required for cell wall protein anchoring and virulence in Staphylococcus aureus. J. Am. Chem. Soc. 2004;126:3404–3405. doi: 10.1021/ja0390294. [DOI] [PubMed] [Google Scholar]
- 39.Ton-That H., Liu G., Mazmanian S.K., Faull K.F., Schneewind O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA. 1999;96:12424–12429. doi: 10.1073/pnas.96.22.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhulenkovs D., Jaudzems K., Zajakina A., Leonchiks A. Enzymatic activity of circular sortase A under denaturing conditions: An advanced tool for protein ligation. Biochem. Eng. J. 2014;82:200–209. doi: 10.1016/j.bej.2013.11.018. [DOI] [Google Scholar]
- 41.Huang X., Aulabaugh A., Ding W., Kapoor B., Alksne L., Tabei K., Ellestad G. Kinetic mechanism of Staphylococcus aureus sortase SrtA. Biochemistry. 2003;42:11307–11315. doi: 10.1021/bi034391g. [DOI] [PubMed] [Google Scholar]
- 42.Schillaci D., Petruso S., Raimondi M.V., Cusimano M.G., Cascioferro S., Scalisi M., La Giglia M.A., Vitale M. Pyrrolomycins as potential anti-staphylococcal biofilms agents. Biofouling. 2010;26:433–438. doi: 10.1080/08927011003718673. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.