

Abstract
By imitating the scaffold of lithocholic acid (LCA), a natural steroidal compound displaying Protein Tyrosine Phosphatase 1B (PTP1B) inhibitory activity, a series of stilbene derivatives containing phenyl-substituted isoxazoles were designed and synthesized. The structures of the title compounds were confirmed by 1H-NMR, 13C-NMR and HRMS. Activities of the title compounds were evaluated on PTP1B and the homologous enzyme TCPTP by using a colorimetric assay. Most of the target compounds had good activities against PTP1B. Among them, compound 29 (IC50 = 0.91 ± 0.33 μM), characterized by a 5-(2,3-dichlorophenyl) isoxazole moiety, exhibited an activity about 14-fold higher than the lead compound LCA and a 4.2-fold selectivity over TCPTP. Compound 29 was identified as a competitive inhibitor of PTP1B with a Ki value of 0.78 μM in enzyme kinetic studies.
Keywords: protein tyrosine phosphatase 1B (PTP1B), lithocholic acid, stilbene, inhibitor
1. Introduction
Protein tyrosine phosphatases (PTPs) are a superfamily of enzymes involved in crucial cellular signalling mechanisms by controlling the phosphorylation levels of specific tyrosine residues in peptides and proteins which regulate many cellular functions, such as cell proliferation, survival, metabolism, adhesion and migration. Among the PTPs so far identified, the cytosolic protein tyrosine phosphatase 1B (PTP1B) was the first phosphatase to be isolated and is therefore the most widely studied [1,2,3,4]. A large body of experimental data acquired from in vivo studies on humans and PTP1B-knockout mice has identified PTP1B as a major negative regulator of both insulin and leptin signals. Consequently, inhibiting of PTP1B was considered to be a potential therapeutics for treating Type 2 diabetes and obesity by enhance insulin sensitivity and resistance to obesity [5,6,7,8]. Moreover, it is worth noting that PTPs share a high degree of structural conservation in the active site, especially for T-cell protein tyrosine phosphatase (TCPTP), which has a sequence identity of about 74% in the catalytic domains with PTP1B [9]. There is always a challenge for the potent PTP1B inhibitors to keep good selectivity over TCPTP in the same time [10].
In the past decades, several steroids and their derivatives were found to possess moderate to good PTP1B inhibitory activities, such as lithocholic acid (LCA, 1, Figure 1) [11], claramine (3) [12] and trodusquemine (2) [13], the first PTP1B inhibitor for the treatment of diabetes in clinical trials. However, risk of the hormonal effect of steroids and their derivatives always cannot be neglected. Years ago, diethylstilbestrol (4) was developed as a mimic and alternative drug for estradiol (5) [14], which indicated the fact that the stilbene moiety could be an ideal replacement of the steroid skeleton in drug design when use the “scaffold hopping” method [15]. Additionally, stilbene is a common moiety found in many natural products endowed with antibacterial, antioxidative, antiproliferative or antidiabetic activities, such as resveratrol (6), pinosylvin (7) and piceatannol (8) [16,17,18,19].
Figure 1.

Steroids 1~3 reported as PTP1B inhibitors and stilbene derivatives 4~8.
In a previous report we disclosed that the introduction of phenyl-substituted heterocyclic rings, such as pyrazole, oxazole, isoxazole and thiazoles, into the ring A on LCA would improve the inhibitory activity against PTP1B. On the other hand, it has also been verified that the 23-COOH of LCA was irreplaceable for maintaining the activities against PTP1B [11]. Based on the views above, we tentatively designed a class of stilbene derivatives, as shown in Figure 2, which was characterized by a phenyl-substituted isoxazole linked to the stilbene scaffold by an ether bond. In order to search for the optimized phenyl isoxazole moiety, at the very beginning, we designed and synthesized four mimics (compounds 15, 16, 17 and 18), containing 3-phenyl-4-isoxazolyl-, 5-methyl-3-phenyl-4-isoxazolyl-, 5-phenyl-4-isoxazolyl and 5-phenyl-3-isoxazolyl groups, respectively, and evaluated their inhibitory effects against PTP1B. As a result, compound 18 was found to have the highest activity to PTP1B (IC50 = 6.33 ± 1.02 μM). Then a series of its derivatives 19~30 with different groups on the phenyl ring C was synthesized and evaluated.
Figure 2.

The design of the title compounds.
2. Results and Discussion
2.1. Chemistry
As shown in Scheme 1, target compounds 15~30 were all prepared by condensation of the stilbene intermediate 14 and the corresponding chloromethyl isoxazole 15c~30c in DMSO with Cs2CO3 as the acid-binding agent, followed by hydrolysis in KOH solution and acidification with hydrochloric acid. Starting from 3-methylbenzoate (9), the key intermediate 14 was synthesized in five steps including bromination [20], Arbuzov reaction, Wittig-Horner reaction [21], demethylation [22] and selective methylation. At the beginning, by using PPh3, a Wittig reaction was employed to synthesize the compound 12, giving a mixture of cis- and trans-products with a ratio of about 1:2. Alternatively, the Wittig-Horner reaction gave a satisfactory result, unexpectedly accompanied by the hydrolysis of methyl ester due to the strong basicity of t-BuOK. In addition, intermediate 10 was obtained conveniently through a routine method using NBS as the brominating agent and AIBN as the radical initiator. Since deprotection of the methyl of 12 in BBr3/DCM led to a mass of by-products, the reaction was carried out successfully in a mixture of AlCl3 and Et3N. Before the condensation of phenolic hydroxyl and the chlorinated intermediates 15c~30c, the carboxyl group on the phenyl A ring of 13 was selectively protected by MeI. Slight methylation of the phenolic hydroxyl group occured in the same time.
Scheme 1.

Synthesis of compounds 10~30. Reagents and conditions: (a) CCl4, NBS, reflux, 6 h; (b) P(OEt)3, 160 °C; (c) t-BuOK, THF, 0 °C~r.t., overnight; (d) AlCl3, Et3N, 60 °C~90 °C, 18 h; (e) MeI, K2CO3, acetone, 60 °C, 15 h; (f) Cs2CO3, DMSO, 65 °C, 8 h; (g) KOH, H2O, 65 °C, 6 h; (h) LiAlH4, 0 °C~r.t., 4 h; (i) SOCl2, DCM, 45 °C, 8 h.
