

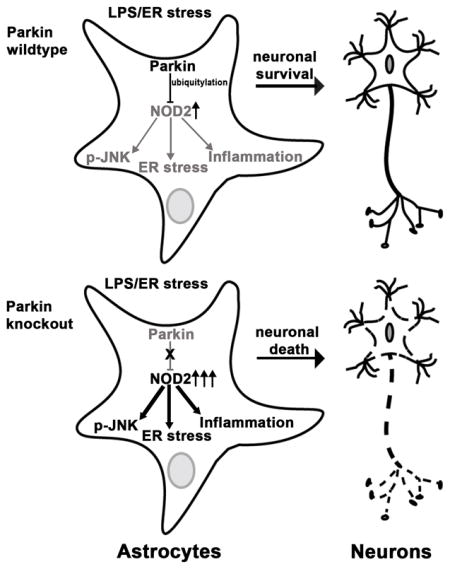
Abstract
Loss of substantia nigra dopaminergic neurons results in Parkinson disease (PD). Degenerative PD usually presents in the seventh decade whereas genetic disorders, including mutations in PARK2, predispose to early-onset PD. PARK2 encodes the parkin E3 ubiquitin ligase which confers pleotropic effects on mitochondrial and cellular fidelity and as a mediator of endoplasmic reticulum (ER) stress signaling. Although the majority of studies investigating ameliorative effects of parkin focus on dopaminergic neurons we found that astrocytes are enriched with parkin. Furthermore, astrocytes deficient in parkin display stress-induced elevation of nucleotide-oligomerization domain receptor 2 (NOD2), a cytosolic receptor integrating ER stress and inflammation. Given the neurotropic and immunomodulatory role of astrocytes we reasoned that parkin may regulate astrocyte ER stress and inflammation to control neuronal homeostasis. We show that, in response to ER stress, parkin knockdown astrocytes exhibit exaggerated ER stress, JNK activation and cytokine release, and reduced neurotropic factor expression. In coculture studied we demonstrate that dopaminergic SHSY5Y cells and primary neurons with the presence of parkin depleted astrocytes are more susceptible to ER stress and inflammation-induced apoptosis than wildtype astrocytes. Parkin interacted with, ubiquitylated and diminished NOD2 levels. Additionally, the genetic induction of parkin ameliorated inflammation in NOD2 expressing cells and knockdown of NOD2 in astrocytes suppressed inflammatory defects in parkin deficient astrocytes and concurrently blunted neuronal apoptosis. Collectively these data identify a role for parkin in modulating NOD2 as a regulatory node in astrocytic control of neuronal homeostasis.
Keywords: Parkin, Astocytes, Endoplasmic reticulum stress, Inflammation, NOD2
Graphical Abstract
1 INTRODUCTION
The E3-ubiquitin ligase parkin has been a prime target in investigating the pathophysiology of Parkinson Disease (PD), given that mutations in PARK2 are the most common genetic defects associated with Early Onset PD (EOPD) (Kitada et al. 1998). At the molecular level, the major focus on the role of parkin has focused on its role in regulating mitochondrial homeostatic programs including mitochondrial autophagy (Youle and Narendra 2011) and dynamics (Tanaka et al. 2010). Less well characterized functions of parkin include roles in diverse cellular functions spanning from the regulation of gene transcription, protein stability, redox stress control, and the regulation of endoplasmic reticulum (ER) stress (Han et al. 2017; Imai et al. 2000; Johnson et al. 2012; Muller-Rischart et al. 2013; Shin et al. 2011).
The genetic disruption of parkin in mice have resulted in a broad spectrum of phenotypes spanning from no apparent detrimental effects to robust increased susceptibility to PD, dependent on effects of aging, or on concurrent stressors including mitochondrial genomic perturbations and inflammation (Frank-Cannon et al. 2008; Palacino et al. 2004; Perez et al. 2005; Pickrell et al. 2015; Thomas et al. 2007). As the loss of substantia nigra dopaminergic neurons is pathognomonic of PD, the majority of studies into this pathophysiology has been directed at the understanding of the role of Parkin in neurons. However, given that lipopolysaccharide (LPS) administration enhances substantia nigra neuronal loss in parkin knockout (KO) mice (Frank-Cannon et al. 2008) and neuroinflammation is increasingly recognized as a contributory factor in PD neurodegeneration (Hirsch et al. 2012; Tansey and Goldberg 2010), a question arising is whether parkin has a role as a glial ‘neurotropic’ mediator to confer protection against neuronal cell degeneration.
The predominant glial cells within the substantia nigra pars compacta include microglia and astrocytes. Microglia are the resident ‘macrophages’ in the brain, possess neuronal repair and maintenance functions, and play a role in the innate immune response (Kofler and Wiley 2011; Ransohoff 2016). Astrocytes are critical supporters of neuronal integrity and homeostasis and function to control processes including antioxidant protection, glutamate clearance and signaling release through gliotransmitters, cytokines and metabolic enzymes (Hamby and Sofroniew 2010; Mamczur et al. 2015; Volterra and Meldolesi 2005).
Concurrently, mechanistic pathways are being identified that link ER stress to sterile inflammation (Garg et al. 2012; Keestra-Gounder et al. 2016). Given that LPS augments neuronal injury in Parkin KO mice (Frank-Cannon et al. 2008) and that parkin expression is higher in astrocytes as compared to microglia, we have begun to explore whether astrocytic ER stress and inflammation work in concert, in a parkin-dependent manner to modulate neuronal homeostasis. In this study we find that parkin plays a critical role in the maintenance of astrocyte neurotropic function in response to ER stress and inflammation. This was shown in ER stressed primary coculture studies where the restricted genetic disruption of parkin in astrocytes resulted in greater apoptotic cell death in neurons compared to their coculture with wildtype (WT) astrocytes. Consistent with this phenotype, parkin KO astrocytes exhibited exaggerated ER stress defects and had decreased expression of neurotropic factors compared with the WT astrocytes. Additionally, we identify NOD2 as a parkin substrate and show that parkin mediates NOD2 ubiqutylation and degradation. This proteasomal degradation of NOD2 is critical for the maintainance of normal astrocyte neurotropic function. Collectively these data identify a role for parkin in modulating NOD2 as a regulatory node in astrocytic control of neuronal homeostasis and support an emerging concept that the neuroprotective effects of parkin may be mediated, in part, through paracrine effects of parkin in glial cells.
2 MATERIALS AND METHODS
2.1 Animals
Parkin KO (B6.129S4-Park2-tm1shn/J) and WT (c57bl/6) mice were obtained from the Jackson Laboratory and backcrossed (>10 generations) into the C57BL/6J strain. All animal procedures were conducted using an animal protocol approved by the National Heart Lung, and Blood Institute (NHLBI) Animal Care and Use Committee.
