

Abstract
A single-step procedure to simultaneously load and protect high concentrations of siRNA in porous silicon nanoparticles (pSiNPs) is presented. Treatment of pSiNPs with an aqueous solution containing siRNA and calcium chloride generates core-shell nanostructures consisting of an siRNA-loaded pSiNP core infiltrated with an insoluble shell of calcium silicate (Ca-pSiNPs). The source of silicate in the shell derives from local dissolution of the pSi matrix, and in solutions containing high concentrations of calcium (II) ion, Ca2SiO4 formation occurs primarily at the nanoparticle surface and is self-limiting. The insoluble calcium silicate shell slows the degradation of the pSiNP skeleton and prolongs delivery of the siRNA payload, resulting in more effective gene knockdown in vitro. Formation of the calcium silicate shell results in an increase in the external quantum yield of photoluminescence from the porous silicon core from 0.1 to 21 %, presumably due to the electronically passivating nature of the silicate shell. Attachment of two functional peptides that incorporate a sequence derived from the rabies virus glycoprotein (RVG) as a neuronal targeting peptide and myristoylated transportan (mTP) as a cell penetrating moiety to the Ca-pSiNPs yields a construct that shows improved gene silencing in vitro and improved delivery in vivo.
Keywords: targeted nanoparticles, injured brain, gene knockdown, photoluminescence, RVG peptide
Graphical Abstract
A significant limitation in efficacy of small molecule, protein, and nucleic acid-based therapeutics is bioavailability. Molecules with low solubility may not enter the blood stream or other bodily fluids at therapeutically effective concentrations,[1–3] and more soluble therapeutics may undergo rapid clearance from the circulatory system by various biological processes before reaching the intended tissues.[4–6] Loading of therapeutics into porous or hollow nanostructures has emerged as a means to control the concentration-time relationship of drug delivery and improve therapeutic efficacy.[7, 8] Much work in nanostructured carriers for drugs has been based on “soft” particles such as liposomes and polymer conjugates,[9, 10] or more rigid porous inorganic materials such as mesoporous silicon or silicon oxide.[11–13] Mesoporous silicon and silicon oxide are inorganic and biodegradable materials that have been well studied for drug delivery applications.[8, 14–26]
The mechanism of degradation of porous silicon (pSi) involves oxidation of the silicon skeleton to form silicon oxide, followed by hydrolysis of the resulting oxide phase to water-soluble orthosilicic acid (Si(OH)4) or its congeners.[25] To prevent rapid degradation of pSi nanoparticles, various “core-shell” types of structures have been synthesized, where an inner core of a pSi skeleton is surrounded by a shell of more stable silicon oxide,[27, 28] titanium oxide,[29–31] carbon[32–34] or other kinetically stable substances.[35] Core-shell structures are attractive platforms for slow releasing drug delivery formulations because the synthesis of the shell can be performed in concert with drug loading in order to more effectively trap the therapeutic in the nanostructure.[36] Furthermore, the ability of core-shell structures to enhance intensity and persistence of photoluminescence from the luminescent silicon domains in pSi has been demonstrated,[27] which adds imaging and self-reporting drug delivery features to the nanomaterial.
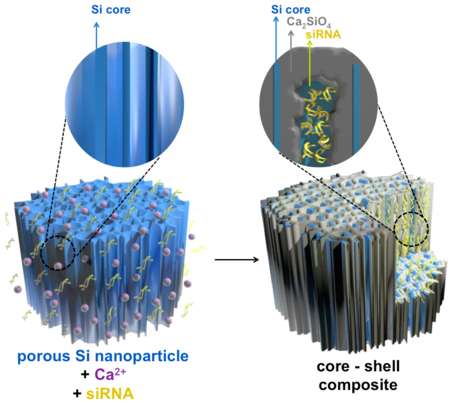
We report here a single-step procedure to simultaneously load and protect high concentrations of siRNA in pSi nanoparticles (pSiNPs) by precipitating an insoluble shell of calcium silicate simultaneous with drug loading (Figure 1). The source of silicate in the shell derives from local dissolution of the pSi matrix, and in solutions containing high concentrations of calcium (II) ion, we find that Ca2SiO4 formation occurs primarily at the nanoparticle surface and is self-limiting. If the calcium ion solution also contains siRNA, the oligocucleotide becomes trapped in the porous nanostructure during shell formation. The insoluble calcium silicate shell slows the degradation of the porous silicon skeleton and the release of siRNA. The porous Si core displays intrinsic photoluminescence due to quantum confinement effects, and we find that the shell formation process leads to an increase in the external quantum yield from 0.1 to 21 %, presumably due to the electronically passivating nature of the silicate shell. To demonstrate the potential for gene delivery with this system, we modify the calcium silicate-coated pSiNPs (Ca-pSiNPs) via silanol chemistry to conjugate two functional peptides, one for neuronal targeting and the other for cell penetration. The resulting construct shows significantly improved gene silencing efficacy in vitro, and it can be delivered to targeted tissues in vivo.
Figure 1.

Schematic illustration of the mechanistic steps involved in preparation of siRNA-loaded, calcium silicate-coated porous silicon nanoparticles (Ca-pSiNP-siRNA). Mild oxidation (in aqueous media) of porous Si particles generates a thin oxide layer on the Si skeleton. As it forms, the oxide layer becomes hydrated and solublized, releasing Si(OH)4 into solution. High concentrations of Ca2+ and siRNA present in the aqueous solution diffuse into the pores, where the Ca2+ ions react with the locally high concentration of Si(OH)4, forming a precipitate that traps the siRNA payload within the nanostructure.
