

Abstract
Cell-matrix and cell-cell interactions influence intracellular signalling and play an important role in physiologic and pathologic processes. Detachment of cells from the surrounding microenvironment alters intracellular signalling. Here, we demonstrate and characterise an integrated microfluidic device to culture single and clustered cells in tuneable microenvironments and then directly analyse the lysate of each cell in situ, thereby eliminating the need to detach cells prior to analysis. First, we utilise microcontact printing to pattern cells in confined geometries. We then utilise a microscale isoelectric focusing (IEF) module to separate, detect, and analyse lamin A/C from substrate-adhered cells seeded and cultured at varying (500, 2000, and 9000 cells per cm2) densities. We report separation performance (minimum resolvable pI difference of 0.11) that is on par with capillary IEF and independent of cell density. Moreover, we map lamin A/C and β-tubulin protein expression to morphometric information (cell area, circumference, eccentricity, form factor, and cell area factor) of single cells and observe poor correlation with each of these parameters. By eliminating the need for cell detachment from substrates, we enhance detection of cell receptor proteins (CD44 and β-integrin) and dynamic phosphorylation events (pMLCS19) that are rendered undetectable or disrupted by enzymatic treatments. Finally, we optimise protein solubilisation and separation performance by tuning lysis and electrofocusing (EF) durations. We observe enhanced separation performance (decreased peak width) with longer EF durations by 25.1% and improved protein solubilisation with longer lysis durations. Overall, the combination of morphometric analyses of substrate-adhered cells, with minimised handling, will yield important insights into our understanding of adhesion-mediated signalling processes.
Introduction
Studying and understanding human biology and disease has benefited from systems biology, including efforts to unify proteomics, transcriptomics, and genomics. High-resolution subcellular maps, such as the Human Protein Atlas, have identified single-cell, intracellular variations that drive disease.1 Extracellularly, cells interact dynamically with the surrounding microenvironment, and these cell-cell and cell-matrix interactions regulate key biological processes such as cell survival,2 stem cell differentiation,3 cell migration,4,5 and cancer metastasis.6 For example, cancer cells differ in adhesive properties,7 with cell adhesion molecules such as integrins and selectins known to play a significant role in cancer progression and metastasis.8–10 Other cell receptor proteins, such as CD44, possess multiple alternatively spliced variants, of which certain isoforms promote resistance to apoptosis and mediate the migratory behaviour of cancer cells.11–13 These cell receptor proteins influence the adhesive strength of the cell, which correlates with the metastatic capability.14–16 Various other adhesive proteins, such as N-cadherin and β-integrin, have important roles in matrix mechanosensing, highlighting the importance of microenvironmental cues on cell behaviour.5,10,17–21 Other biophysical features, such as geometry,22 topography,23 and substrate stiffness,4 also influence cell signalling. Thus, understanding the mechanisms underlying adhesion and downstream protein-mediated signalling is necessary for understanding disease progression and for identifying therapeutic targets.
It is unclear whether contemporary protein analyses accurately recapitulate the cellular environment. Flow cytometry offers high throughput analysis of multiple targets, but adherent cells are removed from the culture environment, often using enzymatic digestion, which proteolytically cleaves important cell-matrix receptors.24,25 Moreover, the long handling times may influence protein expression and modification, especially for rapid dynamic events such as phosphorylation.26
Immunocytochemistry (ICC) is the workhorse immunoassay that detects cell-matrix interactions based on antibody specificity. Recent work has demonstrated multiplexed detection of intracellular and secreted cytokines in microfluidic systems.27,28 However, relying solely on antibody specificity for protein detection can be misleading, as nonspecific antibody binding to other cellular constituents can occur.29 Moreover, chemical fixation of cells has been demonstrated to cause artefacts in the dynamic detection of transcription factors.30 Live-cell imaging can visualise the movement of select proteins dynamically, but is limited to transgenic cell lines, which limits multiplexing (number of targets measured concurrently).
Assays such as the western blot31 offer multiplexing capability and high selectivity by inclusion of an electrophoretic size separation step. However, conventional western blots require large numbers of cells and also entail disruption of the cell-matrix interactions by proteolytic and/or harsh mechanical conditions. Efforts to append an electrophoretic (capillary) separation to adherent cell lysis have shown promise;32 however, serial cell analysis limits throughput. Moreover, in all the aforementioned assays, protein isoforms that have small molecular mass differences are not separable.
In isoelectric focusing (IEF), proteins are resolved electrophoretically by isoelectric point (pI), the pH at which a protein is net neutrally charged. Since proteoforms of similar mass often differ in pI, IEF confers extraordinary selectivity. During IEF, a linear pH gradient is established along a gel or capillary. Proteins carry charge depending on the local pH of the environment, and as such, electromigrate along the pH gradient until neutrally charged.33 In capillary IEF, protein isoforms were identified from the lysates of <25 cells and from tumour aspirates.34,35 Our recently reported work has demonstrated that proteins with just a single charge difference are resolvable in single-cell lysates.36 However, all of these techniques require cell dissociation from the microenvironment, which can alter protein signalling7,11,24,25,37 and limit the ability to monitor spatial, morphometric, and dynamic signalling information.
Given the high selectivity of IEF and the importance of cell environment on signalling processes, we designed a tool that analyses proteins from cells in situ. We combine the selectivity of IEF with controlled micropatterning of cells. We measure proteins from single and clustered cells and correlate areas of individual cells with protein expression. Importantly, we identify protein targets from intact cells that are otherwise undetectable in detached cells. Finally, we discuss and optimise assay parameters to improve protein solubilisation in our system.
Materials and methods
Microcontact printing of fibronectin templates
PDMS stamps for microcontact printing were made by casting PDMS (Dow-Corning) at a 10 : 1 ratio onto a silicon wafer with SU-8 3050 (Microchem) features 40 μm in height. The wafer moulds were created using standard SU-8 soft lithography; photomasks with 30–400 μm features were created in AutoCAD and printed onto Mylar positive masks (CAD/Art services). After PDMS casting and polymerisation, the stamps were peeled off, and incubated with 10 μg mL−1 rhodamine-fibronectin (Cytoskeleton Inc.) at room temperature for 30 min. The fibronectin was then rinsed away with deionised water, and the mould dried under an air stream. PDMS stamps were inverted onto ethanol-cleaned glass slides (VWR International) and pressed down with forceps until contact was made for ~1 min. Stamps were then peeled off, leaving printed features on the glass.
Device fabrication
Substrate gel for cell patterning.
6% T, 3.3% C polyacrylamide gels containing 5 mM N-[3-[(3-benzoylphenyl) formamido]propyl] methacrylamide (BPMAC, Pharm-Agra Laboratories) were fabricated by pipetting the precursor solution between an acrylate-silanized microscope slide and the fibronectin-patterned glass slide described above. 0.08% (w/v) APS (Sigma-Aldrich) and 0.08% (v/v) TEMED (Sigma-Aldrich) were used as the initiator and catalyst. Prior to peeling the fibronectin-patterned slide, the gels were incubated in distilled water for 30 min at room temperature. The process transferred the fibronectin pattern from the imprinted glass to the surface of the gel, as has been previously described (Fig. 1A).22,38 Prior to cell seeding, gels were sterilised in 70% ethanol for 10 min in a biosafety cabinet, then washed in PBS.
Fig. 1.
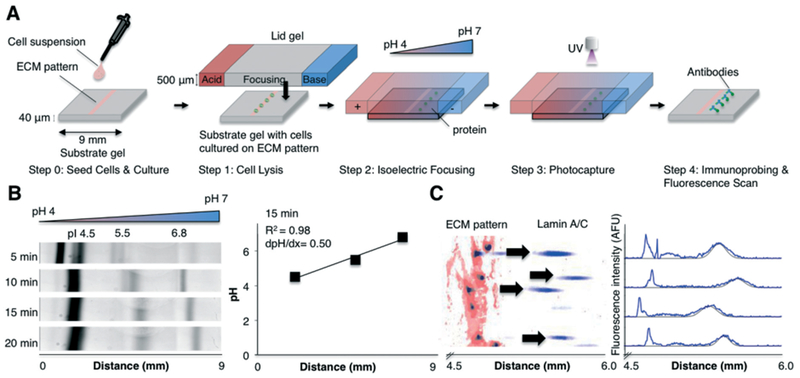
In situ IEF measures protein peaks from single and clustered cells adhered to a substrate gel. (A) Concept schematic of in situ IEF (not-to-scale). Step 0: Cells are cultured on pre-determined regions by seeding cells on the ECM-patterned substrate gel. Step 1: A chemically-imprinted lid gel, which contains the lysis reagents and carrier ampholytes, is interfaced with the substrate gel, which contains the cells attached to the ECM-patterned region, for 30 s. Step 2: An electric potential (600 V) is then applied for 6 min, establishing a stable, linear pH gradient. Step 3: After focusing, proteins are covalently immobilised in the substrate gel via UV activation of a benzophenone moiety. Step 4: Gels are then washed and immunoprobed for targets of interest. Individual protein peaks are then quantified. (B) Inverted fluorescence kymograph of pI markers incorporated into the substrate gel and focused. The established pH gradient is linear and stable for >20 min. (C) Left: False-colour inverted fluorescence micrograph of micropatterned rhodamine-fibronectin, onto which cells were cultured, lysed, separated via IEF, photocaptured, and immunoprobed for lamin A/C. Right: Fluorescence intensity profile of lamin A/C protein peaks that have passed the SNR > 3 and Gaussian fit R2 > 0.7 threshold. Black line indicates Gaussian fit. IEF, isoelectric focusing. pI, isoelectric point. ECM, extracellular matrix. AFU, arbitrary fluorescence units.