2.2 Cell cultures and ELISA
HEK293T, HeLa and Neu7 astrocyte (Fidler et al. 1999) cell lines were used in parkin over-expression studies. Coimmunoprecipitation was performed in HEK293T cell extracts. Primary astrocyte cultures were prepared from postnatal littermates (age range: day 2 (P2) – day 4 (P4)) as described previously (Yu et al. 2012). Briefly, cortical tissues were dissected from the brain and meninges were removed. Tissues were mechanically dissociated with Trypsin and plated on T75 flasks (cortical tissues of two littermates/flask). The cells were cultured for 14–16 days in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and antibiotic/antimycotic solution (Gibco), in a 37°C, 5% CO2 incubator. Upon confluency, the plates were shaken on a rotor for 2 hrs to remove microglia. Astrocytes were dissociated with TrypLE (Thermofisher) and transferred to 6 or 24 well plates. Primary cortical neurons from the brain of wild type embryonic (E18.5) mice were cultured as described (Foo et al. 2011; Tilve et al. 2015). Briefly, cortical tissues were freed from meninges and digested with 0.25% Trypsin. Primary neurons were plated on a confluent layer of primary astrocytes of either genotype. The cocultures were incubated for 7 days in Neurobasal media supplemented with 2% B27 (Gibco), 10% DMEM, 1% FBS and 1% penicillin/streptomycin (Gibco). HEK293T, HeLa, Neu7, and SHSY5Y cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin (Gibco). For ELISA, astrocytes were exposed to LPS (10 ng/ml, Enzo Life Sciences) alone or combination of LPS (10 ng/ml) and thapsigargin (TG, 0.5–1 μg/ml) for 12–16 hrs. TNFα (R&D systems) and IL6 (R&D systems) ELISA was performed on cell culture media following manufacturer’s protocol. The cytokine released was normalized to the protein concentration of respective cell sample and was reported as fold change compared to the wildtype or control as indicated.
2.3 Plasmids, lentiviruses, and transfection
NOD2-HA in pcDNA3 plasmid was a gift from Dr. Gabriel Nunez lab, Univeristy of Michigan, Ann Arbor, MI. Parkin-Flag construct generated previously (Han et al. 2017). NOD2 cDNA from the pcDNA3 plasmid was subcloned into a p3×Flag (Sigma) vector using BamHI and KpnI restriction enzymes. Parkin cDNA was subcloned into the pLVX (Clontech) lentivirus expression plasmid. Parkin shRNA (TRCN0000283 and TRCN0000284) and NOD2 shRNA (TRCN0068813, TRCN0068814, and TRCN0362622) lentivirus plasmids were obtained from Sigma. Lentiviruses were amplified by transfecting HEK293T cells with transfer plasmids, pMDG.2 (Addgene #12259) and psPax2 (Addgene #12260). The lentivirus from the cell culture media was filtered using 45 micron filther and concentrated by ultracentrifugation. Lentiviral transduction was performed in the presence of polybrene and the media was changed 24 hr post transduction. Transient transfection was performed on cells at ~80% confluency using PolyJet (Signagen).
2.4 Immunoprecipitation and immunoblot analysis
Total proteins from cells were extracted using RIPA buffer (50 mM Tris-HCl, pH 8.0, 0.5% deoxycholic acid, 1% NP-40, 0.1% sodium dodecyl sulfate and 0.5 M NaCl) supplemented with protease inhibitor cocktails (Sigma). The lysates were separated by 4–12% Bis-Tris gel (Invitrogen) and transferred to nitrocellulose membrane. Antibodies were purchased from Cell signaling (Parkin (2132), PARP (9542), and NOD1 (3545)), Sigma (Flag (F1804), HA (H3663), and Myc (M4439)), Santa Cruz Biotechnology (CHOP (sc-7351) and Actin (sc-1616)) and Novus (NOD2 (NB100–524)). For immunoprecipitation (IP), cells were extracted using the lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 0.5% NP-40 and 300 mM NaCl) containing protease inhibitor cocktails. Protein extracts were first incubated with antibodies against either Myc, Flag, or HA (Roche,Clone 12CA5) overnight at 4°C. This was followed by incubation with Sera-Mag speedbead Protein A/G (Sigma) for 4 hrs. The agarose beads were washed with the lysis buffer and boiled with loading buffer. The supernantent was used for immunoblot analysis. Immunoblots were scanned in Odyssey Clx imaging system. Densitometric analysis of protein bands was performed using the Odyssey imaging software. Protein band intensity was normalized to actin and reported as fold chage compared to wildtype or control as indicated.
2.5 Primers and quantitative real-time polymerase chain reaction (qRT-PCR)
PARK2, NOD1, NOD2, BDNF, GDNF and 18s (used as control) primers were purchased from Quantitect primer assays (Qiagen). RNA was extracted from cell samples using TriPure (Roche). cDNA was amplified using the SuperScript III (Invitrogene). The transcript read was measured using FastStart Essential DNA Green Master (Roche). The normalized gene expression was reported as fold change compared to the wildtype sample or control as indicated.
2.6 Cocultures and survival assays
Cocultures of dopaminergic SHSY5Y neurons and mouse primary astrocytes were performed in 0.4-micron transwell dishes (Sigma). The astrocytes of either genotype were plated in the transwell and cocultured with SHSY5Y cells where the media and the secretory components were shared between the two cell types. The cocultures were then exposed to 6-hydroxydopamine (6-OHDA, 50μM), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP, 100μM), thapsigargin (0.05 μg/ml) or combination with thapsigargin and LPS (5 ng/ml). In isolated SHSH5Y cultures the doses were adjusted and the dose of 6-OHDA used was 5μM and the toxic metabolite of MPTP, i.e. MPP+ was used (10μM). WT primary cortical neurons were isolated and plated on a confluent bed of either parkin WT or KO astrocytes. The astrocyte-neuron cocultures were incubated in neurobasal media for 5 days (Foo et al. 2011). Astrocytes in the absence of neurons were cultured in neurobasal media served as controls. The thapsigargin dose used in primary coculture experiments was reduced to 0.01(1x) and 0.02 μg/ml (2x) for 24–48 hrs. The thapsigargin dose used in SHSY5Y-astrocytes coculture experiments was 0.05 μg/ml for 24–48 hrs. In contrast 0.5–1 μg/ml of thapsigargin was employed in isolated primary astocytes studies. Cell death was monitored using lactate dehydrogenase (LDH) cytotoxicity assay kit following manufacturer’s protocol (Pierce). The LDH level was normalized to the protein concentration of respective cell sample and was reported as fold change compared to the wildtype or control as indicated.
2.7 Statistical Analysis
Data are expressed as means ± SD for the indicated number of observations. Statistical significance between groups was determined using two-tailed Student’s test when analyzing the response between groups. Multiple comparison analysis was performed using ANOVA. P value < 0.05 was considered statistically significant.