Compounds 15c~30c were synthesized from the corresponding methyl carboxylates 15a~30a via reduction by LiAlH4 and chlorination with SOCl2 in DCM. Intermediates from the above two steps were used directly without any purification. Compounds 15a, 16a and 17a were prepared according to the reported methods [23,24,25]. By reference to Kamal’s report, compounds 18a~30a were synthesized from the corresponding substituted acetophenones [26].
2.2. In Vitro Biological Evaluation
All of the title compounds were assayed on the enzyme PTP1B in vitro. In compounds 15~18, the phenyl position on the isoxazole ring caused huge differences in the inhibition against PTP1B. As shown in Table 1, both compounds 15 and 17, characterized by a 3-phenyl-4-isoxazole moiety, exhibit moderate activities against the enzyme. Meanwhile, the 5-phenyl-4-isoxazole compound 16 (IC50 = 16.2 ± 4.52 μM) also has similar activity, while another 5-phenyl compound, 18, exhibits a much higher activity (IC50 = 6.33 ± 1.02 μM) due to the different position of the isoxazole ring linked to the phenyl B ring. This result inspired us to further investigate the substituents on phenyl C and their effects on the activities against PTP1B. On the other hand, as expected, methylation of the carboxyl of 18 led to an obvious decrease on PTP1B inhibition.
Table 1.
The structures of compounds 15, 16, 17, 18, 18d and their IC50 values against PTP1B.
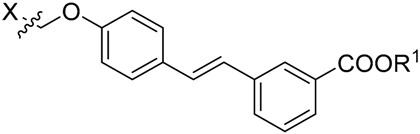
| Compound | X | R1 | IC50 (μM) a |
|---|---|---|---|
| 15 | 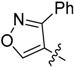 |
H | 20.7 ± 3.83 |
| 16 | 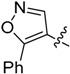 |
H | 16.2 ± 4.52 |
| 17 | 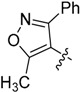 |
H | 15.7 ± 2.15 |
| 18 | 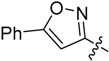 |
H | 6.33 ± 1.02 |
| 18d | 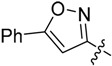 |
Me | 23.7 ± 3.62 |
| Oleanolic acid b | 2.96 ± 0.35 |
a IC50 values were determined by regression analyses and expressed as means ± SD of three replications; b Positive control.
Derivatives of 18 and their activities against PTP1B and TCPTP are listed in Table 2. For substituents at the 2-position of phenyl C, the electron-withdrawing chlorine atom (compound 19) was beneficial to the activity when compared to an electron-donating methoxy (compound 20). 3-subtituents also affect the activities remarkably. Compounds containing 3-Cl (21) and 3-Br (23) exhibit obvious inhibition to PTP1B, whereas a 3-F leads to a dramatic decrease in activity. NO2 (25, IC50 = 2.05 ± 0.54 μM) seems to be the optimal 3-substituent among the synthesized compounds, giving an activity 3-fold higher than compound that of 18. In addition, when the 3-position is occupied by a methyl group, the resulting compound 24 still shows good inhibitory effect against the enzyme (IC50 = 3.68 ± 1.12 μM).
Table 2.
The structures of compounds 18~30 and their IC50 values against PTP1B and TCPTP.
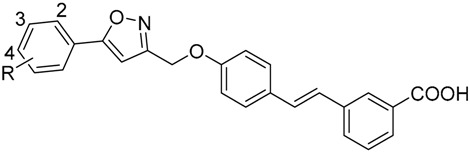
| Compound | R | IC50 (μM) a | TCPTP/PTP1B b | |
|---|---|---|---|---|
| PTP1B | TCPTP | |||
| 18 | H | 6.33 ± 1.33 | 9.58 ± 0.33 | 1.5 |
| 19 | 2-Cl | 1.75 ± 0.82 | 36.15 ± 3.82 | 20.7 |
| 20 | 2-OMe | 7.66 ± 1.05 | >40 | 5.3 |
| 21 | 3-Cl | 5.37 ± 1.53 | 7.58 ± 1.32 | 1.4 |
| 22 | 3-F | >20 | >40 | - c |
| 23 | 3-Br | 9.85 ± 2.04 | >40 | 4.1 |
| 24 | 3-Me | 3.68 ± 1.12 | 18.91 ± 1.18 | 5.1 |
| 25 | 3-NO2 | 2.05 ± 0.54 | 13.25 ± 1.70 | 6.5 |
| 26 | 4-Me | 3.56 ± 0.28 | 7.67 ± 0.35 | 2.2 |
| 27 | 4-OMe | 2.23 ± 0.51 | 8.02 ± 0.99 | 3.6 |
| 28 | 4-F | 3.34 ± 0.81 | 5.19 ± 0.31 | 1.6 |
| 29 | 3,4-Cl2 | 0.91 ± 0.33 | 3.78 ± 0.22 | 4.2 |
| 30 | 3,4-F2 | 1.22 ± 0.48 | 6.02 ± 1.13 | 4.9 |
| Lithocholic acid | 12.54 ± 2.51 | 20.95 ± 3.66 | 1.7 | |
| Oleanolic acid d | 2.71 ± 0.19 | 6.12 ± 0.15 | 2.3 | |
a IC50 values were determined by regression analyses and expressed as means ± SD of three replications; b TCPTP/PTP1B, the ratio of IC50 of TCPTP and PTP1B; c “-”, none accurate value was calculated; d Positive control.
The derivatives present good tolerance of 4-substituents on the phenyl C ring, since compounds 26 (4-Me), 27 (4-OMe) and 28 (4-F) have similar activities against PTP1B. Amazingly, the di-substituted compounds, either dichloro (29) or difluoro (30) exhibited the highest inhibitory activities against the enzyme among all the title compounds. Most of all, compound 29 has an activity (IC50 = 0.91 ± 0.33 μM) about 14-fold higher than the lead compound LCA, which prompts us to further study on its structure modification and biological activity. However, as shown in Table 2, it is unfortunate that most of the compounds also exhibit some activities against TCPTP, except for compounds 20, 22 and 23. The selectivities against PTP1B and TCPTP vary a lot, from 1.4 to 22.9. Among the five compounds most potent against PTP1B (19, 25, 27, 29 and 30), compound 19 has the best selectivity (TCPTP/PTP1B = 20.7) while compound 29 has about a 4-fold selectivity over TCPTP. These two compounds are promising for further study as anti-diabetes drugs.
