

Abstract
Ampakine compounds have been shown to reverse opiate-induced respiratory depression by activation of amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors. However, their pharmacological exploitations are hindered by low blood-brain barrier (BBB) permeability and limited brain distribution. Here, we explored whether thiamine disulfide prodrugs with the ability of “lock-in” can be used to solve these problems. A series of thiamine disulfide prodrugs 7a–7f of ampakine compound LCX001 was synthesized and evaluated. The trials in vitro showed that prodrugs 7e, 7d, 7f possessed a certain stability in plasma and quickly decomposed in brain homogenate by the disulfide reductase. In vivo, prodrug 7e decreased the peripheral distribution of LCX001 and significantly increased brain distribution of LCX001 after i.v. administration. This compound showed 2.23- and 3.29-fold greater increases in the AUC0-t and MRT0-t of LCX001 in brain, respectively, than did LCX001 itself. A preliminary pharmacodynamic study indicated that the required molar dose of prodrug 7e was only one eighth that of LCX001 required to achieve the same effect in mice. These findings provide an important reference to evaluate the clinical outlook of ampakine compounds.
Keywords: opiate, respiratory depression, ampakines, thiamine disulfide prodrugs, brain distribution
1. Introduction
The use of opioid analgesics is still the most effective way to manage moderate to severe pain in the clinic [1]. Nevertheless, opiate-induced respiratory depression is a life-threatening condition and a leading cause of death that may arise not only from overdoses, but also during routine procedures supervised by clinicians, including surgical anaesthesia, post-operative analgesia, and as a result of routine out-patient management of pain from cancer, accidents, or other illnesses [2,3,4,5]. Naloxone and related opioid antagonists are currently used to counter respiratory depression in an emergency setting [6]. However, those agents reverse analgesia, so selective antagonism of the respiratory depressive effects of opioids without decreasing their analgesic effects would be a major step toward improving opioid safety.
Recent studies demonstrated that AMPA receptor was a critical component for the generation of respiratory rhythm within the pre-Botzinger complex [7,8,9,10], and was becoming an ideal target for reversing respiratory depression. Ampakine compounds, developed by Cortex Pharmaceuticals, such as CX717, CX1739 and CX1942 etc., had been proved to modulate respiratory drive and rhythmogenesis positively by activation of AMPA receptor [11,12,13,14,15,16,17,18]. Most advanced ampakine compounds are CX717 and CX1739 [19], both of which are currently in Phase II clinical trials. Studies demonstrated that a single oral dose of 1500 mg of CX717 had positive effects on respiratory depression induced by pain relieving opiates, without affecting the pain relieving effects [14]. The reversal degree of the basal respiratory rate was similar to that obtained with the opioid antagonist, naloxone. Similar studies in respiratory depression with CX1739 at a high dose of 900 mg have also been planned [20].
Structurally these compounds belong to the benzamide derivative type 1 (Figure 1), but their specific structures remained unknown. According to Cortex’s patents [21,22], we found that compound 2 (Figure 1, LCX001) was one of the most active compounds, with the best electrophysiological effect giving a 21% increase in amplitude of the field excitatory postsynaptic potential (EPSP) in the rat dentate gyrus and producing a 100% inhibition of damphetamine- stimulated locomotion in the behavioral testing [21], so we infered that LCX001 was probably an analogue of the clinical compounds or even one of them. To date, no further developments for these compounds were reported for this indication. We assumed that besides poor absorption the low blood-brain barrier permeability (tPSA of LCX001: 74.49) and limited brain distribution might hinder the development of the compounds. As a result, high-dose use of drugs and non-targeting distribution could cause serious safety problems. In 2007, Food and Drug Administration (FDA) even rejected an investigational new drug (IND) application for CX717 as a treatment for attention-deficit hyperactivity disorder (ADHD) because of its toxicity after chronic dosing at very high dose levels in animals.
Figure 1.

Structure of benzamide derivatives, LCX001, thiamine disulfide, DOPA prodrug, naproxen prodrug, ibuprofen prodrug and the designed prodrugs of LCX001.
The thiamine disulfide system (TDS), has been widely applied as a classic brain-targeted prodrug strategy to a variety of drugs such as DOPA, naproxen, ibuprofen (Figure 1) etc. [23,24,25]. In this approach, the increased lipophilic nature of the prodrugs could facilitate passive diffusion across the BBB; subsequently, TDS was reduced and ring-closed to be a thiazolium by disulfide reductase once across the BBB, and then the prodrugs with this system were “locked” in the brain where they can provide sustained release of the active drug via hydrolysis (Figure 2) [23].
Figure 2.

Sequential metabolism and brain “lock-in” pathways of brain-targeted thiamine disulfide prodrugs of LCX001.
In this paper, we introduced the lipophilic TDS with “lock-in” ability to mediate the penetration of LCX001 and designed some brain-targeted prodrugs 7a–7f (Figure 1), which were expected to have synergism and attenuation effects and also provide an important reference value to evaluate the clinical outlook of ampakine compounds. These designed compounds were sequentially synthesized. The in vitro stabilities of these prodrugs were discussed. Furthermore, this account also included the biodistribution and pharmacodynamic study of these prodrugs in mice after i.v. administration.
2. Results and Discussion
2.1. Chemistry
The synthetic routes for the prodrugs 7a–7f are shown in Scheme 1. As shown, N,4-dimethyl-5-(2-(hydroxy)ethyl)thiazolium iodide (9) can be obtained through methylation of 5-(2-hydroxyethyl)-4-methylthiazole (8). Halohydrocarbons 10a–10f and sodium thiosulfate pentahydrate were mixed and refluxed for 7 h to afford intermediates 11a–11f. Thiamine disulfide derivatives 12a–12f were obtained by the reaction of N,4-dimethyl-5-(2-(hydroxy)ethyl) thiazolium iodide (9) with intermediates 11a–11f in the presence of NaOH at room temperature. Thiamine disulfide derivatives 12a–12f further react with glutaric anhydride to give the requisite intermediate 13a–13f. These compounds were conjugated with LCX001 in the presence of EDCI, HOBT, DMAP and triethylamine to afford the prodrugs 7a–7f. The structures of all the synthesized prodrugs 7a–7f have been confirmed by NMR, and MS, and all the corresponding data can be found in the Experimental Section.
