

Abstract
Two novel series of diaryl urea derivatives 5a–i and 13a–l were synthesized and evaluated for their cytotoxicity against H-460, HT-29, A549, and MDA-MB-231 cancer cell lines in vitro. Therein, 4-aminoquinazolinyl-diaryl urea derivatives 5a–i demonstrated significant activity, and seven of them are more active than sorafenib, with IC50 values ranging from 0.089 to 5.46 μM. Especially, compound 5a exhibited the most active potency both in cellular (IC50 = 0.15, 0.089, 0.36, and 0.75 μM, respectively) and enzymatic assay (IC50 = 56 nM against EGFR), representing a promising lead for further optimization.
Keywords: diaryl urea, 4-aminoquinazolinyl, synthesis, cytotoxicity, EGFR inhibitors
1. Introduction
Recently, small molecular multiple targeted drugs have played a crucial role in cancer therapy due to their high efficiency and low toxicity. Diaryl urea derivative sorafenib [1], the first oral multikinase inhibitor targeted Raf and receptor tyrosine kinases (RTKs), has been applied for the treatment of advanced renal cell carcinoma (RCC) [2], unresectable hepatocellular carcinoma (HCC) [3], and differentiated thyroid carcinoma (DTC) [4]. It is reported that the lipophilic diaryl urea moiety served as a key structural fragment for binding with the hydrophobic pocket of the kinase domain through hydrogen bonds and hydrophobic interactions [5]. Subsequently, diverse diaryl urea derivatives were developed successively in the past decades, such as linifanib [6], tivozanib [7], and ki8751 [8] (Figure 1). Therein, thieno[3,2-d]pyrimidine derivative S1 bearing diaryl urea moieties at the C-2 positon exhibited significant inhibition of tyrosine kinases, including c-Kit, orphan receptor tyrosine kinase (RET), and FLT3 for its prominent framework [9]. Similarly, the 4-aminoquinazolinyl skeleton, which competitively binds to the ATP binding pocket of intracellular kinase domains and blocks the conduction of downstream signaling networks mediated by tyrosine kinase, is also extensively used in the design of RTKs inhibitors (e.g., gefitinib, erlotinib and S2) [10,11].
Figure 1.

Structures of diaryl urea derivatives.
In light of the abovementioned considerations, and as ongoing efforts to identify new potent antitumor agents, a novel series of 4-aminoquinazolinyl-diaryl urea derivatives 5a–i were designed according to bioisosterism theory to achieve synergistic antitumor effects (Figure 2). In addition, a variety of substituents and aliphatic amino were introduced into the terminal phenyl group and C-4 position on the quinazoline ring to explore the influence of electronic and steric hindrance effect. Furthermore, another novel series of diaryl urea derivatives 13a–l bearing a 5-pyridyl-4-aminopyrimidinyl motif were designed based on scaffold hopping principle, in the hope of attaining structural diversity and optimal cellular potency via improving water solubility. All compounds were synthesized and evaluated for their cytotoxicity against HT-29, H-460, A549, and MDA-MB-231 cancer cell lines. Additionally, enzymatic assay of the most active compound 5a was also presented in this study.
Figure 2.

Design of target compounds.
2. Results and Discussion
2.1. Chemistry
The synthesis of target compounds 5a–i is depicted in Scheme 1. The synthesis of intermediates 3a–b have been described in detail in our previous work [12], so the synthetic method is not listed here. [12]. A Suzuki reaction of 3a,b with commercially available 4-aminophenylboronic acid pinacol ester under nitrogen protection provided the key intermediates 4a,b [13]. A series of substituted aromatic isocyanates 6 and isothiocynates 7 were subsequently prepared by treating the corresponding arylamine with triphosgene (BTC) or 1,4-diazabicyclo[2.2.2]octane (DABCO) without further purification [14,15]. Eventually, target compounds 5a–i were successfully obtained via the reaction of 4a,b with corresponding 6 or 7 in THF at 30 °C, respectively.
Scheme 1.

Synthesis of target compounds 5a–i. Reagents and conditions: (a) urea, 160 °C, 12 h; (b) POCl3, DIPEA, 90 °C, 6 h; (c) NH2(CH2)nR1, THF, TEA, 30 °C, 15 min; (d) 4-aminophenylboronic acid pinacol ester, Na2CO3, Pd(PPh3)2Cl2, THF, N2, r.f., 5 h; (e) 6a–d or 7a–b, THF, 30 °C, 6 h; (f) BTC, 1,4-dioxane, r.f., 8–12 h; (g) (i) DABCO, CS2, toluene, r.t., 12 h; (ii) BTC, CHCl3, r.f., 1 h.
The synthetic route of target compounds 13a–l is outlined in Scheme 2. Dry HCl was bubbled into the mixture of 4-aminobenzonitrile in EtOH and 1,4-dioxane in an ice bath to generate compound 8, which was further replaced with dry NH3 to afford intermediate 9 [16]. 10 was prepared via reaction of 2-(pyridin-2-yl)acetonitrile and excess N,N-dimethylformamide dimethyl acetal (DMF-DMA) at 30 °C for 24 h [17]. Condensation reaction of 9 and 10 in MeOH-H2O solution in the presence of Na2CO3 gave rise to intermediate 11 [18]. Subsequently, 11 condensed with corresponding aromatic isocyanate 6c–n in a similar manner as described for compounds 5a–i to furnish 12a–l. Finally, 12a–l were treated with 20%–30% hydrochloride ethanol solution to provide target compounds 13a–l.
Scheme 2.

Synthesis of target compounds 5a–i. Reagents and conditions: (a) HCl, EtOH-1,4-dioxane, 0 °C, 6 h, r.t., 48 h; (b) NH3, EtOH, 0 °C, 6 h, r.t., 24 h; (c) DMF-DMA, MeOH, 30 °C, 24 h; (d) Na2CO3, MeOH-H2O, 70 °C, 24 h; (e) 6c–n, THF, 30 °C, 6 h; and (f) HCl-EtOH, CHCl3, 1 h.
2.2. Biological Results and Discussion
For summarizing structure-activity relationships (SARs), all target compounds (5a–i and 13a–l) were evaluated for their cytotoxicity against HT-29 (human colon cancer), H-460 (human lung cancer), A549 (human lung cancer), and MDA-MB-231 (human breast cancer) cell lines by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, taking sorafenib as the positive control. The results expressed as IC50 values (μM) are presented in Table 1 and Table 2.
