

Abstract
Objective:
Injury severity after traumatic brain injury (TBI) is a well-established risk factor for the development of post-traumatic epilepsy (PTE). However, whether lesion location influences the susceptibility of seizures and development of PTE longitudinally has yet to be defined. We hypothesized that lesion location, specifically in the temporal lobe, would be associated with an increased incidence of both early seizures and PTE. As secondary analysis measures, we assessed the degree of brain atrophy and functional recovery, and performed a between-group analysis, comparing patients who developed PTE with those who did not develop PTE.
Methods:
We assessed early seizure incidence (n = 90) and longitudinal development of PTE (n = 46) in a prospective convenience sample of patients with moderate-severe TBI. Acutely, patients were monitored with prospective cEEG and a high-resolution Magnetic Resonance Imaging (MRI) scan for lesion location classification. Chronically, patients underwent a high-resolution MRI, clinical assessment, and were longitudinally monitored for development of epilepsy for a minimum of 2 years post-injury.
Results:
Early seizures, occurring within the first week post-injury, occurred in 26.7% of the patients (n = 90). Within the cohort of subjects who had evidence of early seizures (n = 24), 75% had a hemorrhagic temporal lobe injury on admission. For longitudinal analyses (n = 46), 45.7% of patients developed PTE within a minimum of 2 years post-injury. Within the cohort of subjects who developed PTE (n = 21), 85.7% had a hemorrhagic temporal lobe injury on admission and 38.1% had early (convulsive or non-convulsive) seizures on cEEG monitoring during their acute ICU stay. In a between-group analysis, patients with PTE (n = 21) were more likely than patients who did not develop PTE (n = 25) to have a hemorrhagic temporal lobe injury (p < 0.001), worse functional recovery (p = 0.003), and greater temporal lobe atrophy (p = 0.029).
Conclusion:
Our results indicate that in a cohort of patients with a moderate-severe TBI, 1) lesion location specificity (e.g. the temporal lobe) is related to both a high incidence of early seizures and longitudinal development of PTE, 2) early seizures, whether convulsive or non-convulsive in nature, are associated with an increased risk for PTE development, and 3) patients who develop PTE have greater chronic temporal lobe atrophy and worse functional outcomes, compared to those who do not develop PTE, despite matched injury severity characteristics. This study provides the foundation for a future prospective study focused on elucidating the mechanisms and risk factors for epileptogenesis.
Keywords: Brain trauma, Post-traumatic epilepsy, Coma, Seizures, Brain atrophy, Status epilepticus
1. Introduction
Traumatic brain injury (TBI) is an inherently heterogeneous condition, which can result in debilitating secondary injuries, such as seizures and post-traumatic epilepsy (PTE) (Englander et al., 2003; Asikainen et al., 1999). TBI is one of the primary causes of acquired epilepsy (Delorenzo et al., 2005; Herman, 2002; Forsgren, 1990), with brain injury severity a leading risk factor for PTE development (Annegers et al., 1998; Frey, 2003; Christensen et al., 2009). Specific injury types and admission injury characteristics, related to injury severity, have been linked to an increased incidence of seizures after brain injury: penetrating injury, hemorrhagic lesion, skull fracture, dural injury, surgical treatment, and prolonged impaired consciousness (Asikainen et al., 1999; Frey, 2003; Xu et al., 2017; Temkin, 2003). Beyond severity, few established etiological risk factors for PTE exist. Although demographic factors, such as age (Chen et al., 2017) and sex (Xu et al., 2017), are associated with an increased risk for PTE, no well-established brain or clinical biomarkers exist for prognostic risk assessment of PTE after a moderate to severe brain injury.
Although severity of injury is a well-established risk factor for PTE, whether lesion location differentially influences the propensity of seizure development has yet to be defined. Animal studies may provide insight to the importance of injury location in epileptogenesis. For example, lesions to the perirhinal, entorhinal, and postrhinal cortices have a lower seizure threshold (Kharatishvili and Pitkanen, 2010). Additionally, interictal and ictal-like discharges may originate from medial temporal lobe structures (Barbarosie et al., 2000; de Guzman et al., 2004; Wozny et al., 2005; Sudbury and Avoli, 2007). These region-specific findings suggest that damage to temporal lobe structures could disrupt specific neural networks that have a higher propensity to elicit seizures. However, in humans, there has been limited assessment of the correspondence between cortical injury location and seizure development (Angeleri et al., 1999), making it an important point of investigation.
Onset of seizures after brain injury is quite variable, defined as manifesting either early (within 7 days) or late (after 7 days) post-injury. Early seizures, whether convulsive or non-convulsive in nature, can be identified utilizing intensive care unit (ICU) based physiological monitoring, such as continuous electroencephalography (cEEG). ICU cEEG monitoring has revealed a high incidence of both clinical seizures and subclinical interictal epileptiform activity early after injury, with associated metabolic consequences (Vespa et al., 1999). Late seizures develop after a latent period, which can temporally range from 7 days post-injury up to 30 years post-injury, although most seizures manifest by 2 years post-injury (Agrawal et al., 2006; Pitkanen and Immonen, 2014; Gupta et al., 2014). Latency of seizure onset may be an important marker of future epileptogenesis, as emerging evidence indicates that the timing of initial seizure onset is associated with PTE development (Chen et al., 2017). However, it remains unclear what role early seizures signify in the epileptogenic process.
