

Abstract
In this study, we newly sequenced five mitogenomes of representatives of phytophagous scarab beetles (Coleoptera: Scarabaeidae) by using next-generation sequencing technology. Two species have complete (or nearly complete) mitogenome sequences, namely Popillia mutans Newman (Coleoptera: Scarabaeidae) and Holotrichia oblita Faldermann (Coleoptera: Scarabaeidae). The remaining three species have the partial mitogenomes, and the missing genes are mainly located adjacent to the control region. The complete (or nearly complete) mitogenomes have the same genome structure as most of the existing Scarabaeidae mitogenomes. We conducted phylogenetic analyses together with 24 published mitogenomes of Scarabaeoidea. The results supported a basal split of coprophagous and phytophagous Scarabaeidae. The subfamily Sericinae was recovered as sister to all other phytophagous scarab beetles. All analyses supported a non-monophyletic Melolonthinae, which included two different non-sister clades. The Cetoniinae was recovered as sister to a clade including Rutelinae and Dynastinae. Although the Rutelinae was rendered paraphyletic by Dynastinae in the Bayesian trees inferred under the site-heterogeneous CAT-GTR or CAT-MTART model, discordant patterns were given in some of ML trees estimated using the homogeneous GTR model.
Keywords: Scarabaeidae, mitochondrial genome, next-generation sequencing, phylogeny
The scarab beetles comprise a large group of insects classified as the family Scarabaeidae (Insecta: Coleoptera), with over 30,000 species of beetles worldwide. From the point of view of feeding habits, these insects can be divided into coprophagous and phytophagous (including saprophagous) lineages. The species of coprophagous Scarabaeidae are often found in dung and in excrement, which are also called as dung beetles with various nesting behaviors (e.g., the rolling and tunneling behaviors). Due to their significant economic and ecological importance, dung beetles have been one of the most intensively studied coleopteran groups (Nichols et al. 2008, Brown et al. 2010). In comparison, researches on the phytophagous Scarabaeidae are limited. In particular, there remain many controversies on the taxonomy and phylogeny of the phytophagous clade of Scarabaeidae.
The phytophagous scarab beetles as an independent lineage have been recognized since Erichson (1847). The majority of phytophagous scarabs are traditionally grouped into four subfamilies such as Melolonthinae, Cetoniinae, Dynastinae, and Rutelinae (Smith 2006, Smith et al. 2006, Eberle et al. 2014). Moreover, several other smaller groups are proposed to be incorporated into this clade (e.g., the Sericinae and Trichiinae, Péringuey 1904, Browne and Scholtz 1998). All phytophagous scarabs are also called as Pleurosticti (Erichson 1847). The monophyly of Pleurosticti is often supported by morphological studies (Ritcher 1958, Balthasar 1963, Browne and Scholtz 1998). Based on the partial nuclear 18S and 28S ribosomal DNA sequences, Smith et al. (2006) recovered the phytophagous scarabs as a monophyletic group, which contained the following groups: Melolonthinae, Cetoniinae, Rutelinae, Dynastinae, Anomalini, and Adoretini.
Despite the great progress made in recent years, many issues on the higher-level phylogeny of Scarabaeidae remain to be addressed (Sanmartín and Mmartín-Piera 2003, Smith et al. 2006). Ahrens (2005) recovered the Melolonthinae as a paraphyletic group based on the analysis of morphological characters. Ahrens et al. (2014) retrieved the Melolonthinae as paraphyletic again, based on a multi-locus sequence analysis. On the contrary, other authors considered the Melolonthinae as a separate clade and to be elevated to the rank of family. Cherman and Morón (2014) included the Rutelinae, Dynastinae, and Melolonthinae to form the family Melolonthidae. Moreover, the family status of Melolonthinae was favored by the subsequent study of Cherman et al. (2016). Apart from the monophyly of Melolonthidae, the phylogenetic position of Sericinae was also problematic. Machatschke (1959) recovered the monophyletic Sericinae, including the tribes Diphucephalini, Ablaberini, and Sericini. However, the sericines were more often regarded as a tribe of Melolonthinae in recent literature (Ahrens 2005, Smith 2006, Ahrens et al. 2014). Elucidating the status of subgroups included in the family Scarabaeidae may contribute to the understanding of the evolution of this group, which further improves the conservation strategies of insects and the control measures of associated pests. Thus, it is necessary to investigate the constitution and the phylogeny of Scarabaeidae using additional types of data and increased taxon sampling.