Table 1.
Structures and cytotoxicity of compounds 5a–i against HT-29, H-460, A-549, and MDA-MB-231 cells in vitro.
| Compd. | X | n | R2 | R1 | IC50 (μM) a | |||
|---|---|---|---|---|---|---|---|---|
| HT-29 | H-460 | A-549 | MDA-MB-231 | |||||
| 5a | O | 2 | 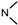 |
2-Cl, 6-CH3 | 0.15 ± 0.14 | 0.089 ± 0.11 | 0.36 ± 0.27 | 0.75 ± 0.65 |
| 5b | O | 2 | |
3,4-(CH3)2 | 1.09 ± 0.21 | 1.50 ± 0.13 | 2.33 ± 1.52 | 2.42 ± 0.28 |
| 5c | O | 3 | 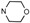 |
2-Cl, 6-CH3 | 0.36 ± 0.72 | 0.27 ± 0.12 | 0.82 ± 0.43 | 1.08 ± 0.15 |
| 5d | O | 3 | |
3,4-(CH3)2 | 1.72 ± 0.37 | 2.93 ± 0.62 | 5.46 ± 0.38 | 3.92 ± 1.24 |
| 5e | O | 3 | |
4-F | 2.64 ± 0.53 | 1.19 ± 0.96 | 3.41 ± 1.27 | 2.72 ± 0.42 |
| 5f | S | 2 | |
4-Cl | 0.24 ± 0.17 | 0.77 ± 0.22 | 2.30 ± 0.18 | 2.59 ± 0.36 |
| 5g | S | 2 | |
4-OCF3 | 1.05 ± 0.012 | 1.28 ± 0.68 | 2.81 ± 0.73 | 3.02 ± 0.51 |
| 5h | S | 3 | |
4-Cl | 0.65 ± 0.33 | 1.02 ± 0.74 | 1.44 ± 0.39 | 3.13 ± 0.85 |
| 5i | S | 3 | |
4-OCF3 | 2.41 ± 0.21 | 2.58 ± 0.74 | 4.86 ± 0.56 | 4.07 ± 1.02 |
| Sorafenib | 3.27 ± 0.32 | 2.15 ± 0.43 | 4.47± 0.28 | 3.81± 0.50 | ||||
a Results are expressed as means ± SD (standard deviation) of three independent experiments.
Table 2.
Structures and cytotoxicity of compounds 13a–l against HT-29, H-460, A-549, and MDA-MB-231 cells in vitro.
| Compd. | R1 | IC50 (μM) a | |||
|---|---|---|---|---|---|
| HT-29 | H-460 | A549 | MDA-MB-231 | ||
| 13a | H | 30.52 ± 2.56 | 28.64 ± 1.08 | 44.83 ± 2.42 | 32.16 ± 1.59 |
| 13b | 2,4-diCH3 | 45.08 ± 1.37 | NA | NA | NA |
| 13c | 2-Cl, 6-CH3 | 12.95 ± 0.68 | 24.66 ± 0.80 | 30.71 ± 1.73 | 25.42 ± 1.14 |
| 13d | 2,6-diCH3 | 18.37 ± 0.45 | 24.62 ± 2.75 | 15.94 ± 0.95 | 28.12 ± 2.44 |
| 13e | 2,6-diF | 35.10 ± 2.81 | ND | NA | 20.94 ± 1.63 |
| 13f | 3-Cl, 4-F | 6.33 ± 0.93 | 15.28 ± 1.19 | 10.49 ± 2.26 | 12.75 ± 2.97 |
| 13g | 3-Cl | 10.07 ± 0.32 | 5.86 ± 1.34 | 15.43 ± 1.02 | 21.95 ± 2.58 |
| 13h | 3-CF3 | ND | ND | ND | ND |
| 13i | 4-OCF3 | 26.58 ± 1.27 | 23.46 ± 2.29 | 16.54 ± 0.70 | 30.65 ± 3.06 |
| 13j | 4-F | 17.95 ± 0.94 | 32.28 ± 2.87 | ND | NA |
| 13k | 3-F | 30.50 ± 3.16 | ND | NA | NA |
| 13l | 2-F | 24.18 ± 1.35 | 32.29 ± 2.63 | NA | 35.56 ± 2.85 |
| Sorafenib | 3.27 ± 0.32 | 2.15 ± 0.43 | 4.47 ± 0.28 | 3.81 ± 0.50 | |
a Results are expressed as means ± SD (standard deviation) of three independent experiments. NA: compound showing IC50 value > 50 μM. ND: Not determined.
As shown in Table 1, compounds 5a–i demonstrated excellent activity towards four tested cell lines with IC50 values ranging from 0.089 to 5.46 μM, and seven of them were more active against tested cell lines than the positive control sorafenib. It was noticeable that all compounds exhibited prominent cytotoxicity against HT-29 superior to sorafenib, reflecting good selectivity for colon cancer. In general, antitumor potency was related to the amino group R1 on side chain, and dimethylamino was more effective than morpholinyl (5a vs. 5c, 5b vs. 5d, 5g vs. 5i), which might be due to its smaller bulk and stronger hydrophilia. Additionally, the substituent R2 on the terminal phenyl ring exerted a major influence on pharmacological activity. Electrophilic groups are beneficial to the improvement of antitumor potency, as might be the reason of compounds 5a, 5c, 5f, and 5h with the Cl group showing eminent cytotoxicity. Conversely, a certain decrease in activity was observed once 3,4-(CH3)2 were introduced, such as compounds 5b and 5d. Interestingly, 2-CH3 and 6-Cl analogue 5a showed best activity, with IC50 values of 0.15, 0.089, 0.36, and 0.75 μM, which were five- to 24-fold more potent than sorafenib respectively. The results indicated that steric hindrance in ortho-position was preferred.
To further verify the optimized structural skeleton and explore the SARs, pharmacological data of compounds 13a–l bearing 4-aminopyrimidinyl moiety are illustrated in Table 2. Unexpectedly, the results were unsatisfied although multiple hydrophilic modifications were performed by introducing pyridine group and further salifying with hydrochloride. The contrast of antitumor potency between 4-aminoquinazoline and 4-aminopyrimidine derivatives suggested that further structural modification might have broken the binding affinity to intracellular kinase domains or switched the binding affinity to other targets, while the 4-aminoquinazolinyl moiety was an excellent skeleton for EGFR inhibitors. The activity toward EGFR kinase of compound 5a was surprised with the IC50 value of 56 nM, indicating that 4-aminoquinazolinyl-diaryl urea derivatives 5a–i were potent EGFR inhibitors (Table 3). Efforts to identify its mechanisms of action and further optimization are ongoing and will be reported in due course.