Lid gel.
The lid gel is chemically-imprinted, using the protocol described previously.36 Briefly, we fabricated 500 μm-thick polyacrylamide gels with three adjacent compartments. The outermost regions define the pH boundary conditions (pH 4 and pH 10) for the IEF assay. The centre focusing gel serves two functions: (i) delivery of lysis reagents to attached cells and (ii) delivery of carrier ampholytes required to establish the pH gradient between the pH boundary gel sections. We used different carrier ampholytes (ZOOM®, 1% (v/v), Thermo Fischer Scientific), as the previously described carrier ampholytes36 were discontinued. We also increased the denaturing and lysis reagents—81 mM CHAPS (Amresco), 2.5% (v/v) Triton X-100 (Sigma-Aldrich), 7 M urea (Sigma-Aldrich) and 2 M thiourea (Sigma-Aldrich) to enhance solubilisation (Fig. S1†).
Cell culture and cell patterning
Human BJ fibroblasts were provided by the Dillin laboratory (UC Berkeley) and validated by short tandem repeat identification (UC Berkeley Cell Culture Facility). These cells were mycoplasma-negative and cultured using DMEM + GlutaMAX media (ThermoFisher Scientific) supplemented with 1% penicillin/streptomycin (Life Technologies), 1× MEM non-essential amino acids (Life Technologies), and 10% foetal bovine serum (Gemini Bio-products). Cells were cultured at 37 °C with 5% CO2 in a humidified incubator. All cells were between passage number 8 and 13.
For cell patterning, cells were detached using 0.25% trypsin-EDTA (Gemini Bio-products), re-suspended, and applied to the fibronectin-patterned gel slide and incubated at 37 °C for 1 h to allow for cell attachment. The gels were then rinsed with PBS to remove unadhered cells and cultured in culturing medium overnight (to allow for recovery of receptor proteins) or up to four days (to obtain 2000 cell per cm2 density). For studies comparing GFP expression, BJ fibroblasts were first transduced with GFP using a BacMam GFP Transduction kit (Thermo Fischer Scientific) at a 5% (v/v) concentration. Cells were then used 24 h after transduction.
For fluorescence-labelling of cells, BJ fibroblasts were stained using Cytopainter Cell Tracking Staining Kit (ABCAM) according to the manufacturer’s instructions.
In situ and microwell IEF
For in situ IEF, cell-patterned substrate gels were incubated in PBS for 10 min, then transferred to a buffer solution of 1% carrier ampholytes in Milli-Q water for ~30 s prior to lysis. Substrate gels were then placed in a custom-built electrophoresis chamber. To initiate lysis and IEF, the chemically-imprinted lid gel (centre focusing gel region) was interfaced with the substrate gel for 30 s, or, for solubilisation studies, for 45 or 70 s. A glass slide was placed on top of the lid gel to minimise evaporation. Next, an electric potential of 600 V was applied using a PowerPac high-voltage power supply (Bio-Rad) for 6 or 8 min. Following IEF, the proteins were immobilised into the substrate gel upon UV light exposure (Hamamatsu LC5) for 45 s, as previously described.39,40 The slide was then washed with 1× TBS + Tween 20 (TBST, Santa Cruz Biotechnology) for 30 min prior to antibody probing.
For microwell IEF, the same procedure as above was performed, except the substrate gel was replaced with a bottom gel layer stippled with a single row of microwells, as previously described.36 Cells were trypsinized and seeded into wells via gravity sedimentation for 10 min. Excess cells were then washed with PBS and quickly rinsed with carrier ampholytes diluted 1 : 100 in MilliQ water prior to lysis. IEF was then performed immediately after washing.
Antibodies
Primary and secondary antibodies were diluted in 2% (w/v) BSA in 1× TBST. Mouse anti-CD44 antibody (Pierce, MA5-15462), rabbit anti-pMLCS19 (Cell Signaling, 3671 T), goat anti-GFP (ABCAM, ab6673), mouse anti-β-integrin (ABCAM, ab30394), and rabbit anti-β-tubulin (ABCAM, ab6046) antibodies were diluted at 1 : 10, and mouse anti-lamin A/C (Pierce, mab636) antibody was diluted at 1 : 5 concentration. Secondary antibodies, AlexaFluor 488-,594-, and 647-conjugated donkey anti-mouse, anti-goat, and anti-rabbit antibodies (Life Technologies), were diluted 1 : 50. Immunoprobing was conducted as previously described.41
Image acquisition and analysis
Brightfield images were obtained on an Olympus IX-71 inverted microscope with an Olympus UPlanFi 4× (numerical aperture (NA) 0.13) or 10× (NA 0.3) objectives and an EMCCD Camera iXon2 (Andor). Fluorescence images for ICC were obtained with the 10× objective and an X-Cite Exacte mercury arc lamp illumination source.
Immunoprobed gels were imaged on a microarray scanner (Genepix, Molecular Devices) under the 488, 535, and 635 excitation wavelength channels. Images were then analysed using an in-house MATLAB (R2017a) script (Fig. S2†). The custom MATLAB algorithm segments peaks by first applying a threshold to identify protein peaks and centroids of each peak. A Gaussian is then fit laterally to each peak to generate a custom region of interest “lane” containing the entire peak (8σ). Intensity profiles of each peak were obtained by summing the intensities of each pixel along the separation axis and dividing by the number of pixels. The intensity profiles were fit to Gaussian curves with a minimum signal-to-noise ratio (SNR) of 3 and R2 threshold of 0.7. Protein mass of focused protein peaks was calculated by summing the area under the curve (AUC) of the fit Gaussian curve. Protein mass of protein retained on the fibronectin pattern was calculated by summing the background-subtracted intensities along the fibronectin pattern (integrated density, ID).
Immunocytochemistry (ICC)
For ICC, BJ fibroblasts were seeded at a concentration of 5 × 103 cells per mL onto fibronectin-patterned gels and allowed to adhere overnight. Slides were then washed in PBS, fixed in 4% PFA (Electron Microscopy Sciences) for 10 min, then washed in PBS three times for 5 min. Cells were permeabilized in 0.25% Triton-X 100 for 10 min, washed in PBS, and blocked with 1% (w/v) BSA in PBST for 30 min. Primary antibodies were diluted at 1 : 100 (anti-lamin A/C) or 1 : 200 (anti-β-tubulin) in PBST and incubated overnight at 4 °C on a shaker. Cells were then washed in PBS and incubated with fluorescent secondary antibody at 1 : 500 dilution in 1% (w/v) BSA in PBST for 1 h in the dark. Cells were then washed and imaged as described above.
Statistical analyses
Statistical analyses were performed in MATLAB. To determine significance between protein masses, we first tested for normality (QQ plot) and homoscedasticity (Levene’s test). For normal distributions with equivalent variance (Levene’s test, p > 0.05), comparison of two-sample groups was conducted using a Student’s t-test. For non-normal or non-homoscedastic samples, we performed a Mann-Whitney U-test for comparison of two groups, or a Kruskal-Wallis test, followed by Dunn’s post-hoc test for comparison of multiple groups. To detect correlation in protein expression with cell area, a Spearman’s rank correlation was performed. The correlation of protein expression with cell area was expected to be monotonic. Correlations with p < 0.05 were considered significant.
Results and discussion
In situ IEF performs rapid protein analysis of single and clustered cells
We designed an assay that measures protein signalling from cultured, adherent cells. The in situ IEF assay couples a substrate gel with cells cultured on an extracellular matrix (ECM) pattern and a microfluidic, lid-gel delivery system (Fig. 1A). The assay is designed for (i) control of cell patterning and attachment to a defined ECM environment, (ii) rapid lysis of attached cells and (iii) separation of proteins via IEF. IEF retains high protein concentrations due to electrophoretic restoring forces that limit diffusion to two dimensions.33,36 Microfluidic integration is key to minimising the time for diffusion-driven dilution and losses of the lysate from each cell.