The pSiNPs of average size 180 ± 20 nm (by dynamic light scattering) were prepared as described previously.[37] The siRNA payload was loaded and sealed into the porous nanostructure in one step, by stirring the pSiNPs in an aqueous solution containing the oligonucleotide in the presence of a high concentration (3M) of CaCl2. Control experiments where the same quantity of free siRNA was mixed in a solution 3M in CaCl2, but without added pSiNPs, showed no evidence of precipitate under the reaction conditions. To avoid the possibility of undetectable precipitates of siRNA with Ca2+, the pSiNPs were isolated after reaction by centrifugation and washing three times, first using DI water, then 70% ethanol, and finally absolute ethanol. The mass loading of siRNA was typically 20%, as determined by difference (free siRNA remaining in the supernatant) and by direct measurement of the quantity of siRNA released from the oligonucleotide-loaded Ca-pSiNPs in RNAse free DI water. The presence of silicon, calcium, and oxygen in the resulting siRNA-loaded, calcium silicate-capped pSiNPs (Ca-pSiNP-siRNA) was confirmed by energy dispersive x-ray (EDX) analysis (Figure S1). No residual chloride was detected. The quantity of oxygen in the pSiNPs increased measurably upon reaction with the Ca2+ solution, demonstrating that pSiNPs are oxidized during the reaction. Transmission electron microscope (TEM) images (Figure 2a-c) of empty pSiNP prior to calcium ion treatment, pSiNP after treatment with Ca2+ (Ca-pSiNP), and pSiNP after loading of siRNA and treatment with Ca2+ (Ca-pSiNP-siRNA) indicated that the reaction with Ca2+ generated a distinctive coating (Figure 2b and 2c). Based on the elemental analysis and considering the low solubility of calcium silicate,[38] we propose the capping material to be dicalcium orthosilicate (Ca2SiO4) or a mixed phase of calcium orthosilicate, metasilicate, and silicon oxides. No crystalline calcium silicate or silicon oxide phases were observed by powder X-ray diffraction (XRD), but residual crystalline Si was detected in the XRD spectrum (Figure S2a) and in the Raman spectrum (Si-Si lattice mode at 520 cm−1, Figure S2b). The characteristic band for surface Si-O (1020 cm−1) was observed in the FTIR spectrum both before and after Ca2+ treatment (Figure S2c). Nitrogen adsorption-desorption isotherm analysis indicated that the total pore volume decreased by 80% (1.36 ± 0.03 cm3/g to 0.29 ± 0.04 cm3/g) upon conversion of pSiNP to Ca-pSiNP (Figure 2d). Prior work has shown that oxidation of pSi results in reduction of the pore volume due to swelling of the pore walls as oxygen is incorporated into the silicon skeleton, and this process can result in effective trapping of a payload in the pores.[25, 36]
Figure 2.

Transmission electron microscope (TEM) images of (a) pSiNP, (b) Ca-pSiNP, and (c) Ca-pSiNP-siRNA formulations. Scale bar is 200 nm. (d) Cryogenic nitrogen adsorption-desorption isotherms for pSiNP and Ca-pSiNP formulations. (e) Photoluminescence emission spectra (λex: 365nm) obtained during reaction of pSiNP with 3M aqueous CaCl2 solution, used to prepare the Ca-pSiNP formulation. Typical of quantum confinement, as the silicon skeleton becomes thinner the emission spectrum shifts to the blue. The growth of an electronically passivating surface layer and suppression of nonradiative recombination centers is evident in the strong increase in photoluminescence intensity observed as the reaction progresses.
Optical absorbance measurements, used to measure the amount of elemental silicon in the solution, showed that ~40% of the pSiNPs were degraded within 80 min in a pH 9 buffer when no calcium ion was present. However, in 3M CaCl2 solution (also at pH 9), only ~10% degradation was observed in the same time period (Figure S3a). The calcium silicate shell also impeded release of the siRNA cargo; the Ca-pSiNP-siRNA formulation showed ~5-fold slower release under physiologic conditions (pH 7.4 buffer, 37 °C), compared to a formulation in which siRNA was held in the pSiNPs by electrostatic means (pSiNPs modified with surface amine groups, pSiNP-NH2, Figure S3b). The Ca-pSiNP-siRNA formulation also showed substantially greater siRNA loading efficiency compared to electrostatically loaded particles (20–25% vs 5–8%, respectively). Thus the calcium silicate trapping chemistry effectively encapsulated and slowed release of the siRNA payload, and it protected the pSi skeleton from subsequent oxidation and hydrolysis in aqueous media.
The photoluminescence spectrum obtained at different times during the course of the reaction between pSiNPs and CaCl2 solution showed a gradual increase in intensity (Figure 2e). Additionally, the peak wavelength of photoluminescence blue shifted as the reaction progressed. Both of these phenomena (increase in photoluminescence intensity and blue shift of the photoluminescence spectrum) are indicative of the growth of a passivating surface layer on the silicon nanocrystallites.[27, 39, 40] The observed blue shift is typical of a quantum-confined silicon nanoparticle, whose emission wavelength is strongly dependent on size and exhibits a blueshift as the quantum-confined silicon domains become smaller.[41] The photoluminescence emission quantum yield (external) for the pSiNP-calcium silicate core-shell structure (Ca-pSiNP) was 21% (λex = 365 nm, Figure S4).
A preliminary in vitro cytotoxicity screen on cultured Neuro-2a (mouse neuroblastoma) cells showed no significant cytotoxicity of the Ca-pSiNP formulation at nanoparticle concentrations up to 50 μg/mL (Figure S5), and so the system was loaded with a targeting and a therapeutic payload for gene silencing studies (the loading procedure is described schematically in Figure S6). A small interfering RNA (siRNA) capable of silencing the endogenous gene (peptidylprolyl isomerase B, PPIB) was chosen to test the ability of the calcium silicate chemistry to retain, protect, and deliver a therapeutic payload for in vivo studies. The pSiNPs were loaded with siRNA against PPIB (siPPIB) in the presence of 3M CaCl2, which resulted in ~20 wt% siRNA content in the resulting nanoparticle (Ca-pSiNP-siRNA). The morphology of the Ca-pSiNP-siRNA construct appeared similar to the drug-free Ca-pSiNP preparation by TEM (Figure 2c), although the surface charge (zeta potential, Figure S7a) of Ca-pSiNP-siRNA was negative instead of positive. The positive zeta potential of the drug-free Ca-pSiNP preparation is attributed to an excess of Ca2+ ions at the particle surface, and the negatively charged siRNA payload neutralizes these charges to the extent that it results in an overall negative zeta potential in the Ca-pSiNP-siRNA construct.