Table 3.
EGFR and VEGFR2/KDR kinases inhibitory activity of compound 5a in vitro.
| Compd. | IC50 (nM) | |
|---|---|---|
| VEGFR2/KDR | EGFR | |
| 5a | >3000 | 56 |
| Sorafenib | 93 | - |
3. Experimental Section
3.1. Chemistry
Melting points were obtained on a Büchi Melting Point B-540 apparatus (Büchi Labortechnik, Flawil, Switzerland) and were uncorrected. Mass spectra (MS) were taken in electrospray ionization (ESI) mode on an Agilent 1100 LC-MS (Agilent, Palo Alto, CA, USA). Proton (1H) nuclear magnetic resonance spectroscopy were performed using a Bruker ARX-300, 300 or 400 MHz spectrometers (Bruker Bioscience, Billerica, MA, USA) with tetramethylsilane (TMS) as an internal standard. Thin-layer chromatography (TLC) analysis was carried out on silica gel plates GF254 (Qingdao Haiyang Chemical, Qingdao, China). Unless otherwise noted, all common reagents and solvents were used as obtained from commercial suppliers without further purification.
3.2. General Procedure for Preparation of 2-(4-Aminophenyl)-4-aminoquinazolines (4a–b)
Dioxane (100 mL) was added to a solution of sodium carbonate (3.2 g, 0.03 mol) in water (25 mL). Under argon, 2-chloro-4-aminoquinazoline 3 (0.01 mol), 4-aminophenylboronic acid pinacol ester (2.4 g, 0.011 mol) and Pd(PPh3)2Cl2 (0.7 g, 1 mmol) were added to the mixture successively. After refluxing for 6 h, water (50 mL) was added to the reaction mixture. The mixture was extracted by dichloromethane (50 mL × 3). The organic phase was washed by brine (50 mL × 1), dried, and evaporated to yield 4a–b.
N1-(2-(4-aminophenyl)quinazolin-4-yl)-N2,N2-dimethylethane-1,2-diamine (4a). Yield: 73.5%; MS (ESI) m/z: 308.2 [M + H+]. 2-(4-Aminophenyl)-N-(3-morpholinopropyl)quinazolin-4-amine (4b). Yield: 78.1%; MS (ESI) m/z: 364.5 [M + H+].
3.3. General Procedure for Preparation of Componds 5a–i
A mixture of intermediate 4 (1 mmmol) and corresponding aromatic isocyanate 6 or isothiocyanate 7 (1.1 mmol) in dry THF (15 mL) was stirred at 30 °C for 6 h and monitored by TLC. The precipitate was collected by filtration, washed with ether, and purified by silica gel chromatography (MeOH:CH2Cl2 = 20:1) to afford target compounds 5a–i.
1-(2-Chloro-6-methylphenyl)-3-(4-(4-((2-(dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)urea (5a). Yield: 62%; m.p.: 144.5–147.0 °C; MS (ESI) m/z: 474.9 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.08 (s, 1H), 8.2.4 (d, J = 8.8 Hz, 2H ), 8.12 (t, J = 5.2, 4.8 Hz, H), 8.04 (d, J = 8.0 Hz, 1H), 7.95 (s, 1H), 7.56 (m, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.29 (m, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.11 (d, J = 7.6 Hz, 1H), 7.05 (m, 1H), 3.17 (q, 2H), 2.47 (t, J = 8.0 Hz, 2H), 2.38 (s, 3H), 2.21 (s, 6H). Anal. Calcd for C26H27ClN6O (%): C, 65.74; H, 5.73; N, 17.69; Found (%): C, 65.71; H, 5.76; N, 17.68.
1-(4-(4-((2-(Dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)-3-(3,4-dimethylphenyl)urea (5b). Yield: 68%; m.p.: 132.0–134.0 °C; MS (ESI) m/z: 455.2 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.04 (s, 1H), 8.39 (t, J = 7.6 Hz, 1H), 8.24 (d, J = 7.2 Hz, 3H), 8.10 (d, J = 8.4 Hz, 1H), 7.55 (m, 3H), 7.46 (d, J = 8.4 Hz, 2H), 7.28 (t, J = 7.2 Hz, 1H), 6.89 (d, J = 7.2 Hz, 1H), 6.60 (d, J = 7.2 Hz, 1H), 3.17 (q, 2H), 2.47 (t, J = 8.0 Hz, 2H), 2.23 (s, 3H), 2.22 (s, 3H), 2.19 (s, 6H). Anal. Calcd for C27H30N6O (%): C, 71.34; H, 6.65; N, 18.49; Found (%): C, 71.36; H, 6.62; N, 18.47.
1-(2-Chloro-6-methylphenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)urea (5c). Yield: 70%; m.p.: 152.5–154.5 °C; MS (ESI) m/z: 531.1 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.20 (s, 1H), 8.38 (d, J = 8.4 Hz, 2H ), 8.28 (t, J = 4.8, 5.2 Hz, H), 8.18 (d, J = 8.4 Hz, 1H), 8.07 (s, 1H), 7.71 (m, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.42 (m, 1H), 7.35 (d, J = 7.6 Hz, 1H), 7.24 (d, J = 7.6 Hz, 1H), 7.19 (m, 1H), 3.70 (m, 2H), 3.57 (t, J = 8.4 Hz, 4H), 2.40 (m, 6H), 2.27 (s, 3H), 1.87(m, 2H). Anal. Calcd for C29H31ClN6O2 (%): C, 65.59; H, 5.88; N, 15.83; Found (%): C, 65.61; H, 5.85; N, 15.88.