The assay consists of two gels that are interfaced together (Fig. 1A). The substrate gel, which also comprises the analytical unit, houses cells that are cultured on pre-determined, fibronectin-patterned regions (Fig. 1A, step 0). For the lid gel, we employ our previously reported single-cell IEF technology (Fig. 1A, step 1).36 The lid gel consists of three regions—a central focusing region that contains denaturing lysis reagents (Triton-X 100, thiourea, CHAPS, and urea) and carrier ampholytes, flanked by acidic and basic regions, which establish the pH boundary conditions (Fig. 1A). The lid gel is denser than the substrate gel to reduce diffusive losses of cell lysate proteins into the lid. To maximise separation resolution, we chose a shallow pH gradient of 4–7.
IEF is conducted in situ, i.e., at the site of the cells, by contacting the lid gel to the cell-patterned substrate gel (Fig. 1A, step 1) for 30 s. During this time, the carrier ampholytes and lysis reagents diffuse into the substrate gel layer. Upon application of an electric potential (600 V), the pH gradient is established, and the proteins migrate along the pH gradient until neutrally charged (at the pI, Fig. 1A, step 2). As previously utilised carrier ampholytes36 have been discontinued, we prepared and characterised the formation of the pH gradient over time (Fig. 1B). The pH gradient was linear and stable for at least 20 min; thus, we achieved similar pH gradient characteristics. After focusing for 6 min, the electric field is removed and the focused proteins are covalently immobilised to the gel via UV excitation of a benzophenone moiety41 in the gel (Fig. 1A, step 3). After washout, the gels are immunoprobed to measure proteins of interest (Fig. 1A, step 4 and C).
The substrate gel, onto which cells are adhered and cultured, is fabricated by patterning a 400 μm-wide line of fibronectin along the centre of the substrate gel, as described above (Fig. S3†). Cells are seeded onto the patterns, and non-adhered cells, i.e., in the unpatterned regions, are washed away. We utilised 400 μm-wide ECM features for facile fabrication and increased cell occupancy; however, varying geometries and size of patterns can be achieved by tuning photomask design. For example, we patterned a 200 μm-wide line, as well as circular features as small as 30 μm, approximately the diameter of a suspended cell (Fig. 2A). The 30 μm fibronectin pattern had a SNR of 55, which implies that patterning of even smaller features is possible (Fig. 2A). Indeed, various groups have previously demonstrated features smaller than 200 nm using a similar nanocontact printing approach.42–45
Fig. 2.
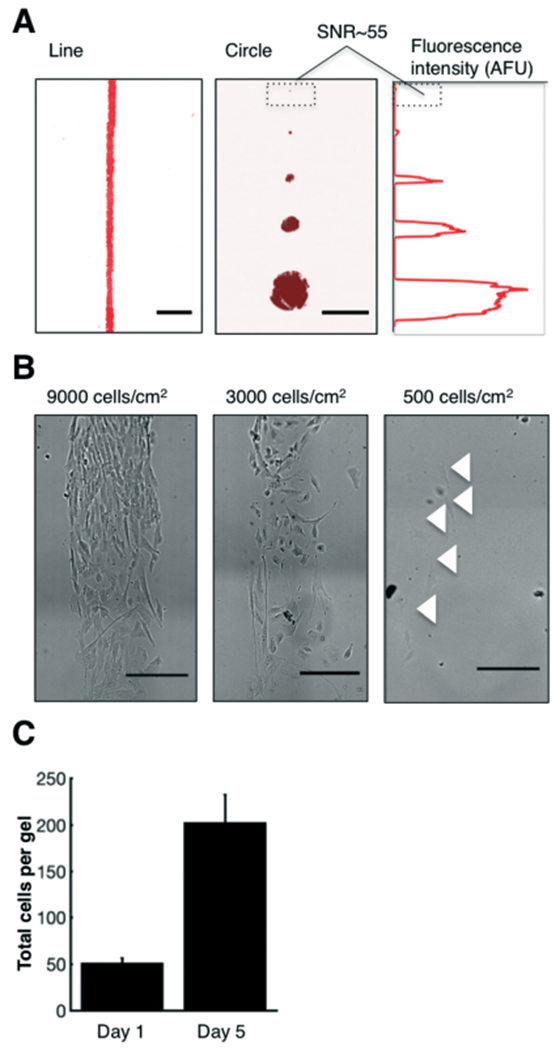
The substrate gel contains a tuneable, ECM-patterned region onto which cells adhere. (A) False-colour inverted micrographs of gels patterned with varying geometries and pattern sizes of rhodamine-fibronectin. The SNR of the smallest circular feature (d = 30 μm) was 55. (B) Brightfield images of cells seeded at varying densities. The density of cells controls the cell-to-cell spacing. (C) Cells cultured at a starting concentration of 500 cells per cm2 on the fibronectin-patterned gels proliferate over a period of four days, quadrupling in cell number. Error bars represent the standard deviation. Scalebars, 500 μm. SNR, signal-to-noise ratio.
In situ IEF analysis of intracellular proteins of single and clustered cells
We first sought to tune cell-to-cell spacing and verify cell proliferation on our patterned gels. We seeded human BJ fibroblasts at varying cell densities (500, 3000, or 9000 cells per cm2) to control the spacing between the cells (Fig. 2B). Notably, even for the highest seeding density, the cells remained constrained to the pattern. We also verified cell culture on fibronectin-patterned substrate gels by seeding BJ fibroblasts at a low density (500 cells per cm2) and monitoring cell growth over four days. The total number of cells per device quadrupled over this time period (n = 3 devices, Fig. 2C), a growth rate that is similar to those previously reported for fibroblasts seeded on fibronectin substrates.46
Next, we characterised separation performance of in situ IEF in comparison to the gold-standard technique of capillary IEF. First, we assessed whether higher cell densities would negatively impact separation performance. We patterned cells at two densities (500 and 9000 cells per cm2) and cultured the cells for 18 h prior to analysis. In addition, we also assessed the effects of culture duration on assay performance by seeding cells at a density of 500 cells per cm2 and culturing for four days on the fibronectin pattern (final density ~2000 cells per cm2, Fig. S4†). We then performed in situ IEF, followed by photocapture and immunoprobing for lamin A/C, a nuclear protein (Fig. 3A). We quantified separation performance by measuring peak width (4σ, where σ is the standard deviation of the Gaussian fit to the fluorescence intensity profile) and ΔpI, the minimum resolvable pI difference.
Fig. 3.
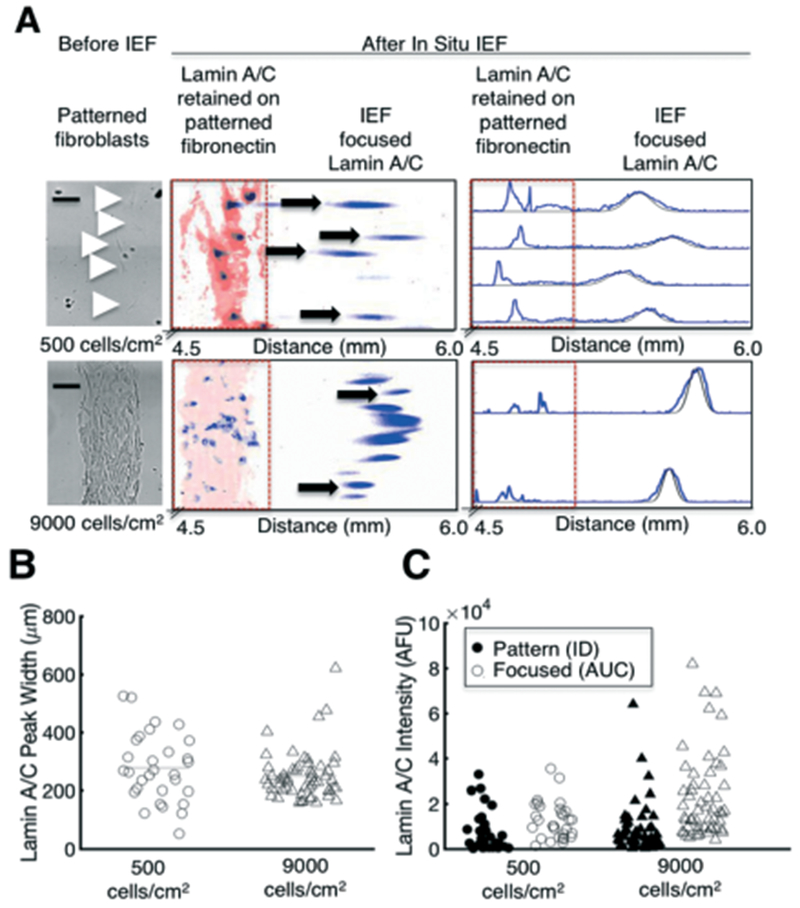
In situ IEF separates and detects proteins from single and clustered cells. (A) Brightfield images of single cells and a ~80% confluent pattern of cells prior to in situ IEF (left), and false-colour inverted fluorescence micrographs of lamin A/C that is retained on the fibronectin pattern and focused into a Gaussian peak (middle). The protein peaks are segmented and fluorescence intensity profiles are generated for each peak (right). Black line indicates Gaussian fit. (B) Separation performance, as measured by lamin A/C peak width, does not significantly change with higher seeding density, indicating that protein is not overloaded. (C) Protein mass measurements of lamin A/C that is retained on the fibronectin pattern (ID) and focused (AUC). Bars represent the mean. Scalebars, 200 μm. AFU, arbitrary fluorescence units. ID, integrated density. AUC, area under curve. pI, isoelectric point.