To achieve targeted delivery and intracellular trafficking of the siRNA therapeutic, a tissue targeting peptide and a cell penetrating peptide were then grafted to the calcium silicate shell of the Ca-pSiNP-siRNA construct. A PEG linker was used to attach both of these peptides to improve systemic circulation (Figure S6). First, the chemical coupling agent 2-aminopropyldimethylethoxysilane (APDMES) was grafted to the nanoparticle surface, generating pendant primary amine groups (Ca-pSiNP-siRNA-NH2).[25] The zeta potential became more positive after the APDMES reaction for either Ca-pSi-NH2 or Ca-pSiNP-siRNA-NH2 formulations due to the primary amine groups on the outermost surface of the nanoparticles (Figure S7a). Functional polyethyleneglycol (PEG) species were then grafted to Ca-pSiNP-siRNA-NH2 via these primary amines, using a maleimide-poly(ethylene-glycol)-succinimidyl carboxy methyl ester (MAL-PEG-SCM) species.[41] The succinimidyl carboxymethyl ester forms an amide bond with primary amines, and thus provides a convenient means to attach PEG to the aminated nanoparticle. The distal end of the PEG chain contained a second functional group, maleimide. Maleimide forms covalent bonds to thiols, allowing attachment of targeting and cell penetrating peptides. Two peptide species, myr-GWTLNSAGYLLGKINLKALAALAKKIL(GGCC), a myristoylated transportan referred to here as “mTP,” and the rabies virus-derived peptide 5FAM-(CCGG)YTIWMPENPRPGTPCDIFTNSRGKRASNG, referred to as “FAM-RVG,” were prepared and conjugated to the Ca-pSiNP-siRNA-PEG formulation via reaction of the maleimide group with a cysteine thiol of the relevant peptides. Here, “5FAM” is the fluorescent label 5-carboxyfluorescein, an amine-reactive fluorophore commonly used to label biomolecules (λex/ λem = 495/518 nm).
Cell-penetrating peptides (CPP) such as transportan (TP) have been found to be promising auxiliaries for siRNA delivery. When CPPs are incorporated into nanoparticles, they can increase endocytic escape after internalization to increase the siRNA knockdown efficiency. However, CPPs lack cell-type specificity. To overcome this shortcoming, CPPs have been combined with cell-specific targeting peptides to generate what is known as tandem peptides, and these constructs have been shown to be very efficient siRNA delivery agents.[42] In the present work, the cell-penetrating transportan peptide was attached to a myristoyl group, which contains a hydrophobic 13 carbon aliphatic chain, to enhance the hydrophobic interaction between the peptide and the lipid bilayer of the cell membrane (mTP).[43] The cell targeting function was accomplished with a peptide sequence from the rabies virus glycoprotein (RVG) that has demonstrated effective neuronal cell targeting efficiency in vitro and in vivo.[44–46] Attachment of both RVG and mTP peptides to a Ca-pSiNP resulted in a dual peptide nanocomplex, referred to here as “Ca-pSiNP-DPNC.” Control nanoparticles containing only mTP or RVG peptides were also prepared, herein designated as Ca-pSiNP-mTP or Ca-pSiNP-RVG, respectively.
Approximately 0.1mg of RVG was conjugated with 1 mg of Ca-pSiNP-siRNA-PEGs, determined by relative fluorescence of the FAM label. In the case of the Ca-pSiNP-siRNA-DPNC construct, approximately 0.04mg of RVG and a comparable amount of mTP was conjugated. The Fourier transform infrared (FTIR) spectrum of Ca-pSiNP-DPNC displayed all the characteristic peaks of Ca-pSiNP-mTP and Ca-pSiNP-RVG (Figure S8). The mean diameter of the Ca-pSiNP-siPPIB-DPNC construct was 220 nm (DLS Z-average, intensity based), representing an increase over the pSiNP starting material of 40 nm. No significant aggregates were observed in the DLS data (Figure S7b).
The Ca-pSiNP-siPPIB-DPNC construct effected knockdown of 52.8% of PPIB gene activity in Neuro-2a cells relative to untreated controls (Figure 3). To eliminate the possibility that gene silencing was caused by toxicity of the nanocomplexes, a similar formulation loaded with a negative control siRNA against the luciferase gene (siLuc) was tested, and it showed no statistically significant difference relative to the untreated control. As additional controls, gene silencing efficiencies of nanoparticles containing only a cell-penetrating or only a cell-targeting peptide were tested (Ca-pSiNP-siPPIB-mTP and Ca-pSiNP-siPPIB-RVG, respectively). Both of these constructs showed some observable knockdown of PPIB gene expression (27.1–28.9% relative to untreated controls), but the silencing effect was greater with the dual peptide nanoparticle Ca-pSiNP-siPPIB-DPNC (p < 0.03) compared with either peptide system individually. In the case of Ca-pSiNP-siPPIB-mTP, the gene knockdown observed in vitro is not expected to translate to in vivo activity, because the cell penetrating effect of mTP lacks cell-type specificity. On the other hand, silencing by Ca-pSiNP-siPPIB-RVG is attributed to more effective cellular localization in vitro due to specific binding of the RVG sequence to Neuro-2a cells. Additional controls using free siPPIB (not contained in a nanoparticle) and siPPIB loaded into bare pSiNPs (no Ca capping chemistry, no targeting peptides, no cell-penetrating peptides) showed no statistically significant knockdown. Furthermore, nano-constructs isolated and stored in ethanol for 7 days at 4 °C still retained their PPIB gene knockdown efficiency (Figure 3).