2.3. Enzyme Kinetic Study
In order to determine the inhibition mode of the title compounds, the most active compound 29 was selected for a PTP1B enzyme kinetic study using the Michaelis-Menten equation. As shown in Figure 3, the Vmax value remained constant while the Km value increased with the mounting compound concentration, indicating that 29 was a competitive PTP1B inhibitor, which was further confirmed by a Lineweaver–Burk plot (Figure 3c). The calculated inhibitory constant Ki of compound 29 was 0.78 μM.
Figure 3.

Characterization of 29 to PTP1B. (a,b) At various fixed concentrations of 29 the initial velocity was determined with various concentrations of pNPP; (c) Typical competitive inhibition of 29 shown by Lineweaver–Burk plot; (d) Linear replot of Km versus the inhibitor (29) concentration [I].
3. Experimental Section
3.1. Chemsitry
3.1.1. General Information
All reagents were chemically pure and solvents were dried according to standard methods. The 1H-NMR and 13C-NMR spectra were obtained on an AV400 spectrometer (400 MHz, 1H; 100 MHz, 13C, Bruker, Billerica, MA, USA) in CDCl3 with tetramethylsilane as the internal standard. The melting points were determined on an X-4 binocular microscope melting point apparatus (Beijing Tech Instrument Co., Beijing, China) and are uncorrected. High-resolution mass data were obtained on a Micromass TOF II spectrometer (Micromass, Cary, NC, USA). The reactions were monitored by analytical thin-layer chromatography (TLC) carried out on silica gel GF254 plates with ultraviolet (UV) light detection.
3.1.2. Synthesis of Methyl 3-((Diethoxyphosphoryl)methyl)benzoate (11)
A solution of methyl 3-methylbenzoate (47 g, 0.31 mol) in CCl4 (200 mL) was heated to reflux followed by addition of AIBN (2.6 g) in one portion. After that, NBS (67 g, 0.38 mol) was added carefully to the mixture during 2 h, and then the reaction was refluxed for an additional 5 h. After cooling to r.t. the mixture was filtered and evaporated under vacuum to give methyl 3-(bromomethyl)benzoate (10, 67 g, Yield: 94%) as a yellowish oil. The obtained oil was then dissolved in triethyl phosphate (130 g, 0.71 mol), heated to 160 °C and kept at this temperature for an additional 10 h. The mixture was concentrated on a rotary evaporator and then purified by flash chromatography (PE:EA = 5:1~PE:EA = 1:3) to give 11 as a yellow oil, which was used without any further purification.
3.1.3. Synthesis of (E)-3-(4-Methoxystyryl)benzoic Acid (12)
To an ice-cooled solution of compound 11 (5 g, about 17.5 mmol) in THF (30 mL) was added a mixture of t-BuOK (3.9 g, 34.8 mmol) and THF (40 mL) during 30 min. After stirring for additional 30 min, a solution of 4-methoxybenzaldehyde (17.5 mmol) in THF (20 mL) was added and then the reaction mixture was warmed to room temperature, and stirred for another 8 h before quenching with 2 N HCl. The mixture was then evaporated to remove the THF and precipitation occurred meanwhile. The precipitate was then filtered and dried to give 12 as a yellow solid without any additional purification. Yield: 4.8 g (46%); 1H-NMR (DMSO-d6) δ: 12.93 (brs, 1H), 8.12 (s, 1H), 7.81 (d, J = 7.6 Hz, 2H), 7.58 (d, J = 8.6 Hz, 2H), 7.49 (t, J = 7.8 Hz, 1H), 7.28 (d, J = 16.5 Hz, 1H), 7.18 (d, J = 16.5 Hz, 1H), 6.96 (d, J = 8.6 Hz, 2H), 3.78 (s, 3H).
3.1.4. Synthesis of (E)-3-(4-Hydroxystyryl)benzoic Acid (13)
Anhydrous aluminium trichloride (30 g) was added to triethylamine (200 mL) in portions during 30 min and then heated to 60 °C for 3 h before adding compound 12 (16.5 g, 65.0 mmol) in one portion. The resultant mixture was then heated to 80 °C and kept for another 16 h. After evaporating most of the triethylamine, the reaction was quenched by addition of iced hydrochloric acid. The water phase was extracted by ethyl acetate (80 mL × 3), dried over anhydrous Na2SO4, filtered and then evaporated to give crude 13, which was then recrystallized from ethyl acetate to give 13 as a yellowish solid. Yield: 7.8 g (48%); 1H-NMR (DMSO-d6) δ: 12.98 (brs, 1H), 9.63 (s, 1H), 8.09 (s, 1H), 7.78~7.81 (m, 2H), 7.48~7.49 (m, 3H), 7.13~7.15 (m, 2H), 6.78 (d, J = 8.7 Hz, 2H).
3.1.5. Synthesis of Methyl (E)-3-(4-Hydroxystyryl)benzoate (14)
Anhydrous K2CO3 (0.58 g, 4.2 mmol) was added to a solution of 13 (1.68 g, 7 mmol) in acetone (15 mL), and the mixture was then stirred for 2 h at room temperature before adding MeI (1.07 g, 4.2 mmol) in one portion. The reaction was then stirred for an additional 16 h. After that, the mixture was filtered, evaporated and purified by column chromatography (PE:EA = 3:1) to give 14 as a yellowish solid. Yield: 0.29 g (51%); 1H-NMR (DMSO-d6) δ: 9.64 (s, 1H), 8.10 (s, 1H), 7.79~7.85 (m, 2H), 7.46~7.50 (m, 3H), 7.25 (d, J = 16.5 Hz, 1H), 7.12 (d, J = 16.5 Hz, 1H), 6.78 (d, J = 8.5 Hz, 2H), 3.88 (s, 3H); 13C-NMR (DMSO-d6) δ: 166.2, 157.6, 138.2, 130.3, 130.1, 129.7, 129.1, 128.1 (2C), 127.7, 127.4, 126.6, 123.9, 115.5 (2C), 52.1.