Scheme 1.

General synthetic route for synthesis of compounds 7a–7f. Reagents and conditions: (i) iodomethane, 50 °C, 2 h, 96.2%; (ii) Na2S2O3·5H2O , ethanol/H2O, 110 °C, 7 h; (iii) NaOH, H2O, rt, 1 h, two steps, 13.3%–70.5%; (iv) glutaric anhydride, DMAP, DCM, 50 °C, 5 h; (v) LCX001, DMAP, HOBT, EDCI, TEA, DCM, 50 °C, 5 h, 38.3%–57.5%.
2.2. Metabolism Studies of the Prodrugs
2.2.1. Metabolic Stability
To determine the plasma stability and brain stability of these prodrugs, we assayed the pharmacokinetics of the prepared compounds 7a–7f in mice plasma extract and brain homogenate at 37 °C. The pseudo-first order rate constants of disappearance (Ke) of the prodrugs and their half lives (t1/2) which were calculated by linear regression of Ln of peak area against time in minutes are listed in Table 1.
Table 1.
Metabolic stability of compounds 7a–7f in mice plasma extract and brain homogenate at 37 °C.
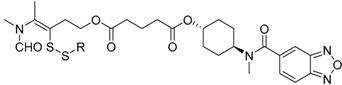
| Compound | R | Biological Matrix | Kinetic Constants | |
|---|---|---|---|---|
| Ke (min−1) (×10−2) | t1/2 (min) | |||
| 7a | 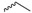 |
Plasma | 31.1 | 2.23 |
| Brain | 19.9 | 3.48 | ||
| 7b |  |
Plasma | 7.06 | 9.82 |
| Brain | 6.82 | 10.17 | ||
| 7c |  |
Plasma | 6.77 | 10.24 |
| Brain | 4.05 | 17.10 | ||
| 7d |  |
Plasma | 4.50 | 15.41 |
| Brain | 6.34 | 10.93 | ||
| 7e | 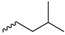 |
Plasma | 4.34 | 15.97 |
| Brain | 3.69 | 18.78 | ||
| 7f | 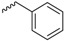 |
Plasma | 4.48 | 15.47 |
| Brain | 5.78 | 11.99 | ||
In plasma, compounds 7a–7f decomposed at a high rate. This instability is due to the existence of ester and disulfide linkages in 7a–7f. The stability of these prodrugs had a descending trend of 7d (t1/2 = 15.41 min) > 7c (t1/2 = 10.24 min) > 7b (t1/2 = 9.82 min) > 7a (t1/2 = 2.23 min), indicating that the longer the carbon chain, the better the plasma stability. Larger steric hinderance produced by longer carbon chain hindered the reduction of the disulfide bond. The half-lives of 7d and 7e were similar, suggesting that substitution in the side chain had little influence on the plasma stability. We tried to further increase the plasma stability of the compounds by introducing the benzyl group, but no effect was obvious. In spite of a relatively high metabolic rate, it is reasonable to give enough time (t1/2 = 15.41 min–15.97 min) for the compounds 7d, 7e, 7f to be distributed and reach the brain before complete decomposition.
In brain homogenate, all the compounds decomposed swiftly (t1/2 = 3.48 min–18.78 min) probably due to the abundance of disulfide reductase in brain. They underwent the expected reduction and cyclization of the thiamine disulfide component to the corresponding thiazolium in brain homogenate. These compounds could be reduced to strongly polar products with high rates, and thus, contributed to the “lock-in” effect in the brain before they were effluxed to the periphery.
2.2.2. Release of the Parent Drug by Prodrugs in Brain Homogenate
To study the release of LCX001 by the prodrugs in brain, the peak in the LC-MS/MS corresponding to the parent compound was also analyzed. A certain increase in peak area over time was observed, indicating that the prodrugs were indeed converted to the parent drug by esterases in mice brain homogenate. Although the deliverance of LCX001 seemed not fast, the concentration increased steadily as time went by, which might indicate sustained release of LCX001 by the prodrugs in the brain. The release rates of different produgs were similar, which indicated that the release process of parent drug was unlikely to be seriously affected by the formation of a thiazolium cation. Peak integrals were normalized by dividing by the area for the peak of each prodrug itself at 5 min. The ratios are shown in Figure 3.
Figure 3.

Deliverence of LCX001 by the prodrugs in brain homogenate.
Several conclusions could be drawn from the above results: first, the half-lives of the prodrugs were mainly influenced by the length of the carbon chains linked to the disulfide functional group. Second, disulfide reductase produced reduction and ring closure of compounds 7a–7f predominated over their clearance in brain with high efficiency, since the decomposition of the prodrugs consisted of reduction and hydrolysis, and the hydrolysis occurs at a relatively low rate. Finally, the release process of the parent drug was unlikely to be seriously affected by the formation of a thiazolium ion. As the above in vitro studies demonstrated that prodrugs 7e, 7d, 7f possess preliminary and favorable physicochemical properties, we selected compound 7e as a model compound to proceed with an in vivo study.
2.3. In Vivo Studies
2.3.1. Pharmacokinetics in Plasma of the Prodrug 7e
To understand the in vivo behavior of the prodrug, we assessed the plasma pharmacokinetics of LCX001 and prodrug 7e in mice (Figure 4).
Figure 4.

Concentration curve of LCX001 in plasma versus time after administration of LCX001 and prodrug 7e. Error bars show the value of SD (n = 3).
The compounds were injected through the caudal vein of the mice with a single dose equivalent to 1 mg/kg body weight of LCX001, and then blood was collected to analyze the concentration of LCX001 at different intervals by an LC-MS/MS method. The pharmacokinetic parameters of LCX001 in blood are reported in Table 2.
Table 2.