Figure 3.

The silencing of relative PPIB gene expression in Neuro-2a cells after treatment with siRNA against the PPIB gene (siPPIB), aminated porous Si nanoparticle (pSiNP) loaded with siPPIB (pSiNP-siPPIB), pSiNP-siPPIB construct prepared with a calcium silicate shell and containing both cell-targeting and cell-penetrating peptides on the outer shell in a dual peptide nanocomplex (Ca-pSiNP-siPPIB-DPNC), the pSiNP-siPPIB-calcium silicate shell construct containing only a cell-penetrating peptide on the outer shell (Ca-pSiNP-siPPIB-mTP), the pSiNP-siPPIB-calcium silicate shell construct containing only the cell-targeting peptide on the outer shell (Ca-pSiNP-siPPIB-RVG), and the pSi nanoparticle-calcium silicate shell construct containing a negative control siRNA sequence against luciferase, and containing both the cell-targeting and the cell-penetrating peptides on the outer shell (Ca-pSiNP-siLuc-DPNC). The “7 days” designations indicate that the nanoparticle construct was stored in ethanol at 4°C for 7 days prior to the experiment. The cell penetrating peptide is a myristoylated transportan, and the cell targeting peptide is a domain derived from the rabies virus glycopeptide (RVG) as described in the text. Statistical analyses were performed with Student’s t test (* p < 0.01, ** p < 0.03).
The present results show better cellular affinity and gene knockdown when the dual peptide nanocomplex (DPNC, containing both mTP and RVG) was used compared with the single peptide conjugated nanoparticles (Ca-pSiNP-siRNA-mTP or Ca-pSiNP-siRNA-RVG). Transmission electron microscope (TEM) data on Ca-pSiNP-DPNC-treated Neuro-2a cells (Figure S9) established that the nanoparticles were indeed internalized and dispersed in the cell cytoplasm after 1 hr incubation. The images are consistent with endosomal uptake of the nanoparticles, although the present studies did not assess the intracellular trafficking or endosomal uptake mechanism(s).
Consistent with its greater knockdown efficiency, confocal microscope images indicated that the Ca-pSiNP-siPPIB-DPNC formulation had greater affinity for Neuro-2a cells than the Ca-pSiNP-siPPIB-RVG formulation (Figure S10). The Ca-pSiNP-siPPIB-DPNC formulation had approximately half the number of fluorescent FAM marker molecules on its surface compared to Ca-pSiNP-siPPIB-RVG. Even with the lower FAM fluorescence signal per particle, Neuro-2a cells treated with Ca-pSiNP-siPPIB-DPNC showed a larger FAM signal because of the greater cellular affinity of this dual peptide construct relative to the RVG-only formulation. The Ca-pSiNPs are visible in the fluorescence microscope images due to the intrinsic photoluminescence from the quantum-confined Si domains of the nanoparticle. In the case of cells treated with Ca-pSiNP-siPPIB-DPNC, the Si signal is colocalized with the signal from the FAM label on the RVG targeting peptide, and the combined signal is seen in the cytosol, indicative of cellular internalization. The cellular affinity of these two nanoparticle constructs was more accurately quantified by fluorescence-activated cell sorting (FACS) analysis (Figure S11), and the data show that the dual peptide nanoparticle was more efficient at targeting Neuro-2a cells than the nanoparticle that contained only the RVG peptide (51.4 ± 5.6% vs 36.4 ± 5.6 % for Ca-pSiNP-siPPIB-DPNC and Ca-pSiNP-siPPIB-RVG, respectively (P < 0.04). Separate fluorescent labels on the RVG peptide and on the siPPIB in the Ca-pSiNP-siPPIB-DPNC established that 65.9 ± 8.7% of the cells contained both RVG and siPPIB (Figure S11d). The results support the hypothesis that conjugating both RVG and mTP to the nanoparticle yields greater cellular affinity, which in turn generates a stronger gene knockdown effect.
Often in vitro targeting results are not replicated in vivo due to active clearance by the MPS organs and other physiologic factors associated with the complex in vivo environment. In order to demonstrate the feasibility of in vivo delivery of siRNA by the nanoconstructs, we performed a pilot study involving a penetrating brain injury model in mice. As having both cell-penetrating and cell-targeting peptides on the same nanoparticle (Ca-pSiNP-siPPIB-DPNC) yielded the strongest gene knockdown in vitro, we selected this construct for the in vivo gene delivery experiments.
Significant quantities of siRNA accumulated in the site of the brain injury in the mice injected with Ca-pSiNP-siRNA-DPNC (Figure 4). The mice (n = 3) showed 2-fold greater intensity of fluorescence associated with the siRNA payload relative to the fluorescence background in saline-injected control mice, and the ratio of fluorescence at the injured site relative to the uninjured hemisphere was 5.2. There was statistically greater observed efficacy of targeting by the dual peptide Ca-pSiNP-siRNA-DPNC relative to the untargeted nanoparticles Ca-pSiNP-siRNA-PEG (p < 0.02). Mice injected with the untargeted Ca-pSiNP-siRNA-PEG construct showed some siRNA fluorescence signal in the brain compared to the uninjected control mice, presumably due to passive leakage into the injury site. Correspondingly, these Ca-pSiNP-siRNA-PEG-injected mice displayed greater fluorescence intensity in the kidney and (to a lesser extent) the liver relative to mice injected with the Ca-pSiNP-siRNA-DPNC constructs. The biodistribution data are thus consistent with the greater ability of the dual-targeted nanoparticles to accumulate in the brain. Mice injected with any of the nanoparticle formulations displayed no overt short-term health effects.