3.1.6. Synthesis of (3-Phenylisoxazol-4-yl)methanol (15b)
On an ice bath, to a solution of 15a (5 mmol) in anhydrous THF (20 mL) was added LiAlH4 (5 mmol) in portions during 30 min. After that, the mixture was stirred for 2 h and quenched by 1 mL H2O added dropwise. After adding anhydrous Na2SO4 (5 g), the reaction was filtered and evaporated to give 15b, which was used in the next step without any purification. Intermediates 16b~30b were synthesized according to methods described above for the synthesis of 15b.
3.1.7. Synthesis of 4-(Chloromethyl)-3-phenylisoxazole (15c)
To a solution of 15b (3 mmol), DCM (20 mL) and DMF (1~2 drops), SOCl2 (10 mmol) was added and heated to 45 °C. After keeping at this temperature for 8 h, the solution was evaporated to give 15c as yellow oil, which was used in the next step without any purification. Intermediates 16c~30c were synthesized according to the above method for the synthesis of 15c and purified by column chromatography (PE:EA = 5:1~10:1) if necessary.
3.1.8. Synthesis of (E)-3-(4-((3-Phenylisoxazol-4-yl)methoxy)styryl)benzoic Acid (15)
A mixture of 14 (1 mmol), 15c (1 mmol) and Cs2CO3 (2 mmol) in DMSO (5 mL) was heated to 85 °C and kept at this temperature for 8 h, then the reaction mixture was cooled to 60 °C followed by the addition of 20% KOH solution (2 mL). After stirring at 60 °C for 4 h, the reaction mixture was cooled, acidified by 1 N hydrochloric acid diluted with water to give a mass of precipitate of the crude product, which was then recrystallized from methanol to give the purified 15. Yellow solid; Yield: 63%; 1H-NMR (DMSO-d6) δ: 13.12 (brs, 1H), 9.22 (s, 1H), 8.07 (s, 1H), 7.77~7.79 (m, 2H), 7.48~7.59 (m, 7H), 7.27 (t, J = 7.6 Hz, 1H), 7.18 (d, J = 16.5 Hz, 1H), 7.13 (d, J = 16.5 Hz, 1H), 7.04 (d, J = 8.8 Hz, 2H), 5.13 (s, 2H); 13C-NMR (DMSO-d6) δ: 169.2, 161.3, 159.2, 157.7, 136.7, 131.0, 130.6, 129.6 (2C), 129.4, 128.7, 128.6, 128.4 (2C), 128.3 (2C), 128.0, 127.8, 127.4, 127.3, 115.6 (2C), 114.6, 59.1; HRMS (ESI) calcd for C25H18NO4 [M − H]− 396.1236, found 396.1265 (see Supplementary Materials). Compounds 16~30 were synthesized according to the above method for the synthesis of 15.
(E)-3-(4-((5-Phenylisoxazol-4-yl)methoxy)styryl)benzoic acid (16): Yellow solid; Yield: 47%; 1H-NMR (DMSO-d6) δ: 13.15 (brs, 1H), 9.22 (s, 1H), 8.13 (s, 1H), 7.77~7.80 (m, 4H), 7.60 (d, J = 8.7 Hz, 2H), 7.52~7.54 (m, 4H), 7.31 (d, J = 16.5 Hz, 1H), 7.22 (d, J = 16.5 Hz, 1H), 7.07 (d, J = 8.7 Hz, 2H), 5.14 (s, 2H); 13C-NMR (DMSO-d6) δ: 169.5, 161.3, 159.1, 158.1, 138.4, 131.0, 130.6, 130.5, 129.7, 129.6, 129.5 (2C), 129.4, 128.6 (2C), 128.4 (2C), 128.2, 127.8, 127.3, 125.9, 115.6 (2C), 114.6, 59.1; HRMS (ESI) calcd for C25H18NO4 [M − H]− 396.1236, found 396.1212.
(E)-3-(4-((5-Methyl-3-phenylisoxazol-4-yl)methoxy)styryl)benzoic acid (17): Yellow solid; Yield: 42%; 1H-NMR (DMSO-d6) δ: 13.04 (brs, 1H), 8.13 (s, 1H), 7.77 (d, J = 7.2 Hz, 1H), 7.69~7.72 (m, 2H), 7.62 (d, J = 7.8 Hz, 1H), 7.58 (d, J = 8.6 Hz, 1H), 7.49~7.52 (m, 3H), 7.36 (t, J = 7.7 Hz, 1H), 7.23 (d, J = 16.5 Hz, 1H), 7.12 (d, J = 16.5 Hz, 1H), 7.02 (s, 1H), 7.10 (d, J = 8.6 Hz, 2H), 5.00 (s, 2H), 2.53 (s, 3H); 13C-NMR (DMSO-d6) δ: 170.4, 168.3, 162.6, 158.1, 137.3, 130.9, 130.4, 129.5 (2C), 129.1, 129.0, 128.7, 128.6, 128.4, 128.3 (2C), 128.2 (2C), 127.4, 126.9, 115.7 (2C), 109.9, 59.5, 11.3; HRMS (ESI) calcd for C26H20NO4 [M − H]− 410.1392, found 410.1388.
(E)-3-(4-((5-Phenylisoxazol-3-yl)methoxy)styryl)benzoic acid (18): Yellow solid; Yield: 42%; 1H-NMR (DMSO-d6) δ: 13.15 (brs, 1H), 8.12 (s, 1H), 7.90 (dd, J1 = 8.0 Hz, J2 = 1.8 Hz, 2H), 7.80 (t, J = 7.5 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.52~7.58 (m, 3H), 7.47 (t, J = 7.7 Hz, 1H), 7.29 (d, J = 16.4 Hz, 1H), 7.22 (d, J = 16.4 Hz, 1H), 7.20 (s, 1H), 7.10 (d, J = 8.8 Hz, 2H), 5.29 (s, 2H); 13C-NMR (DMSO-d6) δ: 170.0, 168.2, 161.8, 158.1, 138.0, 131.1, 130.8, 130.3, 129.8 (2C), 129.3, 129.2, 128.5 (2C), 128.4, 127.4, 127.1, 126.3, 126.1 (2C), 115.6 (2C), 100.5, 61.8; HRMS (ESI) calcd for C25H18NO4 [M − H]− 396.1236, found 396.1219.
