

Abstract
Regioselectively, ethyl propiolate reacted with 1-(propergyl)-pyridinium-3-olate to give two regioisomers; ethyl 4-oxo-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-2,6-diene-6-carboxylate 4, ethyl 2-oxo-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-3,6-diene-6-carboxylate 5 as well as ethyl 2,6-dihydro-6-(prop-2-ynyl)furo(2,3-c)pyridine-3-carboxylate 6. The obtained compounds were identified by their spectral (IR, mass and NMR) data. Moreover, DFT quantum chemical calculations were used to study the mechanism of the cycloaddition reaction. The regioselectivity was explained using transition state calculations, where the calculations agreed with the formation of products 4 and 5 in almost the same ratio. The reaction was also extended for diphenylaceylene as dipolarophile to give only two products instead of three.
Keywords: ultrasonic synthesis; 1,3-dipolar cycloaddition; molecular structure; reaction mechanism
1. Introduction
Nowadays, improving the traditional methods for organic synthesis is an important issue. Ultrasound is widely used to avoid using expensive reagents, highly acidic solutions, long reaction times and high temperatures as well as to improve reaction yields and incompatibility with other functional groups [1].
The use of ultrasound irradiation in the organic synthesis offers many benefits, which include milder reaction conditions, decrease in the reaction times, and increase in selectivity, thus enhancing the purity and reaction efficiency [2,3,4]. Ultrasound irradiation has been used for the synthesis of simple heterocyles [5,6,7], various ionic liquids [8], and poly functionalized heterocyclic compounds [9] and has been reported to promote the reaction rates and yields. One of the fascinating chemical reactions of pyridin-3-ol is the exhibition of 1,3-dipolar characters.
Based on the spectroscopic data [10,11,12,13,14], pyridin-3-ol Ia in polar solvents exists to a considerable extent in the unstable mesomeric form, pyridinium-3-olate Ib, via prototropic rearrangement [15] (which is being considered as a resonance hybrid between the indicated canonical structures) (Scheme 1).
Scheme 1.

Resonating structures of 1-(propergyl)-pyridinium-3-olate.
The aim of this work focused on the use of ultrasound to promote 1,3-dipolar cycloaddition reaction of the, 1-(propergyl)-pyridinium-3-olate 1 with acetylene derivatives.
2. Results and Discussion
2.1. Chemistry
Treatment of pyridin-3-ol with propargylchloride in ethanol afforded directly the corresponding quaternary pyridinium salt as a pale yellow crystalline solid. Upon treatment of the salt with triethylamine, the mixture was subjected to ultrasound at 40 °C for 1 h. The reaction yielded a product with molecular formula of C16H14O2N2 The product was assigned as the dimeric form of the salt formed via the intermediate pyridinium-3-olate 1 (Scheme 2). The IR spectrum of the dimer revealed the characteristic absorption bands of conjugated carbonyl groups at 1734 and 1690 cm−1. The 1H-NMR spectrum revealed a singlet signal at δ = 2.05 for two acetylenic-H and 2 AB system at δ = 4.23 ppm for four methine protons along with the doublets of olefenic and the multiplet of cycloalkanes protons.
Scheme 2.

Formation of 1-(propergyl)-pyridinium-3-olate and its dimerization.
The Reaction of Pyridinium-3-olate (1) with Ethyl Propiolate (2) as 2π-1,3-Dipolarophiles
The cycloaddition reaction between 1-(propergyl)pyridinium-3-olate 1(4π-1,3-dipole across the 2,6-posions as well as O-C4 region of the pyridine ring) and asymmetric 2π-acetylene 1,3-dipolarophiles, ethyl propiolate 2, proceeded effectively in a regioselective manner to give two regioisomers, ethyl 4-oxo-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-2,6-diene-6-carboxylate 4 and ethyl 2-oxo-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-3,6-diene-6-carboxylate 5, as the major products (32% and 27%, respectively) and ethyl 2,6-dihydro-6-(prop-2-ynyl)furo(2,3-c)pyridine-3-carboxylate 6 as a minor product (11%). It is worth noting that the cycloaddition product 7 is not obtained (Scheme 3).
Scheme 3.

Cycloaddition of ethyl propiolate with 1-(propergyl)-pyridinium-3-olate.