Pharmacokinetic parameters of LCX001 in plasma after administration of LCX001 and prodrug 7e (n = 3).
| Compound | Cmax (ng/mL) | AUC0-t (h·ng/mL) | Tmax (h) | t1/2 (h) | MRT0-t (h) |
|---|---|---|---|---|---|
| LCX001 | 833.67 ± 79.85 | 951.66 ± 115.52 | 0.08 | 0.53 ± 0.17 | 1.08 ± 0.04 |
| 7e | 590.36 ± 99.90 * | 384.16 ± 39.21 * | 0.08 | 0.48 ± 0.20 | 0.83 ± 0.03 * |
* p < 0.05 with respect to naked LCX001.
Compared with the concentration curve of LCX001 after administration of underivatized LCX001, prodrug 7e exhibited significantly lower concentrations at different time intervals. Free LCX001 of prodrug 7e and LCX001 presented with an area under the concentration-time profile (AUC0-t) ratio of 0.40 and the mean residence times (MRT) of LCX001 in plasma after i.v. administration of 7e was 0.77 times that of LCX001, indicating that 7e could increase brain distribution because of its increased lipophilicity and be cleared more quickly than LCX001 due to the strongly polar thiazolium cation produced by reduction of the corresponding thiamine disulfides.
2.3.2. Brain Distribution of Prodrug 7e
To further evaluate the possibility of prodrug 7e being transported across the BBB, the brain distribution of prodrug 7e and LCX001 after injection were detected at fixed time intervals. The concentrations of LCX001 in brain versus time curves are displayed in Figure 5. The curves show that prodrug 7e exhibited much higher distribution in brain. The pharmacokinetic parameters of LCX001 in brain are reported in Table 3. The data showed that Cmax of LCX001 in brain after i.v. administration of prodrug 7e was not significantly improved, but the AUC0-t and MRT0-t of LCX001 was significantly higher than that after the injection of the parent compound LCX001. The AUC0-t for prodrug 7e was enhanced to 2.23 times that of LCX001. The MRT0-t was increased to 3.29 times that of LCX001. The curves displayed that LCX001 could be quickly metabolized or effluxed while prodrug 7e showed a certain stability. This agreed well with the fact that the “lock-in” effect and sustained release effect of the prodrug 7e could effectively delay the clearance of LCX001.
Figure 5.

Concentration curve of LCX001 in brain versus time after administration of LCX001 and prodrug 7e. Error bars show the value of SD (n = 3).
Table 3.
Pharmacokinetic parameters of LCX001 in brain after administration of LCX001 and prodrug 7e (n = 3).
| Compound | Cmax (ng/g) | AUC0-t (h·ng/g) | Tmax (h) | t1/2 (h) | MRT0-t (h) |
|---|---|---|---|---|---|
| LCX001 | 609.40 ± 73.66 | 504.32 ± 204.71 | 0.08 | 0.59 ± 0.08 | 0.70 ± 0.22 |
| 7e | 454.53 ± 74.69 | 1125.49 ± 336.57 * | 0.17 | 2.50 ± 0.42 * | 2.30 ± 0.73 * |
* p < 0.05 with respect to naked LCX001.
2.4. Pharmacodynamic Study of Prodrug 7e
To investigate the effect of 7e on opiate-induced respiratory depression, we proceeded with a preliminary pharmacodynamic study in mice. The potent opiate receptor agonist 030418 (Ki = 0.91 nM for μ-opioid receptor) [26] was administered to induce fatal apneas. Varying doses of prodrug 7e were administered 10 min before 030418 administration. The effect of prodrug 7e and LCX001 on opiate-induced respiratory depression was evaluted by the mortality of mice. The results are reported in Table 4. The data suggested that prodrug 7e reduced the death rate in a dose-dependent manner. Moreover, 3 mg/kg prodrug 7e gave the same effect as 10 mg/kg LCX001 on reducing mortality, which indicated that the required molar dose of 7e was only one eighth of that of LCX001 to achieve the same effect. Further pharmacodynamic studies are currently underway.
Table 4.
Mortality of opiate poisoned mice after administration of LCX001 and prodrug 7e.
| Experiment | Treatment | Test Material | n | ROUTE | Dose (mg/kg) | Death Number (n) | Death Rate |
|---|---|---|---|---|---|---|---|
| RD | 030418, 15 mg/kg, sc | Saline | 10 | iv | 8 | 80% | |
| RD | 030418, 15 mg/kg, sc | LCX001 | 10 | iv | 10 | 2 | 20% |
| RD | 030418, 15 mg/kg, sc | 7e | 10 | iv | 30 | 0 | 0% |
| RD | 030418, 15 mg/kg, sc | 7e | 10 | iv | 10 | 0 | 0% |
| RD | 030418, 15 mg/kg, sc | 7e | 20 | iv | 3 | 4 | 20% |
| RD | 030418, 15 mg/kg, sc | 7e | 10 | iv | 1 | 3 | 30% |
| RD | 030418, 15 mg/kg, sc | 7e | 10 | iv | 0.5 | 5 | 50% |
RD—Respiratory depression; sc—subcutaneous injection, iv—intravenous injection.
3. Experimental Section
3.1. General Information
TLC was performed using precoated silica gel plates (Yantai dexin Bio-Technology Co., Ltd., Yantai, China). Column chromatography was performed using silica gel (200–300 mesh; Yantai Chemical Industry Research Institute, Yantai, China). Melting points were measured on a RY-1 apparatus (Tianda Tianfa Technology Co., Ltd., Tianjin, China). NMR spectra were recorded on a JNM-ECA-400 400 MHz spectrometer (JEOL Ltd., Tokyo, Japan) using CDCl3, DMSO-d6 and D2O as solvent. Chemical shifts are expressed in δ (ppm), with tetramethylsilane (TMS) functioning as the internal reference, coupling constants (J) were expressed in Hz. Mass spectroscopy (MS) was carried out on an API 3000 triple-quadrupole mass spectrometer (AB Sciex, Concord, ON, Canada) equipped with a Turbo IonSpray electrospray ionization (ESI) source was used for mass analysis and detection. Analyst 1.4 software (AB Sciex) was used for data acquisition.