(E)-3-(4-((5-(2-Chlorophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (19): Yellow solid; Yield: 51%; 1H-NMR (DMSO-d6) δ: 13.02 (s, 1H), 8.12 (s, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.97 (d, J = 5.5 Hz, 1H), 7.78~7.85 (m, 3H), 7.62 (d, J = 8.4 Hz, 2H), 7.46~7.51 (m, 2H), 7.40 (t, J = 8.7 Hz, 2H), 7.30 (d, J = 16.4 Hz, 1H), 7.22 (d, J = 16.4 Hz, 1H), 7.18 (s, 1H), 7.09 (d, J = 8.4 Hz, 2H), 5.28 (s, 2H); 13C-NMR (DMSO-d6) δ: 168.6, 167.3, 161.3, 157.6, 137.7, 131.2, 130.2, 128.9, 128.2, 128.1, 128.1, 128.0 (2C), 127.9, 126.9, 124.1, 125.6, 116.5, 116.3, 115.5, 115.1 (2C), 99.9, 61.3; HRMS (ESI) calcd for C25H17ClNO4 [M − H]− 430.0846, found 430.0831.
(E)-3-(4-((5-(2-Methoxyphenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (20): Yellow solid; Yield: 47%; 1H-NMR (DMSO-d6) δ: 13.05 (brs, 1H), 8.13 (s, 1H), 7.79~7.83 (m, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.46~7.52 (m, 3H), 7.26 (d, J = 16.4 Hz, 1H), 7.20 (d, J = 16.4 Hz, 1H), 7.10~7.14 (m, 2H), 6.99 (s, 1H), 6.79 (d, J = 8.8 Hz, 2H), 5.29 (s, 2H), 3.96 (s, 3H); 13C-NMR (DMSO-d6) δ: 167.8, 166.2, 161.5, 158.2, 138.4, 138.2, 131.7, 130.7, 130.5, 130.0, 129.4, 129.4, 128.6 (2C), 128.2, 127.4, 127.2, 126.1, 124.6, 121.3, 115.6 (2C), 103.6, 61.8, 56.3; HRMS (ESI) calcd for C26H20NO5 [M − H]− 426.1341, found 426.1376.
(E)-3-(4-((5-(3-Chlorophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (21): Yellow solid; Yield: 40%; 1H-NMR (DMSO-d6) δ: 13.12 (brs, 1H), 8.13 (s, 1H), 8.01 (s, 1H), 7.78~7.82 (m, 2H), 7.62 (d, J = 8.6 Hz, 2H), 7.58~7.59 (m, 2H), 7.46~7.50 (m, 2H), 7.33 (s, 1H), 7.30 (d, J = 16.4 Hz, 1H), 7.22 (d, J = 16.4 Hz, 1H), 7.10 (d, J = 8.6 Hz, 2H), 5.30 (s, 2H); 13C-NMR (DMSO-d6) δ: 168.5, 167.9, 161.9, 158.1, 138.2, 134.5, 131.7, 130.8, 130.6, 130.0, 129.4, 129.0, 128.5, 127.4, 126.2 (2C), 125.8, 124.7, 124.6, 115.6 (2C), 101.7; HRMS (ESI) calcd for C25H17ClNO4 [M − H]− 430.0846, found 430.0857.
(E)-3-(4-((5-(3-Fluorophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (22): Yellow solid; Yield: 48%; 1H-NMR (DMSO-d6) δ: 13.05 (brs, 1H), 8.13 (s, 1H), 7.76~7.84 (m, 4H), 7.58~7.64 (m, 3H), 7.49 (t, J = 7.7 Hz, 1H), 7.29~7.40 (m, 3H), 7.22 (d, J = 16.4 Hz, 1H), 7.10 (d, J = 8.7 Hz, 2H), 5.30 (s, 2H); 13C-NMR (DMSO-d6) δ: 168.7, 167.7, 162.9 (d, J = 145.6 Hz), 161.9, 158.1, 138.2, 132.1, 132.0, 131.7, 130.7, 129.4, 129.1, 129.0, 128.5 (2C), 128.4, 127.4, 126.2, 122.3, 118.2 (d, J = 21.2 Hz), 115.6 (2C), 113.0 (d, J = 23.7 Hz), 101.6, 61.7; HRMS (ESI) calcd for C25H17FNO4 [M − H]− 414.1142, found 414.1133.
(E)-3-(4-((5-(3-Bromophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (23): Yellow solid; Yield: 51%; 1H-NMR (DMSO-d6) δ: 13.13 (brs, 1H), 8.14 (s, 1H), 8.12 (s, 1H), 7.89~7.93 (m, 1H), 7.79 (d, J = 7.6 Hz, 1H), 7.71~7.73 (m, 1H), 7.61 (d, J = 8.7 Hz, 2H), 7.49~7.58 (m, 2H), 7.43 (t, J = 7.6 Hz, 1H), 7.27 (d, J = 16.5 Hz, 1H), 7.20 (d, J = 16.5 Hz, 1H), 7.19 (s, 1H), 7.09 (d, J = 8.7 Hz, 2H), 5.29 (s, 2H); 13C-NMR (DMSO-d6) δ:170.0, 168.4, 161.8, 158.0, 137.8, 133.7, 131.9, 131.0, 130.9, 129.8, 129.1, 128.9, 128.7, 128.5 (2C), 127.4, 126.6, 126.1, 125.0, 123.0, 115.6 (2C), 100.5, 61.7; HRMS (ESI) calcd for C25H17BrNO4 [M − H]− 474.0341, found 474.0371.