3 RESULTS
3. 1 Astrocyte restricted depletion of parkin augments neuronal ER stress and inflammation-induce injury
To assess the role of parkin in astrocytic neurotropic function, primary astrocytes were cultured from parkin WT and KO mice brains. The absence of parkin expression in KO astrocytes was confirmed by quantitative RT-PCR and immunoblot analysis (Supporting Information, Figure S1a,b). To test if parkin loss impacts astrocyte neurotropic function, primary astrocyte and SHSY5Y cocultures were established in transwells and cell death was monitored by measuring lactate dehydrogenase (LDH) secreted into the coculture media. In the absence of stressors, coculturing dopaminergic SHSY5Y cells with either WT or parkin KO astrocytes did not impact cell survival (Figure 1a). Additionally, exposure to dopaminergic neurotoxins including 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) under these conditions resulted in similar levels of cell death in SHSY5Y cells cocultured with either WT or parkin KO astrocytes (Figure 1a). In contrast, these neurotoxins significantly reduced survival in parkin depleted SHSY5Y cells compared to scrambled controls (Figure 1b). This result is consistent with prior data suggesting that parkin reduces neurotoxicity (Bian et al. 2012; Jiang et al. 2004),
Figure 1.

Astrocyte restricted depletion of Parkin impairs neuronal survival in cocultures. (a) Quantitation of LDH released in response to 6-OHDA and MPP+ (a toxic metabolite of MPTP) from SHSY5Y cells cocultured with WT or parkin KO primary astrocytes. (b) Quantitation of LDH released from SHSY5Y cells transduced with control (cont) or parkin shRNA lentivirus and exposed to indicated drugs. (c) Quantitation of LDH released in response to ER stress induced by thapsigargin (TG) or in response to TG and lipopolysaccharide (LPS) exposure in SHSY5Y cells cocultured with either WT or parkin KO primary astrocytes. (d) Representative immunoblot showing levels of total and cleaved PARP levels in SHSY5Y cells extracts in the same coculture conditions described in panel 1c. (e) Quantitation of LDH released in response to increasing doses of TG from primary cortical neurons cocultured with either WT or parkin KO astrocytes. (f) A representative immunoblot showing the TG dose-mediated effects on total and cleaved PARP in the primary neurons cocultured with either WT or parkin KO astrocytes. Statistical difference was assessed by student’s t-test. *p<0.05 and **p<0.01, compared to the corresponding controls. n.s = not significant. All experiments were repeated ≥ 3 times.
To explore our hypothesis pertaining to a possible parkin role in response to ER stress and inflammation these cocultured cells were then exposed to thapsigargin (TG, as a trigger of ER stress) in the presence or absence of LPS. When SHSY5Y and astrocyte cocultures were exposed to TG or a combination of TG and LPS (LPS+TG), significantly higher level of LDH secretion was measured from dopaminergic SHSY5Y cells cocultured with parkin KO astrocytes compared to WT (Figure 1c). These data support that parkin KO astrocytes have impaired neurotropic function and have impaired ability to protect SHSY5Y cells from ER stress and ER-stress-induced inflammation compared to WT astrocytes. As excessive ER stress can induce apoptosis, we explored this cell death pathway in the SHSY5Y cells. In the SHSY5Y cells cocultured with parkin KO astrocytes, excess apoptosis was evident in response to TG and TG+LPS as detected by increased cleavage of poly (ADP-ribose) polymerase (PARP) compared to the response in SHSY5Y cells cocultured with WT astrocytes (Figure 1d). Of note, these triggers had no effect on the viability of isolated WT and parkin KO astrocyte cultures as they released similar levels of LDH and did not express cleaved PARP (Supporting Information, Figure S1c and data not shown).
To further confirm the impaired neurotropic abilities of parkin KO astrocytes, primary mouse cortical neurons were isolated from WT brains and plated onto monolayers of astrocytes. The contact cocultures were then exposed to two different concentrations of TG. Consistent with the findings in the SHSY5Y and astrocytes cocultures, cell death as measured by LDH release, and apoptosis as measured by PARP cleavage, were exacerbated when primary cortical neurons were cultured with parkin KO versus WT astrocytes after exposure to TG (Figure 1e,f). Similarly, the levels of cleaved PARP was higher in the neurons cocultured with parkin KO astrocytes (Supporting Information, Figure S1d). Of note, to exclude the potential for differential immunomodulatory effects between primary neurons and astrocytes isolated from the same versus different mice, primary cortical neurons and astrocytes were not harvested from the same mice.
3.2 Lack of parkin alters expression level of neurotropic factor and ER stress genes in astrocytes
The impaired neuroprotection conferred by parkin KO astrocytes is likely due to the alteration in neurotropic signaling in these cells. To address this possibility, targeted gene expression profiling using qRT-PCR was performed in primary astrocytes in response to the ER stressor TG. Consistent with the phenotype described above, transcripts encoding canonical astrocyte neurotropic growth factors including brain and glial derived neurotropic factors (BDNF and GDNF) were found to be expressed at significantly reduced level in parkin KO astrocytes after exposure to TG (Figure 2a). In rescue experiments, BDNF and GDNF mRNA levels were restored when parkin KO astrocytes were transduced with parkin cDNA lentivirus (Figure 2b), and the introduction of BDNF into the culture media abolished LDH release after exposure to TG in both parkin replete and deficient SHSY5Y cells (Supporting Information, Figure S2a). Additionally, canonical ER stress regulated genes including the expression of spliced Xbp1, ATF6, ATF4, CHOP and Ccl2 were expressed at significantly higher levels in parkin KO astrocytes in response to TG (Figure 2c). The increased ER stress encoding gene transcript levels in parkin KO astrocytes was similarly rescued following reconstitution of parkin in these KO astrocytes (Figure 2d).
Figure 2.

Parkin KO astrocytes have altered expression levels of neurotropic factor genes and ER stress genes. (a) Quantitation of BDNF and GDNF mRNA transcript levels in WT and parkin KO astrocytes in response to TG. (b) Quantitation of BDNF and GDNF mRNA transcript levels in parkin KO astrocytes transduced with either empty (parkin KO) or parkin cDNA lentivirus (Parkin KO+OE) in response to TG. (c) Quantitation of gene expression levels of ER stress regulation encoding genes in WT and parkin KO astrocytes in response to TG. (d) Quantitation of gene expression levels of ER stress regulatory genes in parkin KO astrocytes transduced with either empty (Parkin KO) or parkin cDNA lentivirus (Parkin KO+OE) in response to TG. Statistical difference was assessed by student’s t-test. *p<0.05 and **p<0.01, compared to the corresponding controls. n.s = not significant. All experiments were repeated ≥ 3 times.
3.3 Parkin KO astrocytes display exaggerated ER stress and ER stress-induced inflammation
Consistent with the gene expression data, parkin KO astrocytes also constitutively expressed significantly higher steady-state protein levels of phospho-JNK levels and CHOP in response to ER stress (Figure 3a). Interestingly, parkin overexpression reversed ER stress induced phosphorylation of JNK in Neu7 astrocyte cells (Supporting Information, Figure S3a,b). In addition to the production of neurotropic factors, astrocytes also secrete cytokines as a component of their immunomodulatory repertoire (Farina et al. 2007; Johann et al. 2015). Hence levels of several cytokines were measured in response to LPS ± TG. Interestingly, the activation of astrocytic toll-like receptor signaling (Gorina et al. 2011) by LPS showed a similar induction of inflammatory cytokines, TNFα and IL6 irrespective of the presence or absence of parkin (Figure 3b). Also the administration of TG alone did not evoke cytokine release in either genotype (data not shown). However, when ER stress-induced inflammation (LPS+TG) was concurrently applied, the levels of cytokines were induced to a greater extent in parkin KO astrocytes (Figure 3b). Taken together, these data suggest a possible mechanistic link between ER stress and inflammation in astrocytic parkin-mediated neuroprotection. Recent evidence implicates the nucleotide-binding oligomerization domain receptors NOD1 and NOD2 as a novel mediator of ER-stress-induced inflammation (Keestra-Gounder et al. 2016). We therefore explored the expression and protein levels of NOD1 and NOD2 in WT and parkin KO astrocytes. The transcript levels of NOD1and NOD2 were similar between WT and parkin KO astrocytes at baseline (data not shown), and only NOD2 was more robustly induced in the KO astrocytes in response to TG (Figure 3c). In parallel, NOD1 protein levels were similar in WT and parkin KO astrocytes and were not responsive to TG+LPS, whereas TG and LPS administration signficantly increased NOD2 protein levels in KO astrocytes compared to their induction in WT astrocytes (Figure 3d,e).