The mitochondrial genome (mitogenome) is a useful maker in resolving the phylogeny at different taxonomic levels of Coleoptera (Timmermans et al. 2010, 2016; Coates 2014; Gillett et al. 2014; Crampton-Platt et al. 2015; Breeschoten et al. 2016; Nie et al. 2018). Recent mitochondrial phylogenomic studies have applied the next-generation sequencing (NGS) technologies to reconstruct the insect mitogenomes (e.g., Gillett et al. 2014, Crampton-Platt et al. 2015, Song et al. 2016). Compared with traditional PCR and Sanger sequencing, the approaches implemented by NGS are fast and cost-effective due to their capability to assemble a large number of mitogenomes from mixtures of species samples simultaneously.
Herein, we reconstructed five mitogenomes of representatives of the phytophagous scarab beetles from pooled genomic DNA. Combined with existing Scarabaeoidea mitogenomes, we inferred the phylogeny of Scarabaeidae to 1) test the monophyly of Melolonthinae, 2) to examine the position of Sericinae, and 3) to investigate the relationship between Rutelinae and other scarab beetles.
Materials and Methods
Taxon Sampling and DNA Extraction
Taxa were collected from Zhengzhou, Henan province, China. No specific permits were required for the insect specimens collected for this study. The field studies did not involve endangered or protected species. All sequenced insects are common beetle species in China and not included in the ‘List of Protected Animals in China’.
Two species of Melolonthinae (i.e., Holotrichia oblita and Holotrichia sp.), two of Sericinae (i.e., Serica orientalis and Serica sp.) and one of Rutelinae (i.e., Popillia mutans) were utilized for DNA extraction and further sequencing. The adult specimens were preserved in 100% ethanol. Total genomic DNA was extracted from the thoracic leg muscle tissue, with the TIANamp Micro DNA Kit (Tiangen Biotech Co., Ltd) following the manufacturer’s protocol. Vouchers are deposited at the Entomological Museum of Henan Agricultural University. DNA concentration was measured by Nucleic acid protein analyzer (Quawell Technology Inc.).
Library Construction and Illumina Sequencing
Five scarab beetle species were respectively added into five different sequencing pools. Besides scarab beetle, the separate pool included other 29 unrelated samples. Following Gillett et al. (2014), each sample in the mixture was devised to have a largely identical DNA concentration (1.5 μg) to maximize the efficiency of genome assembling. The Illumina libraries were prepared with Illumina TruSeqTM DNA Sample Prep Kit (Illumina, San Diego, CA) and insert size of 350 bp. Each library was sequenced on the Illumina HiSeq X Ten platform (Beijing Novogene Bioinformatics Technology Co., Ltd, China) generating 150 bp paired-end reads.
Sequence Quality Control and Assembly
FastQC was used for the quality control checks on raw sequence data (Andrews 2010). All reads were filtered with NGS QC Toolkit using default settings (Patel and Jain 2012). In this step, reads containing adapters and ploy-N, and low-quality reads were removed from raw data. At the same time, Q20, Q30, GC-content, and sequence duplication level of the cleaned data were calculated. All the downstream analyses were based on clean data with high quality (avg. Q20 > 90%, and avg. Q30 > 85%). De novo assembly were performed with IDBA-UD v. 1.1.1 (Peng et al. 2012). The assemblies were constructed using 200 for the setting of minimum size of contig, and an initial k-mer size of 40, an iteration size of 10, and a maximum k-mer size of 90.
Mitogenome Identification and Annotation
The mitochondrial scaffolds were identified by mitochondrial baiting, with local-blasting implemented in BioEdit (Hall 1999) against the genome data assembled by IDBA-UD. The bait gene fragments (i.e., mitochondrial cox1, cytb, and rrnS) were amplified using the universal primers identical to those of Song et al. (2016). The preliminary mitogenome annotation were conducted using the MITOS webserver, under default settings and the invertebrate genetic code for mitochondria (Bernt et al. 2013). The gene boundaries were further checked and refined by alignment with the published mitogenome sequences of Scarabaeidae (see details in Supp. Table S1). To check the quality of the mitogenome sequences assembled, the mappings to the mitochondrial contigs were performed using Geneious R11.