2.2. Computational Details
Full geometry optimizations were carried out for the reactants, transition states (TSs) and cycloaddition products (CAs) using the B3LYP [16,17]. DFT function and the 6-31G (d, p) basis set. All calculations were carried out with GAUSSIAN 03 [18]. The stationary points were characterized by frequency calculations in order to verify that minima and transition states have zero and one imaginary frequency, respectively. Each TS gave one negative vibrational mode corresponding to the motion involving the formation of the newly forming C-C bonds. The vibrational modes were assigned appropriately by means of visual inspection and animation using the Gaussview software [19]. The reported total energies include zero point energy (ZPE) corrections are given at 298.15 K. Analysis of the frontier molecular orbital (FMO) interactions, global electrophilicity index (ω), was calculated following the expression [20,21,22,23], ω = (μ2/2η), where μ is the electronic chemical potential, μ = (EH + EL)/2, where EH and EL are FMO energies and η is the chemical hardness, η = (EL − EH). The nucleophilicity index, N [21], which is defined as N = EH(Nu) − EH(TCE), where tetracyanoethylene (TCE) is chosen as the reference [24,25]. The atomic electronic population and DFT-based reactivity indices were computed using natural population analysis (NPA) [26]. The local electrophilicity ωk and nucleophilicity Nk indices of atom k are obtained by the help of Fukui index (fk) using equations:
| (1) |
| (2) |
where Qk(N), Qk(N + 1), Qk(N − 1) are the gross electronic population of site k in neutral, anionic, and cationic systems, respectively [27]. The optimized structures of the reactant molecules are shown in Figure S1 (Supplementary Materials).
We used DFT calculations in order to explain the regioselectivity of the studied reaction system. The reaction goes regioselectively to form the products 4–6 in different yields while 7 is not formed. Table 1 shows the values of the FMO energies (eV), the electronic chemical potentials (μ), the global electrophilicity (ω) and nucleophilicity (N) of the reactants. Figure 1 presents a schematic representation of the possible interactions between the FMOs (HOMO of 1-LUMO of 2) and (HOMO of 2-LUMO of 1). Table 1 and Figure 1 show that the gap between HOMO of 1 and LUMO of 2 is smaller (3.965 eV) than the gap between HOMO of 2 and LUMO of 1 (6.208 eV). It implies that the main interaction occurs between the HOMO of 1 and the LUMO of 2
Table 1.
The FMO energies (E), electronic chemical potential (μ), nucleophilicty (N) and electrophilicity (ω) indices for the reactants.
| Reactant | EH | EL | Μ | ω | N |
|---|---|---|---|---|---|
| 1 | −5.1376 | −1.3783 | −3.2579 | 1.4117 | 3.9830 |
| 2 | −7.5860 | −1.1725 | −4.3793 | 1.4951 | 1.5345 |
Figure 1.

Schematic representation of the possible interactions between the FMOs.
Moreover, the electronic chemical potential of 1 (−3.2579 eV) is higher than that of 2 (−4.3793 eV) indicating that charge transfer will take place from the reactant 1 to 2, which is in agreement with the FMO analysis. The amount of charge transfer from 1 to 2 is calculated to be 0.1427 e, 0.1326 e and 0.2172 e for products 4, 5 and 6, respectively. These results indicate that the studied cycloaddition reactions have polar character. The amount of charge transfer is the highest for TS of 6 so; it has the highest polar cycloaddition (CA) reaction. At the transition state structures, the reactant 1 fragment has positive charge while fragment of reactant 2 have negative charge which confirm that 1 is the nucleophile while 2 is the electrophile in these CA reactions.
Domingo et al. classified electrophiles based on the electrophilicity index as strong (ω > 1.50 eV), moderate (1.50 > ω > 0.80 eV) and marginal (ω < 0.80 eV). In addition, nucleophiles are classified as strong (N > 3.00 eV), moderate (3.00 > N > 2.00 eV) and marginal (N < 2.00 eV) [28,29,30]. According to the classification, 1 is a strong nucleophile and moderate electrophile, whereas 2 is moderate electrophile and marginal nucleophile. Based on the values of nucleohilicity index (N) listed in Table 1, 1 has higher nucleophilicity (3.9830 eV) than 2 (1.5345 eV). On the other hand, the electrophlicity index (ω) of 1 (1.4117 eV) is lower than 2 (1.4951 eV). As a result, 1 is the nucleophile while 2 is the electrophile in this reaction, in agreement with the previous analyses.