1-(3,4-Dimethylphenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)urea (5d). Yield: 56%; m.p.: 140.7–143.0 °C; MS (ESI) m/z: 510.9 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.18 (s, 1H), 8.54 (t, J = 7.2 Hz, 1H), 8.37 (d, J = 7.2 Hz, 3H), 8.23 (d, J = 8.0 Hz, 1H), 7.69 (m, 3H), 7.59 (d, J = 8.0 Hz, 2H), 7.41 (t, J = 7.2 Hz, 1H), 7.01 (d, J = 7.2 Hz, 1H), 6.73 (d, J = 7.2 Hz, 1H), 3.70 (m, 2H), 3.57 (t, J = 7.2 Hz, 4H), 2.42 (t, J = 7.2 Hz, 2H), 2.37 (t, J = 7.2 Hz, 4H), 2.24 (s, 3H), 2.23 (s, 3H), 1.86 (m, 2H). Anal. Calcd for C30H34N6O2 (%): C, 70.56; H, 6.71; N, 16.46; Found (%): C, 70.55; H, 6.76; N, 16.42.
1-(4-Fluorophenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)urea (5e). Yield: 52%; m.p.: 137.0–139.5 °C; MS (ESI) m/z: 501.2 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 9.91 (s, 2H), 8.51 (t, J = 5.2 Hz, 1H), 8.39 (d, J = 8.8 Hz, 2H), 8.30 (d, J = 8.4 Hz, 1H), 7.72 (m, 4H), 7.60 (d, J = 8.8 Hz, 2H), 7.43 (m, 1H), 7.36 (d, J = 8.4 Hz, 2H), 4.15 (t, J = 6.8 Hz, 2H ), 3.72 (t, J = 6.4 Hz, 4H), 2.43 (t, J = 6.8 Hz, 2H), 2.38 (t, J = 6.4 Hz, 4H), 1.86 (m, 2H). Anal. Calcd for C28H29FN6O2 (%): C, 67.18; H, 5.84; N, 16.79; Found (%): C, 67.15; H, 5.88; N, 16.75.
1-(4-Chlorophenyl)-3-(4-(4-((2-(dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)thiourea (5f). Yield: 65%; m.p.: 131.5–134.0 °C; MS (ESI) m/z: 477.1 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.75 (s, 2H), 8.31 (d, J = 8.4 Hz, 3H), 8.07 (d, J = 8.1 Hz, 1H), 7.68–7.59 (m, 4H), 7.45 (d, J = 8.4 Hz, 2H), 7.37–7.26 (m, 3H), 3.86–3.77 (q, 2H), 2.73 (t, J = 6.4 Hz, 2H), 2.33 (s, 6H). Anal. Calcd for C25H25ClN6S (%): C, 62.95; H, 5.28; N, 17.62; Found (%): C, 62.94; H, 5.27; N, 17.64.
1-(4-(4-((2-(Dimethylamino)ethyl)amino)quinazolin-2-yl)phenyl)-3-(4-(trifluoromethoxy)phenyl)thiourea (5g). Yield: 60%; m.p.: 150.8–153.0 °C; MS (ESI) m/z: 527.2 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.12 (s, 1H), 10.03 (s, 1H), 8.25 (d, J = 8.4 Hz, 2H), 8.18 (t, J = 6.8 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 7.58 (m, 3H), 7.47 (d, J = 8.8 Hz, 2H), 7.31 (m, 3H), 6.96 (d, J = 8.4 Hz, 1H), 3.38 (q, 2H), 2.56 (t, J = 7.2 Hz, 2H), 2.23 (s, 6H). Anal. Calcd for C26H25F3N6OS (%): C, 59.30; H, 4.79; N, 15.96; Found (%): C, 59.32; H, 4.76; N, 15.94.
1-(4-Chlorophenyl)-3-(4-(4-((3-morpholinopropyl)amino)quinazolin-2-yl)phenyl)thiourea (5h). Yield: 63%; m.p.: 145.5–148.0 °C; MS (ESI) m/z: 533.8 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.90 (s, 2H), 8.42 (d, J = 8.8 Hz, 3H), 8.24 (d, J = 8.4 Hz, 1H), 7.73 (m, 4H), 7.65 (d, J = 8.4 Hz, 2H), 7.44 (m, 1H), 7.38 (d, J = 8.8 Hz, 2H), 3.71 (m, 2H), 3.58 (t, J = 8.4 Hz, 4H), 2.43 (t, J = 7.2 Hz, 2H), 2.38 (t, J = 8.4 Hz, 4H), 1.88 (m, 2H). Anal. Calcd for C28H29ClN6OS (%): C, 63.09; H, 5.48; N, 15.76; Found (%): C, 63.05; H, 5.47; N, 15.79.
1-(4-(4-((3-Morpholinopropyl)amino)quinazolin-2-yl)phenyl)-3-(4-(trifluoromethoxy)phenyl)thiourea (5i). Yield: 59%; m.p.: 164.5–166.5 °C; MS (ESI) m/z: 583.9 [M + H+]; 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.25 (s, 1H), 10.16 (s, 1H), 8.45 (d, J = 8.8 Hz, 2H), 8.35 (t, J = 7.2 Hz, 1H), 8.22 (d, J = 8.4 Hz, 1H), 7.74 (m, 3H), 7.64 (d, J = 8.8 Hz, 2H), 7.48 (m, 3H), 7.11 (d, J = 8.0 Hz, 1H), 3.73 (m, 2H), 3.59 (t, J = 4.4 Hz, 4H), 2.44 (t, J = 7.2 Hz, 2H), 2.39 (t, J = 4.4 Hz, 4H), 1.89 (m, 2H). Anal. Calcd for C29H29F3N6O2S (%): C, 59.78; H, 5.02; N, 14.42; Found (%): C, 59.75; H, 5.01; N, 14.47.
3.4. General Procedure for Preparation of Aromatic Isocyanates (6a–n)
Appropriate aromatic amine (0.02 mol) was added slowly to a stirred solution of BTC (3.0 g, 0.01 mol) in 1,4-dioxane (30 mL) at room temperature. After refluxing for 8-12 h, excess 1,4-dioxane was evaporated. The residue was distilled under reduced pressure to afford compounds 6a–n.
1-Chloro-4-isocyanatobenzene (6a). Purity: 78.2%; b.p.: 110–114 °C (30–40 mmHg).
4-Isocyanato-1,2-dimethylbenzene (6b). Purity: 80.5%; b.p.: 59–65 °C (30–40 mmHg).
1-Chloro-2-isocyanato-3-methylbenzene (6c). Purity: 80.8%; b.p.: 116–118 °C (30–40 mmHg).
1-Fluoro-4-isocyanatobenzene (6d). Purity: 84.2%; b.p.: 94–95 °C (30–40 mmHg).