Figure 4.

Ex vivo fluorescence images of harvested organs after intravenous injection of (1) saline as a control, (2) Ca-pSiNP-siRNA-PEG, and (3) Ca-pSiNP-siRNA-DPNC. All siRNA constructs contained covalently attached dy677 fluorophore. (a) Fluorescence image of injured brains obtained using infra-red imaging system Pearl Trilogy (Li-Cor). Green channel in the images corresponds to 700 nm emission from dy677, and the bright field image of the brain tissues is merged with the 700 nm emission. (b) Fluorescence image of whole major organs taken with IVIS (xenogen) imaging system in the Cy5.5 channel (λex/em: 675/694 nm).
In summary, this work demonstrates a self-sealing chemical procedure that can load oligonucleotides in a biodegradable and intrinsically photoluminescent nanoparticle. Substantial quantities of siRNA can be loaded (> 20% by mass), and the payload is retained for therapeutically relevant timescales. The calcium silicate shell is readily modified with cell targeting (RVG peptide from rabies virus glycoprotein) and cell-penetrating (myristolated transportan) peptides, and the combination of the two peptides, along with the ability of the calcium silicate chemistry to retain and protect the siRNA payload, yields improved cellular targeting and gene knockdown in vitro. The multivalent core-shell nanoparticles circulate to deliver an siRNA payload to a brain injury in live mice, and the dual targeted nanoparticles show improved delivery of siRNA in the in vivo brain injury model relative to non-targeted nanoparticles.
Experimental Section
Preparation of porous silicon nanoparticles:
The pSiNPs were prepared following the published “perforation etching” procedure.[37] A highly boron-doped p++-type silicon wafer (~ 1 mΩ-cm resistivity, 100 mm diameter, Virginia Semiconductor, Inc.) was anodically etched in an electrolyte composed of 3:1 (v:v) of 48% aqueous HF:ethanol. The etching waveform consisted of a square wave in which a lower current density of 46 mA cm−2 was applied for 1.818 sec, followed by a higher current density pulse of 365 mA cm−2 applied for 0.363 sec. This waveform was repeated for 140 cycles, generating a stratified porous silicon (pSi) film with thin, high porosity “perforations” repeating approximately every 200 nm through the porous layer. The film was removed from the silicon substrate by applying a current density of 3.4 mA cm−2 for 250 sec in a solution containing 1:20 (v:v) of 48% aqueous HF:ethanol. The freestanding pSi film was fractured into nanoparticles of mean (Z-average, intensity based) diameter 180 nm (Figure S7b) by immersion in deionized water and ultrasonication for ~12 hr.
Preparation of calcium silicate-coated, siRNA-loaded porous silicon nanoparticles (Ca-pSiNP-siRNA):
A stock solution 4 M in calcium chloride (CaCl2) were prepared by adding 2.25 g of solid CaCl2 (MW: 110.98, Anhydrous, Spectrum chemicals) to 5 mL of RNAse-free water. The solution was centrifuged to remove any precipitates and stored at 4 °C before use. For oligonucleotide loading, three kinds of duplexed siRNA constructs for the knockdown of PPIB(1), PPIB(2), and Luciferase was synthesized by Dharmacon Inc. with 3’-dTdT overhangs.[47, 48]: For PPIB gene against siRNA (siPPIB), siPPIB(1) and siPPIB(2) were obtained, respectively, and used 1:1 mixture of siPPIB(1):siPPIB(2) to cover broad range of PPIB gene on the siRNA sequence sense 5’-CAA GUU CCA UCG UGU CAU C dTdT-3’ and antisense 5’- GAU GAC ACG AUG GAA CUU G dTdT-3’ for siPPIB(1) and sense 5’-GAA AGA GCA UCU AUG GUG A dTdT-3’ and antisense 5’- UCA CCA UAG AUG CUC UUU C dTdT-3’ for siPPIB(2). Luciferase gene against siRNA (siLuc) was obtained on the siRNA sequence sense 5’-CUU ACG CUG AGU ACU UCG A dTdT-3’ and antisense 5’- UCG AAG UAC UCA GCG UAA G dTdT-3’. The pSiNPs (100 μL DI, 1 mg) were mixed with the oligonucleotide solution (150 μL DI, 150 μM in siRNA) and added to the 4 M CaCl2 stock solution (750 μL). The mixture was agitated for 60 min and purified by successive dispersion in/centrifugation from RNAse free deionized water, 70% ethanol, and 100% ethanol. To analyze siRNA loading efficiency, supernatants from each centrifugation step were collected and assayed for free siRNA using a NanoDrop 2000 spectrophotometer (Thermo Scientific, ND-2000). As a control, Ca-pSiNPs without siRNA were prepared in the same manner as described above, but excluding the added siRNA. Mass loading of siRNA was verified by quantification of siRNA released from the oligonucleotide-loaded Ca-pSiNPs, performed in RNAse-free deionized water and measured using a NanoDrop 2000 spectrophotometer. The two determinations varied by < 10% in the quantity of siRNA loaded.