The secondary structures of mitochondrial rrnL and rrnS were predicted, mainly with reference to the models of Apis mellifera (Gillespie et al. 2006) and of Drosophila virilism (Cannone et al. 2002), using the method of ‘Comparative sequence analysis’ in the study of Ouvrard et al. (2000).
Multiple Alignment
For the mitochondrial protein-coding genes, we first removed the stop codon of each sequence. Then, the nucleotide matrix of each protein-coding gene was translated into amino acids under the invertebrate mitochondrial genetic code in MEGA 6 (Tamura et al. 2013) and aligned based on their amino acid sequences with Muscle as implemented in MEGA. The amino acid matrix was back-translated into the corresponding nucleotide matrix. For the mitochondrial tRNA and rRNA genes, each of them was aligned using MAFFT (version 7) (Katoh and Standley 2013) under the iterative refinement method incorporating the most accurate local pairwise alignment information (E-INS-i). The resulting alignments were checked in MEGA. Gaps were striped by Gap Strip/Squeeze v2.1.0 with 40% Gap tolerance (http://www.hiv.lanl.gov/content/sequence/GAPSTREEZE/gap.html). Finally, all alignments were concatenated using FASconCAT_v1.0 (Kuck and Meusemann 2010) to construct the full dataset of PCGRNA.
To reduce the bias effect introduced by synonymous change on the phylogenetic analysis, protein-coding genes were degenerated by using the Perl script Degen v1.4 to construct the dataset of PCGDegen (Regier et al. 2010, Zwick et al. 2012). The degenerated protein-coding genes were combined with RNA genes to compile the dataset of PCGDegenRNA.
Possible sequence saturation at each codon position and at each gene partition of the concatenated alignment was individually examined by DAMBE 5 (Xia 2013). Estimates of nonsynonymous (dN) and synonymous (dS) substitution rates of protein-coding genes were obtained by the method of Yang and Nielsen (Yang and Nielsen 2000) using the program yn00 as implemented in PAML 4.9 (Yang 2007). The Excel 2016 was used to perform one-way analysis of variance (ANOVA) analysis in order to test for significant differences of substitution rates between different lineages, and the significance level was set to be 0.05.
Phylogenetic Analysis
We estimated the phylogeny of Scarabaeidae with two different approaches: maximum likelihood (ML) analysis under homogeneous models and Bayesian analysis under heterogeneous models. The following datasets were utilized in the phylogenetic analysis: 1) PCG: the nucleotide sequences of 13 protein-coding genes; 2) PCG_AA: the deduced amino acid sequences of the protein-coding genes; 3) PCGDegen: the nucleotide sequences of 13 protein-coding genes degenerated by Degen script; 4) PCGRNA: the nucleotide sequences of 13 protein-coding genes combined with RNA genes; and 5) PCGDegenRNA: the sequences of PCGDegen combined with RNA genes.
Prior to ML analyses based on the datasets of PCG, PCGRNA and PCG_AA, the PartitionFinder (Lanfear et al. 2012) was run to select the optimal partitioning strategies and best-fitting models for each dataset under the Bayesian Information Criterion (BIC), using a greedy search with RAxML (Stamatakis 2015). ML tree searches were performed in IQ-TREE (Nguyen et al. 2015), with various data partition schemes and best-fitting models determined by PartitionFinder. The detailed information on the partitions and the best models selected are listed in Supp. Table S2. Branch support analysis was conducted using 1,000 ultrafast bootstrap replicates. For the datasets of PCGDegen and PCGDegenRNA, un-partitioned analyses were performed, due to the altered variation of alignments by Degeneracy recoding scheme.
The parallel version of PhyloBayes (pb_mpi1.5a implemented on a HP server) was used for Bayesian tree calculation (Lartillot and Philippe 2004, Lartillot et al. 2009). The CAT-GTR model was used for nucleotide analyses, while the CAT-MTART model for amino acids. Two independent chains were run in parallel and started from a random topology. The ‘bpcomp’ program contained in the package of PhyloBayes was used to calculate the largest (maxdiff) and mean (meandiff) discrepancy observed across all bipartitions. The program ‘tracecomp’ was also used to summarize the discrepancies and the effective sizes estimated for each column of the trace file. When the maximum ‘maxdiff’ value was lower than 0.1 and minimum effective size was higher than 100, the Bayesian runs were recognized to be reached good convergence. A consensus tree was calculated from the saved trees by bpcomp program after checking for stationarity, with the default options.