Increasing the cell density from single cells (500 cells per cm2) to an ~80% confluent “line” (9000 cells per cm2) did not increase the median peak width (Fig. 3B, Mann-Whitney, p > 0.05, n500 = 30 cells, n9000 = 54 peaks). The equivalent peak widths suggest that even for a highly confluent “line” of cells, protein was not overloaded, which would saturate buffering capacity of the carrier ampholytes, and thus increase the peak width.47,48 We further estimate that the total local protein concentration of the highest density condition (9000 cells per cm2) is ~1.6 × 10−5 mg per cm, which is 5 orders of magnitude lower than the predicted buffering capacity of 1–10 mg per cm;49 thus, our system can assay millions of cells without reducing separation performance. Moreover, we calculated a ΔpI value of 0.11 pH units (ΔpI = the pH gradient slope × peak width), which is similar to the separation performance of capillary IEF35 and on the order of a single phosphorylation event.50
In addition to characterising separation performance as a function of cell density, we also measured the pI and protein mass (AUC) of each lamin A/C peak (Fig. 3C and S5†). We only analysed peaks that were minimally resolved laterally, (i.e., perpendicular to the IEF axis), to ensure that each peak had minimal (<4%) crosstalk with other peaks. For the 500 cells per cm2) density, ~70% of lamin A/C peaks were minimally resolved and could thus be indexed back to a single, adherent cell (Fig. 3A and S6†). Importantly, for the high (9000 cells per cm2) density condition, only ~56% of peaks were resolvable laterally and passed our minimum Gaussian fit threshold (R2 > 0.7). Thus, peaks that contained the highest protein content were excluded from analysis. The lamin A/C protein mass of the excluded peaks can be found in Fig. S7.†
As expected, the highest cell density (9000 cells per cm2) yielded the highest quantity of focused protein, as each peak corresponds to the greatest number of cells (Fig. 3C, Levene’s test, p = 1.2 × 10−7, Kruskal-Wallis and Dunn’s test, p = 1.4 × 10−7). The measured lamin A/C content was independent of starting cell-cluster geometry (Fig. S8†). Interestingly, the focused protein mass was lowest for the medium (2000 cells per cm2) density, the only cells that were cultured for multiple (four) days prior to analysis (Fig. S4†). Furthermore, we observed that lamin A/C was partially retained on the fibronectin pattern (which did not electromigrate) after cell lysis, regardless of the cell density (Fig. 3A and S9†). We hypothesize that longer culture times of adherent cells result in a more difficult-to-lyse phenotype. Adhered fibroblasts produce and secrete ECM proteins, such as fibronectin, and become more strongly adhered to the substrate with time.51,52 These secreted ECM proteins limit biomolecular diffusion extracellularly,53 whereas the network of transmembrane proteins and the cytoskeleton act as a “fence” that limits diffusion of intracellular contents.54,55 Altogether, we hypothesize that adhered cells possess a tethered network of ECM–transmembrane–cytoskeletal proteins that act as a protein “barrier” that diminishes cell lysis and protein solubilisation. We present improvements to protein solubilisation below.
Since the protein retained on the fibronectin pattern was not electrophoretically separated and thus not Gaussian, we chose to analyse the protein content similarly to a reverse phase protein assay (RPPA).56 We thus measured the ID of the immunoprobed signal retained in the fibronectin-patterned area (Fig. 3C). The ratio of focused protein (AUC) to total protein (AUC + ID) was similar for all densities (n500 = 30 cells; n2000 = 40 peaks; n9000 = 54 peaks, non-normal distribution from QQ plot, not shown, Kruskal-Wallis p > 0.05), further suggesting that IEF performance in our system is independent of the starting density of cells.
Cell morphology and protein expression
Using in situ IEF, we next correlated individual protein peak measurements back to single-cell morphometric information. We hypothesized that protein expression would increase linearly with cell area, as previous reports have suggested.57–59 Most of these studies, however, were conducted in bulk populations of yeast cells or for multiple cell lines, in which the cell areas were averaged.60 Similar studies using transcriptomics have also linked RNA content with cell size.61
We first seeded BJ fibroblasts at single-cell density (500 cells per cm2) overnight. We then imaged BJ fibroblasts under brightfield microscopy, then performed the in situ IEF and probed for lamin A/C and β-tubulin (Fig. 4A). We quantified the total lamin A/C and β-tubulin content by summing the protein focused into a peak (AUC) and the protein retained on the pattern (ID). Interestingly, our measured lamin A/C and β-tubulin protein mass did not correlate well with cell area (Fig. 4B, Spearman’s correlation test, p = 0.05 for lamin A/C and p = 0.21 for β-tubulin, n = 16 cells). Since image segmentation and accurate identification of cell borders is challenging using brightfield microscopy, we further assessed cell area using fluorescently-labelled cells, which also yielded poor correlation between cell area and lamin A/C protein mass (Fig. S10,† Spearman’s test, ρ = 0.33, p = 0.08, n =28 cells).
Fig. 4.
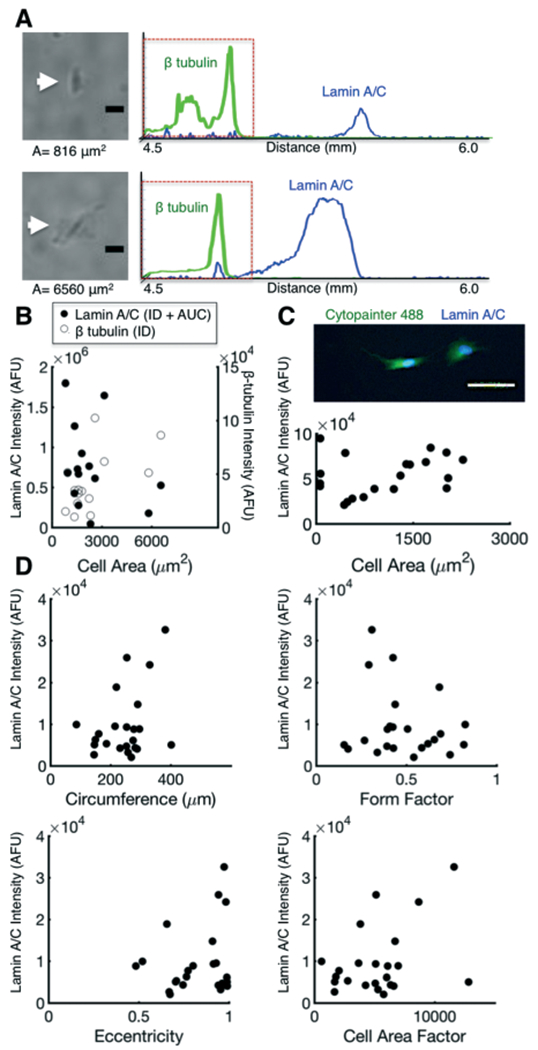
Protein expression does not correlate with cell morphology. (A) Brightfield micrographs of cells of varying area and corresponding fluorescence intensity profiles of lamin A/C and β-tubulin. (B) Comparison of lamin A/C and β-tubulin protein content (AUC and ID), which correlate poorly with cell area, as measured via brightfield. (C) ICC fluorescence micrograph of fluorescently-labelled (Cytopainter) cells seeded on patterned gels. Lamin A/C protein expression does not correlate with cell area. (D) Scatter plots of lamin A/C protein mass as a function of cell circumference, form factor, eccentricity, and cell area factor (cell area/form factor). Lamin A/C protein mass did not correlate with these morphometric parameters. Scalebars, 100 μm. ICC, immunocytochemistry. AFU, arbitrary fluorescence units. ID, integrated density. AUC, area under curve.