Figure 3.

Parkin KO astrocytes have exaggerated stress and ER stress induced inflammation levels. (a) Representative immunoblot of the endogenous induction of Phospho-JNK in parkin KO astrocytes and the exaggerated induction of CHOP in parkin KO astrocytes compared to WT astrocytes in response to TG. (b) Fold induction of TNFα and IL6 secretion comparing WT to parkin KO astrocytes in response to LPS or LPS+TG. (c) Quantitation of the TG-induced expression of genes encoding NOD1 and NOD2 genes in WT and KO astrocytes. (d) Representative immunoblot of NOD1, NOD2, Parkin, and CHOP proteins in WT and KO astrocytes cultured in the presence or absence of LPS and TG. (e) Quantitation of NOD2, CHOP, and Parkin steady-state protein levels in WT and KO astrocytes at baseline and in response to LPS and TG exposure. Statistical difference was assessed by student’s t-test or 2 way ANOVA followed by multiple comparisons tests. *p<0.05, compared to the corresponding controls. n.s = not significant. All experiments were repeated ≥ 3 times.
3.4 NOD2 is a Parkin substrate and degradates in a proteasome-dependent manner
Potential interactions between NOD2 and parkin were tested using immunoprecipitation studies. Following cotransfection of HEK293T cells with parkin-Flag and NOD2-HA, an anti-Flag antibody was employed to immunoprecipitate parkin versus a non-specific IgG antibody as a control. An additional control included cells cotransfected with empty-Flag and NOD2-HA with subsequent immunoprecipitation with anti-Flag antibody. Immunoblot analysis revealed a significant enrichment of NOD2-HA protein level in cell extracts coexpressing parkin-Flag and NOD2-HA compared to the controls, supporting the interaction between parkin and NOD2 (Figure 4a). Given, the induction of NOD2 levels in parkin KO astrocytes, and as parkin mediates protein ubiquitylation and proteasome mediated degradation, we assayed Flag-tagged NOD2 levels in response to dose-dependent overexpression of myc-tagged parkin in cotransfection studies in HeLa cells. Western blot analysis of cell extracts revealed a significant parkin-dosage dependent reduction in exogenous NOD2 levels supporting that parkin may directly degrade NOD2 (Figure 4b and Supporting Information, Figure S4a). To evaluate this further, we assayed NOD2 protein electrophoretic mobility in the presence of parkin with and without the proteasome inhibitor MG132. Following overexpression of parkin-Myc and NOD2-Flag, the cells were incubated with the proteasome inhibitor MG132 or vehicle (control). NOD2-Flag immunoprecipitate analyzed by immunoblotting showed numerous lower mobility bands with higher molecular weight in the presence of MG132 compared to the absence of MG132 (Figure S4b). To validate whether parkin ubiquitylated NOD2, additional immunoprecipitation studies were performed in the presence of HA-labeled ubiquitin. Firstly, NOD2-flag and HA-ubiquitin with or without parkin-myc were contransfected into HEK293T cells and immunoprecipitation studies were performed in the presence and absence of MG132. Immunoprecipitation with a Flag antibody and subsequent immunoblot analysis using HA antibody showed a significantly greater extent of NOD2 ubiquitylation in cells overexpressing parkin in the presence of MG132 (Figure 4c and Supporting Information, Figure S4c). Additionally, cell extracts were immunoprecipitated with an antibody recognizing the HA tag to isolate ubiquitylated proteins and then immunoblotted with antibodies directed against NOD2 and Myc antibodies to detect ubiquitylated NOD2 and parkin, respectively. Consistent with the protein degradation data and the prior immunoprecipitation studies, ubiquitinated NOD2 was highly enriched in the cell extracts expressing parkin when MG132 was present (Figure 4d). In the absence of MG132, there was no evidence of NOD2 ubiquitination in the HA antibody immunoprecipitates (data not shown). Collectively these data support ubiquitylation-mediated proteasome degradation of NOD2 by parkin. To explore this further, the kinetics of parkin-mediated NOD2 degradation was assayed in HEK293T cells co-transfected with NOD2 and either an empty plasmid or different doses of parkin in the presence of the protein synthesis inhibitor cycloheximide. Consistent with the data presented in Figure 4b, NOD2 protein levels decreased over time, and the rate of decrease was dependent on the parkin concentration (Figure 4e). Densitometric analysis of the NOD2 protein bands revealed that parkin co-expression significantly increased NOD2 degradation when compared with the relatively stable levels of NOD2 in the absence of exogenous parkin (Figure 4f). Together, these data support that parkin, in a proteasome-dependent manner promotes NOD2 degradation.
Figure 4.

NOD2 is a parkin substrate. (a) Representative immunoblot of a coimmunoprecipitation studies where HEK293T cells were transfected with expression constructs encoding Flag-Empty, Flag-tagged parkin, and HA-tagged NOD2. The immunoprecipitation was performed using an anti-Flag antibody and an anti-HA and anti-Flag antibody was employed for subsequent immunoblot analysis of the NOD2 and parkin, respectively. (b) Immunoblot showing the degradation of NOD2-Flag in response to the dose-dependent increase in parkin-Myc overexpression in HeLa cells. The concentration of the plasmids used are indicated on top of the gel. (c) HEK293T cells were transfected with expression constructs encoding Myc-Empty, Myc-tagged parkin, Flag-tagged NOD2, and HA-tagged ubiquitin (Ub) as indicated. The immunoprecipitation was performed using an anti-Flag antibody and anti-HA and anti-Myc antibodies were employed for subsequent immunoblot analysis to detect ubiquitination of NOD2 and parkin, respectively. Representative immunoblot showing evidence of ubiquitination of NOD2 (depicted with the vertical line) when NOD2 was coexpressed with parkin and ubiquitin, in the presence of the proteasome inhibitor MG132. * refers to nonspecific protein band. (d) HEK293T cells were transfected with expression constructs encoding Myc-tagged parkin, Flag-tagged NOD2, and HA-tagged ubiquitin (Ub) as indicated. The immunoprecipitation was performed using an anti-HA antibody and anti-NOD2 and anti-Myc antibodies were employed for subsequent immunoblot analysis to detect ubiquitinated NOD2 and parkin, respectively. Representative immunoblot showing evidence of ubiquitination of NOD2 (depicted with the vertical line) when NOD2 was coexpressed with parkin in the presence of the proteasome inhibitor MG132. (e) Representative immunoblot of the temporal degradation of NOD2 protein levels in cells cotransfected with NOD2-Flag and parkin-Myc (or Empty-Myc, as indicated) in the presence of cycloheximide (CHX, 5 μg/ml) for indicated time. (f) Quantitation of temporal NOD2 protein levels in the presence of cycloheximide for indicated time. Statistical difference was assessed by student’s t-test or 2 way ANOVA followed by multiple comparisons tests. *p<0.05 and **p<0.01, compared to the corresponding controls. All experiments were repeated ≥ 3 times. IP: immunoprecipitation. WB: western blot.