3.2. Liquid Chromatographic Conditions
An Agilent 1100 system (Agilent Technologies, Palo Alto, CA, USA) consisting of a vacuum degasser, a quaternary pump, and an autosampler was used for HPLC solvent and sample delivery. Chromatographic separation was achieved using a Poroshell 120 C18 column (50 mm × 2.1 mm, 2.7 μm, Agilent) and a C18 guard column (4 × 3.0 mm, 5 μm; Phenomenex, Torrance, CA, USA). The column temperature was maintained at 25 °C. Gradient elution was performed using purified water as solvent A and acetonitrile as solvent B. Two elution gradient programs were used. The program applied for in vitro samples was as follows: 0.0–0.5 min 10% B to 80% B; 0.5–1.5 min 80% B; 1.5–1.6 min 80% B to 10% B; 1.6–6.0 min 10% B. And the program applied for in vivo samples was as follows: 0.0–0.5 min 10% B to 80% B; 0.5–1.3 min 80% B; 1.3–1.5 min 80% B to 10% B; 1.5–7.0 min 10% B. The flow rate was set at 0.3 mL/min. The injection volume was 10 μL.
3.3. Mass Spectrometric Conditions
The mass spectrometer was operated in the positive-ion mode. Ultrapure nitrogen was used as a nebulizer, curtain, and collision-activated dissociation (CAD) gas at 8, 10, and 6 instrument units, respectively. The optimized Turbo IonSpray voltage and temperature were set to 4500 V and 450 °C, respectively. Quantification was performed with multiple reaction monitoring. Glipizide was chosen as internal standard. The transitions and the optimized collision energies (CE) for each compound were listed in Table 5. Each transition was monitored with a 150-ms dwell-time.
Table 5.
Mass spectrometric conditions for the compounds.
| [M + H]+ | Precursor ion | Product ion | CE (V) |
|---|---|---|---|
| LCX001 | 276.2 | 178.2 | 20 |
| Glipizide | 446.0 | 321.0 | 20 |
| 7a | 607.5 | 258.2 | 25 |
| 7b | 621.5 | 258.2 | 26 |
| 7c | 635.5 | 258.2 | 27 |
| 7d | 649.5 | 258.2 | 29 |
| 7e | 649.5 | 258.2 | 29 |
| 7f | 669.5 | 258.2 | 29 |
3.4. Stability in Plasma Extract and Brain Homogenate
Blood was drawn from mice though the orbital sinus and was collected in a heparinized tube paved with heparin sodium. Samples were centrifuged at 10,000 rpm for 16 min to separate plasma which was diluted with four volumes of water. The brain was removed and homogenized. Samples were then placed on ice and used immediately. Ten μL acetonitrile solution (10 μg/mL) of the compound was added to 50 μL of plasma or brain homogenate and gently vortexed. Samples were incubated at 37 °C and aliquots were removed after 0, 5, 15, 30, 60 and 90 min, respectively. One hundred μL acetonitrile was added to each and vortexed. Samples were centrifuged for 10 min to remove proteins and the supernatants were analyzed by LC-MS/MS method as described.
3.5. Release of the Parent Drug by Prodrugs in Brain Homogenate
The prodrugs were treated in brain homogenate according to the procedure in Section 3.4 except that the intervals were changed to 0, 5, 15, 30 and 60 min. The peak of LCX001 was analyzed by LC-MS/MS described in Section 3.2 and Section 3.3. Peak integrals were normalized by dividing by the area for the peak of each prodrug itself at 5 min.
3.6. Biodistribution Studies in Vivo
3.6.1. Test Animals
Adult Kunming mice weighing 20–22 g were obtained from the animal center of academy of military medical sciences. The animals were left for two days to acclimatize to animal room conditions and were maintained on standard pellet diet and water ad libitum. Food was withdrawn on the day before the experiment, but free access to water was allowed. Since the experiments could be completed within 24 h, there was no significant change in the mice body weight during the experiment. All animals received human care, and the study protocols complied with the guidelines of the animal center of academy of military medical sciences. Throughout the experiments, the animals were handled according to the international ethical guidelines for the care of laboratory animals.
3.6.2. Biodistribution Studies of the Prodrug and LCX001
Mice were randomly divided into two groups, 27 in each group, for different sampling times and housed in one cage. Each animal was injected with 7e or LCX001 in 20% w/v hydroxypropyl-β-cyclodextrin and saline (0.45%) solution through the tail vein at a single dose equivalent to 1 mg/kg body weight of LCX001. At appropriate time intervals (5, 10, 30, 45, 60, 90, 120, 240 and 480 min), the animals were sacrificed and 1 mL blood samples withdrawn from the orbital sinus were collected in heparinized tubes. Fifty μL plasma was immediately separated by centrifugation, diluted with 200 μL methanol and stored at −20 °C until assay. Meanwhile, the brain was removed, weighed, homogenized and diluted with methanol to 1:5 (g/mL). The homogenates were also stored at −20 °C until assay. Internal standard (200 ng/mL) was added to the samples. The samples were vortexed for 3 min and then centrifuged at 9500 rpm for 15 min. Fifty μL supernatants were withdrawn, then 50 μL of water was added to each aliquot and vortexed. Ten μL samples were analyzed by LC-MS/MS method as described.
3.7. Statistical Analysis
The Cmax, AUC0-t, Tmax, MRT0-t and t1/2 were calculated by WinNonlin software (version 5.2.1, Pharsight, Mountain View, CA, USA). Statistical evaluation was performed using analysis of variance followed by t-test. A value of p < 0.05 was considered significant.
3.8. Pharmacodynamic Study of Prodrug 7e
3.8.1. Test Animals
A total of 80 male Kunming mice weighing 20–22 g were used in this study. Animals were fed ad libitum with a commercial rodent feed and had free access to drinking water. All animals received human care, and the study protocols complied with the guidelines of the animal center of academy of military medical sciences. Throughout the experiments, the animals were handled according to the international ethical guidelines for the care of laboratory animals.
3.8.2. Model of Opiate-Induced Respiratory Depression
Respiratory depression in conscious mice was induced by hypodermic (15 mg/kg) administration of 030418. Ten min prior to 030418 administration, animals were randomly assigned to the experimental groups and received an intravenous injection of either saline solution (control), LCX001 (10 mg/kg), prodrug 7e (30 mg/kg, 10 mg/kg, 3 mg/kg, 1 mg/kg or 0.5 mg/kg) (Table 4).