(E)-3-(4-((5-(m-Tolyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (24): Yellow solid; Yield: 44%; 1H-NMR (DMSO-d6) δ: 13.05 (brs, 1H), 8.12 (s, 1H), 7.96 (s, 1H), 7.80 (t, J = 7.0 Hz, 2H), 7.61~7.73 (m, 4H), 7.41~7.49 (m, 2H), 7.33 (d, J = 16.5 Hz, 1H), 7.25 (d, J = 16.5 Hz, 1H), 7.41 (s, 1H), 7.10 (d, J = 8.8 Hz, 2H), 5.28 (s, 2H), 2.39 (s, 3H); 13C-NMR (DMSO-d6) δ: 170.2, 168.1, 161.7, 158.1, 139.2, 138.0, 131.7, 130.7, 130.3, 129.7, 129.3, 129.2, 128.5 (2C), 128.4, 127.4, 127.0, 126.5, 126.3, 123.3, 115.6 (2C), 100.4, 61.8, 21.3; HRMS (ESI) calcd for C26H20NO4 [M − H]− 410.1392, found 410.1388.
(E)-3-(4-((5-(3-Nitrophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (25): Yellow solid; Yield: 38%; 1H-NMR (DMSO-d6) δ: 13.17 (brs, 1H), 8.35 (s, 1H), 8.13 (s, 1H), 7.78~7.83 (m, 3H), 7.63 (d, J = 8.8 Hz, 2H), 7.46~7.49 (m, 4H), 7.28 (d, J = 16.5 Hz, 1H), 7.22 (d, J = 16.5 Hz, 1H), 7.11 (d, J = 8.8 Hz, 2H), 5.33 (s, 2H); 13C-NMR (DMSO-d6) δ: 167.7, 162.2, 158.0, 148.9, 138.5, 138.2, 131.7, 130.7, 130.5, 130.0, 129.4, 128.6 (2C), 128.4, 127.4, 127.2, 126.2, 125.4, 124.6, 120.7, 115.6 (2C), 102.5, 61.7; HRMS (ESI) calcd for C25H17N2O6 [M − H]− 441.1087, found 441.1072.
(E)-3-(4-((5-(p-Tolyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (26): Yellow solid; Yield: 55%; 1H-NMR (DMSO-d6) δ: 12.98 (brs, 1H), 8.13 (s, 1H), 7.82~7.88 (m, 2H), 7.79 (d, J = 8.1 Hz, 1H), 7.62 (d, J = 8.6 Hz, 2H), 7.49~7.54 (m, 1H), 7.35 (d, J = 8.1 Hz, 2H), 7.47 (t, J = 7.7 Hz, 1H), 7.28 (d, J = 16.5 Hz, 1H), 7.23 (d, J = 16.5 Hz, 1H), 7.10 (d, J = 8.6 Hz, 2H), 7.09 (s, 1H), 5.27 (s, 2H), 2.37 (s, 3H); 170.2, 166.7, 161.7, 158.2, 141.0, 138.4, 131.0, 130.6, 130.3 (2C), 129.7, 129.6, 128.6 (2C), 127.3, 126.1 (2C), 125.9, 124.4, 115.6 (2C), 99.9, 61.8, 21.5; HRMS (ESI) calcd for C26H20NO4 [M − H]− 410.1392, found 410.1373.
(E)-3-(4-((5-(4-Methoxyphenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (27): Yellow solid; Yield: 52%; 1H-NMR (DMSO-d6) δ: 13.04 (brs, 1H), 8.12 (s, 1H), 7.77~7.86 (m, 5H), 7.62 (d, J = 8.4 Hz, 2H), 7.46~7.54 (m, 2H), 7.30 (d, J = 16.5 Hz, 1H), 7.22 (d, J = 16.5 Hz, 1H), 7.09 (d, J = 8.4 Hz, 2H), 7.04 (s, 1H), 5.26 (s, 2H), 3.83 (s, 3H); 13C-NMR (DMSO-d6) δ: 167.7, 166.1, 161.5, 158.2, 138.4, 138.2, 131.7, 130.7, 130.7, 130.0, 129.4, 128.6 (2C), 127.7, 127.5, 127.4, 127.2, 126.1, 124.6, 120.0, 115.2 (2C), 100.0, 61.8, 55.7; HRMS (ESI) calcd for C26H20NO5 [M − H]− 426.1341, found 426.1365.
(E)-3-(4-((5-(4-Fluorophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (28): Yellow solid; Yield: 37%; 1H-NMR (DMSO-d6) δ: 13.10 (brs, 1H), 8.13 (s, 1H), 7.83~7.95 (m, 5H), 7.64 (d, J = 8.5 Hz, 2H), 7.48~7.55 (m, 2H), 7.31 (d, J = 16.5 Hz, 1H), 7.24 (d, J = 16.5 Hz, 1H), 7.21 (s, 1H), 7.11 (d, J = 8.4 Hz, 2H), 5.33 (s, 2H); 13C-NMR (DMSO-d6) δ: 168.1, 166.7, 161.5, 158.8 (d, J = 154.0 Hz, 2C), 158.1, 139.4, 132.5, 132.4 (2C), 130.9,129.9, 128.7 (2C), 128.5, 128.2, 127.9, 127.3, 126.2 (d, J = 26.5 Hz, 2C), 124.6, 115.5 (2C), 104.8, 61.8; HRMS (ESI) calcd for C25H17FNO4 [M − H]− 414.1142, found 414.1159.
(E)-3-(4-((5-(3,4-Dichlorophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (29): Yellow solid; Yield: 52%; 1H-NMR (DMSO-d6) δ: 13.03 (brs, 1H), 8.22 (s, 1H), 8.12 (s, 1H), 7.80~7.83 (m, 3H), 7.62 (d, J = 8.7 Hz, 2H), 7.45~7.52 (m, 2H), 7.37 (s, 1H), 7.30 (d, J = 16.4 Hz, 1H), 7.23 (d, J = 16.4 Hz, 1H), 7.09 (d, J = 8.7 Hz, 2H), 5.30 (s, 2H); 13C-NMR (DMSO-d6) δ: 167.3, 167.1, 161.5, 157.5, 137.7, 133.1, 132.2, 131.6, 131.3, 130.5, 130.3, 130.2, 130.1, 129.1, 128.9, 128.1, 128.0 (2C), 127.5, 127.0, 126.9, 125.7, 115.5, 115.1 (2C), 101.6, 61.2; HRMS (ESI) calcd for C25H16Cl2NO4 [M − H]− 464.0456, found 464.0438.