3.5 NOD2 depletion rescues inflammatory/neurotropic defects in parkin KO astrocytes
To further evaluate the effect of the parkin-NOD2 regulatory axis on astrocyte function, NOD2 was depleted by shRNA in astrocytes. WT and parkin KO astrocytes were transduced with control or various NOD2 shRNA lentiviruses and then exposed to endotoxin (LPS) and TG. Parkin KO astrocytes transduced with control shRNA lentivirus displayed significantly greater secretion of inflammatory cytokines, TNFα and IL6 compared to WT astrocytes (Figure 5a,b). Moreover and consistent with our hypothesis, transduction with NOD2 shRNA lentiviruses reduced cytokines secretion from astrocytes of both genotypes (Figure 5a,b). At the same time knockdown of NOD2 achieved a > 50% reduction in steady-state NOD2 levels which was sustained after exposure to LPS+TG (Figure 5c,d). In parallel, immunoblot analysis showed that the knockdown of NOD2 significantly blunted phospho-JNK and CHOP protein levels in response to LPS and TG (Figure 5c,d) and showed that the depletion of NOD2 reduced the levels of these stress responsive genes, even in the absence of parkin. Since NOD2 shRNA knockdown reduced inflammation and stress in parkin KO astrocytes (Fig 5a–c), the impact of NOD2 knockdown was tested on the neurotropic capacity of parkin KO astrocytes. Here, transwell cocultures were established with SHSY5Y cells and KO astrocytes which were transduced with either control or NOD2 shRNA lentivirus. Upon exposure to TG or TG+LPS, NOD2 shRNA transduced KO astrocytes exhibited significantly increased neuroprotection as shown by decreased LDH release from cocultured SHSY5Y cells when compared with the SHSY5Y cells cocultured with control shRNA transduced parkin KO astrocytes (Figure 5e). To validate these findings, primary neurons were plated on top of a confluent layer of primary KO astrocytes (transduced with either control or NOD2 shRNA lentivirus). Consistent with the SHSY5Y coculture data, primary neurons released significantly lower levels of LDH when cocultured with NOD2-depleted parkin KO astrocytes compared to coculturing with control-shRNA transduced parkin KO astrocytes in response to TG (Figure 5f). The primary neuron and astrocyte cocultures similarly revealed reduced levels of cleaved PARP protein following NOD2 shRNA knockdown in the parkin KO astrocytes (Figure 5g,h). Together these data support that NOD2 downregulation in astrocytes suppresses ER stress and inflammation and thereby positively impacts astrocyte neurotropic functions; and that the parkin-NOD2 regulatory axis is critical for normal astrocyte neurotropic function, inflammatory signaling and neuronal survival.
Figure 5.

NOD2 depletion ameliorates the inflammatory and neurotropic defects in parkin KO astrocyte. (a) Histograms showing the effect of three different shRNA sequences (shRNA1, shRNA2, and shRNA3) knockdown of NOD2 on TNFα secretion in response to LPS and TG in WT and parkin KO astrocytes. (b) Histograms showing the effect of different shRNA knockdown of NOD2 on IL6 secretion in response to LPS and TG in WT and parkin KO astrocytes. (c) Representative immunoblot of NOD2, CHOP, and p-JNK levels in response to LPS and TG following NOD2 knockdown by three different shRNAs in WT and parkin KO astrocytes. (d) Histogram showing the densitometric quantitation of steady-state protein levels of LPS and TG induced regulatory proteins in response to NOD2 knockdown in WT and parkin KO astrocytes. (e) Quantitation of LDH released from SHSY5Y cells cocultured with parkin KO astrocytes transduced with control (cont) or NOD2 shRNA after exposure to either TG or LPS+TG. (f) Quantitation of LDH released in response to increasing doses of TG from primary cortical neurons cocultured with parkin KO astrocytes transduced with either control (cont) or NOD2 shRNA. (g) Representative immunoblot showing total and cleaved PARP in the primary neurons cocultured with either WT or parkin KO astrocytes. (h) Histogram showing the quantitation of cleaved PARP protein level from cortical neuron/astrocyte coculture studies with or without the knockdown of NOD2. Statistical difference was assessed by student’s t-test or 2 way ANOVA followed by multiple comparisons tests. *p<0.05, compared to the corresponding controls. n.s = not significant. All experiments were repeated ≥ 3 times.
4 DISCUSSION
In this study, we show that parkin deficient astrocytes have an exaggerated ER stress signature, increased JNK activation and blunting of neurotropic factor transcript induction following ER stress. The consequence of these defective neurotropic programs in primary astrocytes are manifest by increased injury and apoptosis in cocultured SHSY5Y cells and primary cortical neurons. We identify NOD2 as a functional target of parkin in astrocytes and show that in the absence of parkin, NOD2 levels are induced. Moreover, we show that parkin is upregulated by ER stress in primary astrocytes and this correlated with reduced NOD2 levels. We demonstrated that parkin interacted with NOD2, enhanced NOD2 ubiquitylation and that parkin, in a dose-dependent manner facilitated proteasome-dependent NOD2 degradation. Finally, we demonstrated that the rescue of parkin or the knockdown of NOD2 rescued astrocytic neurotropic effects. Together these findings support that NOD2 is an additional ER stress and inflammatory target controlled by parkin and show that parkin plays a role in modulating ER stress and inflammation in astrocytes to regulate their neurotropic functions.
Numerous studies have shown that ER stress upregulates parkin (Bouman et al. 2011; Wang et al. 2007), and that parkin is implicated in the modulation of ER stress protein folding (Imai et al. 2002; Imai et al. 2001; Imai et al. 2000). Additionally, mutations in parkin and its genetic depletion activate CHOP-mediated ER stress triggered apoptosis (Han et al. 2017) and the depletion of CHOP protects against neurotoxin-induced substantia nigra dopaminergic neuron cell death (Silva et al. 2005). Moreover, emerging data support the integration of ER stress signaling with immune cell activation (Keestra-Gounder et al. 2016; Martinon et al. 2010; Shenderov et al. 2014). Studies find that ER stress activates innate immune signaling (Deng et al. 2004; Wu et al. 2004) and conversely that the activation of innate immune receptors amplify inflammation via ER stress signaling (Martinon et al. 2010). Taken together these findings implicate a coordinate and amalgamated interaction between ER stress and inflammatory signaling pathways.