The complexity of PTE, resulting from the heterogeneous nature TBI and variable onset of seizure manifestation, has made identifying early biomarkers and mechanisms of epileptogenesis difficult (Diaz-Arrastia et al., 2009). To address these hurdles, it remains imperative to identify potential acute clinical biomarkers of epileptogenesis, by longitudinally assessing a robust and well-powered sample of patients with moderate-severe TBI. As an initial first step, our study sought to 1) map the correspondence between injury location and seizure incidence, 2) identify potential acute clinical predictors of PTE, and 3) identify the statistical power needed for a future multi-center prospective study on PTE. We analyzed a large cohort of moderate-severe TBI patients, who were assessed both acutely post-injury and chronically through long-itudinal follow-up assessments. Acutely, patients were monitored with prospective cEEG and underwent a high-resolution Magnetic Resonance Imaging (MRI) scan for lesion location classification. Chronically, patients underwent a high-resolution MRI, clinical assessment, and were longitudinally monitored for development of epilepsy for a minimum of 2 years post-injury.
We hypothesized that injury location, specifically the temporal lobe, would be associated with a higher incidence of 1) early seizures and 2) development of post-traumatic epilepsy (PTE) longitudinally. Additionally, we hypothesized that early seizures (either convulsive or non-convulsive in nature), assessed with ICU cEEG, would be associated with an increased risk for development of PTE. As secondary analysis measures, we sought to characterize the functional recovery and degree of brain atrophy between those patients who developed PTE and those who did not.
2. Methods
2.1. Patient inclusion & exclusion
From January 2001 through December 2011, a prospective sequential series of acute moderate-severe TBI patients at the UCLA Brain Injury Research Center (BIRC) was enrolled in the study. Inclusion criteria included TBI with admission Glasgow Coma Scale (GCS) score ≤ 8 or a GCS of 9–12 with evidence of a mass lesion demonstrating an intracranial bleed. Patients were excluded if they had a GCS > 12 with a non-significant computed tomography (CT) brain scan, brain death, pre-existing neurological disease, previous seizures, or a previous TBI. Informed consent was obtained from a surrogate family member or legal authorized representative (LAR), using IRB approved consent methods.
2.2. General TBI continuous EEG monitoring protocol and quantification of epileptiform activity
Intensive care unit (ICU) surface continuous electroencephalography (cEEG) monitoring was prospectively performed to evaluate for early seizures for the duration of their acute ICU stay, according to standardized neurocritical care guidelines (Brophy et al., 2012). Patients were monitored for at least the first 7 days after TBI. Early seizures were prospectively determined either by a neurointensivist or electroencephalographer. Seizure detection was performed using manual review of the raw EEG and review of quantitative trends including total power and spectral edge envelope using a standardized protocol (Vespa et al., 1999). Occurrence of early seizures and status epilepticus was documented in the patient’s medical record. All patients were routinely placed on seizure prophylaxis medication, phenytoin, for 7 days post injury. Phenytoin was discontinued at 7 days if no seizures occurred during that period. Patients demonstrating early seizures had anticonvulsants continued as per standard of care. Patients demonstrating status epilepticus received standard of care treatment including lorazepam 0.1 mg/kg bolus followed in stepwise fashion by continuous midazolam infusion 0.5–2 mg/kg/h to induce burst suppression and secondary anticonvulsants were added to phenytoin. Patients then failing to be controlled were advanced to pentobarbital infusion to induced burst suppression (Vespa et al., 1999). cEEG monitoring was performed for a minimum of 7 days in all cases. Con-firmatory review of EEG from this cohort was undertaken retrospectively to confirm the initial clinical EEG interpretation. For the current study, we characterized ICU cEEGs based on evaluation criteria based on 1) presence/absence of seizures, 2) interictal spikes, or 3) the interictal continuum.
2.3. Longitudinal follow up and post-traumatic epilepsy (PTE) assessment
Patients were longitudinally monitored in the study from time of hospital admission to discharge, and prospectively seen in research clinic at 3, 6 and 12 months. Scheduled structured follow-up clinic visits documented presence/absence of epilepsy, neurocognitive function (Glasgow Outcome Score Extended; GOSE), and clinical exam. Attrition over time occurred, where 82% of patients completed the 6 month follow up, 48% of patients completed the 1 year in-person follow-up. In September 2013 (2–10 year follow-up), in a retrospective study for the incidence of PTE, these same 90 subjects were contacted for telephonic follow-up. 46/90 (51%) completed the telephonic follow-up which included the Extended Glasgow Outcome Scale (GOSE) and a structured epilepsy interview questionnaire (Ottman et al., 2010) to document presence of PTE, seizure onset, latency, prevalence, and seizure type. Subsequently, three clinicians (PV, RDA, PVN) were provided blinded copies of the telephonic interview questionnaire and independently adjudicated the diagnosis of post-traumatic epilepsy including epilepsy subtype. Only those subjects meeting consensus agreement on PTE among the panel were considered to have PTE. Demographic, clinical and neuropsychological data were retrospectively collected on the respondents. The inclusive attrition rate was 49% with 35 (38.8%) patients lost to follow-up. 6 (6.7%) patients refused the follow-up interview and clinical evaluation. Three (3.3%) patients were reported deceased. Subjects not available for a follow-up interview were excluded from the remaining analysis.