1-Fluoro-3-isocyanatobenzene (6e). Purity: 83.6%; b.p.: 85–91 °C (30–40 mmHg).
1-Fluoro-2-isocyanatobenzene (6f). Purity: 84.3%; b.p.: 76–80 °C (30–40 mmHg).
1-Chloro-3-isocyanatobenzene (6g). Purity: 80.1%; b.p.: 125–128 °C (30–40 mmHg).
2-Chloro-1-fluoro-4-isocyanatobenzene (6h). Purity: 85.2%; b.p.: 93–98 °C (30–40 mmHg).
1,3-Difluoro-2-isocyanatobenzene (6i). Purity: 80.9%; b.p.: 67–71 °C (30–40 mmHg).
2-Isocyanato-1,3-dimethylbenzene (6j). Purity: 76.9%; b.p.: 67–71 °C (30–40 mmHg).
1-Isocyanato-2,4-dimethylbenzene (6k). Purity: 77.5%; b.p.: 71–77 °C (30–40 mmHg).
1-Isocyanato-3-(trifluoromethyl)benzene (6l). Purity: 83.2%; b.p.: 105–111 °C (30–40 mmHg).
1-Isocyanato-4-(trifluoromethoxy)benzene (6m). Purity: 81.0%; b.p.: 113–116 °C (30–40 mmHg).
Isocyanatobenzene (6n). Purity: 80.2%; b.p.: 45–50 °C (30–40 mmHg).
3.5. General Procedure for Preparation of Aromatic Isothiocyanates (7a,b)
Appropriate aromatic amine (0.02 mol) and DABCO (6.7 g, 0.06 mol) were dissolved in toluene (25 mL) at room temperature. Under stirring, CS2 (4.6 g, 0.06 mol) was added dropwise to the solution within 20 min. After stirring at room temperature for 12 h, the precipitate was collected by filtration and washed with toluene. The residue was dried and suspended in CHCl3 (25 mL), and BTC (19.6 g, 0.066 mol) in CHCl3 (25 mL) was added dropwise to the mixture slowly in an ice bath. Then the mixture was stirred for 1 h at room temperature and refluxed for 1 h. The mixture was filtered and evaporated to give compounds 7a,b.
1-Chloro-4-isothiocyanatobenzene (7a). Purity: 77.5%.
1-Isothiocyanato-4-(trifluoromethoxy)benzene (7b). Purity: 79.8%.
3.6. Ethyl 4-aminobenzimidate Dihydrochloride (8)
Dry hydrogen chloride was bubbled into the mixture of 4-aminobenzonitrile (15 g, 0.127 mol), 1,4-dioxane (100 mL) and anhydrous ethanol (100 mL) for 6 h in an ice bath. Then the mixture was sealed and stirred at room temperature for 48 h. After completion, the solution was concentrated under reduced pressure and the semisolid residue was diluted with anhydrous ether (200 mL). The suspension was filtered, washed with anhydrous ether and dried to give the title compound 8 as a white solid (29.3 g, 97.7%). MS (ESI) m/z: 164.9 [M + H+].
3.7. 4-Aminobenzimidamide Hydrochloride (9)
Dry ammonia was bubbled into the mixture of compound 8 (29.3 g, 0.123 mol) and anhydrous ethanol (200 mL) for 6 h in an ice bath. Then the mixture was sealed and stirred at room temperature for 24 h. Then the solution was concentrated under a reduced pressure and the semisolid residue was diluted with anhydrous ether (200 mL). The suspension was filtered, washed with anhydrous ether, and dried to give the title compound 9 as a white solid (20.2 g, 95.3%). MS (ESI) m/z: 135.9 [M + H+].
3.8. 3-(Dimethylamino)-2-(pyridin-2-yl)acrylonitrile (10)
A mixture of 2-(pyridin-2-yl)acetonitrile (15.0 g, 0.127 mol), DMF-DMA (30 mL, 0.229 mol) and methanol (36 mL) was stirred at 30 °C for 24 h. The solvent was evaporated, moderate methanol and ethyl acetate was added in. The solution was concentrated under a reduced pressure to remove excess DMF-DMA completely. The residue was poured into petroleum ether (100 mL) and stirred for 6 h. The suspension was separated by filtration and washed with ether to give compound 10 as a dark red solid (18.3 g, 83.2%). MS (ESI) m/z: 174.0 [M + H+].
3.9. 2-(4-Aminophenyl)-5-(pyridin-2-yl)pyrimidin-4-amine (11)
A mixture of compound 9 (20.2 g, 0.118 mol ), compound 10 (17.0 g, 0.098 mol), sodium carbonate (12.5 g, 0.0118 mol), methnol (200 mL) and water (60 mL) was heated to 70 °C for 24 h. The solution was concentrated and added to water (200 mL). The precipitate was collected by filtration, washed with ether, and recrystallized with ethnol to afford compound 11 as a pale white solid (13.9 g, 53.8%). MS (ESI) m/z: 264.1 [M + H+].