To validate protein mass and cell area measurements, we performed conventional ICC of fluorescently labelled BJ fibroblasts seeded on the fibronectin-patterned gels (Fig. 4C and S10†). The calculated lamin A/C and β-tubulin protein mass (ID) demonstrated similar variability (CVlaminAC = 41%, CVβtubulin = 60%, n = 20 cells) as in situ IEF (CVlaminAC = 60%, CVβtubulin = 67%, n = 16 cells). Moreover, we again observed poor correlation between cell area and lamin A/C protein mass (Fig. 4C, Spearman’s correlation test, p = 0.22, n = 20 cells). Interestingly, we observed a positive correlation between lamin A/C expression and cell area when β-tubulin was used for segmentation (Fig. S10,† Spearman’s test, ρ = 0.36, p = 0.0027, n = 68 cells). The correlation of lamin A/C to β-tubulin-segmented area, but not to cell area itself, is possibly attributable to the association of lamin proteins with the cellular cytoskeletal network, which in turn regulates cytoskeletal reorganisation.62
We next hypothesized that cell area could be an indicator of cell division stage, and that normalising the protein expression values to the pre-measured cell area would thus account for different cell cycle stages and decrease the variation. We chose to examine endogenous proteins, without the use of chemicals or serum starvation, which may alter cell biology.63 Both proteins demonstrated similar levels of variability (CVlaminAC = 60%, CVβtubulin = 67%, n = 16 cells). After normalisation to cell area, lamin A/C yielded a CV = 56%; in comparison, the CV for most proteins in a human cell, when normalised in silico to cell cycle, is 12–28%.64 Since the variation of our area-normalised protein expression is not representative of the CV of protein normalised to cell cycle, we conclude that cell area alone is not a proxy for cell division. The higher variance of protein expression in our system is possibly attributed to the protein types we assayed. For example, proteins involved in ECM interactions exhibit higher cell-to-cell variability.65 Lamin A/C and β-tubulin are both mechanosensitive;66,67 thus, some of the variation may arise from the cells’ differential interaction with the substrate.
In addition to scrutinising the relationship between the area of each cell and protein mass of lamin A/C expressed in each same cell, we assessed the relationship between lamin A/C expression and a set of morphometric parameters, including: cell circumference (perimeter), form factor (circularity), eccentricity, and cell area factor (cell area normalised to form factor). We observed no appreciable dependence of lamin A/C protein mass (AUC + ID) on any of the morphometric parameters investigated (Fig. 4D). Interestingly, a predictive model of Src and MAPK1/2 expression using cell membrane eccentricity shows a dependence on Src and MAPK1/2 expression on eccentricity.68 However, these approaches were modelled in 3-D and excluded consideration of cytoskeletal-mediated membrane signalling events. Thus, integration with flow cytometry or 3-D imaging modalities may yield complementary data to further elucidate how 3-D morphology influences or is predictive of intracellular protein expression.
In situ IEF detects distinct, adherent-cell biomarkers
Since signalling can be quickly disrupted by detachment of cells (minute timescales),11,12,26 we compared the protein expression of single BJ fibroblasts that were detached and seeded into individual microwells (single-cell IEF)36 with that of BJ fibroblasts patterned at single-cell density on a substrate gel and cultured overnight (in situ IEF). In both cases, we used the same lid-based IEF system. We compared expression of three markers: β-integrin, mono-phosphorylated myosin light chain (pMLCS19), and CD44 (Fig. 5A). β-Integrin mediates cell attachment to fibronectin, pMLCS19 is involved in the adhesion-dependent Rho GTPase pathway and is diminished upon detachment,69 and CD44 is a cell surface receptor protein subject to proteolytic cleavage.11 The BJ fibroblasts were transduced with GFP to serve as a loading control.
Fig. 5.
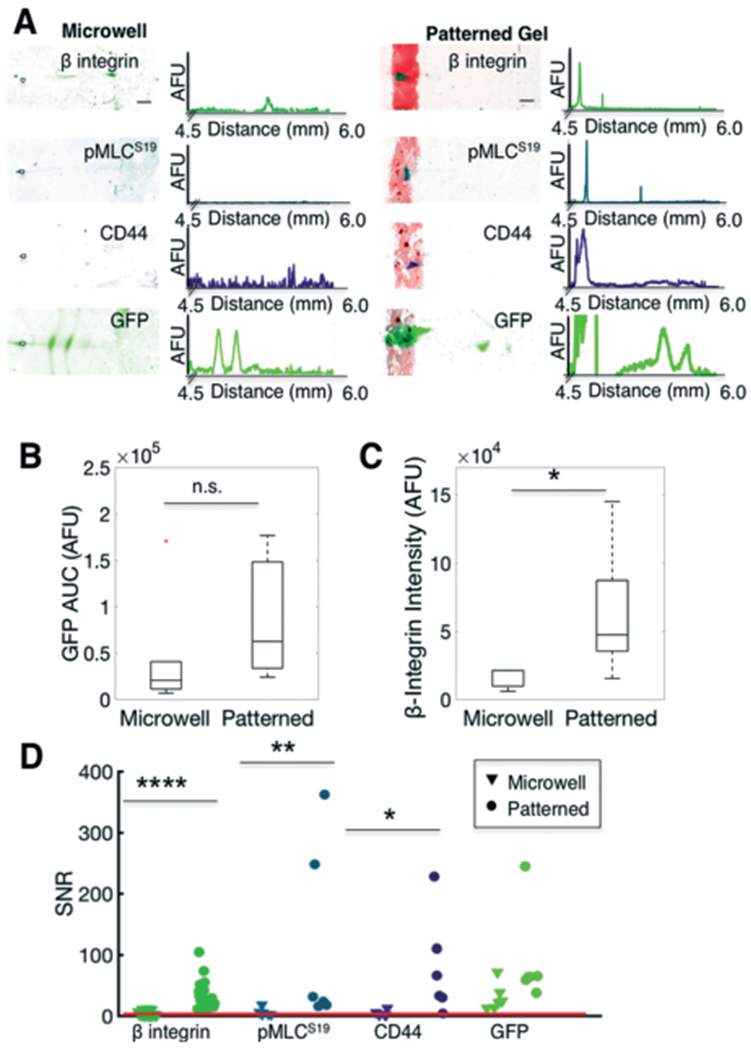
In situ IEF of adherent cells assays protein profiles that differ from trypsin-treated (detached) cells. (A) False-colour inverted fluorescence micrographs and intensity profiles of IEF of single cells detached using trypsin and seated into microwells (left) or analysed via in situ IEF of fibronectin-patterned (attached) cells (right). (B) Box plot of GFP control protein content (AUC) of focused peaks indicates comparable expression between microwells and fibronectin-patterned cells. Line in the box represents the median and box ends represent the 25th and 75th percentiles. Red dots indicate outliers. (C) Box plot of β-integrin indicates higher expression in patterned cells. (D) Comparison of measured SNR of various targets. Compared to microwell IEF, in situ (adhered cells on fibronectin pattern) IEF results in higher SNR of β-integrin, pMLCS19, and CD44, targets that are adversely affected by trypsin treatment. The red line indicates SNR threshold of 3. Scalebars, 200 μm. AUC, area under curve. SNR, signal-to-noise ratio. AFU, arbitrary fluorescence units. * p < 0.05, ** p < 0.01, **** p < 0.0001.
Characterisation of focused GFP protein content indicated no significant difference in the median measured amount of protein in the microwell (detached) cells, as compared to the fibronectin-patterned (attached) cells (Fig. 5B, Mann-Whitney p = 0.16, n = 6 cells in microwells, n = 3 cells on fibronectin-patterned substrate gels), indicating that detection sensitivity of both systems are comparable. For β-integrin, pMLCS19, and CD44, all of the detectable protein (SNR > 3) remained on the fibronectin pattern (Fig. 5A). We hypothesize that the amount of protein that is retained on the fibronectin pattern may be a proxy for cell attachment, i.e., proteins that tether to the cytoskeleton and focal adhesion complexes are more highly retained on the fibronectin pattern. Accordingly, β-tubulin and pMLCS19, cytoskeletal proteins, remained on the fibronectin pattern, as did transmembrane proteins CD44 and β-integrin (Fig. 4A and 5A). In contrast, lamin A/C and GFP, a nuclear membrane and cytosolic protein, respectively, focused into protein peaks (Fig. 4A and 5A).