The local electrophilicity indices ωk and Nk of atom k were used to explain the regioselectivity of the studied CA reaction. The values of the Fukui indices ( and ) and local electrophilicity indices (ωk and Nk) are reported in Table 2. The atom numbering Scheme of the reactants are given in Figure S1 (Supplementary Materials). For better visualization, we have depicted these interactions in Scheme 4. It is slightly more favorable if C1 and C2 of reactant 2 attack on C5 and C1 of 1, respectively rather than the C1 and C5 of 1, respectively. Hence, one could predict that 4 will be formed slightly more than 5. Experimentally, we observed the formation of product 6 while 7 was not formed at all. Based on the magnitude of local descriptors shown in Scheme 4, it is clear that in the [2 + 3] cycloaddition reaction, the C1 and C2 of reactant 2 favor the attack on C3 and O10 of 1, giving product 6 as the only furan product.
Table 2.
Electrophilic and nucleophilic Fukui indices and local electrophilicities for the reactive atoms of 1 and 6.
| Reactant | 1 | 2 | |||||
|---|---|---|---|---|---|---|---|
| Parameter | C1 | C5 | C3 | O10 | Parameter | C1 | C2 |
| 0.1840 | 0.1738 | 0.1378 | 0.2951 | −0.0661 | −0.2783 | ||
| 0.1044 | 0.1977 | 0.1761 | 0.0936 | −0.0395 | −0.3229 | ||
| Nk | 0.7330 | 0.6922 | 0.5487 | 1.1754 | ωk | 0.0591 | 0.4827 |
Scheme 4.

Illustration of the favorable interactions using local electrophilicity indices.
In order to explain the formation of 4 and 5 as the major products, 6 as a minor product, and no formation of 7, we performed transition state calculations for the four regioisomeric pathways. The four transitions states of the reactions leading to the formation of products 4, 5, 6 and 7 will be abbreviated TS4, Ts5, Ts6 and TS7, respectively. The geometries of these four TSs are given in Figure 2 together with the newly forming bond lengths. Table 3 reports the energies (a.u.) and relative energies (kcal/mol). The potential energy surfaces (PESs) corresponding to all the reaction channels, are illustrated in Figure 3. Based on the calculated energy difference between the product and reactants, all products are stable thermodynamically where the most stable product is 5. On the other hand, the calculated activation energies of the different reaction pathways between 1 and 2 showed that adduct 4 is the most favored kinetically in comparison with the other approaches. The small energy difference (0.984 Kcal/mol) between the two TSs of adducts 4 and 5 indicated that why these two products are formed in almost the same proportions. The high activation energy of product 7 TS (21.849 Kcal/mol) suggested that 7 could not be formed while 6 is formed although in lower amount than the others (4 and 5). These results agree with what we observed experimentally.
Figure 2.

The four transition state structures of the studied CA reaction.
Table 3.
Energies and relative energies (∆E) of the reactants, transition states, and products.
| System | E (a.u.) | ∆E (Kcal/mol) a | ∆G (Kcal/mol) a |
|---|---|---|---|
| 1 | −438.804 | ||
| 2 | −344.429 | ||
| TS4 | −783.212 | 13.213 | 26.498 |
| TS5 | −783.210 | 14.197 | 27.013 |
| TS6 | −783.208 | 15.539 | 27.231 |
| TS7 | −783.198 | 21.849 | 34.320 |
| 4 | −783.282 | −30.691 | −16.514 |
| 5 | −783.284 | −32.179 | −18.097 |
| 6 | −783.280 | −29.502 | −16.561 |
| 7 | −783.273 | −25.174 | −11.936 |
a The energies of the TSs and products are referred to the sum (E1 + E2).
Figure 3.

Energy profiles, in kcal/mol for the two pathways of the CA reactions (red; 4, violet 5, brown 6 and black 7).
It is worth noting that product 6 could undergo rearrangement via proton transfer leading to two other possible structures (8 and 9) shown in Figure 4. These products could not be easily differentiated experimentally using routine NMR measurements as the H and C atoms of the three adducts have very close chemical shifts. Form this point of view, theoretical calculations on the three suggested products of 6 were performed in order to predict the most stable adduct based on the energy analysis of these compounds. The energies and thermodynamic parameters of the equilibrium reactions shown in Scheme 5 are given in Table 4. The relative energy and relative Gibbs free energy are given in Kcal/mol.
Figure 4.

The possible regioisomers of 6.