3.10. General Procedure for Preparation of Compounds 12a–l and 13a–l
A mixture of intermediate 11 (1 mmmol) and corresponding aromatic isocyanate 6 (1.1 mmol) in dry THF (15 mL) was stirred at 30 °C for 6 h and monitored by TLC. The precipitate was collected by filtration, washed with ether, and dried to afford compounds 12a–l without further purification. At room temperature, 20%–30% hydrochloride ethanol solution (1 mL) was added dropwise to a stirred solution of compound 12 dissolved in CHCl3 (10 mL). The suspension was stirred for 1 h, filtered and dried to give the corresponding compounds 13a–l.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-phenylurea dihydrochloride (13a). Yield: 79%; m.p.: 303.0–306.0 °C; MS (ESI) m/z: 383.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.41 (s, 1H), 9.11 (s, 1H), 8.93 (s, 2H), 8.77 (s, 1H), 8.68 (d, J = 4.4 Hz, 1H), 8.32 (d, J = 8.4 Hz, 2H), 8.10 (d, J = 8.0 Hz, 1H), 7.94 (t, J = 7.9 Hz, 1H), 7.58 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.4 Hz), 7.38–7.32 (m, 1H), 7.30 (t, J = 7.7 Hz, 2H), 7.00 (t, J = 6.9 Hz). Anal. Calcd for C22H20Cl2N6O (%): C, 58.03; H, 4.43; N, 18.46; Found (%): C, 58.07; H, 4.46; N, 18.42.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2,4-dimethylphenyl)urea dihydrochloride (13b). Yield: 65%; m.p.: 286.0–288.5 °C; MS (ESI) m/z: 411.3 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.77 (s, 1H), 8.89 (s, 1H), 8.74 (d, J = 4.6 Hz, 1H), 8.25(d, J = 8.6 Hz, 2H), 8.22 (d, J = 7.8 Hz, 1H), 8.05 (t, J = 7.9 Hz, 1H), 7.72(d, J = 8.6 Hz, 2H), 7.66 (d, J = 8.1 Hz, 1H), 7.52 (t, J = 4.8 Hz, 1H), 7.02 (s, 1H), 6.98 (d, J = 8.2 Hz, 2H), 2.24 (s, 6H). Anal. Calcd for C24H24Cl2N6O (%): C, 59.63; H, 5.00; N, 17.39; Found (%): C, 59.60; H, 5.06; N, 17.34.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(6-chloro-2-methylphenyl)urea dihydrochloride (13c). Yield: 72%; m.p.: 252.0–254.0 °C; MS (ESI) m/z: 431.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.78 (s, 1H), 8.85 (s, 1H), 8.74 (d, J = 4.2 Hz, 1H), 8.56 (s, 1H), 8.26 (d, J = 8.7 Hz, 2H), 8.22 (d, J = 8.1 Hz, 1H), 8.04 (t, J = 6.7 Hz, 1H), 7.72 (d, J = 8.9 Hz, 2H), 7.54–7.50 (m, 1H), 7.36 (d, J = 5.4 Hz, 1H), 7.27–7.18 (m, 2H), 2.28 (s, 3H). Anal. Calcd for C23H21Cl3N6O (%): C, 54.83; H, 4.20; N, 16.68; Found (%): C, 54.84; H, 4.23; N, 16.65.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2,6-dimethylphenyl)urea dihydrochloride (13d). Yield: 76%; m.p.: 271.0–274.0 °C; MS (ESI) m/z: 411.2 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.83 (s, 1H), 8.85 (s, 1H), 8.74(d, J = 4.2 Hz, 1H), 8.34 (s, 1H), 8.23 (d, J = 9.3 Hz, 2H), 8.21 (d, J = 8.1 Hz, 1H), 8.05(t, J = 7.8 Hz, 1H), 7.72 (d, J = 9.0 Hz, 2H), 7.54–7.50 (m, 1H), 7.08 (s, 3H), 2.23 (s, 6H). Anal. Calcd for C24H24Cl2N6O (%): C, 59.63; H, 5.00; N, 17.39; Found (%): C, 59.67; H, 5.02; N, 17.36.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2,6-difluorophenyl)urea dihydrochloride (13e). Yield: 68%; m.p.: 279.5–281.0 °C; MS (ESI) m/z: 419.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.78 (s, 1H), 8.88 (s, 1H), 8.73 (d, J = 4.7 Hz, 1H), 8.53 (s, 1H), 8.40 (s, 2H), 8.24 (d, J = 8.8 Hz, 2H), 8.20 (d, J = 9.3 Hz, 1H), 8.04 (t, J = 7.8 Hz, 1H), 7.72 (d, J = 8.6 Hz, 2H), 7.54–7.49 (m, 1H), 7.32–7.25 (m, 3H). Anal. Calcd for C22H18Cl2F2N6O (%): C, 53.78; H, 3.69; N, 17.10; Found (%): C, 53.75; H, 3.72; N, 17.08.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(3-chloro-4-fluorophenyl)urea dihydrochloride (13f). Yield: 80%; m.p.: 289.5–291.5 °C; MS (ESI) m/z: 435.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.69 (s, 1H), 9.55 (s, 1H), 8.88 (s, 1H), 8.74 (d, J = 4.4 Hz, 1H), 8.25 (d, J = 8.8 Hz, 2H), 8.21 (d, J = 8.2 Hz, 1H), 8.05 (t, J = 7.8 Hz, 1H), 7.85–7.82 (m, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.54–7.49 (m, 1H), 7.40–7.34 (m, 2H). Anal. Calcd for C22H18Cl3FN6O (%): C, 52.04; H, 3.57; N, 16.55; Found (%): C, 52.05; H, 3.52; N, 16.56.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(3-chlorophenyl)urea dihydrochloride (13g). Yield: 71%; m.p.: 270.0–272.5 °C; MS (ESI) m/z: 417.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.89 (s, 1H), 9.71 (s, 1H), 8.86 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.25 (d, J = 8.7 Hz, 2H), 8.21 (d, J = 8.2 Hz, 1H), 8.05 (t, J = 7.9 Hz, 1H), 7.73 (d, J = 8.8 Hz, 2H), 7.72 (s, 1H), 7.55–7.51 (m, 1H), 7.33–7.30 (m, 2H), 7.04 (d, J = 5.8 Hz, 1H). Anal. Calcd for C22H19Cl3N6O (%): C, 53.95; H, 3.91; N, 17.16; Found (%): C, 53.94; H, 3.92; N, 17.14.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(4-(trifluoromethyl)phenyl)urea dihydrochloride (13h). Yield: 65%; m.p.: 267.0–269.5 °C; MS (ESI) m/z: 451.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.78 (s, 1H), 9.74 (s, 1H), 8.89 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.26 (d, J = 8.8 Hz, 2H), 8.21 (m, 2H), 8.05 (s, 2H), 7.74 (d, J = 8.8 Hz, 2H), 7.61–7.50 (m, 4H), 7.35 (d, J = 7.6 Hz, 1H). Anal. Calcd for C23H19Cl2F3N6O (%): C, 52.79; H, 3.66; N, 16.06; Found (%): C, 52.75; H, 3.69; N, 16.05.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(4-(trifluoromethoxy)phenyl)urea dihydrochloride (13i). Yield: 78%; m.p.: 308.5–310.5 °C; MS (ESI) m/z: 467.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.50 (s, 1H), 9.35 (s, 1H), 8.89 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.24 (d, J = 8.7 Hz, 2H), 8.19 (d, J = 8.4 Hz, 1H), 8.04 (t, J = 6.7 Hz, 1H), 7.71 (d, J = 8.7 Hz, 2H), 7.59 (d, 2H), 7.60–7.51 (m, 1H), 7.32 (d, J = 8.1 Hz, 2H). Anal. Calcd for C23H19Cl2F3N6O2 (%): C, 51.22; H, 3.55; N, 15.58; Found (%): C, 51.24; H, 3.59; N, 15.54.