3.8.3. Assessment of the Prodrug 7e Effect
During the experiment, the poisoning time, poisoning symptoms and death time were recorded. The death rate (death number/sample number) was used to evaluate the effect of 7e on opiate-induced respiratory depression.
3.9. Synthesis
N,4-Dimethyl-5-(2-(hydroxy)ethyl)thiazolium iodide (9). 5-(2-Hydroxyethyl)-4-methylthiazole (100.0 g, 698.2 mmol) and methyl iodide (100.0 mL, 1500.0 mmol) were mixed and refluxed for 2 h. After evaporation of excess methyl iodide, to the residual brown syrup was added ether (100 mL) and stirred for 30 min, the precipitate was filtered to yield 9 as a pale-yellow solid (191.4 g, 96.2%). Mp: 82–84 °C. 1H-NMR (DMSO-d6, ppm): 2.43 (s, 3H), 3.03 (t, 2H, J = 5.5 Hz), 3.63 (t, 2H, J = 5.6 Hz), 4.09 (s, 3H), 9.96 (s, 1H); MS m/z [M]+ calculated for C7H12NS+: 158.1; found: 158.1.
General procedure for synthesis of S-substituted sodium thiosulfates 11a–11f. A solution of sodium thiosulfate pentahydrate (24.8 g, 100.0 mmol) in water (60 mL) was added to the solution of alkyl bromide (100 mmol) in ethanol (30 mL) under stirring. The reation mixture was heated to 110 °C and maintained for 7 h. After evaporation of the solvent, the residual white solid was dried in a vacuum desiccator to yield S-substituted sodium thiosulfates 11a–11f.
General procedure for synthesis of compounds 12a–12f. Under argon protection, to a 30 mL aqueous of quaternary ammonium salt 9 (14.3 g, 50.0 mmol) and sodium hydroxide (4.0 g, 100.0 mmol) was added S-substituted sodium thiosulfates 11a–11f (120.0 mmol). The reaction mixture was stirred for 1 h at room temperature. The resultant oily substance was extracted with ethyl acetate (450 mL) and the organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification of the crude product by column chromatography with dichloromethane-methanol (100:1) as eluent gave a yellow oil.
N-(3-(Ethyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (12a, Figure S1). 40.9% yield as a yellow oil. 1H-NMR (CDCl3, ppm): 1.19 (t, 3H, J = 7.3 Hz), 1.96, 1.86 (2s, 3H), 2.64 (q, 2H, J = 7.3 Hz), 2.70 (t, 2H, J = 6.7 Hz), 2.98, 2.83 (2s, 3H), 3.55–3.43 (m, 2H), 7.96, 7.84 (2s, 1H). MS m/z [M + H]+ calculated for C9H17NO2S2: 236.1; found: 236.2.
N-(3-(Propyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (12b, Figure S2). 64.8% yield as a yellow oil. 1H-NMR (CDCl3, ppm): 0.97 (t, 3H, J = 7.3 Hz), 1.69–1.63 (m, 2H), 2.01, 1.96 (2s, 3H), 2.47 (br, 1H), 2.59 (t, 2H, J = 7.6 Hz), 2.87 (t, 2H, J = 6.7 Hz), 3.06, 2.95 (2s, 3H), 3.81 (t, 3H, J = 6.4 Hz), 7.96, 7.92 (2s, 1H); MS m/z [M + H]+ calculated for C10H19NO2S2: 250.1; found: 250.1.
N-(3-(Butyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (12c, Figure S3). 70.5% yield as a yellow oil. 1H-NMR (CDCl3, ppm): 0.91(t, 3H, J = 7.3 Hz), 1.43–1.33 (m, 2H), 1.65–1.57 (m, 2H), 2.01, 1.96 (2s, 3H), 2.51 (br, 1H), 2.62 (t, 2H, J = 7.3 Hz), 2.87 (t, 2H, J = 6.7 Hz), 3.06, 2.95 (2s, 3H), 3.81 (t, 2H, J = 6.8 Hz), 7.96, 7.92 (2s, 1H). MS m/z [M + H]+ calculated for C11H21NO2S2: 264.1; found: 264.2.
N-(3-(Amyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (12d, Figure S4). 52.0% yield as a yellow oil. 1H-NMR (CDCl3, ppm): 0.90 (t, 3H, J = 7.3 Hz), 1.36–1.29 (m, 4H), 1.65–1.60 (m, 2H), 2.01, 1.97 (2s, 3H), 2.49 (br, 1H), 2.61 (t, 2H, J = 7.3 Hz), 2.87 (t, 2H, J = 6.7 Hz), 3.06, 2.95 (2s, 3H), 3.80 (t, 2H, J = 6.8 Hz), 7.96, 7.92 (2s, 1H). MS m/z [M + H]+ calculated for C12H23NO2S2: 278.1; found: 278.0.
N-(3-Isoamyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (12e, Figure S5). 76.8% yield as a yellow oil. 1H-NMR (CDCl3, ppm): 1.19 (t, 3H, J = 7.3 Hz), 1.53–1.47 (m, 2H), 1.68–1.61 (m, 1H), 2.01, 1.96 (2s, 3H), 2.49 (br, 1H), 2.62 (t, 2H, J = 7.6 Hz), 2.88 (t, 2H, J = 6.8 Hz), 3.06, 2.95 (2s, 3H), 3.80(t, 3H, J = 6.4 Hz), 7.96, 7.92(2s, 3H). MS m/z [M + H]+ calculated for C12H23NO2S2: 278.1; found: 278.0.
N-(3-(Benzyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (12f, Figure S6). 44.1% yield as a yellow oil. 1H-NMR (d6-DMSO, ppm): 1.91, 1.85 (2s, 3H), 2.62 (t, 2H, J = 6.7 Hz), 2.82, 2.81 (2s, 3H), 3.52–3.47 (m, 2H), 3.92 (s, 2H), 4.74–4.70 (m, 1H), 7.35–7.25 (m, 5H), 7.96, 7.81 (2s, 1H). MS m/z [M + H]+ calculated for C14H19NO2S2: 298.1; found: 298.2.