2.4. MRI acquisition and analysis methods
High-resolution MRI was acquired acutely, within 14 days post-injury, and chronically, at 6 months post-injury. For all subject’s (n = 90) acute volumetric MRI, a radiologist (PV) reviewed the following brain injury types for lesion location classification analysis: intraparenchymal hematoma, intraventricular hemorrhage (IVH), subdural hematoma (SDH), epidural hematoma (EDH), traumatic subarachnoid hemorrhage (tSAH), contusion, diffuse axonal injury (DAI), skull fracture, and temporal lobe lesion.
For the longitudinal cohort (n = 46), we performed volumetric analysis, using T1-weighted three-dimensional gradient echo sequence, on 42 patients. Each cortical lobe was identified and masked using the MNI structural atlas (Mazziotta et al., 2001; Collins et al., 1995). Next, a nonlinear registration was performed using ANTs (Avants et al., 2011) to align each individual subject’s T1 whole-head structural MRI to the atlas standard space, confirmed by visual inspection to ensure accurate alignment. The transform was then inverted to calculate the registration from the atlas to each individual subject’s space, and again visually inspected. The volume of each mask (cortical lobe), estimated using FSL utilities (Smith et al., 2004), was used to determine within-subject volume changes (i.e. atrophy) from acute to chronic time points. For each cortical lobe (e.g. frontal, parietal, temporal, occipital lobe), we evaluated the amount of atrophy over 6 months. We subtracted the chronic cortical volumetric measurement from the acute volumetric measurement, then divided by the initial acute volume to obtain a percent decrease in volume.
2.5. Statistical analyses
Clinical data was acquired using SQL Server 2012 version 11.0.3128.0 and subsequently integrated into Microsoft office products 2003 (Microsoft Corp., Redmond WA). All statistical analyses were performed with R version 3.2.2 (University of Auckland, New Zealand) (R Foundation for Statistical Computing, 2010).
Count data were analyzed by test of proportions. Univariate analyses and the Yuen-Welch t-test were performed for the primary group comparisons, using analysis of variance and mixed effects model statistics as necessary.
The longitudinal cohort (n = 46) was verified as representative of the entire cohort (n = 90) by performing a two-group Yuen-Welch t-test across demographic and clinical variables. No statistically significant differences were found between subjects who responded and subjects who did not respond to the longitudinal (2–10 year) epilepsy interview.
For between-group analysis, we corrected for multiple comparisons, using False Discovery Rate (FDR) methods. Predictive modeling was conducted using a generalized linear model (GLM) and results of such modeling were further studied using multi-model inference techniques, which mathematically examine the relative strengths of competing submodels to determine how close solutions might be when terms and/ or interactions are selectively entered or dropped (Burnham and Anderson, 2010).
3. Results
3.1. Acute assessment: early cEEG seizure evaluation (n = 90)
The cohort was comprised of 73 males and 17 females, with a mean age of 37.72 ± 17.83 and admission GCS of 5.76 ± 3.63. The main injury characteristics are shown in Table 1. The primary mechanism of injury was falls (28.9%) followed by auto accidents (26.7%).
Table 1.
Main injury characteristics (n = 90).
| Summary (n = 90) | |||
|---|---|---|---|
| Demographics | Injury characteristics | ||
| Age | 37.72 ± 17.83 | Surgical intervention | 55.6% |
| GCS | 5.76 ± 3.63 | Temporal lobe injury | 71.1% |
| % male | 81.10% | Mortality | |
| Ethnicity | 12 month mortality | 0.0% | |
| Asian | 2.2% | 2–12year post-injury | 3.3% |
| Black | 4.4% | Outcome measure | |
| Hispanic | 21.1% | 3 month GOSE | 3.98 ± 1.48 |
| White | 71.1% | 6 month GOSE | 4.64 ± 1.68 |
| Other | 1.1% | 12 month GOSE | 5.33 ± 1.58 |
| Mechanism | |||
| Bike vs. Auto | 4.4% | ||
| Blunt | 10.0% | ||
| Fall | 28.9% | ||
| GSW | 2.2% | ||
| MCA | 14.4% | ||
| MVA | 26.7% | ||
| Peds vs Auto | 13.3% | ||
Abbreviations: GCS — Glasgow Coma Score, Bike vs. Auto — bicycle struck by motor vehicle, GSW — gunshot wound, MCA — motorcycle accident, MVA — motor vehicle accident, Peds vs. Auto — pedestrian struck by automobile, GOSE — Glasgow Outcome Scale Extended.
In an ICU cEEG classification analysis, derived from the first 7 days of cEEG monitoring after injury, 63.3% of patients had no evidence of epileptiform activity, 26.7% had epileptic seizures, and 10% had evidence of interictal spikes or cEEG on the interictal continuum. Early cEEG classification is shown in Table 2. Of the 26.7%, who had early epileptic seizures (n = 24), 75% also had evidence of a hemorrhagic lesion to the temporal lobe on MRI.