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(4-fluorophenyl)urea dihydrochloride (13j). Yield: 75%; m.p.: 264.0–267.0 °C; MS (ESI) m/z: 401.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.80 (s, 1H), 9.47 (s, 1H), 8.87 (s, 1H), 8.75 (d, J = 4.6 Hz, 1H), 8.26 (d, J = 8.7 Hz, 2H), 8.22 (d, J = 7.8 Hz, 1H), 8.07 (t, J = 7.9 Hz, 1H), 7.73 (d, J = 8.8 Hz, 2H), 7.56–7.52 (m, 2H), 7.49 (t, J = 4.8 Hz, 1H), 7.15 (t, J = 8.8 Hz, 2H). Anal. Calcd for C22H19Cl2FN6O (%): C, 55.82; H, 4.05; N, 17.76; Found (%): C, 55.84; H, 4.01; N, 17.79.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(3-fluorophenyl)urea dihydrochloride (13k). Yield: 67%; m.p.: 228.5–231.0 °C; MS (ESI) m/z: 401.1 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 9.89 (s, 1H), 9.73 (s, 1H), 8.86 (s, 1H), 8.74 (d, J = 4.5 Hz), 8.26 (d, J = 8.9 Hz, 2H), 8.22 (d, J = 8.4 Hz, 1H), 8.05 (t, J = 7.8 Hz, 1H), 7.72 (d, J = 8.9 Hz, 2H), 7.55 (s, 1H), 7.53–7.50 (m, 1H), 7.37–7.29 (m, 1H), 7.14 (d, J = 8.1 Hz, 1H), 6.82 (t, J = 8.4 Hz, 1H). Anal. Calcd for C22H19Cl2FN6O (%): C, 55.82; H, 4.05; N, 17.76; Found (%): C, 55.81; H, 4.06; N, 17.72.
1-(4-(4-Amino-5-(pyridin-2-yl)pyrimidin-2-yl)phenyl)-3-(2-fluorophenyl)urea dihydrochloride (13l). Yield: 73%; m.p.: 258.5–261.5 °C; MS (ESI) m/z: 401.0 [M + H+]; 1H-NMR (300 MHz, DMSO-d6) δ (ppm): 10.01 (s, 1H), 8.94 (s, 1H), 8.87 (s, 1H), 8.74 (d, J = 4.5 Hz, 1H), 8.27 (d, J = 9.0 Hz, 2H), 8.22 (d, J = 8.1 Hz, 1H), 8.16–8.03 (m, 1H), 7.74 (d, J = 8.7 Hz, 2H), 7.55–7.50 (m, 1H), 7.29–7.03 (m, 3H). Anal. Calcd for C22H19Cl2FN6O (%): C, 55.82; H, 4.05; N, 17.76; Found (%): C, 55.86; H, 4.03; N, 17.75.
3.11. Evaluation of the Biological Activity
The antitumor activity of compounds 5a–i and 13a–l was evaluated with HT-29, H-460, A549, and MDA-MB-231 by the MTT method in vitro, with sorafenib as positive control. The cancer cells were cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS).
Approximately 4 × 103 cells, suspended in MEM medium, were plated onto each well of a 96-well plate and incubated in 5% CO2 at 37 °C for 24 h. The test compounds were added to the culture medium at the indicated final concentrations and the cell cultures were continued for 72 h. Fresh MTT was added to each well at a final concentration of 5 μg/mL and incubated with cells at 37 °C for 4 h. The formazan crystals were dissolved in 100 μL DMSO per each well, and the absorbency at 492 nm (for the absorbance of MTT formazan) and 630 nm (for the reference wavelength) was measured with the ELISA reader. All of the compounds were tested twice in each of the cell lines. The results expressed as IC50 (inhibitory concentration of 50%) were the averages of two determinations and were calculated by using the Bacus Laboratories Incorporated Slide Scanner (Bliss) software.
3.12. EGFR and VEGFR2/KDR Kinases Assay In Vitro
The target compound 5a was tested for its activity against EGFR and VEGFR2/KDR kinases through the mobility shift assay. All kinase assays were performed in 96-well plates in a 50 μL reaction volume. The kinase buffer contains 50 mM HEPES, pH 7.5, 10 mM MgCl2, 0.0015% Brij-35 and 2 mM DTT. The stop buffer contains 100 mM HEPES, pH 7.5, 0.015% Brij-35, 0.2% Coating Reagent #3, and 50 mM ethylene diamine tetraacetic acid (EDTA). Compounds were diluted to 500 μM by 100% DMSO, then 10 μL of the compounds were transfered to a new 96-well plate as the intermediate plate, and 90 μL kinase buffer was added to each well. Five microliters of each well of the intermediate plate was transferred to a 384-well plate. The following amounts of enzyme and substrate were used per well: kinase base buffer, FAM-labeled peptide, ATP, and enzyme solution. Wells containing the substrate, enzyme, and DMSO without compound were used as the DMSO control. Wells containing just the substrate without enzyme were used as the low control. The compounds were incubated at room temperature for 10 min. Ten microliters of peptide solution was added to each well and incubated at 28 °C for a specified period of time and the reaction stopped by 25 μL of stop buffer. Finally, data was collected using the Caliper program, which converted conversion values to inhibition values.
| Percent inhibition = (max − conversion)/(max − min) × 100, | (1) |
where “max” stands for DMSO control; “min” stands for low control.
4. Conclusions
Taken as a whole, two novel series of diaryl urea derivatives were synthesized and evaluated for their cytotoxicity against four human cancer cell lines (H-460, HT-29, A549, and MDA-MB-231). Our strategy that 4-aminoquinazolinyl moiety was incorporated into the diaryl urea scaffold conveyed cellular potency and resulted in analogues 5a–i demonstrating excellent activity in the single-digit μM range, superior to sorafenib. The preliminary SARs showed that electrophilic groups on terminal phenyl ring and steric hindrance in ortho-position were beneficial to improved antitumor potency. The exploration of detailed SARs led to the identification of EGFR inhibitor 5a as a valuable lead, which possessed prominent activity both in cellular (IC50 = 0.15, 0.089, 0.36, and 0.75 μM, respectively) and enzymatic assay (IC50 = 56 nM against EGFR). Further optimization and investigation based on these structures are in progress.