Conjugation of peptides to Ca-pSiNP:
As-prepared Ca-pSiNP-siRNA, Ca-pSiNP or pSiNP samples (1 mg) were suspended in absolute ethanol (1 mL), an aliquot (20 μL) of aminopropyldimethylethoxysilane (APDMES) was added, and the mixture was agitated for 2h. The aminated nanoparticles (Ca-pSiNP-siRNA-NH2, Ca-pSiNP-NH2, or pSiNP-NH2) were then purified three times by centrifugation from absolute ethanol to eliminate unbound APDMES. The solutions (200 μL) of the hetero-functional linkers maleimide-PEG-succinimidyl carboxy methyl ester (MAL-PEG-SCM, MW: 5,000, Laysan Bio Inc., 5 mg/mL in ethanol) or methoxy-PEG-succinimidyl α-methylbutanoate (mPEG-SMB, Mw: 5,000, NEKTAR, 5 mg/mL in ethanol) were added to the aminated nanoparticles (1 mg in 100 μL) and agitated for 2h. Unbound PEG linker molecules were eliminated from the PEGylated nanoparticles (Ca-pSiNP-siRNA-PEG or Ca-pSiNP-PEG) by centrifugation from ethanol three times. For the peptide-conjugated formulations, one of two peptide constructs was used: either mTP, which consists of a myristoyl group (myr) covalently attached by amide bond to the N-terminal glycine residue on the peptide sequence myr-GWTLNSAGYLLGKINLKALAALAKKIL(GGCC), or FAM-RVG, which consists of 5-carboxyfluorescein (5-FAM) attached by amide bond to the N-terminal cysteine residue on the peptide sequence 5-FAM(CCGG)YTIWMPENPRPGTPCDIFTNSRGKRASNG. Both of these constructs were obtained from CPC Scientific Inc. (1 mg/mL in RNAse free water). For Ca-pSiNP-dual peptide nanocomplex (Ca-pSiNP-DPNC or Ca-pSiNP-siRNA-DPNC) synthesis, 50 μL of each peptide solution (mTP and FAM-RVG) was added to 100 μL of Ca-pSiNP-PEG in ethanol, incubated at 4 °C for 4 hours, purified three times by centrifugation, immersed in ethanol and stored at 4 °C before use. For synthesis of the single peptide conjugated Ca-pSiNP (Ca-pSiNP-siRNA-mTP or Ca-pSiNP-siRNA-RVG) control samples, 100 μL of peptide solution (mTP or FAM-RVG) was added to 100 μL of Ca-pSiNP-siRNA-PEG in ethanol, respectively. The subsequent workup was the same as described above for the Ca-pSiNP-siRNA-DPNC constructs.
Characterization:
Transmission electron microscope (TEM) images were obtained with a JEOL-1200 EX II instrument. Scanning electron microscope (SEM) images and energy dispersed x-ray (EDX) data were obtained using an FEI XL30 field-emission instrument. The hydrodynamic size and zeta potential was measured by dynamic light scattering (DLS, Zetasizer ZS90, Malvern Instruments). An Ocean Optics QE-Pro spectrometer was used to obtain steady-state photoluminescence spectra (λex: 365 nm) with a 460 nm long-pass emission filter. Quantum yield measurements were performed relative to a Rhodamine 6G in ethanol standard (Q.Y. 95%). All solutions used for quantum yield measurements had optical absorbance values < 0.1 at λ = 365 nm. The photoluminescence intensity in the wavelength range 500 – 980 nm was integrated and plotted vs absorbance (Figure S4). Nitrogen adsorption-desorption isotherms were obtained on dry particles at a temperature of 77 K with a Micromeritics ASAP 2020 instrument. Fourier transform infrared (FTIR) spectra were recorded using a Thermo Scientific Nicolet 6700 FTIR instrument. Raman spectra were obtained using a Renishaw inVia Raman microscope with 532 nm laser excitation source.
In vitro experiments:
Murine Neuro-2a neuroblastoma cells (ATCC, CCL-131) were cultured in Eagle’s Minimum Essential Medium (EMEM) containing 10% fetal bovine serum (FBS). Cytotoxicity of the synthesized nanoparticles was assessed using the Molecular Probes Live/Dead Viability/Cytotoxicity Kit (Molecular Probes, Invitrogen).[49] This kit uses 2 probes, Calcein AM for live cell staining (λex/λem = 494/517 nm) and Ethidium homodimer-1 (EthD-1) for dead cell staining (λex/λem = 528/617 nm). Neuro-2a cells (3000 cells/well) were treated with nanoparticles in triplicate in a 96-well plate. After 48 hrs, each well was washed and treated with the assay solution consisting of 4 μM EthD-1 and 2 μM Calcein AM in Dulbecco’s phosphate buffered saline. After 45 min incubation at room temperature in the assay solution, well plates were read with a fluorescence plate reader (Gemini XPS spectrofluorometer, Molecular Devices, inc.) using excitation, emission, and cutoff wavelengths 485/538/515 nm and 544/612/590 nm, respectively. A total of 15 wells per treatment group were evaluated, and plotted as a percentage of untreated control fluorescence intensity.
Neuro-2a cells treated with nanoparticles were visualized with a confocal microscope (Zeiss LSM 710 NLO), using a 40x oil immersion objective. Cells were seeded onto the coverslips (BD Biocoat Collagen Coverslip, 22 mm), incubated with nanoparticles for 2 hrs, washed three times with PBS, fixed with 4% paraformaldehyde, nucleus stained with DAPI and mounted (Thermo Fisher Scientific, Prolong Diamond Antifade Mountant with DAPI). Neuro-2a cells treated with nanoparticles were quantified to demonstrate cellular affinity and siRNA delivery efficiency by FACS analysis (LSR Fortessa).
In order to investigate knockdown efficiency in vitro, real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR, Stratagene Mx3005P qPCR system) analysis was performed to examine PPIB mRNA expression. Neuro-2a cells were seeded in 24-well plates (4 × 104 cells per well), and incubated with siRNA-loaded nanoparticles, with concentration corresponding to 100 nM of siRNA. After 48 hrs, cells were harvested and total RNA was isolated following the manufacturer’s protocol (Qiagen, Vlencia, CA). Isolated RNA was transcribed into cDNA following the manufacturer’s protocol (Bio-Rad, iScript cDNA Synthesis Kit). Synthesized cDNA was subjected to qPCR analysis using SYBR Green PCR Master Mix. Primer sequences for PPIB as a target mRNA amplification and HPRT as a reference mRNA amplification are described below. PPIB forward: GGAAAGACTGTTCCAAAAACAGTG, PPIB reverse: GTCTTGGTGCTCTCCACCTTCCG; HPRT forward: GTCAACGGGGGACATAAAAG, HPRT reverse: CAACAATCAAGACATTCTTTCCA. All procedures were performed in triplicate.