To measure the differences in protein expression of attached versus detached cells, we measured AUC of focused peaks and ID of protein retained on the fibronectin pattern. For β-integrin, we observed a significant difference in the protein expression of the focused peaks (AUC) from the microwells as compared to the protein retained on the fibronectin pattern (ID, Fig. 5C, Mann-Whitney, p = 0.01, nmicrowell = 3 cells and npatterned = 29 cells). We attribute the lower expression and detection of β-integrin to trypsin cleavage of the receptor protein.70 Similarly, for both pMLCS19 and CD44, no protein peaks were detectable (SNR < 3, or Gaussian fit R2 < 0.7) in the microwells, but detectable signal was measured for the attached cells (Fig. 5D, nmicrowell = 6 cells and npatterned = 6 cells). These results corroborate previous research demonstrating that proteolytic cleavage and/or cell detachment alters detection of surface proteins and downstream signalling.11,69 Altogether, our results implicate that minimised handling, obviation of enzymatic treatments, and direct in situ analysis of cell lysates analyses proteins that are otherwise undetectable or disrupted by the sample preparation of conventional single-cell studies, such as flow cytometry. Using our in situ IEF assay, we detect protein expression from substrate-adhered cells and thus assay different protein signatures compared to detached cells. This unique capability is important in assaying rapidly changing events (e.g., phosphorylations) or various adhesion-mediated signalling pathways. Overall, our results highlight the critical importance of sample preparation parameters for single-cell analysis.
Tuning assay parameters for improved separation performance and protein solubilisation
Given the notable intracellular protein adhesion to the fibronectin pattern (31 ± 20% lamin A/C retained on fibronectin pattern, n = 30 cells) that was observed under the single-cell condition, and variation of the peak width (CVlaminAC = 39.8%, n = 30 cells), we sought to improve protein solubilisation and separation performance by systematically increasing the lysis or electrofocusing (EF) duration (Fig. 6). We hypothesized that the high variation in peak width (Fig. 3B) arose from proteins continuing to focus, i.e., equilibrium had not been reached within the 6 min. Increasing the assay duration presents trade-offs, as longer durations may improve separation performance but concomitantly increase diffusive losses of the cell lysate.41,71 We thus increased the EF duration modestly, from 6 min to 8 min, and immunoprobed for lamin A/C and β-tubulin (Fig. 6A and S11†).
Fig. 6.
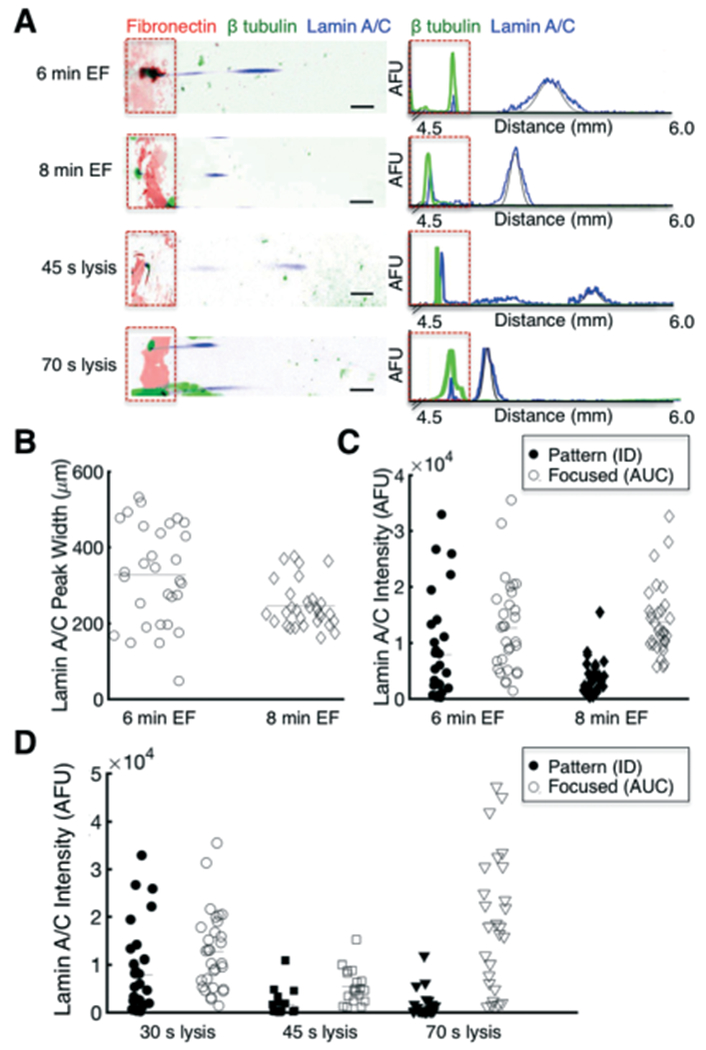
Separation performance, as measured by lamin A/C peak width, and protein solubilisation are enhanced by tuning the EF and lysis durations, respectively. (A) False-colour inverted fluorescence micrographs and intensity profiles of single cells lysed for 30 s and electrofocused for 6 min, compared to either a longer EF duration of 8 min, or longer lysis durations of 45 s or 70 s. Contrast adjusted for improved visualisation. Black line indicates Gaussian fit. (B) Analysis of peak width for longer EF duration indicates improved separation performance (lower peak width). Bars represent the mean. (C) Comparison of lamin A/C protein mass retained on the fibronectin pattern or focused to lamin A/C pI as a function of EF duration. (D) Comparison of lamin A/C protein mass retained on the fibronectin pattern or focused to lamin A/C pI as a function of lysis duration. The longest lysis duration (70 s) improved lamin A/C solubilisation, as evidenced by higher focused protein content. Data from 30 s lysis duration are the same as the 6 min EF duration. Scalebars, 200 μm. AFU, arbitrary fluorescence units. ID, integrated density. AUC, area under curve. EF, electrofocusing.
The longer EF duration improved separation performance without decreasing lateral resolution (Fig. 6B and S12†). The average peak width of lamin A/C decreased by 25.1%, from 327 ± 130 μm (mean ± s.d., n6min = 30 cells) to 245 ± 60 μm (n8min = 31 cells), with lower variation (Fig. 6B, CV6min = 39.8%, CV8min = 24.3%, Levene’s test, p = 4.2 × 10−5). These results imply that equilibrium is not yet reached for in situ IEF within 6 min, and that longer EF durations enhance separation performance.
Next, to assess how EF duration influences solubilisation, we assessed the protein content both remaining on the fibronectin pattern (ID) and focused into a peak (AUC) for the longer EF durations (Fig. 6C). The longer EF duration increased the detected amount of β-tubulin, likely due to increased denaturation and thus photocapture efficiency (Fig. S11†).72 For lamin A/C, the median protein retained on the fibronectin pattern (ID) did not significantly change for a longer EF duration (Mann-Whitney, p > 0.05); however, the distribution was narrower (σ6min2 = 9090, n6min = 30 cells; σ8min2 = 3115, n8min = 32 cells, Levene’s test, p = 2.9 × 10−5), suggesting that more protein was solubilised (and migrated to the pI) with the longer EF duration. However, for the focused protein peak, there was no significant difference in the mean protein content, (normal distribution from QQ plot, not shown, Levene’s test, p > 0.05, two sample t-test, p = 0.69). Although longer focusing duration may have increased protein solubilisation, the longer assay duration concomitantly increased diffusive losses of the lamin A/C. Thus, EF duration improves separation performance, but with the trade-off of higher diffusive losses.
We next analysed whether longer lysis durations could improve protein solubilisation (Fig. 6D). We assayed modest increases in lysis duration, from 30 s to 45 s or 70 s, and maintained a 6 min focusing duration. For β-tubulin, we observed no difference in the protein content from the longer lysis (Figure S11†). For lamin A/C, we observed a narrower distribution of protein content retained on the fibronectin pattern for longer lysis durations (Fig. 6D, σ30s2 = 9090, n30s = 30; σ45s2 = 2673, n45s = 19, σ70s2 = 2629, n70s = 26 cells, Levene’s test, p = 8.5 × 10−7). Longer lysis durations also decreased the mean amount of protein retained on the fibronectin pattern (non-normal distribution from QQ plot, not shown, Kruskal-Wallis and Dunn’s test, p = 2.5 × 10−5), suggesting increased protein solubilisation of lamin A/C with lysis duration. Interestingly, for the focused lamin A/C peak, we noticed a biphasic trend (Levene’s test, p = 7.9 × 10−6; Kruskal-Wallis and Dunn’s test, p = 4.2 × 10−6). It is possible that the intermediate lysis duration (45 s) was insufficient for protein solubilisation, and thus the increased duration only increased diffusive losses of the cell lysate, whereas the longest lysis duration (70 s) increased solubilisation sufficiently, thus resulting in higher protein content in the focused peak. Altogether, these results suggest that optimising EF and lysis duration can significantly improve separation performance and protein solubilisation. Further improvements of solubilisation may be achievable via chemical modulation, as has previously been reported in conventional preparatory IEF,73,74 or perhaps by modulating the substrate to allow for laser-based lift off of adherent cells75 prior to lysis and IEF.