Hypothesis Testing
For all five datasets the alternative hypotheses were tested, using the Shimodaira-Hasegawa (SH) test (Shimodaira and Hasegawa 1999) and the approximately unbiased (AU) test (Shimodaira 2002). The specific hypotheses concerning four phylogenetic issues were assessed: 1) the basal divergence between coprophagous and phytophagous Scarabaeidae, 2) the Sericinae as sister to all other phytophagous taxa, 3) the monophyly of Melolonthinae, 4) the monophyly of Rutelinae. Additionally, we compared the tree topologies resulting from phylogenetic inferences using different datasets under ML and Bayes criteria. The site-log-likelihood values were calculated under the GTR+G model using TREE-PUZZLE (Version 5.3) (Schmidt et al. 2002). And then, the obtained values were used as input for the software CONSEL (Shimodaira and Hasegawa 2001). All constraint likelihood trees were generated by IQ-TREE (Nguyen et al. 2015) using the settings described above.
Results
Mitogenome Assembly
Five Illumina HiSeq runs resulted in 72,395,049, 72,720,510, 74,104,974, 81,772,168, and 89,829,307 raw paired reads, respectively. After filtering, 66,451,916, 67,980,632, 72,278,440, 81,189,879, and 89,122,956 clean paired reads were generated for corresponding libraries. For each species, at least one Sanger sequence was obtained and utilized to identify the potential mitochondrial contig (Supp. Table S3). Local blast searching results showed that each baiting fragment had its best blast match with the targeted mitochondrial contig (i.e., Identities ≥ 99% and E-value = 0.00). In contrast, other contigs assembled had the lower identities and the higher e-values (Supp. Table S3). Moreover, the mitogenome identified was represented by a single large-contig (contig length > 13 kb in most cases). The sequencing coverages corresponding to five mitogenomes ranged from 171-fold to 370-fold.
Mitogenome Organization
The mitogenome organization of five scarab beetle species sequenced are summarized in Table 1. P. mutans and H. oblita had the complete or nearly complete mitogneomes, which contained the full 37 mitochondrial genes and the complete or partial control region. The other three mitogenomes (i.e., Holotrichia sp., S. orientalis and Serica sp.) were partial, and the missing fragments were located in the putative control region and the adjacent regions.
Table 1.
The organization of the newly sequenced mitogenomes
| Gene region | Popillia mutans | Holotrichia oblita | Serica sp. | Serica orientalis | Holotrichia sp. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start | Stop | Start | Stop | Start | Stop | Start | Stop | Start | Stop | |
| Partial control region | - | - | - | - | 1 | 657 | 1 | 658 | 1 | 179 |
| trnI | 1 | 65 | 1 | 65 | 658 | 721 | 659 | 723 | 180 | 242 |
| trnQ | 69 | 137 | 63 | 131 | 719 | 787 | 721 | 789 | 240 | 309 |
| trnM | 137 | 205 | 131 | 200 | 787 | 855 | 789 | 857 | 319 | 387 |
| nad2 | 206 | 1213 | 201 | 1208 | 856 | 1860 | 858 | 1862 | 388 | 1395 |
| trnW | 1229 | 1299 | 1226 | 1292 | 1864 | 1931 | 1884 | 1951 | 1409 | 1473 |
| trnC | 1292 | 1354 | 1285 | 1347 | 1924 | 1985 | 1944 | 2005 | 1466 | 1527 |
| trnY | 1355 | 1419 | 1353 | 1417 | 1985 | 2048 | 2005 | 2069 | 1527 | 1589 |