In vivo experiments:
All animal experiments were performed under protocols approved by the MIT Institutional Animal Care and Use Committees (IACUC) and the Sanford Burnham Prebys Medical Discovery Institute Committee on Animal Use and Care. All housing and care of laboratory animals used in this study conformed to the NIH Guide for the Care and Use of Laboratory Animals in Research (see document 180F22) and all requirements and regulations issued by the USDA, including regulations implementing the Animal Welfare Act (P.L. 89–544) as amended (see document 18-F23). The in vivo model involved a penetrating brain injury in mice. First, a 5 mm diameter portion of the skull on the right hemisphere of the mouse was removed. Wounds were induced using a 21 gauge needle in a 3×3 grid for a total of 9 wounds, each 3mm deep. After induction of injuries, the skull was replaced (Figure S12). The mice were injected with nanoparticle constructs 6 hours post-injury via the tail vein. To quantify delivery efficiency of the siRNA cargo to the targeted injury site, Dy677-labeled (λem = 700 nm) siRNA was loaded into Ca-pSiNP-PEG and Ca-pSiNP-DPNC and each of these formulations were injected into separate mice. After 1 hour of circulation the mice were perfused and the organs harvested.
Fluorescence images of harvested organs were obtained using conventional IVIS 200, xenogen, and Pearl Trilogy, Li-Cor imaging systems.
Statistical analysis:
All data in this article are expressed as the means ± standard error of the mean. Significance testing was conducted using two-tailed Student’s t test. Unless otherwise indicated, p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
The authors thank Jack Kyte for helpful discussions. This work supported in part by the National Institutes of Health, through contract # R24 EY022025–01, by the National Science Foundation, under Grant No. DMR-1210417, and by the Defense Advanced Research Projects Agency (DARPA) under Cooperative Agreement HR0011–13–2–0017. The content of the information within this document does not necessarily reflect the position or the policy of the Government. This work was supported in part by the Marie-D. & Pierre Casimir-Lambert Fund (MIT). E. Kwon acknowledges support from the Ruth L. Kirschstein National Research Service Award (1F32CA177094–01). S. Bhatia is an HHMI Investigator.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Jinyoung Kang, Department of Nanoengineering, University of California San Diego, La Jolla, California, 92093-0358, USA.
Dr. Jinmyoung Joo, Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California, 92093-0358, USA Current address: Biomedical Engineering Research Center, Asan Institute for Life Sciences, Asan Medical Center, University of Ulsan College of Medicine, Seoul 05505, Rep. of Korea..
Dr. E. Ester Kwon, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, Massachusetts, 02139, USA
Matthew Skalak, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, Massachusetts, 02139, USA.
Dr. Sazid Hussain, Cancer Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California, 92037, USA
Dr. Zhi-gang She, Cancer Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California, 92037, USA
Prof. Erkki Ruoslahti, Cancer Center, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California, 92037, USA and Center for Nanomedicine and Department of Molecular, Cellular, and Developmental Biology, University of California, Santa Barbara, Santa Barbara, California, 93106-9610, USA
Prof. Sangeeta N. Bhatia, Harvard-MIT Division of Health Sciences and Technology, Department of Electrical Engineering and Computer Science, and David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, Massachusetts, 02139, USA, Division of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts, 02115, USA, Howard Hughes Medical Institute, Cambridge, Massachusetts, 02139, USA
Prof. Michael J. Sailor, University of California, San Diego, Department of Chemistry and Biochemistry, Department of Nanoengineering, Department of Bioengineering, Materials Science and Engineering Program, 9500 Gilman Drive, m/c 0358, La Jolla, CA 92093-0358, USA, msailor@ucsd.edu
References
- [1].Muller RH, Jacobs C, Kayser O, Adv Drug Deliver Rev 2001, 47, 3. [DOI] [PubMed] [Google Scholar]
- [2].Kataoka M, Sugano K, Mathews CD, Wong JW, Jones KL, Masaoka Y, Sakuma S, Yamashita S, Pharm Res-Dordr 2012, 29, 1485. [DOI] [PubMed] [Google Scholar]
- [3].Kipp JE, Int J Pharm 2004, 284, 109. [DOI] [PubMed] [Google Scholar]
- [4].Chonn A, Semple SC, Cullis PR, J Biol Chem 1992, 267, 18759. [PubMed] [Google Scholar]
- [5].Pirollo KF, Chang EH, Trends Biotechnol 2008, 26, 552. [DOI] [PubMed] [Google Scholar]
- [6].Gabizon A, Papahadjopoulos D, P Natl Acad Sci USA 1988, 85, 6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lou XW, Archer LA, Yang ZC, Adv Mater 2008, 20, 3987. [Google Scholar]
- [8].Anglin EJ, Cheng LY, Freeman WR, Sailor MJ, Adv Drug Deliver Rev 2008, 60, 1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gu Z, Biswas A, Zhao MX, Tang Y, Chem Soc Rev 2011, 40, 3638. [DOI] [PubMed] [Google Scholar]
- [10].Nishiyama N, Kataoka K, Pharmacol Therapeut 2006, 112, 630. [DOI] [PubMed] [Google Scholar]
- [11].Park JH, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ, Nat Mater 2009, 8, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wu EC, Park JH, Park J, Segal E, Cunin F, Sailor MJ, Acs Nano 2008, 2, 2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Godin B, Gu JH, Serda RE, Bhavane R, Tasciotti E, Chiappini C, Liu XW, Tanaka T, Decuzzi P, Ferrari M, J Biomed Mater Res A 2010, 94a, 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Meng HA, Xue M, Xia TA, Zhao YL, Tamanoi F, Stoddart JF, Zink JI, Nel AE, J Am Chem Soc 2010, 132, 12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Meng HA, Liong M, Xia TA, Li ZX, Ji ZX, Zink JI, Nel AE, Acs Nano 2010, 4, 4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Patel K, Angelos S, Dichtel WR, Coskun A, Yang YW, Zink JI, Stoddart JF, J Am Chem Soc 2008, 130, 2382. [DOI] [PubMed] [Google Scholar]
- [17].Lu J, Liong M, Zink JI, Tamanoi F, Small 2007, 3, 1341. [DOI] [PubMed] [Google Scholar]
- [18].Shabir Q, Pokale A, Loni A, Johnson DR, Canham LT, Fenollosa R, Tymczenko M, Rodriguez I, Meseguer F, Cros A, Cantarero A, Silicon-Neth 2011, 3, 173. [Google Scholar]
- [19].Wang MJ, Coffer JL, Dorraj K, Hartman PS, Loni A, Canham LT, Mol Pharmaceut 2010, 7, 2232. [DOI] [PubMed] [Google Scholar]
- [20].Kashanian S, Harding F, Irani Y, Klebe S, Marshall K, Loni A, Canham L, Fan DM, Williams KA, Voelcker NH, Coffer JL, Acta Biomater 2010, 6, 3566. [DOI] [PubMed] [Google Scholar]
- [21].Canham LT, Barrett CP, Cox TI, Wright PJ, Bowditch AP, US Patent 20,150,352,211, 2015.