General procedure for synthesis of 4-(N-methylformamido)pent-3-en-1-yl)oxy)-5-oxopentanoic acids 13a–13f. A stirred solution of intermediate 12a–12f (10.0 mmol) in dry dichloromethane (50 mL) was treated with DMAP (0.1 g, 0.8 mmol) and glutaric anhydride (3.4 g, 30.0 mmol). The mixture was refluxed for 5 h. The solvent was evaporated under vacuum and purification of the crude product by column chromatography with dichloromethane-methanol (100:1) as eluent gave 13a–13f as yellow syrup.
General procedure for synthesis of compounds 7a–7f. The intermediate 13a–13f (4.7 mmol), LCX001 (1.0 g, 3.6 mmol), triethylamine (0.5 mL, 3.6 mmol) and HOBT (486 mg, 3.6 mmol) were dissolved in dry dichloromethane (50 mL) in a round-bottomed flask under string for 10 min. After addition of DMAP (44 mg, 0.36 mmol) and EDCI (1.1 g, 5.4 mmol), the mixture was refluxed for 5 h. The resulting mixture was concentrated by a rotary evaporator to afford yellow oleosus residue that was purified by column chromatography on silica gel (ethyl acetate/petroleum ether, (1:1)) to provide 7a–7f as white solids.
S-3-(Ethyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (7a, Figure S7). 43.2% yield as a white solid. Mp: 78.8–79.5 °C; 1H-NMR (CDCl3, ppm): 1.27 (t, 3H, J = 7.3 Hz), 1.99, 1.97 (2s, 3H), 1.94–1.62 (m, 8H), 2.12–1.99 (m, 2H), 2.44–2.35 (m, 4H), 2.63 (q, 2H, J = 7.3 Hz), 3.04–2.87 (m, 8H), 4.70–4.59, 3.51 (m, 1H), 4.24 (t, 2H, J = 6.7 Hz), 7.41 (d, 1H, J = 8.1 Hz); 13C-NMR (CDCl3, ppm): 172.6, 172.2, 168.6, 162.3, 161.2, 148.4, 139.7, 136.5, 130.6, 117.6, 114.5, 113.6, 71.8, 62.0, 51.9, 33.3, 33.0, 31.8, 30.3, 29.6, 29.6, 29.0, 29.0, 28.2, 26.9, 19.8, 18.4, 14.0. HRMS (ESI+) m/z [M + H]+ calculated for C28H38N4O7S2: 607.2182; found: 607.2255.
S-3-(Propyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (7b, Figure S8). 40.7% yield as a white solid. Mp: 78.0–79.3 °C; 1H-NMR (CDCl3, ppm): 0.96 (t, 3H, J = 7.3 Hz), 1.94–1.23 (m, 10H), 2.00, 1.97 (2s, 3H), 2.13–2.11 (m, 2H), 2.42–2.26 (m, 4H), 2.58 (t, 2H, J = 6.2 Hz), 3.04–2.88 (m, 8H), 4.70–4.56, 3.50 (m, 1H), 4.25 (t, 2H, J = 5.6 Hz), 7.42 (d, 1H, J = 8.9 Hz), 7.98–7.83 (m, 3H); 13C-NMR (CDCl3, ppm): 172.7, 172.3, 168.8, 162.3, 161.2, 148.4, 139.7, 136.6, 130.7, 117.5, 114.6, 113.5, 71.8, 62.1, 51.9, 41.1, 33.3, 33.1, 30.4, 29.6, 29.6, 29.0, 29.0, 28.2, 26.9, 21.9, 19.9, 18.5, 12.9. HRMS (ESI+) m/z [M + H]+ calculated for C29H40N4O7S2: 621.2338; found: 621.2411.
S-3-(Butyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (7c, Figure S9). 38.3% yield as a white solid. Mp: 67.6–68.4 °C; 1H-NMR (CDCl3, ppm): 0.91 (t, 3H, J = 7.6 Hz), 2.00–1.25 (m, 12H), 2.03, 2.00 (2s, 3H), 2.15–2.09 (m, 2H), 2.40–2.26 (m, 4H), 2.61 (t, 2H, J = 7.3 Hz), 3.03–2.87 (m, 8H), 4.72–4.53, 3.51 (m, 1H), 4.25 (t, 2H, J = 7.0 Hz), 7.42 (d, 1H, J = 8.7 Hz), 7.94–7.83 (m, 3H); 13C-NMR (CDCl3, ppm): 172.7, 172.3, 168.6, 162.4, 161.2, 148.3, 139.8 136.6, 130.7, 117.5, 114.6, 113.7, 71.6, 62.1, 52.0, 39.0, 33.4, 33.3, 31,7, 30.4, 29.6, 29.6, 29.0, 29.0, 28.3, 26.9, 21.5, 20.0, 18.5, 13.6. HRMS (ESI+) m/z [M + H]+ calculated for C30H42N4O7S2: 635.2495; found: 635.2568.
S-3-(Amyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (7d, Figure S10). 45.5% yield as a white solid. Mp: 69.3–70.3 °C; 1H-NMR (CDCl3, ppm): 0.89 (t, 3H, J = 6.8 Hz), 1.94–1.28 (m, 14H), 2.00, 1.97 (2s, 3H), 2.14–2.09 (m, 2H), 2.41–2.27 (m, 4H), 2.60 (t, 2H, J = 5.8 Hz), 3.04–2.88 (m, 8H), 4.71–4.59, 3.50 (m, 1H), 4.24 (t, 2H, J = 6.8 Hz), 7.42 (d, 1H, J = 8.4 Hz), 7.94–7.84 (m, 3H); 13C-NMR (CDCl3, ppm): 172.7, 172.2, 168.8, 162.4, 161.2, 148.5, 139.8, 136.6, 130.4, 117.4, 114.6, 113.7, 71.9, 62.1, 51.9, 39.3, 33.4, 33.1, 31.9, 30.5, 30.2, 29.6, 29.6, 29.0, 29.0, 28.3, 26.9, 22.2, 19.2, 18.5, 13.9. HRMS (ESI+) m/z [M + H]+ calculated for C31H44N4O7S2: 649.2651; found: 649.2724.