Table 2.
Early seizure subtype (within 7 days post-injury).
| Early seizure subtype (n = 90) | N (%) |
|---|---|
| None | 57 (63.3%) |
| EEG nonconvulsive or clinical seizures | 24 (26.7%) |
| EEG interictal continuum | 9 (10.0%) |
Abbreviations: cEEG — continuous electroencephalography.
3.2. Longitudinal assessment: PTE evaluation (n = 46)
3.2.1. PTE characterization
A subgroup of 46 (51%) patients from the initial acute assessment cohort (n = 90) completed the longitudinal epilepsy assessment (2–10 years post-injury). The subgroup of 46 patients was a representative sample of the larger cohort of 90 patients; the two groups had no significant differences across demographic and injury severity variables. Of the 24 patients who had early seizures during the acute assessment, 10 (41.7%) were available for longitudinal follow-up. The cohort was comprised of 37 males and 9 females, with a mean age of 36.9 ± 17.3 years and admission GCS of 5.5 ± 3.3. 73.9% were Caucasian, 21.7% Hispanic, 2.2% Asian, and 2.2% African American. Skull fractures occurred in 28 (60.9%) patients. Twenty-nine (63.0%) patients underwent a craniotomy or hemicraniectomy surgical intervention. Forty-four (95.7%) of the patients had an external ventricular drain (EVD) placed for intracranial pressure (ICP) management and control.
At the time of the longitudinal assessment (minimum of 2 years post-injury), 45.7% of the patients had developed PTE. Of those with early seizures, who were available at longitudinal follow-up (n = 10), 80% developed PTE. Table 3 shows the seizure subtype for the PTE group. In the cohort of patients who developed PTE, 38.1% had their first seizure early (within 7 days post injury) and 61.9% had a delayed latency in seizure onset, manifesting late (after 7 post-injury days). Overall, the latency between injury and initial seizure onset ranged from 1 day to 6 years post-injury. However, the majority of patients had their first initial seizure within 2 years after injury. Fig. 1 shows the timing of seizure onset.
Table 3.
PTE subtype (assessed at longitudinal follow-up).
| PTE subtype (n = 46) | N (%) |
|---|---|
| None | 26 (56.5%) |
| Focal | 11 (23.9%) |
| Focal with secondary generalized | 6 (13.0%) |
| Generalized | 4 (8.7%) |
Abbreviations: PTE — post-traumatic epilepsy.
Fig. 1.
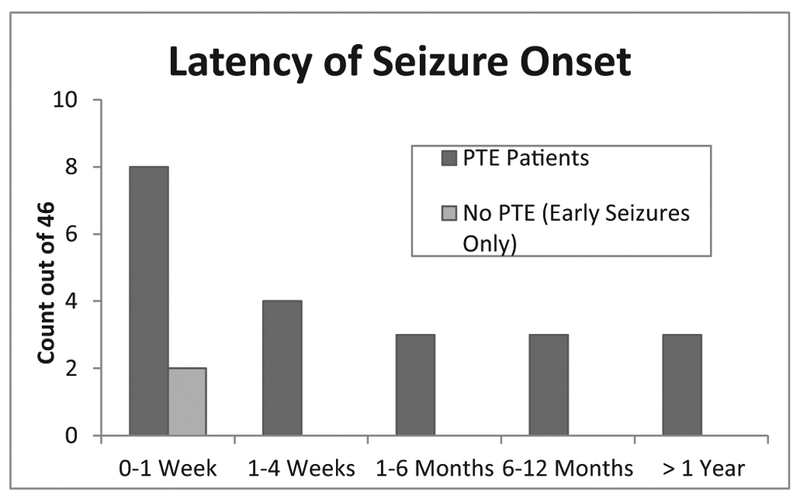
Latency of seizure onset. The timing seizure onset in the longitudinal cohort (n = 46).
3.2.2. PTE vs no-PTE group comparison
3.2.2.1. Injury severity and location classification.
In a between-group analysis, comparing patients who developed PTE (n = 21) and those who did not (n = 25), we established that the groups had similar levels of injury severity; no significant differences in age, initial GCS, or other admission characteristics. However, patients who developed PTE had a significantly higher incidence (85.7%) of acute temporal lobe hemorrhagic injury than patients who never developed PTE (36.0%) (p < 0.001). Additionally, patients who developed PTE had a significantly lower incidence of diffuse axonal injury (DAI) when compared to those patients who did not develop PTE (p = 0.030). Overall, injury to the temporal lobe was the only injury characteristic resulting in a robust (p < 0.001) difference between patients who developed PTE and those who did not (Table 4).
Table 4.