Scheme 5.
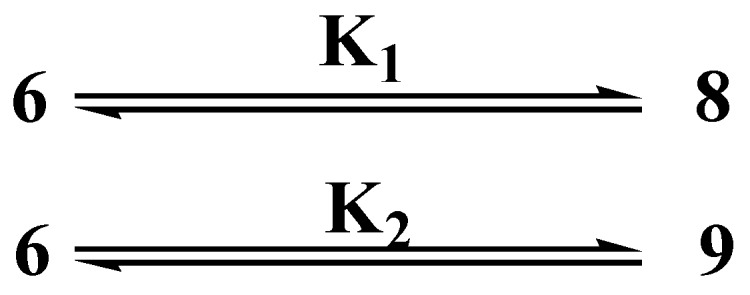
The equilibrium constants among the proton transfer structures of adduct 6.
Table 4.
The energies, thermodynamic parameters and rate constants (k) of the studied reactants.
| Energies | 6 | 8 | 9 |
|---|---|---|---|
| E (a.u) | −783.5544 | −783.5158 | −783.5455 |
| ZPVE (a.u) | 0.2371 | 0.2359 | 0.2368 |
| Ecorr (a.u) | −783.3172 | −783.2799 | −783.3087 |
| ∆E (Kcal/mol) | 0.0000 | −23.4370 | −5.3823 |
| H (a.u) | −783.3003 | −783.2628 | −783.2916 |
| S (Cal/Mol·K) | 130.8760 | 133.5250 | 131.7610 |
| G (a.u) | −783.3625 | −783.3262 | −783.3542 |
| ∆G (Kcal/mol) | 0.0000 | −22.7472 | −5.2121 |
| K | 3.69 × 1016 | 6.25 × 103 |
It is found that 6 has the lowest energy and hence the most stable form. The rearrangement product 6 is more stable by 5.3823 and 23.4370 kcal/mol than 9 and 8, respectively. In addition, the negative Gibbs free energy change indicated that the rearrangement process from 8 and 9 to 6 is spontaneous. Using the relation ΔG = −RTlnK, the equilibrium constants were obtained where ΔG is the difference between the Gibbs free energy of 8 or 9 with respect to the most stable one (6). Using the equilibrium constants obtained from such calculations, the ratio of the three products 8, 9 and 6 were obtained. The results showed that 6 has 99.98% and it is only one that could exist.
2.3. NMR Spectra
The values of isotropic magnetic shielding (IMS) are calculated by the GIAO approach at the 6-31G (d, p) level that were used to predict the 13C- and 1H-NMR chemical shifts (δcalc) for the studied compound, and the results were correlated with the experimental NMR data (δexp) in CDCl3 solvent. Figure 5, Figure 6 and Figure 7 and Table 5, Table 6 and Table 7 showed the correlation between the experimental and theoretical 13C- and 1H-NMR chemical shifts of the studied compounds. According to the results, the calculated chemical shifts were in good agreement with the experimental findings. The correlation coefficients for 13C- and 1H-NMR chemical shifts are up to 0.991 and 0.947, respectively.
Figure 5.

The correlation graphs between the calculated and experimental chemical shifts compound 4.
Figure 6.

The correlation graphs between the calculated and experimental chemical shifts compound 5.
Figure 7.

The correlation graphs between the calculated and experimental chemical shifts compound 6.
Table 5.
Calculated and experimental NMR chemical shifts of compound 4.
| Atom | δcalc | δexp | Atom | δcalc | δexp |
|---|---|---|---|---|---|
| 1 C | 56.47 | 76.21 | 18 H | 4.64 | 4.19 |
| 2 C | 130.20 | 139.98 | 19 H | 7.04 | 6.79 |
| 3 C | 111.94 | 126.40 | 20 H | 5.75 | 6.10 |
| 4 C | 179.58 | 190.89 | 21 H | 4.17 | 4.06 |
| 5 C | 68.39 | 82.50 | 22 H | 7.84 | 7.32 |
| 7 C | 131.65 | 121.11 | 23 H | 1.04 | 1.69 |
| 8 C | 138.18 | 146.87 | 24 H | 1.32 | 1.69 |
| 9 C | 147.91 | 162.71 | 25 H | 1.85 | 1.69 |
| 12 C | 7.88 | 11.24 | 26 H | 5.17 | 4.21 |
| 13 C | 53.13 | 62.71 | 27 H | 3.94 | 4.21 |
| 14 C | 30.78 | 31.78 | 28 H | 3.69 | 3.24 |
| 15 C | 64.85 | 79.89 | 29 H | 4.18 | 3.24 |
| 16 C | 59.16 | 72.5 | 30 H | 1.89 | 2.16 |
Table 6.