S-3-(Isoamyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (7e, Figure S11). 45.0% yield as a white solid. Mp: 74.0–75.4 °C; 1H-NMR (CDCl3, ppm): 0.89 (d, 6H, J = 6.5 Hz), 1.97–1.47 (m, 11H), 2.00,1.97 (2s, 3H), 2.15–2.09 (m, 2H), 2.41–2.26 (m, 4H), 2.62 (t, 2H, J = 6.7 Hz), 3.04–2.88 (m, 8H), 4.71–4.57, 3.51 (m, 2H), 4.24 (d, 2H, J = 6.4 Hz), 7.42 (d, 1H, J = 8.1 Hz), 9.97–7.83 (m, 3H); 13C-NMR (CDCl3, ppm): 172.7, 172.3, 168.6, 162.4, 161.3, 148.5, 139.7, 136.5, 130.8, 117.4, 114.6, 113.6, 71.8, 62.1, 51.9, 37.6, 37.4, 33.4, 33.1, 31.9, 30.2, 29.6, 29.6, 29.0, 29.0, 28.3, 28.3, 26.9, 22.2, 20.0, 18.5. HRMS (ESI+) m/z [M + H]+ calculated for C31H44N4O7S2: 649.2651; found: 649.2723.
S-3-(Benzyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (7f, Figure S12). 57.5% yield as a white solid. Mp: 72.9–73.8 °C; 1H-NMR (CDCl3, ppm): 1.91–1.25 (m, 8H), 1.94, 1.91 (2s, 3H), 2.10–2.03 (m, 2H), 2.41–2.25 (m, 4H), 3.00–2.75 (m, 8H), 4.69–4.59, 3.50 (m, 1H), 3.86 (s, 2H), 4.15 (t, 2H, J = 7.1 Hz), 7.42–7.28 (m, 5H), 7.97–7.83 (m, 3H). 13C-NMR (CDCl3, ppm): 172.6, 172.3, 168.8, 162.4, 161.3, 148.4, 139.7, 136.1, 135.8, 130.4, 129.2, 129.2, 128.5, 128.5, 127.7, 117.7, 114.6, 113.6, 71.8, 62.0, 51.9, 44.2, 33.3, 33.3, 31.9, 30.4, 29.5, 29.5, 28.7, 28.7, 26.9, 19.9, 18.4. HRMS (ESI+) m/z [M + H]+ calculated for C33H40N4O7S2: 669.2338; found: 669.2411.
4. Conclusions
In this paper, a series of brain-targeted prodrugs of LCX001 was synthesized and evaluated. The in vitro trials showed that prodrugs 7e, 7d, 7f possessed a certain stability in plasma which was propitious to get sufficient time to be delivered across the BBB. Moreover, prodrugs 7e, 7d, 7f were quickly decomposed in brain homogenate by the disulfide reductase, indicating the “lock-in” effect of these compounds after they entered the central nervous system (CNS). In vivo, prodrug 7e decreased the peripheral distribution of LCX001 and significantly increased brain distribution of LCX001 after i.v. administration. As a result, a lower dosage of prodrug 7e could give the same effect as the parent drug LCX001 on reversing opiate-induced respiratory depression, which was helpful in promoting the therapeutic efficacy and safety. More significantly, this prodrug also provides an important reference value to evaluate the clinical outlook of ampakine compounds.
Acknowledgments
Funding for this study was provided by the Integrated Drug Discovery Technology Platform (2012ZX09301003-001-006) of National Science and Technology Major Projects for “Major New Drugs Innovation and Development” in China.
Abbreviations
The following abbreviations are used in this manuscript:
| AMPA | Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| BBB | Blood-brain barrier |
| EPSP | Excitatory postsynaptic potential |
| FDA | Food and Drug Administration |
| IND | Investigational new drug |
| ADHD | Attention-deficit hyperactivity disorder |
| TDS | Thiamine disulfide system |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride |
| HOBT | N-Hydroxybenzotriazole |
| DMAP | 4-Dimethylaminopyridine |
| TEA | Triethylamine |
| DCM | Dichloromethane |
| rt | Room temperature |
| MRT | Mean residence times |
| RD | Respiratory depression |
| sc | Subcutaneous injection |
| iv | Intravenous injection |
| NMR | Nuclear Magnetic Resonance |
| LC | Liquid chromatography |
| MS | Mass spectrometry |
| ESI | Electrospray ionization |
| CAD | Collision-activated dissociation |
| CE | Collision energies |
| USA | United States of America |
| TMS | Tetramethylsilane |
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/4/488/s1.
Author Contributions
X.-B.Z., Z.Y. and S.L. conceived the project; D.X. and X.-B.Z. designed the experiments and executed the chemical synthesis; F.-H.M. and J.-Q.L. carried out the pharmacokinetics experiments; W.D. and Z.Y. performed the pharmacodynamic experiments; D.X. wrote the paper. All authors discussed the results and commented on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 7–13 are available from the authors.