No PTE vs. PTE comparison summary table.
| No PTE | PTE | p-Value | ||
|---|---|---|---|---|
| Demographics | Age | 33.64 ± 12.67 | 40.76 ± 21.22 | 0.393 |
| GCS | 5.96 ± 3.55 | 4.70 ± 3.01 | 0.090 | |
| Admission events | Surgery | 56.0% | 71.4% | 0.311 |
| % ICP > 20 | 9.3% | 10.3% | 0.965 | |
| MRI lesion type | Contusions | 44.0% | 57.1% | 0.396 |
| Diffuse axonal injury | 52.0% | 23.8% | 0.061 | |
| Epidural hematoma | 12.0% | 19.0% | 0.526 | |
| Intraparenchymal hemorrhage | 56.0% | 52.4% | 0.808 | |
| Intraventricular hemorrhage | 24.0% | 28.6% | 0.698 | |
| Skull fracture | 48.0% | 76.2% | 0.061 | |
| Subarachnoid hemorrhage | 72.0% | 71.4% | 0.929 | |
| Subdural hematoma | 72.0% | 61.9% | 0.467 | |
| Temporal lobe injury | 36.0% | 85.7% | < 0.001* | |
| GOSE scores | Discharge | 2.67 ± 0.52 | 2.56 ± 0.69 | 0.471 |
| 6 months | 5.39 ± 1.62 | 4.06 ± 1.66 | 0.006* | |
| 12 months | 5.62 ± 1.56 | 4.33 ± 1.66 | 0.057 | |
| 2–10years | 6.64 ± 1.29 | 4.81 ± 2.06 | 0.003* |
Abbreviations: PTE — post-traumatic epilepsy, GCS — Glasgow Coma Score, % ICP > 20 — percentage of time by patient intracranial pressure was above 20, GOSE — Glasgow Outcome Scale Extended.
p-Value is statistically significant after FDR multiple comparisons correction.
3.2.2.2. Atrophy analysis.
Patients who developed PTE, compared to patients who did not develop PTE, had significantly greater temporal lobe atrophy (p = 0.029). Specifically, patients who developed PTE had an average of 7.7 ± 3.9% reduction in temporal lobe volume, whereas patients who did not develop PTE had an average of 4.7 ± 4.3% reduction. No other differences between regional cortical atrophy measures were found between groups.
3.2.2.3. Functional recovery assessment.
At 6 months post-injury, patients with PTE had significantly worse GOSE functional outcomes (4.06 ± 1.66) than patients who did not develop PTE (5.39 ± 1.62) (p = 0.006), which became more robust (p = 0.003) at the time of their longitudinal epilepsy assessment (minimum of 2 years post-injury).
3.2.3. Longitudinal predictive modeling
In the longitudinal cohort (n = 46), we ran two generalized linear models (GLM) to assess potential early predictors of PTE. In the first GLM, based on demographic and early admission factors (e.g. age, gender, GCS, acute neurosurgical intervention, and early seizures), we identified that occurrence of early seizures (p = 0.026) was the only significant predictor of PTE. In the second GLM analysis, based on MRI injury characteristics, we found that skull fractures (p = 0.018) and temporal lobe injury (p < 0.001) were the only significant injury types associated with PTE.
Based on the GLM results, we combined the significant predictors of clinical and lesion location data (e.g. early seizures, temporal lobe injury, and skull fractures) in a multi-model inference analysis to assess the combination of features that was most predictive of PTE development. The most predictive model indicated that the combination of early seizures, skull fractures, and a temporal lobe injury together were most predictive of PTE development. Temporal injury was a significant contributor to the top models. The top three models are shown in Table 5.
Table 5.
Multi-model inference analysis for prediction of PTE.
| Model | Predictors | Estimate | SE | p-Value | AICc | Delta | Weight |
|---|---|---|---|---|---|---|---|
| 1 | Temporal lobe injury | 3.03 | 0.96 | 0.002* | 48.30 | 0.00 | 0.69 |
| + Skull fracture | 1.92 | 0.87 | 0.028* | ||||
| + Early seizure | 2.35 | 1.13 | 0.038* | ||||
| 2 | Temporal lobe injury | 2.83 | 0.86 | 0.001* | 51.40 | 3.04 | 0.15 |
| + Skull fracture | 1.88 | 0.82 | 0.022* | ||||
| 3 | Temporal lobe injury | 2.57 | 0.86 | 0.003* | 51.60 | 3.22 | 0.14 |
| + Early seizure | 2.27 | 1.05 | 0.031* |
Abbreviations: SE — Standard Error, AICc — second order Akaike information criterion, GCS — Glasgow Coma Score.
p-value is statistically significant after running generalized linear models and a multi-model inference analysis.
4. Discussion
This study indicates that after a severe TBI (1) there is a high incidence of both early seizures and PTE after a moderate-severe TBI; (2) The development of PTE is associated with hemorrhagic contusional injury to the temporal lobe, but not to overall injury severity (i.e. GCS) alone; (3) PTE develops in a greater percentage of patients who have early seizures during the ICU course; (4) PTE is associated with greater atrophy in the temporal lobe at 6 months; and (5) PTE is associated with poor functional outcome (i.e. GOSE) longitudinally.
Moderate-severe TBI is a life-threatening condition known to be a risk factor for PTE (Annegers et al., 1998; Frey, 2003; Christensen et al., 2009). ICU cEEG studies have shown a high incidence (18–30%) of early seizures within 7 days after TBI, most of which are nonconvulsive seizures (Vespa et al., 1999; Vespa et al., 2010; Liesemer et al., 2011), which is consistent with our finding that 26.7% of the patients had early seizures. Depth EEG monitoring recently revealed a higher incidence of seizures (Vespa et al., 2016). Early seizures are associated with induced higher intracranial pressure (ICP) (Vespa et al., 2007), worsening brain edema, metabolic crisis (Vespa et al., 2016), and hippocampal atrophy (Vespa et al., 2010). Thus, early seizures have established negative consequences acutely, during the period of critical care. However, the long-term influence of EEG-confirmed early seizures on PTE has not been well established. In this study, we extend our understanding of the relationship between the incidence of early cEEG seizures and the development PTE. Our findings indicate that early seizures not only have acute consequences, but increase the risk of PTE longitudinally.