Calculated and experimental NMR chemical shifts of compound 5.
| Atom | δcalc | δexp | Atom | δcalc | δexp |
|---|---|---|---|---|---|
| 1 C | 62.32 | 72.5 | 18 H | 4.86 | 4.02 |
| 2 C | 134.02 | 139.98 | 19 H | 7.42 | 6.40 |
| 3 C | 126.52 | 121.11 | 20 H | 5.83 | 6.41 |
| 4 C | 184.79 | 190.89 | 21 H | 3.83 | 3.98 |
| 5 C | 76.89 | 82.50 | 22 H | 7.48 | 6.80 |
| 7 C | 96.28 | 139.98 | 23 H | 1.09 | 1.22 |
| 8 C | 129.53 | 146.87 | 24 H | 1.43 | 1.22 |
| 9 C | 169.53 | 162.71 | 25 H | 1.90 | 1.22 |
| 12 C | 13.24 | 11.24 | 26 H | 5.19 | 4.22 |
| 13 C | 64.62 | 62.71 | 27 H | 3.98 | 4.22 |
| 14 C | 32.78 | 31.78 | 28 H | 3.58 | 3.24 |
| 15 C | 72.50 | 79.89 | 29 H | 4.19 | 3.24 |
| 16 C | 68.71 | 76.21 | 30 H | 1.92 | 1.62 |
Table 7.
Calculated and experimental NMR chemical shifts of compound 6.
| Atom | δcalc | δexp | Atom | δcalc | δexp |
|---|---|---|---|---|---|
| 1 C | 93.78 | 113.56 | 18 H | 6.51 | 6.33 |
| 2 C | 121.1 | 143.35 | 19 H | 6.39 | 6.22 |
| 3 C | 94.75 | 144.09 | 20 H | 5.92 | 7.25 |
| 4 C | 146.6 | 149.14 | 21 H | 4.18 | 4.09 |
| 5 C | 124.88 | 142.15 | 22 H | 4.02 | 4.09 |
| 7 C | 38.14 | 42.7 | 23 H | 2.19 | 1.92 |
| 8 C | 62.53 | 73.8 | 24 H | 5.65 | 5.07 |
| 9 C | 61.77 | 82.7 | 25 H | 5.64 | 5.07 |
| 12 C | 72.87 | 69.5 | 26 H | 0.97 | 1.42 |
| 13 C | 87.91 | 104.82 | 27 H | 1.27 | 1.42 |
| 14 C | 147.75 | 173.4 | 28 H | 1.88 | 1.42 |
| 15 C | 8.78 | 13.6 | 29 H | 5.22 | 4.23 |
| 16 C | 50.73 | 60.1 | 30 H | 3.76 | 4.23 |
On the other hand, this reaction was extended for the cycloaddition reaction of 1-(propergyl)pyridinium-3-olate 1 with diphenylacetylene as symmetric dipolarophile to afford only two products, 6,7-diphenyl-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-3,6-dien-2-one 10 as major product and 2,6-dihydro-2,3-diphenyl-6-(prop-2-ynyl)furo(2,3-c)pyridine 11 as a minor product (Scheme 6).
Scheme 6.

Cycloaddition of diphenylacetylene with 1-(propergyl)-pyridinium-3-olate.
3. Materials and Methods
1-(propergyl)-pyridinium-3-olate (1): A mixture of pyridin-3-ol (1.9 g, 0.02 mol) and propargyl chloride (1.45 g, 0.02 mol) in absolute alcohol (30 mL) was heated under reflux in water bath while stirring for 3 h. The reaction mixture was allowed to cool until room temperature; the solvent was removed under reduced pressure. The yellow residue obtained was recrystallized from ethanol to give 1 as pale yellow crystals yield 88%, mp 98–100 °C. IR: ν 3401–3250 (broad, OH), 2364 (acetylenic bond), 1571, 1241 cm−1 (ring vibrations). 1H-NMR (CDCl3): δ = 2.05 (s, 1H, acetylenic-H), 3.25 (s, 1H, OH), 3.52 (s, 1H, H-2), 3.75 (d, 1H, H-5), 5.2 (s, 1H, CH2), 5.92 (dd, 1H, H-3), 6.48 (d, 1H, H-4). Anal. Calcd for C8H8NOCl (169.6): MS (EI): m/z = 133 [M+ − HCl], base peak at m/z = 95 [M+ − Cl-CH2CCH].