Conclusion
The cell microenvironment plays an important role in modulating cell behaviour. Here, we report on a microfluidic device that is designed for direct lysis of cells seeded on a tuneable microenvironment and subsequent analysis of the protein contents in situ, thereby interrogating cell-matrix mediated signalling. We utilise a tuneable cell-patterning technique and demonstrate the separation, detection, and quantitation of protein targets of single and clustered cells. We also assess separation performance of the assay in terms of peak width, measured pI, and ΔpI. Furthermore, we correlate morphometric information with protein expression levels of two commonly used housekeeping genes, demonstrating high variation that is independent of cell area. Finally, we analyse protein expression of cells that are attached to a fibronectin-patterned substrate and compare to those of detached cells, and demonstrate detection of targets that are otherwise masked or cleaved in conventional single-cell analysis approaches.
In future device work, we aim to expand the applications of the assay. Currently, the cell type determines the seeding specificity; seeding U251 cells, a glioblastoma cell line, did not result in specific cell adhesion as was observed for the BJ fibroblasts (results not shown). Since different cell types vary in their adhesive strength and response to extracellular matrix environment,18 special attention to lysis conditions will be necessary to ensure sufficient solubilisation of the intracellular proteins. Moreover, for higher densities of cells, we cannot decouple protein signal from individual cells as of now; nonetheless, we can envision approaches using fluorescently tagged proteins, such as in co-culture systems, to determine the origin of each protein to a specified cell type.
While select microenvironmental cues are discussed herein, the value of the direct lysis IEF technology extends beyond studying individual cell-cell or cell-matrix interactions. Indeed, the platform could be combined with combinatorial soluble factor gradients and ECM microarrays/niches, such as those previously described elsewhere,76–78 to address the role of complex environments on stem cell behaviour or cancer metastasis. Since the analytical module (substrate gel) is grafted onto a conventional microscope slide, our device is particularly amenable to microscopy, which opens possibilities to further corroborate morphometric and spatial information with signalling and also enabling longitudinal measurements. Another important capability of this platform is to study cell dynamics. Minimal handling requirements and direct study of adherent cells lends a unique opportunity to explore rapidly changing events, such as protein phosphorylation, or other applications wherein detachment of the cells would drastically alter signalling. Moreover, IEF is uniquely suited to study protein-signalling events such as phosphorylations because of the ability to physically separate the phospho-forms. Ultimately, our findings move us one step closer to better recapitulating the in vivo environment and has applications to study stem cell and tissue engineering.
Supplementary Material
Acknowledgements
The authors thank members of the Herr lab for insightful technical discussions as well as the QB3 Nanobiotechnology Center at UC Berkeley, the Berkeley Stem Cell Center, and the CNR Biological Imaging Facility for providing additional facilities. This research was supported by the US National Institutes of Health and the National Cancer Institute (R21CA183679 and R01CA203018 to A. E. H.), and the National Institute of Biomedical Imaging and Bioengineering (R21EB019880 to A. E. H.), and NSF Graduate Research Fellowship Program (DGE 1106400 to E. J. S.).
Footnotes
Electronic supplementary information (ESI) available: Fig. S1 compares the effect of two lysis buffer compositions on IEF performance. Fig. S2 illustrates the workflow for image segmentation and generation of intensity profiles in MATLAB. Fig. S3 is a schematic of the fabrication of microcontact-printed substrate gels. Fig. S4 assesses the effect of culture duration on IEF performance. Fig. S5 is a scatter plot of the measured pI of lamin A/C. Fig. S6 discusses lateral resolution of lamin A/C peaks for single-cell (500 cells per cm2) density. Fig. S7 analyses lamin A/C protein content as a function of cell density. Fig. S8 compares the effect of cell-cluster geometry on detected lamin A/C protein mass. Fig. S9 analyses fibronectin fluorescence before and after in situ IEF. Fig. S10 demonstrates the poor correlation between cell area and lamin A/C protein mass in cells segmented via fluorescence of a cell tracker dye, which is not observed when segmenting via β-tubulin-expressing area. Fig. S11 compares measured protein content of β-tubulin under varying lysis and EF durations. Fig. S12 is a scatter plot of lateral peak width of lamin A/C as a function of longer EF durations. See DOI: 10.1039/c7lc01012e
Conflicts of interest
A E. H. has financial interest in intellectual property related to the device and assay described here and has financial interest in commercialisation.
Notes and references
- 1.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson Å, Sjöstedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Pontén F, von Feilitzen K, Lilley KS, Uhlén M and Lundberg E, Science, 2017, 356, eaal3321. [DOI] [PubMed] [Google Scholar]
- 2.Reddig PJ and Juliano RL, Cancer Metastasis Rev., 2005, 24, 425–439. [DOI] [PubMed] [Google Scholar]
- 3.Engler AJ, Sen S, Sweeney HL and Discher DE, Cell, 2006, 126, 677–689. [DOI] [PubMed] [Google Scholar]
- 4.Wang YL and Pelham RJ, Methods Enzymol., 1998, 298, 489–496. [DOI] [PubMed] [Google Scholar]
- 5.Acharya PS, Majumdar S, Jacob M, Hayden J, Mrass P, Weninger W, Assoian RK and Puré E, J. Cell Sci., 2008, 121, 1393–1402. [DOI] [PubMed] [Google Scholar]
- 6.McBeath R, Pirone DM, Nelson CM, Bhadriraju K and Chen CS, Dev. Cell, 2004, 6, 483–495. [DOI] [PubMed] [Google Scholar]
- 7.Khalili AA and Ahmad MR, Int. J. Mol. Sci, 2015, 16, 18149–18184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witz IP, Cancer Metastasis Rev., 2008, 27, 19–30. [DOI] [PubMed] [Google Scholar]
- 9.Paschos KA, Canovas D and Bird NC, Cell. Signalling, 2009, 21, 665–674. [DOI] [PubMed] [Google Scholar]
- 10.Desgrosellier JS and Cheresh DA, Nat. Rev. Cancer, 2010, 10, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biddle A, Gammon L, Fazil B and Mackenzie IC, PLoS One, 2013, 8, e57314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ponta H, Sherman L and Herrlich PA, Nat. Rev. Mol. Cell Biol., 2003, 4, 33–45. [DOI] [PubMed] [Google Scholar]
- 13.Lakshman M, Subramaniam V, Rubenthiran U and Jothy S, Exp. Mol. Pathol, 2004, 77, 18–25. [DOI] [PubMed] [Google Scholar]
- 14.Petri B, Phillipson M and Kubes P, J. Immunol, 2008, 180, 6439–6446. [DOI] [PubMed] [Google Scholar]
- 15.Imhof BA and Aurrand-Lions M, Nat. Rev. Immunol, 2004, 4, 432–444. [DOI] [PubMed] [Google Scholar]
- 16.Muller WA, Trends Immunol., 2003, 24, 327–334. [DOI] [PubMed] [Google Scholar]
- 17.Cosgrove BD, Mui KL, Driscoll TP, Caliari SR, Mehta KD, Assoian RK, Burdick JA and Mauck RL, Nat. Mater, 2016, 15, 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallant ND, Michael KE and García AJ, Mol. Biol. Cell, 2005, 16, 4329–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veiga SS, Chammas R, Cella N and Brentani RR, Int. J. Cancer, 1995, 61, 420–424. [DOI] [PubMed] [Google Scholar]
- 20.Maitra N, Flink IL, Bahl JJ and Morkin E, Cardiovasc. Res, 2000, 47, 715–725. [DOI] [PubMed] [Google Scholar]