Epidemiological studies have documented that the latency between injury time and seizure onset varies greatly (Annegers et al., 1998; Immonen et al., 2013). Recent data suggest that the majority (86%) of patients will develop a subsequent seizure within 2 years of their first late onset seizure (Haltiner et al., 1997). In the current cohort, we found that the occurrence of early seizures, as detected by cEEG, was a predictive factor for PTE. This result is important in itself since as it suggests another negative long-term effect of early post-traumatic seizures. Moreover, this result may inform the design of future longitudinal interventional studies on PTE and specifically suggest that cEEG may be a useful metric to select an enriched population of TBI patients, who are at higher risk for PTE. Because seizures occur early, and those patients have an elevated risk of PTE development, we postulate that epilepto-genesis may be starting during the acute critical care period, despite the long latency to manifest epilepsy. Further experimental studies will be needed to determine if the early seizures signify the onset of PTE or whether they are just markers of future PTE. But, the current data suggest a translational paradigm that can be tested experimentally.
TBI phenotype and location may be important predictors of PTE (Wang et al., 2013). PTE arises most frequently in the temporal lobe (57%), occasionally in the frontal lobe (35%), and rarely from the occipital and parietal lobes (Gupta et al., 2014). Not only is the temporal lobe the region most frequently implicated in PTE, its underlying medial structures have been associated with both medically intractable PTE in humans (Gupta et al., 2014; Diaz-Arrastia et al., 2000), lower seizure threshold and susceptibility in animal models (Trombin et al., 2011; Avoli et al., 2002; Mamad et al., 2018; Abdelmalik et al., 2005), and enhanced glutamatergic excitatory connectivity in-vitro (Li et al., 2011). Additionally, temporal lobe structures are at an increased risk for atrophy, as our past and present findings suggest that seizures are associated with increased temporal lobe and hippocampal atrophy (Vespa et al., 2010). Hence, early seizures may be intertwined with both the genesis and consequences of PTE. In the current study, we found that temporal lobe hemorrhagic injury was independently associated with PTE, and that other trauma phenotypes such as subdural hemorrhage, diffuse axonal injury or parenchymal hemorrhages in other non-temporal lobe regions were not. Interestingly, we found that patients with DAI had a lower incidence of PTE, suggesting that nonspecific axonal disconnection does not play a significant pathophysio-logic role in PTE development (Scheid and von Cramon, 2010). Moreover, these findings may point to a biological mechanism for the genesis of PTE that involves disruption of specific regional networks, such as those implicated in the temporal lobe, rather than diffuse white matter connections. We postulate that disruption of temporal lobe associated circuits could lead to epileptogenesis. Such disruption of circuits, altered by an increase in metabolic load required by the excitation of seizures, may ultimately lead to cell death, explaining the susceptibility to increased atrophy in the temporal lobe (Vespa et al., 2010; Bernasconi et al., 2003). Since abnormalities to the mesial temporal structures have been associated with epileptogenic neural networks, injury to the underlying structures may disrupt normal neural processing, engendering the construction and maturation of an epileptogenic network (Hunt et al., 2013). These findings suggest that the temporal lobe and its underlying structures are an important area of focus for future studies assessing the genesis and progression of PTE.
Patients with PTE had worse long term functional outcome compared with the non-epileptic group. This finding is consistent with previous literature, suggesting that the electrophysiological and structural consequences of PTE and/or the treatment of PTE manifest in depressed functional outcome after TBI (Asikainen et al., 1999). Functional recovery differences between the cohorts were significant even at 6 months post-injury and became robust at the 2 to 10 year follow-up interview. In some cases, this is before the occurrence of PTE. This finding could suggest that early seizures have a detrimental effect on functional recovery and morbidity over time or could suggest comorbidity of epileptogenesis and cognitive function, before manifestation of the epilepsy in the chronic setting. Detailed study of the mechanisms that relate to poor functional outcome and PTE, including functional assessment of neural networks stemming from the temporal lobe could help to illuminate the process of epileptogenesis further.
There appears to be both phenotypic anatomical and electro-physiological indicators that predict the occurrence of PTE. These findings suggest that there may be utility to adopting routine use of cEEG and MRI in moderate-severe TBI patients in order to guide intermediate therapy with regards to anticonvulsants. By isolating specific brain regions and functions, this study will help drive future prospective studies to evaluate mechanistic links between TBI and epileptogenesis.