The canonical mediators linking these two stress responsive pathways include ROS, NF-κB signaling and JNK activation (Zhang and Kaufman 2008). More recently the cytosolic NOD1/2 sensors, which traditionally respond to bacterial peptidoglycan fragments, were found to respond to ER stress to initiate inflammatory signaling (Keestra-Gounder et al. 2016). Interestingly, this ER stress-induced NOD signaling was independent of peptidoglycans, but required the canonical TNF receptor associated factor 2 (TRAF2) signaling pathway for immune activation (Keestra-Gounder et al. 2016). Our study contributes to the further characterization of the role of parkin in the control of ER stress and in the integration of ER stress signaling with inflammatory signaling.
Although, the role of NOD signaling in Parkin has not been extensively explored, an association with polymorphisms of NOD2 could potentially be associated with sporadic PD (Bialecka et al. 2007; Ma et al. 2013), although this finding has also been questioned (Appenzeller et al. 2012). Another potential link between NOD2 with increased susceptibility to PD could hypothetically be through the multifunctional protein kinase LRRK2. Mutations in LRKK2 is a well characterized causative mutations linked to EOPD (Klein and Schlossmacher 2006) and it has recently been recognized that the inflammatory effects of NOD2 are mediated by LRRK2 (Yan and Liu 2017; Zhang et al. 2015). This regulatory link does not appear to have been directly explored in the pathogenesis of PD, but warrants further exploration.
Parkin, through the specificity of the lysine residue and number of covalent ubiquitin modifications, confers either non-degrading signaling effects or promote proteasome-dependent degradation of substrate proteins (Abumrad and Moore 2011). This spectrum of substrate modifications is evident on inflammatory mediators where parkin-dependent ubiquitylation stabilizes the CD36 scavenger receptor (Kim et al. 2011) and NFκB essential modulator (NEMO) (Muller-Rischart et al. 2013) and conversely enhances TRAF2 and TRAF6 degradation (Chung et al. 2013). Despite the identification of these numerous immune regulatory proteins, a role of parkin in the modulation of immune function has not been extensively characterized (de Leseleuc et al. 2013; Manzanillo et al. 2013; Mira et al. 2004; Piquereau et al. 2013). Our findings add additional information to support the role of parkin in immune modulation. Interestingly, as parkin has previously been shown to regulate the stability of TRAF2 (Chung et al. 2013), and as TRAF2 and NOD2 are operational in the same inflammatory signaling pathway (Keestra-Gounder et al. 2016), these data suggest that Parkin modulates multiple signaling intermediates via the control of protein stability in innate immune signal transduction.
In the coculture studies of primary astrocytes and SHSY5Y cells, the combined stressors of thapsigargin and LPS were required to demonstrate the astrocytic parkin effect on neurotropic function, whereas ER stress alone could elucidate this phenotype when primary astrocytes were cocultured with primary neurons. These data may reflect, in part, the stress resilience of the transformed SHSY5Y cells. However, the finding that ER stress alone could uncover this program in the primary cell cocultures concurrently supports that the NOD2 inflammatory regulatory program operates downstream of astrocytic ER stress, a concept that replicates this signaling sequence evident in bone marrow derived macrophages (Keestra-Gounder et al. 2016).
In contrast to the effects on ER stress and endotoxin stress, exposure to the traditional neurotoxic stressors linked to PD, 6-OHDA and MPTP, resulted in a similar level of neuronal cell death when neurons were cocultured with astrocytes of either genotypes. These data support that the astrocyte specific role of parkin that is primarily involved in curbing ER stress to maintain neuronal integrity. These findings are consistent with the growing body of published data that points to an emerging role for glial cells implicated in many neurological disorders (Garden and Campbell 2016; Jansen et al. 2014; Liu et al. 2017). Consistently, significant glial reactions have been reported in the post-mortem PD brains (Banati et al. 1998; Forno et al. 1992). However, it is not clear if the defective glial function causes or results from PD pathology and additional investigations are needed to elucidate which aspect of glial dysfunction leads to reactive glial cell accumulation and neuronal death.
As more mechanistic details of the ER stress signaling emerge, cellular pathways connecting ER stress to mitochondria have been identified. Caspase-2, Bid and the thioredoxin-interacting protein play a critical role in relaying ER stress signaling to mitochondria with the subsequent induction of inflammation (Bronner et al. 2015; Oslowski et al. 2012). Although parkin has been found to modulate mitochondrial homeostasis (Narendra et al. 2012) and ER stress (Han et al. 2017), as further evident in this study, whether parkin orchestrates the coordinate regulation of mitochondria and ER in astrocytes has not been explored. Parkin has also been found to modulate endolysosome mediated clearance of damaged mitochondria (Hammerling et al. 2017) and interestingly other PD associated mutations are linked to the disruption of endolysosome function (MacLeod et al. 2013). Although parkin is now linked to mitochondrial function, ER stress and endo-lysosome functioning whether parkin plays a role in these integrated stress responses needs further validation. It is interesting to note the the combination of these three stress reponses have been identified in neurodegeneration linked to Alzheimer’s disease (Umeda et al. 2011).
Putting this together parkin may play a role in orchestrating multiple intracellular organelles, and stress signaling in conferring protection. Moreover, the demonstration that the restricted depletion of parkin in astrocytes exacerbates neuronal injury highlight that this E3 ligase may have both cell autonomous and paracrine effects on the overall health of dopaminergic neurons. The specific depletion of parkin in astrocytes in-vivo is required to test these neurotropic effects of parkin in substantia nigra pars compacta dopaminergic neuronal homeostasis.
Supplementary Material
Figure S1. (a) Quantitation of parkin mRNA levels in WT and parkin KO astrocytes. (b) Representative immunoblot of parkin and actin in wildtype, parkin knockout, and parkin heterozygous astrocytes. (c) Quantitation of LDH released from astrocytes that were cultured in neurobasal media exposed to 0.02 μg/ml TG and 5 ng/ml LPS. (d) Quantitation of cleaved PARP protein level from primary neurons-astrocyte coculture cell extracts. Statistical difference was assessed by student’s t-test. *p<0.05, compared to the corresponding controls. n.s = not significant. All experiments were repeated ≥ 3 times.
Figure S3. (a) Representative immunoblot of parkin, phospho-JNK, and actin in Neu7 cell line transfected with increasing dose of parkin-Flag overexpression plasmid and thapsigargin (TG) treatment as indicated. (b) Quantitation of phospho-JNK protein level from Neu7 cell extracts after control and TG treatment. Statistical difference was assessed by student’s t-test. *p<0.05 and **p<0.01, compared to the corresponding control. All experiments were repeated ≥ 3 times.