(E)-3-(4-((5-(3,4-Difluorophenyl)isoxazol-3-yl)methoxy)styryl)benzoic acid (30): Yellow solid; Yield: 45%; 1H-NMR (DMSO-d6) δ: 13.09 (brs, 1H), 8.10 (s,1H), 8.03~8.08 (m, 1H), 7.72~7.80 (m, 3H), 7.57~7.67 (m, 3H), 7.43 (t, J = 7.7 Hz, 1H), 7.17~7.29 (m, 3H), 7.08 (d, J = 8.7 Hz, 1H), 6.96 (d, J = 8.5 Hz, 1H), 5.29 (s, 2H); 13C-NMR (DMSO-d6) δ: 167.3, 167.2, 159.1, 157.5, 137.9, 137.7, 130.7 (d, J = 97.3 Hz), 130.6 (d, J = 97.0 Hz), 130.0, 129.5, 128.9, 128.1, 128.0, 127.7, 127.6, 126.9, 126.7, 125.6, 124.1, 115.5, 115.1, 100.9, 61.2; HRMS (ESI) calcd for C25H16F2NO4 [M − H]− 432.1047, found 432.1083.
3.2. Biological Activity against PTP1B and TCPTP
A colorimetric assay to measure inhibition against PTP1B and TCPTP was performed in 96-well plates. The assay was conducted as described by Zhang et al. [27]. Briefly, the tested compounds were solubilized in DMSO and serially diluted into concentrations for the inhibitory test. The assays were carried out in a final volume of 100 mL containing 50 mmol/L MOPS, pH 6.5, 2 mmol/L pNPP, 30 nmol/L GST-PTP1B or GST-TCPTP, and 2% DMSO, and the catalysis of pNPP was continuously monitored on a SpectraMax 340 microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 405 nm for 2 min at 30 °C. The IC50 value was calculated from the nonlinear curve fitting of the percent inhibition [inhibition (%)] vs. the inhibitor concentration [I] using the following equation:
| Inhibition (%) = 100/[1 + (IC50/[I])·κ], |
where κ is the Hill coefficient.
3.3. Characterization of Compound 29
To determine the mode of inhibition of compound 29, an assay was carried out in a 100 μL assay mixture containing 50 mM MOPS at pH 6.5, 30 nM PTP1B, pNPP in 2-fold dilution up to 80 mM, and different concentrations of inhibitor as described by Zhang et al. [27]. In the presence of the competitive inhibitor, the Michaelis–Menten equation is described as 1/ν = (Km/[Vmax[S]])·(1 + [I]/Ki) + 1/Vmax, where ν is the initial rate, Vmax is the maximum rate, and [S] is the substrate concentration. Ki value was obtained by Lineweaver–Burk plot of apparent Km/Vmax (slope) from primary reciprocal plot versus the inhibitor concentration [I] according to the equation Km/Vmax = 1 + [I]/Ki.
4. Conclusions
In this paper we report a series of stilbene derivatives containing phenyl-substituted isoxazoles as inhibitors against protein tyrosine phosphatase 1B (PTP1B). Most of these compounds showed moderate to good inhibitory activity against PTP1B. The most potent compound 29 (IC50 = 0.91 ± 0.33 μM) showed 14-fold improved PTP1B inhibitory activity compared to the lead compound LCA and about a 4-fold selectivity over TCPTP. Our SAR study clarified the effects of the substituents on the phenyl C ring on the corresponding activities. Compound 29 was identified as a competitive inhibitor of PTP1B with a Ki value of 0.78 μM in enzyme kinetic studies. These novel stilbene derivatives could be promising lead compounds for the development of a new class of PTP1B inhibitors. Further SAR research and evaluation of new derivatives are still in progress.
Acknowledgments
This research was financially supported by the Science Fund for Young Scholar of Jiangsu (BK20140425), Initiating Fund for Introduced Talents of Nantong University (03080694), the National Natural Science Foundation of China (81402849), Zhejiang Province Natural Science Foundation (LQ14H300003) and Public Projects of Zhejiang Province (2015C33159 & 20116C31017). Many thanks to Li Jia’s group of National Center for Drug Screening (Shanghai Institute of Materia Medica Academy of Science) for their help in bioassays. We also thank the Analysis & Testing Centre of Nantong University for support.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/12/1722/s1.
Author Contributions
Y.S. designed the research; H.H., J.J., and S.C. performed the research and analyzed the data; Y.G. and F.Y. performed the biological test; H.D. wrote the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 15~30 are available from the authors.