5. Limitations
Considering the heterogeneous nature of TBI, the current study included a small sample size with a high attrition rate. The high attrition rate may contribute a bias, because patients may be more willing to consent to follow-up interviews by clinicians if they are experiencing seizures and impairments in functional outcome. The cohort was primarily comprised of Caucasian males, and consequently, may not be generalizable to other demographic groups. Data were collected retrospectively and observationally, obviating study of causal relationships. Clinical practice such as the use and duration of anticonvulsants was standardized for the initial 7 days after TBI, but the drug selection varied and it is possible that clinical anticonvulsant use beyond the acute hospitalization varied in ways that bias the development of PTE. Since patients self-reported to a clinician, seizure incidence and recovery may be misrepresentative. A prospective longitudinal study is needed to resolve these limitations and to clarify causal and biomarker relationships between traumatic brain injury and the development and progression of epilepsy after injury.
Acknowledgments
Study funding
This manuscript was supported by the following grants: NS080181, NS058489, PT074268, NS049471, K08 NS02089, NS086090. NINDS Center without Walls, U54 NS100064 (EpiBioS4Rx).
References
- Abdelmalik PA, Burnham WM, Carlen PL, 2005. Increased seizure susceptibility of the hippocampus compared with the neocortex of the immature mouse brain in vitro. Epilepsia 46, 356–366. [DOI] [PubMed] [Google Scholar]
- Agrawal A, Timothy J, Pandit L, Manju M, 2006. Post-traumatic epilepsy: an overview. Clin. Neurol. Neurosurg. 108, 433–439. [DOI] [PubMed] [Google Scholar]
- Angeleri F, Majkowski J, Cacchio G, et al. , 1999. Posttraumatic epilepsy risk factors: one-year prospective study after head injury. Epilepsia 40, 1222–1230. [DOI] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA, 1998. A population-based study of seizures after traumatic brain injuries. N. Engl. J. Med. 338, 20–24. [DOI] [PubMed] [Google Scholar]
- Asikainen I, Kaste M, Sarna S, 1999. Early and late posttraumatic seizures in traumatic brain injury rehabilitation patients: brain injury factors causing late seizures and influence of seizures on long-term outcome. Epilepsia 40, 584–589. [DOI] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC, 2011. A reproducible evaluation of ANTs similarity metric performance in brain image registration. NeuroImage 54, 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, D’Antuono M, Louvel J, et al. , 2002. Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog. Neurobiol. 68, 167–207. [DOI] [PubMed] [Google Scholar]
- Barbarosie M, Louvel J, Kurcewicz I, Avoli M, 2000. CA3-released entorhinal seizures disclose dentate gyrus epileptogenicity and unmask a temporoammonic pathway. J. Neurophysiol. 83, 1115–1124. [DOI] [PubMed] [Google Scholar]
- Bernasconi N, Bernasconi A, Caramanos Z, Antel SB, Andermann F, Arnold DL, 2003. Mesial temporal damage in temporal lobe epilepsy: a volumetric MRI study of the hippocampus, amygdala and parahippocampal region. Brain 126, 462–469. [DOI] [PubMed] [Google Scholar]
- Brophy GM, Bell R, Claassen J, et al. , 2012. Guidelines for the evaluation and management of status epilepticus. Neurocrit. Care. 17, 3–23. [DOI] [PubMed] [Google Scholar]
- Burnham KP, Anderson DA, 2010. Model Selection and Multimodel Inference — A Practical Information-Theoretic Approach, 2nd ed. Springer. [Google Scholar]
- Chen W, Li MD, Wang GF, Yang XF, Liu L, Meng FG, 2017. Risk of post-traumatic epilepsy after severe head injury in patients with at least one seizure. Neuropsychiatr. Dis. Treat. 13, 2301–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Pedersen MG, Pedersen CB, Sidenius P, Olsen J, Vestergaard M, 2009. Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 373, 1105–1110. [DOI] [PubMed] [Google Scholar]
- Collins D, Holmes C, Peters T, Evans A, 1995. Automatic 3-D model-based neuroanatomical segmentation. Hum. Brain Mapp. 208, 190–208. [Google Scholar]
- Delorenzo RJ, Sun DA, Deshpande LS, 2005. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol. Ther. 105, 229–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Arrastia R, Agostini MA, Frol AB, et al. , 2000. Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch. Neurol. 57, 1611–1616. [DOI] [PubMed] [Google Scholar]
- Diaz-Arrastia R, Agostini MA, Madden CJ, Van Ness PC, 2009. Posttraumatic epilepsy: the endophenotypes of a human model of epileptogenesis. Epilepsia 50 (Suppl 2), 14–20. [DOI] [PubMed] [Google Scholar]
- Englander J, Bushnik T, Duong TT, et al. , 2003. Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation. Arch. Phys. Med. Rehabil. 84, 365–373. [DOI] [PubMed] [Google Scholar]
- Forsgren L, 1990. Prospective incidence study and clinical characterization of seizures in newly referred adults. Epilepsia 31, 292–301. [DOI] [PubMed] [Google Scholar]