| cox1 | 1412 | 2956 | 1383 | 2951 | 2047 | 3585 | 2068 | 3606 | 1588 | 3123 |
| trnL-UUR | 2952 | 3016 | 2952 | 3018 | 3581 | 3646 | 3602 | 3667 | 3130 | 3194 |
| cox2 | 3017 | 3718 | 3019 | 3738 | 3647 | 4330 | 3668 | 4351 | 3195 | 3896 |
| trnK | 3705 | 3775 | 3704 | 3774 | 4332 | 4402 | 4353 | 4423 | 3883 | 3951 |
| trnD | 3778 | 3844 | 3774 | 3839 | 4403 | 4469 | 4424 | 4487 | 3971 | 4034 |
| atp8 | 3845 | 4000 | 3840 | 3995 | 4469 | 4624 | 4488 | 4643 | 4035 | 4190 |
| atp6 | 3994 | 4668 | 3989 | 4660 | 4618 | 5289 | 4637 | 5308 | 4184 | 4858 |
| cox3 | 4668 | 5454 | 4660 | 5446 | 5289 | 6076 | 5308 | 6095 | 4858 | 5645 |
| trnG | 5455 | 5519 | 5447 | 5510 | 6076 | 6138 | 6095 | 6157 | 5645 | 5707 |
| nad3 | 5520 | 5873 | 5511 | 5864 | 6139 | 6492 | 6158 | 6509 | 5708 | 6061 |
| trnA | 5872 | 5937 | 5863 | 5928 | 6491 | 6556 | 6510 | 6575 | 6061 | 6124 |
| trnR | 5937 | 6001 | 5929 | 5993 | 6556 | 6620 | 6575 | 6639 | 6124 | 6190 |
| trnN | 6004 | 6067 | 5993 | 6056 | 6618 | 6683 | 6637 | 6702 | 6195 | 6259 |
| trnS-AGN | 6068 | 6134 | 6057 | 6124 | 6684 | 6750 | 6703 | 6769 | 6260 | 6326 |
| trnE | 6135 | 6202 | 6125 | 6189 | 6751 | 6814 | 6770 | 6831 | 6327 | 6391 |
| trnF | 6201 | 6265 | 6188 | 6253 | 6813 | 6878 | 6830 | 6896 | 6390 | 6456 |
| nad5 | 6265 | 7980 | 6254 | 7972 | 6878 | 8593 | 6896 | 8611 | 6457 | 8175 |
| trnH | 7981 | 8043 | 7973 | 8037 | 8594 | 8657 | 8612 | 8675 | 8176 | 8239 |
| nad4 | 8043 | 9380 | 8037 | 9374 | 8657 | 9994 | 8675 | 9871 | 8239 | 8967 |
| nad4l | 9374 | 9664 | 9368 | 9658 | 9988 | 10278 | - | - | - | - |
| trnT | 9667 | 9731 | 9661 | 9725 | 10281 | 10345 | - | - | - | - |
| trnP | 9732 | 9796 | 9726 | 9790 | 10346 | 10409 | - | - | - | - |
| nad6 | 9798 | 10301 | 9783 | 10295 | 10411 | 10908 | - | - | - | - |
| cytb | 10301 | 11443 | 10295 | 11437 | 10908 | 12050 | - | - | - | - |
| trnS-UCN | 11442 | 11506 | 11436 | 11501 | 12049 | 12113 | - | - | - | - |
| nad1 | 11523 | 12473 | 11519 | 12469 | 12132 | 13082 | - | - | - | - |
| trnL-CUN | 12475 | 12538 | 12471 | 12532 | 13084 | 13146 | - | - | - | - |
| rrnL | 12539 | 13827 | 12510 | 13804 | 13147 | 13866 | - | - | - | - |
| trnV | 13828 | 13897 | 13803 | 13872 | - | - | - | - | - | - |
| rrnS | 13897 | 14695 | 13873 | 14672 | - | - | - | - | - | - |
| Complete or partial control region | 14696 | 16192 | 14673 | 15968 | - | - | - | - | - | - |
Dash (-) indicates no application or the missing genome region.
The complete or nearly complete mitogenomes reconstructed showed the same gene arrangement proposed as ancestral for insects (Cameron 2014). The nucleotide composition of new mitogenome sequences were similar to those published scarab beetle species, with the significant A + T bias in the major strand. For the protein-coding genes, five new mitogenomes used the typical ATN as start codons, except for the nad3 (using ACA) and nad4 (using TGT) in H. oblita. Ten of 13 protein-coding genes terminated with the conventional stop codons TAG or TAA, while the cox2 (in P. mutans and Holotrichia sp.), cox3 (in P. mutans, S. orientalis, Serica sp., and Holotrichia sp.), and nad3 (in S. orientalis) had an incomplete stop codon T.