References
- 1.Alonso A., Sasin J., Bottini N., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 2.Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/S0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 3.Neel B.G., Tonks N.K. Protein tyrosine phosphatases in signal transduction. Curr. Opin. Cell Biol. 1997;9:193–204. doi: 10.1016/S0955-0674(97)80063-4. [DOI] [PubMed] [Google Scholar]
- 4.Tonks N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 5.Elchebly M., Payette P., Michaliszyn E., Cromlish W., Collins S., Loy A.L., Normandin D., Cheng A., Himms-Hagen J., Chan C.C., et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 6.Klaman L.D., Boss O., Peroni O.D., Kim J.K., Martino J.L., Zabolotny J.M., Moghal N., Lubkin M., Kim Y.B., Sharpe A.H., et al. Increased energy expenditure, decreased adiposity, and tissuc insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 2000;20:5479–5489. doi: 10.1128/MCB.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng A., Uetani N., Simoncic P.D., Chaubey V.P., Lee-Loy A., McGlade C.J., Kennedy B.P., Tremblay M.L. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev. Cell. 2002;2:497–503. doi: 10.1016/S1534-5807(02)00149-1. [DOI] [PubMed] [Google Scholar]
- 8.Zabolotny J.M., Bence-Hanulec K.K., Stricker-Krongrad A., Haj F., Wang Y., Minokoshi Y., Kim Y.B., Elmquist J.K., Tartaglia L.A., Kahn B.B., et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell. 2002;2:489–495. doi: 10.1016/S1534-5807(02)00148-X. [DOI] [PubMed] [Google Scholar]
- 9.Iversen L.F., Moller K.B., Pedersen A.K., Peters G.H., Petersen A.S., Andersen H.S. Structure Determination of T Cell Protein-tyrosine Phosphatase. J. Biol. Chem. 2002;277:19982–19990. doi: 10.1074/jbc.M200567200. [DOI] [PubMed] [Google Scholar]
- 10.Wang W.L., Yang D.L., Gao L.X., Tang C.L., Ma W.P., Ye H.H., Zhang S.Q., Zhao Y.N., Xu H.J., Hu Z., et al. 1H-2,3-Dihydroperimidine Derivatives: A New Class of Potent Protein Tyrosine Phosphatase 1B Inhibitors. Molecules. 2014;19:102–121. doi: 10.3390/molecules19010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He H.B., Gao L.X., Deng Q.F., Ma W.P., Tang C.L., Qiu W.W., Tang J., Li J.Y., Li J., Yang F. Synthesis and biological evaluation of 4,4-dimethyl lithocholic acid derivatives as novel inhibitors of protein tyrosine phosphatase 1B. Bioorg. Med. Chem. Lett. 2012;22:7237–7242. doi: 10.1016/j.bmcl.2012.09.040. [DOI] [PubMed] [Google Scholar]
- 12.Qin Z.H., Pandey N.R., Zhou X. Functional properties of Claramine: A novel PTP1B inhibitor and insulin-mimetic compound. Biochem. Biophys. Res. Commun. 2015;458:21–27. doi: 10.1016/j.bbrc.2015.01.040. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell F.R. MSI-1436 reduces acute food intake without affecting dopamine transporter activity. Pharmacol. Biochem. Behav. 2010;97:138–143. doi: 10.1016/j.pbb.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dodds E.C., Goldberg L., Lawson W., Robinson R. Oestrogenic activity of certain synthetic compounds. Nature. 1938;141:247–248. doi: 10.1038/141247b0. [DOI] [Google Scholar]
- 15.Sun H.M., Tawa G., Wallqvist A. Classification of scaffold-hopping approaches. Drug Discov. Today. 2012;17:310–324. doi: 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inamori Y., Kato Y., Kubo M., Yasuda M., Baba K., Kozawa M. Physiological activities of 3,3′,4,5′-tetrahydroxystilbene isolated from the heartwood of Cassia garrettiana CRAIB. Chem. Pharm. Bull. 1984;32:213–218. doi: 10.1248/cpb.32.213. [DOI] [PubMed] [Google Scholar]
- 17.Belofsky G., Percivili D., Lewis K.G., Tegos P., Ekart J. Phenolic Metabolites of Dalea versicolor that Enhance Antibiotic Activity against Model Pathogenic Bacteria. J. Nat. Prod. 2004;67:481–484. doi: 10.1021/np030409c. [DOI] [PubMed] [Google Scholar]
- 18.López-Nicolás J.M., Rodríguez-Bonilla P., García-Carmona F. Complexation of Pinosylvin, an Analogue of Resveratrol with High Antifungal and Antimicrobial Activity, by Different Types of Cyclodextrins. J. Agric. Food Chem. 2009;57:10175–10180. doi: 10.1021/jf902519d. [DOI] [PubMed] [Google Scholar]
- 19.Bagula P.K., Dindab A.K., Banerjeea S.K. Effect of resveratrol on sirtuins expression and cardiac complications in diabetes. Biochem. Biophys. Res. Commun. 2015;468:221–227. doi: 10.1016/j.bbrc.2015.10.126. [DOI] [PubMed] [Google Scholar]
- 20.Sheng R., Xu Y., Hu C.Q., Zhang J., Lin X., Li J.Y., Yang B., He Q.J., Hu Y.Z. Design, synthesis and AChE inhibitory activity of indanone and aurone derivatives. Eur. J. Med. Chem. 2009;44:7–17. doi: 10.1016/j.ejmech.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Anthony N.G., Breen D., Clarke J., Donoghue G., Drummond A.J., Ellis E.M., Gemmell C.G., Helesbeux J.J., Hunter I.S., Khalaf A.I., et al. Antimicrobial Lexitropsins Containing Amide, Amidine, and Alkene Linking Groups. J. Med. Chem. 2007;50:6116–6125. doi: 10.1021/jm070831g. [DOI] [PubMed] [Google Scholar]
- 22.Vallejos J.C., Schouteeten A., Wilhelm D. Process for the Synthesis of Polyhydroxystilbene Compounds. 8,399,714. US Patent. 2010 Dec 23;
- 23.Yamamoto T., Fujita K., Asari S., Chiba A., Kataba Y., Ohsumi K., Ohmuta N., Iida Y., Ijichi C., Iwayama S., et al. Synthesis and evaluation of isoxazole derivatives as lysophosphatidic acid (LPA) antagonists. Bioorg. Med. Chem. Lett. 2007;17:3736–3740. doi: 10.1016/j.bmcl.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 24.Herrero M.T., Tellitu I., Domínguez E., Hernández S., Moreno I., SanMartín R. A general and efficient PIFA mediated synthesis of heterocycle-fused quinolinone derivatives. Tetrahedron. 2002;58:8581–8589. doi: 10.1016/S0040-4020(02)00903-1. [DOI] [Google Scholar]
- 25.Ramana P.V., Reddy A.R. Synthesis of 1,2,3-triazole substituted isoxazoles via copper(I) catalyzed cycloaddition. J. Heterocycl. Chem. 2012;49:621–627. doi: 10.1002/jhet.837. [DOI] [Google Scholar]
- 26.Kamal A., Tamboli J.R., Vishnuvardhan M.V., Adil S.F., Nayak V.L., Ramakrishna S. Synthesis and anticancer activity of heteroaromatic linked 4β-amido podophyllotoxins as apoptotic inducing agents. Bioorg. Med. Chem. Lett. 2013;23:273–280. doi: 10.1016/j.bmcl.2012.10.099. [DOI] [PubMed] [Google Scholar]
- 27.Zhang W., Hong D., Zhou Y., Zhang Y., Shen Q., Li J.Y., Hu L.H., Li J. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochim. Biophys. Acta. 2006;1760:1505–1512. doi: 10.1016/j.bbagen.2006.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.