- [22].Jiang K, Loni A, Canham LT, Coffer JL, Phys Status Solidi A 2009, 206, 1361. [Google Scholar]
- [23].Fan DM, Loni A, Canham LT, Coffer JL, Phys Status Solidi A 2009, 206, 1322. [Google Scholar]
- [24].Salonen J, Kaukonen AM, Hirvonen J, Lehto VP, J Pharm Sci-Us 2008, 97, 632. [DOI] [PubMed] [Google Scholar]
- [25].Sailor MJ, Park JH, Adv Mater 2012, 24, 3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ruoslahti E, Bhatia SN, Sailor MJ, J Cell Biol 2010, 188, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Joo J, Cruz JF, Vijayakumar S, Grondek J, Sailor MJ, Adv Funct Mater 2014, 24, 5688. [Google Scholar]
- [28].Ray M, Sarkar S, Bandyopadhyay NR, Hossain SM, Pramanick AK, J Appl Phys 2009, 105. [Google Scholar]
- [29].Betty CA, Sasikala R, Jayakumar OD, Sakuntala T, Tyagi AK, Prog Photovoltaics 2011, 19, 266. [Google Scholar]
- [30].Li JL, Sailor MJ, Biosens Bioelectron 2014, 55, 372. [DOI] [PubMed] [Google Scholar]
- [31].Jeong G, Kim JG, Park MS, Seo M, Hwang SM, Kim YU, Kim YJ, Kim JH, Dou SX, Acs Nano 2014, 8, 2977. [DOI] [PubMed] [Google Scholar]
- [32].Tsang CK, Kelly TL, Sailor MJ, Li YY, Acs Nano 2012, 6, 10546. [DOI] [PubMed] [Google Scholar]
- [33].Zhou XT, Zhang RQ, Peng HY, Shang NG, Wang N, Bello I, Lee CS, Lee ST, Chem Phys Lett 2000, 332, 215. [Google Scholar]
- [34].Gao PF, Fu JW, Yang J, Lv RG, Wang JL, Nuli YN, Tang XZ, Phys Chem Chem Phys 2009, 11, 11101. [DOI] [PubMed] [Google Scholar]
- [35].Buriak JM, Chem Rev 2002, 102, 1271. [DOI] [PubMed] [Google Scholar]
- [36].Fry NL, Boss GR, Sailor MJ, Chem Mater 2014, 26, 2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Qin ZT, Joo J, Gu L, Sailor MJ, Part Part Syst Char 2014, 31, 252. [Google Scholar]
- [38].Medinagonzales OA, Fox RL, Bosshart RP, Fert Res 1988, 16, 3. [Google Scholar]
- [39].Petrovakoch V, Muschik T, Kux A, Meyer BK, Koch F, Lehmann V, Appl Phys Lett 1992, 61, 943. [Google Scholar]
- [40].Sa’ar A, J Nanophotonics 2009, 3. [Google Scholar]
- [41].Joo J, Liu XY, Kotamraju VR, Ruoslahti E, Nam Y, Sailor MJ, Acs Nano 2015, 9, 6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ren Y, Hauert S, Lo JH, Bhatia SN, Acs Nano 2012, 6, 8620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ren Y, Cheung HW, von Maltzhan G, Agrawal A, Cowley GS, Weir BA, Boehm JS, Tamayo P, Karst AM, Liu JF, Hirsch MS, Mesirov JP, Drapkin R, Root DE, Lo J, Fogal V, Ruoslahti E, Hahn WC, Bhatia SN, Sci Transl Med 2012, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Alvarez-Erviti L, Seow YQ, Yin HF, Betts C, Lakhal S, Wood MJA, Nat Biotechnol 2011, 29, 341. [DOI] [PubMed] [Google Scholar]
- [45].Lentz TL, J Mol Recognit 1990, 3, 82. [DOI] [PubMed] [Google Scholar]
- [46].Kumar P, Wu HQ, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N, Nature 2007, 448, 39. [DOI] [PubMed] [Google Scholar]
- [47].Ambardekar VV, Han HY, Varney ML, Vinogradov SV, Singh RK, Vetro JA, Biomaterials 2011, 32, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Waite CL, Sparks SM, Uhrich KE, Roth CM, Bmc Biotechnol 2009, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yee RW, Norcom EG, Zhao XPC, Adv Ther 2006, 23, 511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.