Reaction of 1-(propergyl)-pyridinium-3-olate (1) with ethyl propiolate (2): To a mixture of 3-hydroxy prop-2-yne-1-yl-pyridinium chloride (1.69 g, 0.01 mol) and ethyl propiolate (2 mL, 0.026 mol) in the presence of hydroquinone (0.01 g), triethylamine (1 mL) was added dropwise. The reaction mixture was heated in ultrasonic bath, for 6 h at 40 °C. At the end of the reaction, the brown residue obtained was triturated with distilled water and then extracted with chloroform that was separated, dried over anhydrous sodium sulfate, filtered off, and evaporated under reduced pressure to give a brown sticky residue, which gave three spots on TLC. The residue obtained was eluted through a column of alumina with a mixture of ethyl acetate-light petroleum (1:2). Three products 4–6 were obtained in a combined yield of 80% (1.84 g).
Ethyl 4-oxo-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-2,6-diene-6-carboxylate (4): Yellow crystals (0.61 g, 32%), mp 176–177 °C, IR ν = 2929 (CH), 1698 (conjugated CO), 1735 cm−1 (CO ester), C13H13NO3 (231.25), [M+] at m/z = 231, base peak at m/z = 80.
Ethyl 2-oxo-8-(prop-2-ynyl)-8-aza-bicyclo[3.2.1]octa-3,6-diene-6-carboxylate (5): Orange crystals (0.52 g, 27.5%), mp 180–182 °C, IR ν = 2931 (CH), 1686 (conjugated CO), 1726 cm−1 (CO ester), C13H13NO3 (231.25), [M+] at m/z = 231, base peak at m/z = 80.
Ethyl 2,6-dihydro-6-(prop-2-ynyl)furo(2,3-c)pyridine-3-carboxylate (6): Yellow oil (0.20 g, 11.0%), IR ν = 2924 (CH), 1723 (CO ester), 1616 (C=C-N), 1348 cm−1 (furan ring). C13H13NO3 (231.24), [M+] at m/z = 231, base peak at m/z = 180.
3.1. Reaction of 3-Hydroxy-1-prop-2-yne-1-ylpyridinium Chloride with Diphenyl Acetylene
To a mixture of 1-(propergyl)-pyridinium-3-olate (1) (1.69 g, 0.01 mol) and diphenyl acetylene (3.56 mL, 0.02 mol) in the presence of hydroquinone (0.01 g), triethylamine (1 mL) was added dropwise. The reaction mixture was heated in ultrasonic bath, for 6 h at 40 °C. At the end of the reaction, the brown residue obtained was triturated with distilled water and then extracted with chloroform that was separated, dried over anhydrous sodium sulfate, filtered off, and evaporated under reduced pressure to give a brown sticky residue. The residue obtained was eluted through a column of alumina with a mixture of chloroform–light petroleum (3:1). The solvent was removed under reduced pressure to give two products, 10–11 (2.54 g, overall yield 82%).
6,7-Diphenyl-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-3,6-dien-2-one (10): A brown oil, (1.52 g , 60%), IR ν = 2920 (CH), 1690 cm−1 (conjugated CO). C22H17NO (311.37), [M+] at m/z = 311 a.m.u., base peak at m/z = 95 a.m.u 1H-NMR (DMSO) at δ = 2.18 (s, 1H, acetylenic-H); 4.05 (s, 2H, CH2); 4.93 (d, J4,5 = 5 Hz, 1H, H-5); 5.22 (s, 1H, H-1); 6.00 (d, J3,4 = 5 Hz, 1H, H-3); 6.58 (dd, J4,5 = 5 Hz, J4,3 = 5 Hz, 1H, H-4); 7.20–7.91 (m, 10H, arom-H).