- Frey LC, 2003. Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia 44 (Suppl 10), 11–17. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Sayed N, Ding K, et al. , 2014. Subtypes of post-traumatic epilepsy: clinical, electrophysiological, and imaging features. J. Neurotrauma 31, 1439–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guzman P, D’Antuono M, Avoli M, 2004. Initiation of electrographic seizures by neuronal networks in entorhinal and perirhinal cortices in vitro. Neuroscience 123, 875–886. [DOI] [PubMed] [Google Scholar]
- Haltiner AM, Temkin NR, Dikmen SS, 1997. Risk of seizure recurrence after the first late posttraumatic seizure. Arch. Phys. Med. Rehabil. 78, 835–840. [DOI] [PubMed] [Google Scholar]
- Herman ST, 2002. Epilepsy after brain insult: targeting epileptogenesis. Neurology 59, S21–S26. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Boychuk JA, Smith BN, 2013. Neural circuit mechanisms of post-traumatic epilepsy. Front. Cell. Neurosci. 7, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immonen R, Kharatishvili I, Grohn O, Pitkanen A, 2013. MRI biomarkers for post-traumatic epileptogenesis. J. Neurotrauma 30, 1305–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharatishvili I, Pitkanen A, 2010. Association of the severity of cortical damage with the occurrence of spontaneous seizures and hyperexcitability in an animal model of posttraumatic epilepsy. Epilepsy Res. 90, 47–59. [DOI] [PubMed] [Google Scholar]
- Li Y, Fan S, Yan J, et al. , 2011. Adenosine modulates the excitability of layer II stellate neurons in entorhinal cortex through A1 receptors. Hippocampus 21, 265–280. [DOI] [PubMed] [Google Scholar]
- Liesemer K, Bratton SL, Zebrack CM, Brockmeyer D, Statler KD, 2011. Early post-traumatic seizures in moderate to severe pediatric traumatic brain injury: rates, risk factors, and clinical features. J. Neurotrauma 28, 755–762. [DOI] [PubMed] [Google Scholar]
- Mamad O, Islam MN, Cunningham C, Tsanov M, 2018. Differential response of hippocampal and prefrontal oscillations to systemic LPS application. Brain Res. 1681, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazziotta J, Toga A, Evans A, et al. , 2001. A probabilistic atlas and reference system for the human brain: International Consortium for Brain Mapping (ICBM). Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci 356, 1293–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman R, Barker-Cummings C, Leibson CL, Vasoli VM, Hauser WA, Buchhalter JR, 2010. Validation of a brief screening instrument for the ascertainment of epilepsy. Epilepsia 51, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, Immonen R, 2014. Epilepsy related to traumatic brain injury. Neurotherapeutics 11, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Foundation for Statistical Computing, 2010. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Scheid R, von Cramon DY, 2010. Clinical findings in the chronic phase of traumatic brain injury: data from 12 years’ experience in the Cognitive Neurology Outpatient Clinic at the University of Leipzig. Dtsch. Arztebl. Int 107, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, et al. , 2004. Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage 23 (Suppl 1), S208–S219. [DOI] [PubMed] [Google Scholar]
- Sudbury JR, Avoli M, 2007. Epileptiform synchronization in the rat insular and perirhinal cortices in vitro. Eur. J. Neurosci. 26, 3571–3582. [DOI] [PubMed] [Google Scholar]
- Temkin NR, 2003. Risk factors for posttraumatic seizures in adults. Epilepsia 44 (Suppl 10), 18–20. [DOI] [PubMed] [Google Scholar]
- Trombin F, Gnatkovsky V, de Curtis M, 2011. Changes in action potential features during focal seizure discharges in the entorhinal cortex of the in vitro isolated guinea pig brain. J. Neurophysiol. 106, 1411–1423. [DOI] [PubMed] [Google Scholar]
- Vespa PM, Nuwer MR, Nenov V, et al. , 1999. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J. Neurosurg. 91, 750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa PM, Miller C, McArthur D, et al. , 2007. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit. Care Med 35, 2830–2836. [PMC free article] [PubMed] [Google Scholar]
- Vespa PM, McArthur DL, Xu Y, et al. , 2010. Nonconvulsive seizures after traumatic brain injury are associated with hippocampal atrophy. Neurology 75, 792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa P, Tubi M, Claassen J, et al. , 2016. Metabolic crisis occurs with seizures and periodic discharges after brain trauma. Ann. Neurol. 79, 579–590. [DOI] [PubMed] [Google Scholar]
- Wang Huaqing, Xin Tao, Sun Xiubin, Wang Shuzhen, Guo Hua, Holton-Burke Conor, Pang Qi, 2013. Post-traumatic seizures—A prospective, multicenter, large case study after head injury in China. Epilepsy Research 107 (3), 272–278. http://dx.doi.org/10.1016/j.eplepsyres.2013.10.006 . 10.1016/j.eplepsyres.2013.10.006http://www.sciencedirect.com/science/article/pii/092012111300260X. http://www.sciencedirect.com/science/article/pii/092012111300260X ISSN 0920-1211. [DOI] [PubMed] [Google Scholar]
- Wozny C, Gabriel S, Jandova K, Schulze K, Heinemann U, Behr J, 2005. Entorhinal cortex entrains epileptiform activity in CA1 in pilocarpine-treated rats. Neurobiol. Dis. 19, 451–460. [DOI] [PubMed] [Google Scholar]
- Xu T, Yu X, Ou S, et al. , 2017. Risk factors for posttraumatic epilepsy: a systematic review and meta-analysis. Epilepsy Behav. 67, 1–6. [DOI] [PubMed] [Google Scholar]