References
- 1.Swarm R.A., Karanikolas M., Kalauokalani D. Pain treatment in the perioperative period. Curr. Probl. Surg. 2001;38:835–920. doi: 10.1067/msg.20011.118495. [DOI] [PubMed] [Google Scholar]
- 2.Desrosiers G. When opioid analgesia kills. Perspect. Infirm. 2006;4:6–9. [PubMed] [Google Scholar]
- 3.Lötsch J., Dudziak R., Freynhagen R., Marschner J., Geisslinger G. Fatal respiratory depression after multiple intravenous morphine injections. Clin. Pharmacokinet. 2006;45:1051–1060. doi: 10.2165/00003088-200645110-00001. [DOI] [PubMed] [Google Scholar]
- 4.Dahan A., Aarts L., Smith T.W. Incidence, reversal, and prevention of opioid-induced respiratory depression. Anesthesiology. 2010;112:226–238. doi: 10.1097/ALN.0b013e3181c38c25. [DOI] [PubMed] [Google Scholar]
- 5.Jammal W., Gown G. Opioid prescribing pitfalls: Medicolegal and regulatory issues. Aust. Prescr. 2015;38:198–203. doi: 10.18773/austprescr.2015.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis C.S., Southwell J.K., Niehaus V.R., Walley A.Y., Dailey M.W. Emergency medical services naloxone access: A national systematic legal review. Acad. Emerg. Med. 2014;21:1173–1177. doi: 10.1111/acem.12485. [DOI] [PubMed] [Google Scholar]
- 7.Rekling J.C., Feldman J.L. PreBotzinger complex and pacemaker neurons: Hypothesized site and kernel for respiratory rhythm generation. Annu. Rev. Physiol. 1998;60:385–405. doi: 10.1146/annurev.physiol.60.1.385. [DOI] [PubMed] [Google Scholar]
- 8.Shao X.M., Ge Q., Feldman J.L. Modulation of AMPA receptors by cAMP-dependent protein kinase in preBötzinger complex inspiratory neurons regulates respiratory rhythm in the rat. J. Physiol. 2003;547:543–553. doi: 10.1113/jphysiol.2002.031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kc P., Martin R.J. Role of central neurotransmission and chemoreception on airway control. Respir. Physiol. Neurobiol. 2010;173:213–222. doi: 10.1016/j.resp.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pace R.W., Del Negro C.A. AMPA and metabotropic glutamate receptors cooperatively generate inspiratory-like depolarization in mouserespiratory neurons in vitro. Eur. J. Neurosci. 2008;28:2434–2342. doi: 10.1111/j.1460-9568.2008.06540.x. [DOI] [PubMed] [Google Scholar]
- 11.Ren J., Poon B.Y., Tang Y., Funk G.D., Greer J.J. Ampakines alleviate respiratory depression in rats. Am. J. Respir. Crit. Care Med. 2006;174:1384–1391. doi: 10.1164/rccm.200606-778OC. [DOI] [PubMed] [Google Scholar]
- 12.Ren J., Ding X., Funk G.D., Greer J.J. Ampakine CX717 protects against fentanyl-induced respiratory depression and lethal apnea in rats. Anesthesiology. 2009;110:1364–1370. doi: 10.1097/ALN.0b013e31819faa2a. [DOI] [PubMed] [Google Scholar]
- 13.Greer J.J., Ren J. Ampakine therapy to counter fentanyl-induced respiratory depression. Respir. Physiol. Neurobiol. 2009;168:153–157. doi: 10.1016/j.resp.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Oertel B.G., Felden L., Tran P.V., Bradshaw M.H., Angst M.S., Schmidt H., Johnson S., Greer J.J., Geisslinger G., Varney M.A., et al. Selective antagonism of opioid-induced ventilatory depression by an ampakine molecule in humans without loss of opioid analgesia. Clin. Pharmacol. Ther. 2010;87:204–211. doi: 10.1038/clpt.2009.194. [DOI] [PubMed] [Google Scholar]
- 15.Ren J., Ding X., Greer J.J. Respiratory depression in rats induced by alcohol and barbiturate and rescue by ampakine CX717. J. Appl. Physiol. 2012;113:1004–1011. doi: 10.1152/japplphysiol.00752.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren J., Lenal F., Yang M., Ding X., Greer J.J. Coadministration of the AMPAKINE CX717 with propofol reduces respiratory depression and fatal apneas. Anesthesiology. 2013;118:1437–1445. doi: 10.1097/ALN.0b013e318291079c. [DOI] [PubMed] [Google Scholar]
- 17.ElMallah M.K., Pagliardini S., Turner S.M., Cerreta A.J., Falk D.J., Byrne B.J., Greer J.J., Fuller D.D. Stimulation of Respiratory Motor Output and Ventilation in a Murine Model of Pompe Disease by Ampakines. Am. J. Respir. Cell Mol. Biol. 2015;53:326–335. doi: 10.1165/rcmb.2014-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren J., Ding X., Greer J.J. Ampakines enhance weak endogenous respiratory drive and alleviate apnea in perinatal rats. Am. J. Respir. Crit. Care Med. 2015;191:704–710. doi: 10.1164/rccm.201410-1898OC. [DOI] [PubMed] [Google Scholar]
- 19.Partin K.M. AMPA receptor potentiators: From drug design to cognitive enhancement. Curr. Opin. Pharmacol. 2015;20:46–53. doi: 10.1016/j.coph.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lippa Arnold S. “Strategic Initiatives in Respiratory Disorders and Key Objectives”; Proceedings of the Rodman and Renshaw 16th Annual Global Investment Conference; New York, NY, USA. 8–11 September 2014. [Google Scholar]
- 21.Street L., Mueller R., Lee S. Bicyclic Amide Derivatives for the Treatment of Respiratory Depression. 2008143963. International Patent. 2008 Nov 27;
- 22.Mueller R., Street L. Bicyclic Amides for Enhancing Glutamatergic Synaptic Responses. 8263591. U.S. Patent. 2012 Sep 11;
- 23.Toyoaki I., Takashi S., Hiroshi I., Takashi K., Teruomi I. Drug delivery to the brain. DOPA prodrugs based on a ring-closure reaction to quaternary thiazolium compounds. Int. J. Pharm. 1995;116:51–63. [Google Scholar]
- 24.Fanm W., Wu Y., Li X.K., Yao N., Li X., Yu Y.G., Hai L. Design, synthesis and biological evaluation of brain-specific glucosyl thiamine disulfide prodrugs of naproxen. Eur. J. Med. Chem. 2011;46:3651–3661. doi: 10.1016/j.ejmech.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y., Qu B., Wu X., Li X., Liu Q., Jin X., Guo L., Hai L., Wu Y. Design, synthesis and biological evaluation of brain targeting L-ascorbic acid prodrugs of ibuprofen with “lock-in” function. Eur. J. Med. Chem. 2014;82:314–323. doi: 10.1016/j.ejmech.2014.05.072. [DOI] [PubMed] [Google Scholar]
- 26.Quan W. Ph.D. thesis. Academy of Military Medical Sciences; Beijing, China: 2011. [(accessed on 8 April 2016)]. Investigation on Receptor Mechanisms Underlying Powerful Antinociception and Low Addiction of Novel. Available online: http://www.cnki.net/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.