2,6-Dihydro-2,3-diphenyl-6-(prop-2-ynyl)furo(2,3-c)pyridine (11): orange oil, (0.42 g, 14%), IR ν = 2924 (CH), 1628–1348 (furan ring), 1525 cm−1 (C=C-N). C22H17NO (311.37) C (84.84%), H (5.50%), N (4.50%) required C (84.65%), H (5.33%), N (4.42%). 1H-NMR (DMSO) at δ = 2.1 (s, 1H, acetylenic-H); 3.55 (s, 2H, CH2 methylene-H); 4.92(s, 1H, H-1); 5.2(d, 1H, H-2); 6.35 (d, 1H, H-3); 7.90–8.07 (m, 10H, arom-H and 1H, H-4).
4. Conclusions
The cycloaddition reaction between 1-(propergyl)pyridinium-3-olate 1 with either symmetric or asymmetric 2π-acetylene 1,3-dipolarophiles proceeded effectively in a regioselective manner to give bicyclic compounds (8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-3,6-diene derivative) and 2,6-dihydro-6-(prop-2-ynyl)furo(2,3-c)pyridine derivative. The results of the DFT calculations showed that in the cycloaddition reaction between 1-(propergyl)pyridinium-3-olate 1 and ethyl propiolate 2, the reactant 1 is the nucleophile while 2 is the electrophile. The global and local reactivity descriptors were used to study the mechanism of the studied CA reaction. Transition state calculations were used to explain the regioselectivity of the CA reaction. Product 7 could not be formed as it has the highest activation energy. Products 4–6 have lower activation energy where 4 and 5 have very close energy barrier so they are formed almost in the same ratio. Product 6 has higher activation energy; hence, it is formed in a lower amount than the others.
Acknowledgments
The authors highly acknowledge Alexandria University and Ain Shams University to give us the opportunity to do this work.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/21/7/848/s1.
Author Contributions
All authors are equally contributed in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.Yadav J.S., Reddy B.V.S., Reddy K.B., Raj K.S., Prasad A.R. Ultrasound-accelerated synthesis of 3,4-dihydropyrimidin-2(1H)-ones with ceric ammonium nitrate. J. Chem. Soc. Perkin Trans. 1. 2001:1939–1941. doi: 10.1039/b102565c. [DOI] [Google Scholar]
- 2.Suslick K.S. Kirk-othmer encyclopedia of chemical technology. Sonochemistry. 1990;247:1439–1445. doi: 10.1126/science.247.4949.1439. [DOI] [PubMed] [Google Scholar]
- 3.Park Y.S., Han J.H., Yoo B., Choi K.I., Kim J.H., Yoon C.M. Facile Synthesis of beta-Ketoesters by Indium-Mediated Reaction of Acyl Cyanides with Ethyl Bromoacetate under Ultrasonication. Bull. Korean Chem. Soc. 2005;26:878–879. [Google Scholar]
- 4.Soengas R.G. Studies on indium-mediated additions to lactones: Synthesis of 2-deoxy-2-substituted-3-ulosonic acids. Tetrahedron Asymmetry. 2010;21:2249–2253. doi: 10.1016/j.tetasy.2010.07.011. [DOI] [Google Scholar]
- 5.Patil R., Bhoir P., Deshpande P., Wattamwar T., Shirude M., Chaskar P. Relevance of sonochemistry or ultrasound (US) as a proficient means for the synthesis of fused heterocycles. Ultrason. Sonochem. 2013;20:1327–1336. doi: 10.1016/j.ultsonch.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Lahyani A., Trabelsi M. Ultrasonic-assisted synthesis of flavones by oxidative cyclization of 2′-hydroxychalcones using iodine monochloride. Ultrason. Sonochem. 2016;3:626–630. doi: 10.1016/j.ultsonch.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 7.Rabiei K., Naeimi H. Ultrasonic assisted synthesis of gem-dichloroaziridine derivatives using Mg/CCl4 under neutral conditions. Ultrason. Sonochemistry. 2015;24:150–154. doi: 10.1016/j.ultsonch.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Gholap R., Venkatesan K., Daniel T., Lahoti R., Srinivasan K. Ionic liquid promoted novel and efficient one pot synthesis of 3,4-dihydropyrimidin-2-(1H)-ones at ambient temperature under ultrasound irradiation. Green Chem. 2004;6:147–150. doi: 10.1039/b314015f. [DOI] [Google Scholar]