Figure S4. (a) Histogram showing the quantitation of NOD2 protein level normalized to actin in the presence of increasing dosage of parkin-Myc plasmid described in Fig 4b. (b) HEK293T cells were transfected with expression constructs encoding Flag-tagged parkin and HA-tagged NOD2 and incubated with or without the proteasome inhibitor MG132. The immunoprecipitation was performed using an anti-Flag antibody and anti-Flag and anti-Myc antibodies were employed for subsequent immunoblot analysis of the NOD2 and parkin, respectively. Representative immunoblot showing evidence of lower electrophoretic mobility NOD2-Flag protein bands (depicted with the vertical line) when NOD2 was coexpressed with parkin in the presence of the proteasome inhibitor MG132. (c) Quantitation of ubiquitinated NOD2 in the coimmunoprecipitation experiment described in Fig. 4c with indicated plasmids in the presence or absence of MG132. Statistical difference was assessed by student’s t-test. *p<0.05, compared to the corresponding control. All experiments were repeated ≥ 3 times. IP: immunoprecipitation. WB: western blot.
Figure S2. (a) Quantitation of LDH released from SHSY5Y cells that were transduced with control or parkin shRNA lentivirus after exposure to BDNF and thapsigargin (ER stress). Statistical difference was assessed by student’s t-test. *p<0.05, compared to the control. All experiments were repeated ≥ 3 times.
Main Points.
The restricted genetic depletion of parkin in astrocytes exacerbates ER stress induced neuronal apoptosis
Parkin degrades NOD2 to blunt astocytic inflammation and sustain astrocytic neurotropic effects
Acknowledgments
MNS and HG were funded by the NHLBI Division of intramural research and through the Michael J. Fox foundation. We thank Dr. Gabriel Nunez from the University of Michigan, Ann Arbor, MI for the NOD2-HA pcDNA3 plasmid. pMD2.G and psPAX2 was a gift from Didier Trono from EPFL SV Global Health Institute, Switzerland
Footnotes
Conflicts: No conflicts to report
References
- Abumrad NA, Moore DJ. Parkin reinvents itself to regulate fatty acid metabolism by tagging CD36. J Clin Invest. 2011;121:3389–92. doi: 10.1172/JCI59219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appenzeller S, Thier S, Papengut F, Klein C, Hagenah J, Kasten M, Berg D, Srulijes K, Gasser T, Schreiber S, et al. No association between NOD2 variants and Parkinson’s disease. Mov Disord. 2012;27:1191–2. doi: 10.1002/mds.25059. [DOI] [PubMed] [Google Scholar]
- Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord. 1998;13:221–7. doi: 10.1002/mds.870130205. [DOI] [PubMed] [Google Scholar]
- Bialecka M, Kurzawski M, Klodowska-Duda G, Opala G, Juzwiak S, Kurzawski G, Tan EK, Drozdzik M. CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci Res. 2007;57:473–6. doi: 10.1016/j.neures.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Bian M, Liu J, Hong X, Yu M, Huang Y, Sheng Z, Fei J, Huang F. Overexpression of parkin ameliorates dopaminergic neurodegeneration induced by 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. PLoS One. 2012;7:e39953. doi: 10.1371/journal.pone.0039953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouman L, Schlierf A, Lutz AK, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N, Berg D, et al. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ. 2011;18:769–82. doi: 10.1038/cdd.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nunez G, He Y, Yin XM, O’Riordan MX. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity. 2015;43:451–62. doi: 10.1016/j.immuni.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JY, Park HR, Lee SJ, Lee SH, Kim JS, Jung YS, Hwang SH, Ha NC, Seol WG, Lee J, et al. Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Lab Invest. 2013;93:663–76. doi: 10.1038/labinvest.2013.60. [DOI] [PubMed] [Google Scholar]
- de Leseleuc L, Orlova M, Cobat A, Girard M, Huong NT, Ba NN, Thuc NV, Truman R, Spencer JS, Adams L, et al. PARK2 mediates interleukin 6 and monocyte chemoattractant protein 1 production by human macrophages. PLoS Negl Trop Dis. 2013;7:e2015. doi: 10.1371/journal.pntd.0002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol Cell Biol. 2004;24:10161–8. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–45. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Fidler PS, Schuette K, Asher RA, Dobbertin A, Thornton SR, Calle-Patino Y, Muir E, Levine JM, Geller HM, Rogers JH, et al. Comparing astrocytic cell lines that are inhibitory or permissive for axon growth: the major axon-inhibitory proteoglycan is NG2. J Neurosci. 1999;19:8778–88. doi: 10.1523/JNEUROSCI.19-20-08778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo LC, Allen NJ, Bushong EA, Ventura PB, Chung WS, Zhou L, Cahoy JD, Daneman R, Zong H, Ellisman MH, et al. Development of a method for the purification and culture of rodent astrocytes. Neuron. 2011;71:799–811. doi: 10.1016/j.neuron.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forno LS, DeLanney LE, Irwin I, Di Monte D, Langston JW. Astrocytes and Parkinson’s disease. Prog Brain Res. 1992;94:429–36. doi: 10.1016/s0079-6123(08)61770-7. [DOI] [PubMed] [Google Scholar]
- Frank-Cannon TC, Tran T, Ruhn KA, Martinez TN, Hong J, Marvin M, Hartley M, Trevino I, O’Brien DE, Casey B, et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 2008;28:10825–34. doi: 10.1523/JNEUROSCI.3001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Campbell BM. Glial biomarkers in human central nervous system disease. Glia. 2016;64:1755–71. doi: 10.1002/glia.22998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol Med. 2012;18:589–98. doi: 10.1016/j.molmed.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–55. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7:494–506. doi: 10.1016/j.nurt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, Boassa D, Phan S, Thor A, Jimenez RE, et al. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun. 2017;8:14050. doi: 10.1038/ncomms14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Hassanzadeh S, Singh K, Menazza S, Nguyen TT, Stevens MV, Nguyen A, San H, Anderson SA, Lin Y, et al. Parkin regulation of CHOP modulates susceptibility to cardiac endoplasmic reticulum stress. Sci Rep. 2017;7:2093. doi: 10.1038/s41598-017-02339-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S210–2. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol Cell. 2002;10:55–67. doi: 10.1016/s1097-2765(02)00583-x. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–4. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- Jansen AH, Reits EA, Hol EM. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front Mol Neurosci. 2014;7:73. doi: 10.3389/fnmol.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–54. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63:2260–73. doi: 10.1002/glia.22891. [DOI] [PubMed] [Google Scholar]
- Johnson BN, Berger AK, Cortese GP, Lavoie MJ. The ubiquitin E3 ligase parkin regulates the proapoptotic function of Bax. Proc Natl Acad Sci U S A. 2012;109:6283–8. doi: 10.1073/pnas.1113248109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chavez-Arroyo A, Tsai AY, Cevallos SA, Winter MG, Pham OH, Tiffany CR, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. 2016;532:394–7. doi: 10.1038/nature17631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Stevens MV, Akter MH, Rusk SE, Huang RJ, Cohen A, Noguchi A, Springer D, Bocharov AV, Eggerman TL, et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest. 2011;121:3701–12. doi: 10.1172/JCI44736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klein C, Schlossmacher MG. The genetics of Parkinson disease: Implications for neurological care. Nat Clin Pract Neurol. 2006;2:136–46. doi: 10.1038/ncpneuro0126. [DOI] [PubMed] [Google Scholar]