- 21.Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong T-T, Shaw RM and Srivastava D, Dev. Cell, 2009, 16, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribeiro AJS, Ang Y-S, Fu J-D, Rivas RN, Mohamed TMA, Higgs GC, Srivastava D and Pruitt BL, Proc. Natl. Acad. Sci. U. S. A, 2015, 112, 12705–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Downing TL, Soto J, Morez C, Houssin T, Fritz A, Yuan F, Chu J, Patel S, Schaffer DV and Li S, Nat. Mater, 2013, 12, 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abrahamsen I and Lorens JB, BMC Cell Biol., 2013, 14, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavallo D, McLeod RS, Rudy D, Aiton A, Yao Z and Adeli K, J. Biol. Chem, 1998, 273, 33397–33405. [DOI] [PubMed] [Google Scholar]
- 26.Yamashiro S, Totsukawa G, Yamakita Y, Sasaki Y, Madaule P, Ishizaki T, Narumiya S and Matsumura F, Mol. Biol. Cell, 2003, 14, 1745–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu H, Stybayeva G, Macal M, Ramanculov E, George MD, Dandekar S and Revzin A, Lab Chip, 2008, 8,2197. [DOI] [PubMed] [Google Scholar]
- 28.Shen J, Zhou Y, Lu T, Peng J, Lin Z, Huang L, Pang Y, Yu L and Huang Y, Lab Chip, 2012, 12, 317–324. [DOI] [PubMed] [Google Scholar]
- 29.Ellington AA, Kullo IJ, Bailey KR and Klee GG, Clin. Chem, 2010, 56, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teves SS, An L, Hansen AS, Xie L, Darzacq X and Tjian R, eLife, 2016, 5, e22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Towbin H, Staehelin T and Gordon J, Proc. Natl. Acad. Sci. U. S. A, 1979, 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marc PJ, Christopher A, Sims E and Allbritton NL, Anal. Chem, 2007, 79, 9054–9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Righetti PG, Isoelectric Focusing: Theory, Methodology and Application: Theory, Methodology and Application, Elsevier, 2000, vol. 2. [Google Scholar]
- 34.Fan AC, Deb-Basu D, Orban MW, Gotlib JR, Natkunam Y, O’Neill R, Padua R-A, Xu L, Taketa D, Shirer AE, Beer S, Yee AX, Voehringer DW and Felsher DW, Nat. Med, 2009, 15, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Neill RA, Bhamidipati A, Bi X, Deb-Basu D, Cahill L, Ferrante J, Gentalen E, Glazer M, Gossett J, Hacker K, Kirby C, Knittle J, Loder R, Mastroieni C, Maclaren M, Mills T, Nguyen U, Parker N, Rice A, Roach D, Suich D, Voehringer D, Voss K, Yang J, Yang T and Vander Horn PB, Proc. Natl. Acad. Sci. U. S. A, 2006, 103, 16153–16158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tentori AM, Yamauchi KA and Herr AE, Angew. Chem., Int. Ed, 2016, 55, 12431–12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyata S, Koshikawa N, Yasumitsu H and Miyazaki K, J. Biol. Chem, 2000, 275, 4592–4598. [DOI] [PubMed] [Google Scholar]
- 38.Kumar A, Biebuyck HA and Whitesides GM, Langmuir, 1994, 10, 1498–1511. [Google Scholar]
- 39.Hughes AJ, Spelke DP, Xu Z, Kang C-C, Schaffer DV and Herr AE, Nat. Methods, 2014, 11, 749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang C-C, Lin J-MG, Xu Z, Kumar S and Herr AE, Anal. Chem, 2014, 86, 10429–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang C-C, Yamauchi KA, Vlassakis J, Sinkala E, Duncombe TA and Herr AE, Nat. Protoc, 2016, 11, 1508–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee MH, Lin JY and Odom TW, Angew. Chem., Int. Ed, 2010, 49, 3057–3060. [DOI] [PubMed] [Google Scholar]
- 43.MacNearney D, Mak B, Ongo G, Kennedy TE and Juncker D, Langmuir, 2016, 32, 13525–13533. [DOI] [PubMed] [Google Scholar]
- 44.Takulapalli BR, Morrison ME, Gu J and Zhang P, Nanotechnology, 2011, 22, 285302. [DOI] [PubMed] [Google Scholar]
- 45.Li H-W, Muir BVO, Fichet G and Huck WTS, Langmuir, 2003, 19, 1963–1965. [Google Scholar]
- 46.Sottile J, Hocking DC and Swiatek PJ, J. Cell Sci., 1998, 111, 2933–2943. [DOI] [PubMed] [Google Scholar]
- 47.O’Farrell PH, J. Biol. Chem, 1975, 250, 4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 48.Mack S, Cruzado-Park I, Chapman J, Ratnayake C and Vigh G, Electrophoresis, 2009, 30, 4049–4058. [DOI] [PubMed] [Google Scholar]
- 49.Chrambach A, Mol. Cell. Biochem, 1980, 29, 23–46. [DOI] [PubMed] [Google Scholar]
- 50.Zhu Kan, Zhao Jia, Lubman DM, Miller FR and Barder TJ, Anal. Chem, 2005, 77, 2745–2755. [DOI] [PubMed] [Google Scholar]
- 51.Gildner CD, Roy DC, Farrar CS and Hocking DC, Matrix Biol., 2014, 34, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu C-C, Su H-W, Lee C-C, Tang M-J and Su F-C, Biochem. Biophys. Res. Commun, 2005, 329, 256–265. [DOI] [PubMed] [Google Scholar]
- 53.Kihara T, Ito J and Miyake J, PLoS One, 2013, 8, e82382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ritchie K, Iino R, Fujiwara T, Murase K and Kusumi A, Mol. Membr. Biol, 2003, 20, 13–18. [DOI] [PubMed] [Google Scholar]
- 55.Sako Y and Kusumi A, J. Cell Biol., 1994, 125, 1251–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spurrier B, Ramalingam S and Nishizuka S, Nat. Protoc, 2008, 3, 1796–1808. [DOI] [PubMed] [Google Scholar]
- 57.Milo R, BioEssays, 2013, 35, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marguerat S and Bähler J, Trends Genet., 2012, 28, 560–565. [DOI] [PubMed] [Google Scholar]
- 59.Crissman HA and Steinkamp JA, J. Cell Biol., 1973, 59, 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lundberg E, Gry M, Oksvold P, Kononen J, Andersson-Svahn H, Pontén F, Uhlén M and Asplund A, J. Proteomics, 2008, 71, 448–460. [DOI] [PubMed] [Google Scholar]
- 61.Fomina-Yadlin D, Du Z and McGrew JT, J. Biotechnol, 2014, 189, 58–69. [DOI] [PubMed] [Google Scholar]
- 62.Houben F, Ramaekers FC, Snoeckx LHEH and Broers JLV, Biochim. Biophys. Acta, Mol. Cell Res., 2007, 1773, 675–686. [DOI] [PubMed] [Google Scholar]
- 63.Shedden K and Cooper S, Proc. Natl. Acad. Sci. U. S. A, 2002, 99, 4379–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sigal A, Milo R, Cohen A, Geva-Zatorsky N, Klein Y, Liron Y, Rosenfeld N, Danon T, Perzov N and Alon U, Nature, 2006, 444, 643–646. [DOI] [PubMed] [Google Scholar]
- 65.Alemu EY, Carl JW, Corrada Bravo H and Hannenhalli S, Nucleic Acids Res., 2014, 42, 3503–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osmanagic-Myers S, Dechat T and Foisner R, Genes Dev., 2015, 29, 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Méjat A and Misteli T, Nucleus, 2010, 1, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rangamani P, Lipshtat A, Azeloglu EU, Calizo RC, Hu M, Ghassemi S, Hone J, Scarlata S, Neves SR and Iyengar R, Cell, 2013, 154, 1356–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ren X-D, J. Cell Sci., 2004, 117, 3511–3518. [DOI] [PubMed] [Google Scholar]
- 70.Akiyama SK and Yamada KM, J. Biol. Chem, 1985, 260, 4492–4500. [PubMed] [Google Scholar]
- 71.Sinkala E, Sollier-Christen E, Renier C, Rosàs-Canyelles E, Che J, Heirich K, Duncombe TA, Vlassakis J, Yamauchi KA, Huang H, Jeffrey SS and Herr AE, Nat. Commun, 2017, 8, 14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hughes AJ, Lin RKC, Peehl DM and Herr AE, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 5972–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rabilloud T, Adessi C, Giraudel A and Lunardi J, Electrophoresis, 2007, 18, 307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Padula M, Berry I, O’Rourke M, Raymond B, Santos J and Djordjevic SP, Proteomes, 2017, 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shadpour H and Allbritton NL, ACS Appl. Mater. Interfaces, 2010, 2, 1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reticker-Flynn NE, Malta DFB, Winslow MM, Lamar JM, Xu MJ, Underhill GH, Hynes RO, Jacks TE and Bhatia SN, Nat. Commun, 2012, 3, 1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gobaa S, Hoehnel S, Roccio M, Negro A, Kobel S and Lutolf MP, Nat. Methods, 2011, 8, 949–955. [DOI] [PubMed] [Google Scholar]
- 78.Tatárová Z, Abbuehl JP, Maerkl S and Huelsken J, Lab Chip, 2016, 16, 1934–1945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.