Acknowledgments
This work was supported by National Natural Science Foundation of China (21002065), Project of Education Department of Liaoning (L2013382), Development Project of Ministry of Education Innovation Team (IRT1073) and Science and Technology Program of Shenyang (No. F15-139-9-02).
Author Contributions
N.J. and D.Z. conceived and designed the experiments; Y.B. and M.N. performed the experiments; Y.W. analyzed the data; X.Z. contributed reagents/materials/analysis tools; N.J. wrote the paper.
Conflicts of Interest
The authors declare no conflict interest.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.Wilhelm S., Carter C., Lynch M., Lowinger T., Dumas J., Smith R.A., Schwartz B., Simantov R., Kelley S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 2.Kan R.C., Farrell A.T., Saber H., Tang S., Williams G., Jee J.M., Liang C., Booth B., Chidambaram N., Morse D., et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006;12:7271–7278. doi: 10.1158/1078-0432.CCR-06-1249. [DOI] [PubMed] [Google Scholar]
- 3.Keating G.M., Santoro A. Sorafenib. Drugs. 2009;69:223–240. doi: 10.2165/00003495-200969020-00006. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari S.M., Politti U., Spisni R., Materazzi G., Baldini E., Ulisse S., Miccoli P., Antonelli A., Fallahi P. Sorafenib in the treatment of thyroid cancer. Expert Rev. Anticancer Ther. 2015;15:863–874. doi: 10.1586/14737140.2015.1064770. [DOI] [PubMed] [Google Scholar]
- 5.Kim D.H., Sim T. Novle small molecule Raf kinase inhibitors for targeted cancer therapeutics. Arch. Pharm. Res. 2012;35:605–615. doi: 10.1007/s12272-012-0403-5. [DOI] [PubMed] [Google Scholar]
- 6.Aversa C., Leone F., Zucchini G., Serini G., Geuna E., Milani A., Valdembri D., Martinello R., Montemurro F. Linifanib: current status and future potential in cancer therapy. Expert Rev. Anticancer Ther. 2015;15:677–687. doi: 10.1586/14737140.2015.1042369. [DOI] [PubMed] [Google Scholar]
- 7.Jamil M.O., Hathaway A., Mehta A. Tivozanib: Status of development. Curr. Oncol. Rep. 2015;17 doi: 10.1007/s11912-015-0451-3. [DOI] [PubMed] [Google Scholar]
- 8.Kubo K., Shimizu T., Ohyama S., Murooka H., Iwai A., Nakamura K., Hasegawa K., Kobayashi Y., Takahashi N., Takahashi K., Kato S., et al. Novel potent orally active selective VEGFR-2 tyrosine kinase inhibitors: synthesis, structure-activity relationships, and antitumor activities of N-phenyl-N'-{4-(4-quinolyloxy)phenyl}ureas. J. Med. Chem. 2005;48:1359–1366. doi: 10.1021/jm030427r. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z., Wang Y., Lin H., Zuo D., Wang L., Zhao Y., Gong P. Design, synthesis and biological evaluation of novel thieno[3,2-d] pyrimidine derivatives containing diaryl urea moiety as potent antitumor agents. Eur. J. Med. Chem. 2014;85:215–227. doi: 10.1016/j.ejmech.2014.07.099. [DOI] [PubMed] [Google Scholar]
- 10.Chen J.N., Wang X.F., Li T., Wu D.W., Fu X.B., Zhang G.J., Shen X.C., Wang H.S. Design, synthesis, and biological evaluation of novel quinazolinyl-diaryl urea derivatives as potential anticancer agents. Eur. J. Med. Chem. 2016;107:12–25. doi: 10.1016/j.ejmech.2015.10.045. [DOI] [PubMed] [Google Scholar]
- 11.Singh K., Sharma P.P., Kumar A., Chaudhary A., Roy R.K. 4-Aminoquinazoline analogs: A novel class of anticancer agents. Mini Rev. Med. Chem. 2013;13:1177–1194. doi: 10.2174/1389557511313080006. [DOI] [PubMed] [Google Scholar]
- 12.Jiang N., Zhai X., Zhao Y., Liu Y., Qi B., Tao H., Gong P. Synthesis and biological evaluation of novel 2-(2-arylmethylene) hydrazinyl-4-aminoquinazoline derivatives as potent antitumor agents. Eur. J. Med. Chem. 2012;54:534–541. doi: 10.1016/j.ejmech.2012.05.039. [DOI] [PubMed] [Google Scholar]
- 13.Lin Q., Meloni D., Pan Y., Xia M., Rodgers J., Shepard S., Li M., Galya L., Metcalf B., Yue T.Y., et al. Enantioselective synthesis of janus kinase inhibitor INCB018424 via an organocatalytic aza-Michael reaction. Org. Lett. 2009;11:1999–2002. doi: 10.1021/ol900350k. [DOI] [PubMed] [Google Scholar]
- 14.Liu D., Tian Z., Yan Z., Wu L., Ma Y., Wang Q., Liu W., Zhou H., Yang C. Design, synthesis and evaluation of 1,2-benzisothiazol-3-one derivatives as potent caspase-3 inhibitors. Bioorg. Med. Chem. 2013;21:2960–2967. doi: 10.1016/j.bmc.2013.03.075. [DOI] [PubMed] [Google Scholar]
- 15.Du X, Xu X, Fu Y., Lou Y., Xu Z. Overcoming aromatic isothiocyanate synthesis difficulties with a method avoiding use of thiophosgene. Chin. J. Pestic. 2004;43:78–79. [Google Scholar]
- 16.Torkelson S.M., Vojkovsly T. Factor VIIa Inhibitor. WO2005121102. 2005 Dec 22;
- 17.Christopher A.L., John L.L. Bioisosteric prototype design of biaryl imidazolyl and triazolyl competitive histamine H2-Receptor antagonists. J. Med. Chem. 1986;29:2154–2163. doi: 10.1021/jm00161a005. [DOI] [PubMed] [Google Scholar]
- 18.Li M., Guo W., Wen L., Yang H. Synthesis of enaminones and their utility in organic synthesis. Chin. J. Org. Chem. 2006;26:1192–1207. [Google Scholar]