- Kofler J, Wiley CA. Microglia: key innate immune cells of the brain. Toxicol Pathol. 2011;39:103–14. doi: 10.1177/0192623310387619. [DOI] [PubMed] [Google Scholar]
- Liu B, Teschemacher AG, Kasparov S. Astroglia as a cellular target for neuroprotection and treatment of neuro-psychiatric disorders. Glia. 2017;65:1205–1226. doi: 10.1002/glia.23136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, An X, Li Z, Zhang H, Huang W, Cai L, Hu P, Lin Q, Tzeng CM. P268S in NOD2 associates with susceptibility to Parkinson’s disease in Chinese population. Behav Brain Funct. 2013;9:19. doi: 10.1186/1744-9081-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–39. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamczur P, Borsuk B, Paszko J, Sas Z, Mozrzymas J, Wisniewski JR, Gizak A, Rakus D. Astrocyte-neuron crosstalk regulates the expression and subcellular localization of carbohydrate metabolism enzymes. Glia. 2015;63:328–40. doi: 10.1002/glia.22753. [DOI] [PubMed] [Google Scholar]
- Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–6. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–8. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira MT, Alcais A, Nguyen VT, Moraes MO, Di Flumeri C, Vu HT, Mai CP, Nguyen TH, Nguyen NB, Pham XK, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature. 2004;427:636–40. doi: 10.1038/nature02326. [DOI] [PubMed] [Google Scholar]
- Muller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell. 2013;49:908–21. doi: 10.1016/j.molcel.2013.01.036. [DOI] [PubMed] [Google Scholar]
- Narendra D, Walker JE, Youle R. Mitochondrial quality control mediated by PINK1 and Parkin: links to parkinsonism. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, Ishigaki S, Zhu LJ, Hayashi E, Hui ST, et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–73. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–22. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Perez FA, Curtis WR, Palmiter RD. Parkin-deficient mice are not more sensitive to 6-hydroxydopamine or methamphetamine neurotoxicity. BMC Neurosci. 2005;6:71. doi: 10.1186/1471-2202-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron. 2015;87:371–81. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piquereau J, Godin R, Deschenes S, Bessi VL, Mofarrahi M, Hussain SN, Burelle Y. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy. 2013;9:1837–51. doi: 10.4161/auto.26502. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–83. doi: 10.1126/science.aag2590. [DOI] [PubMed] [Google Scholar]
- Shenderov K, Riteau N, Yip R, Mayer-Barber KD, Oland S, Hieny S, Fitzgerald P, Oberst A, Dillon CP, Green DR, et al. Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1beta in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J Immunol. 2014;192:2029–2033. doi: 10.4049/jimmunol.1302549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–86. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–80. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Goldberg MS. Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis. 2010;37:510–8. doi: 10.1016/j.nbd.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, von Coelln R, Mandir AS, Trinkaus DB, Farah MH, Leong Lim K, Calingasan NY, Flint Beal M, Dawson VL, Dawson TM. MPTP and DSP-4 susceptibility of substantia nigra and locus coeruleus catecholaminergic neurons in mice is independent of parkin activity. Neurobiol Dis. 2007;26:312–22. doi: 10.1016/j.nbd.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilve S, Difato F, Chieregatti E. Cofilin 1 activation prevents the defects in axon elongation and guidance induced by extracellular alpha-synuclein. Sci Rep. 2015;5:16524. doi: 10.1038/srep16524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda T, Tomiyama T, Sakama N, Tanaka S, Lambert MP, Klein WL, Mori H. Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res. 2011;89:1031–42. doi: 10.1002/jnr.22640. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–40. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Wang HQ, Imai Y, Kataoka A, Takahashi R. Cell type-specific upregulation of Parkin in response to ER stress. Antioxid Redox Signal. 2007;9:533–42. doi: 10.1089/ars.2006.1522. [DOI] [PubMed] [Google Scholar]
- Wu S, Tan M, Hu Y, Wang JL, Scheuner D, Kaufman RJ. Ultraviolet light activates NFkappaB through translational inhibition of IkappaBalpha synthesis. J Biol Chem. 2004;279:34898–902. doi: 10.1074/jbc.M405616200. [DOI] [PubMed] [Google Scholar]
- Yan R, Liu Z. LRRK2 enhances Nod1/2-mediated inflammatory cytokine production by promoting Rip2 phosphorylation. Protein Cell. 2017;8:55–66. doi: 10.1007/s13238-016-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Wang H, Katagiri Y, HMG . An In Vitro Model of Reactive Astrogliosis and Its Effect on Neuronal Growth. In: Milner R, editor. Astrocytes Methods in Molecular Biology (Methods and Protocols) Humana Press; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–62. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Pan Y, Yan R, Zeng B, Wang H, Zhang X, Li W, Wei H, Liu Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol. 2015;16:918–26. doi: 10.1038/ni.3233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. (a) Quantitation of parkin mRNA levels in WT and parkin KO astrocytes. (b) Representative immunoblot of parkin and actin in wildtype, parkin knockout, and parkin heterozygous astrocytes. (c) Quantitation of LDH released from astrocytes that were cultured in neurobasal media exposed to 0.02 μg/ml TG and 5 ng/ml LPS. (d) Quantitation of cleaved PARP protein level from primary neurons-astrocyte coculture cell extracts. Statistical difference was assessed by student’s t-test. *p<0.05, compared to the corresponding controls. n.s = not significant. All experiments were repeated ≥ 3 times.
Figure S3. (a) Representative immunoblot of parkin, phospho-JNK, and actin in Neu7 cell line transfected with increasing dose of parkin-Flag overexpression plasmid and thapsigargin (TG) treatment as indicated. (b) Quantitation of phospho-JNK protein level from Neu7 cell extracts after control and TG treatment. Statistical difference was assessed by student’s t-test. *p<0.05 and **p<0.01, compared to the corresponding control. All experiments were repeated ≥ 3 times.
Figure S4. (a) Histogram showing the quantitation of NOD2 protein level normalized to actin in the presence of increasing dosage of parkin-Myc plasmid described in Fig 4b. (b) HEK293T cells were transfected with expression constructs encoding Flag-tagged parkin and HA-tagged NOD2 and incubated with or without the proteasome inhibitor MG132. The immunoprecipitation was performed using an anti-Flag antibody and anti-Flag and anti-Myc antibodies were employed for subsequent immunoblot analysis of the NOD2 and parkin, respectively. Representative immunoblot showing evidence of lower electrophoretic mobility NOD2-Flag protein bands (depicted with the vertical line) when NOD2 was coexpressed with parkin in the presence of the proteasome inhibitor MG132. (c) Quantitation of ubiquitinated NOD2 in the coimmunoprecipitation experiment described in Fig. 4c with indicated plasmids in the presence or absence of MG132. Statistical difference was assessed by student’s t-test. *p<0.05, compared to the corresponding control. All experiments were repeated ≥ 3 times. IP: immunoprecipitation. WB: western blot.
Figure S2. (a) Quantitation of LDH released from SHSY5Y cells that were transduced with control or parkin shRNA lentivirus after exposure to BDNF and thapsigargin (ER stress). Statistical difference was assessed by student’s t-test. *p<0.05, compared to the control. All experiments were repeated ≥ 3 times.