- 9.Khurana J.M., Nand B., Kumar S. Rapid synthesis of polyfunctionalized pyrano (2,3-c) pyrazoles via multicomponent condensation in room-temperature ionic liquids. Synth. Commun. 2011;41:405–410. doi: 10.1080/00397910903576669. [DOI] [Google Scholar]
- 10.Zhang H.B., Liu L., Chen Y.J., Wang D., Li C.J. “On Water”-Promoted Direct Coupling of Indoles with 1,4-Benzoquinones without Catalyst. Eur. J. Org. Chem. 2006;2006:869–873. doi: 10.1002/ejoc.200500863. [DOI] [Google Scholar]
- 11.Lown J. 1,3-Dipolar Cycloaddition Chemistry. Wiley; New York, NY, USA: 1984. [Google Scholar]
- 12.Katritzky A.R., Singh S.K. Microwave-assisted heterocyclic synthesis. Arkivoc. 2003;13:68–86. [Google Scholar]
- 13.Katritzky A.R., Lagowski J.M. Prototropic Tautomerism of Heteroaromatic Compounds: IV. Five-Membered Rings with Two or More Hetero Atoms. Adv. Heterocycl. Chem. 1963;18:27–81. doi: 10.1016/s0065-2725(08)60746-1. [DOI] [PubMed] [Google Scholar]
- 14.Grigg R., Hargreves S., Redpath J., Turchi S., Yoganathan G. 1,3-Dipolar Cycloaddition Reactions of Imines of γ-and δ-Dialdehydes: Applications to the Synthesis of Novel Polyfunctional Pyrrolizidines and Indolizidines. Synthesis. 1999:441–446. doi: 10.1055/s-1999-3415. [DOI] [Google Scholar]
- 15.El-Abbady S.A., Ahmed M., Abdulhamid M.G., Shalaby A.A., Moustafa A.H. Regio- and stereoselectivity of 1-(Pyridazin-3-yl)-3-oxidopyridinium Betaines towards 2π- and 4π-conjugated olefins. J. Prakt. Chem. 1989;331:105–110. doi: 10.1002/prac.19893310116. [DOI] [Google Scholar]
- 16.Becke A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988;A38:3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 17.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988;B37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 18.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A. Gaussian 03. Gaussian, Inc.; Wallingford, CT, USA: 2004. [Google Scholar]
- 19.Dennington R., Keith T., Millam J. Gauss View. 4th ed. Semichem Inc.; Shawnee Mission, KS, USA: 2007. [Google Scholar]
- 20.Parr R.G., von Szentpaly L., Liu S. Electrophilicity index. J. Am. Chem. Soc. 1999;121:1922–1924. doi: 10.1021/ja983494x. [DOI] [Google Scholar]
- 21.Parr R.G., Pearson R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983;105:7512–7516. doi: 10.1021/ja00364a005. [DOI] [Google Scholar]
- 22.Parr R.G., Yang W. Density Functional Theory of Atoms and Molecules. Oxford University; New York, NY, USA: 1989. [Google Scholar]
- 23.Kohn W., Sham L. Self-Consistent Equations Including Exchange and Correlation Effects. J. Phys. Rev. 1965;140:1133–A1135. doi: 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- 24.Domingo L.R., Chamorro E., Pérez P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008;72:4615–4624. doi: 10.1021/jo800572a. [DOI] [PubMed] [Google Scholar]
- 25.Domingo L.R., Pérez P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011;9:7168–7175. doi: 10.1039/c1ob05856h. [DOI] [PubMed] [Google Scholar]
- 26.Reed A.E., Weinhold F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983;78:4066–4073. doi: 10.1063/1.445134. [DOI] [Google Scholar]
- 27.Domingo L.R., Aurell M.J., Pérez P.C., Renato A. Quantitative characterization of the local electrophilicity of organic molecules. Understanding the regioselectivity on Diels-Alder reactions. Phys. Chem. A. 2002;106:6871–6875. doi: 10.1021/jp020715j. [DOI] [Google Scholar]
- 28.Domingo L.R., Aurell M.J., Perez P., Contreras R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron. 2002;58:4417–4423. doi: 10.1016/S0040-4020(02)00410-6. [DOI] [Google Scholar]
- 29.Domingo L.R., Perez P. Global and local reactivity indices for electrophilic/nucleophilic free radicals. Org. Biomol. Chem. 2013;11:4350–4358. doi: 10.1039/c3ob40337h. [DOI] [PubMed] [Google Scholar]
- 30.Jaramillo P., Domingo L.R., Chamorro E., Perez P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. Theochem. 2008;865:68–72. doi: 10.1016/j.theochem.2008.06.022. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.