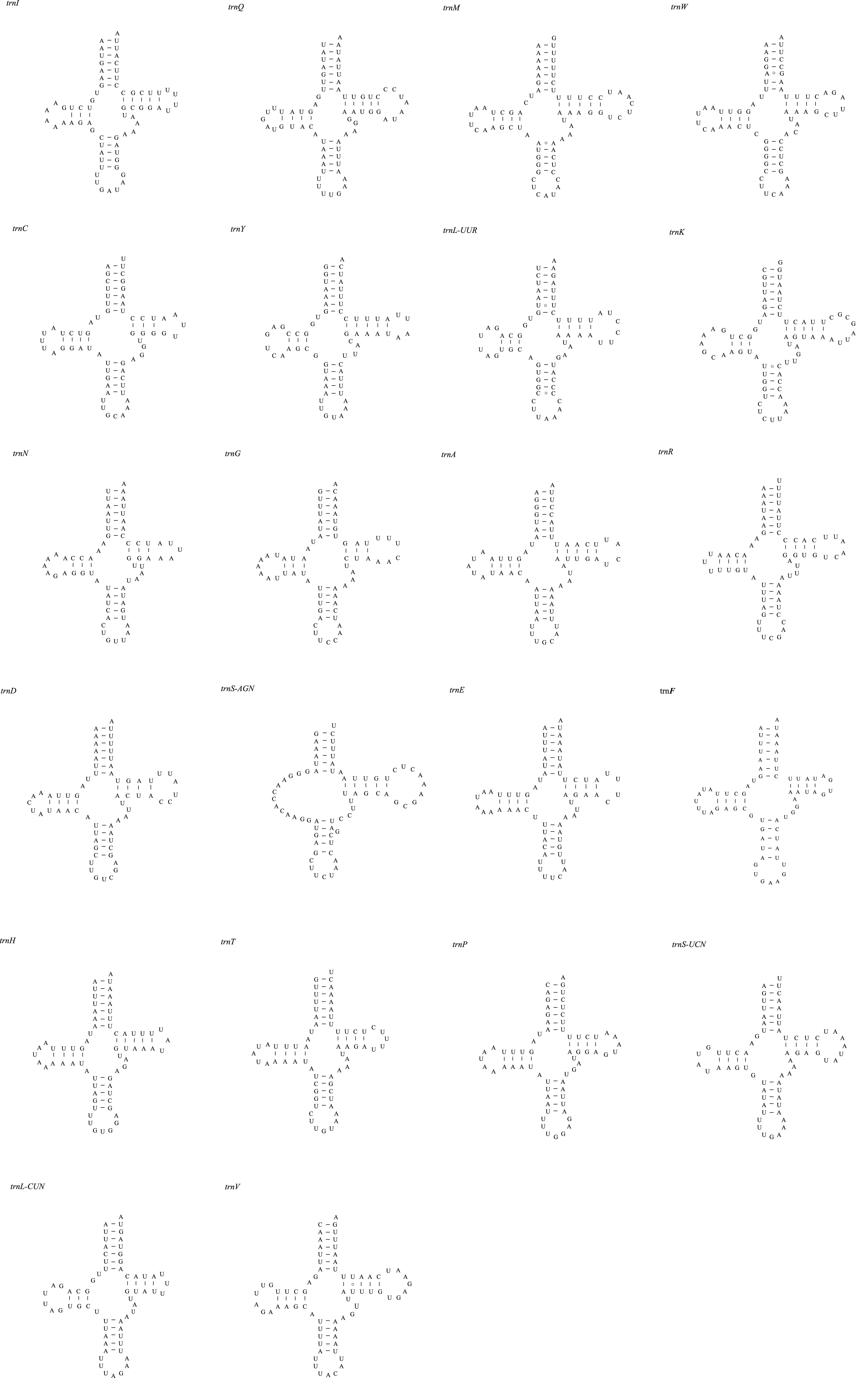
The secondary structures of mitochondrial tRNA genes were predicted. Supp. Fig. S1 illustrates the tRNA secondary structures of the complete mitogenome of P. mutans (Supp. Fig. S1A) and the nearly complete mitogenome of H. oblita (Supp. Fig. S1B). All tRNA genes had the typical clover-leaf structure except for the trnS-AGN. The DHU arm was reduced or missing in the trnS-AGN.
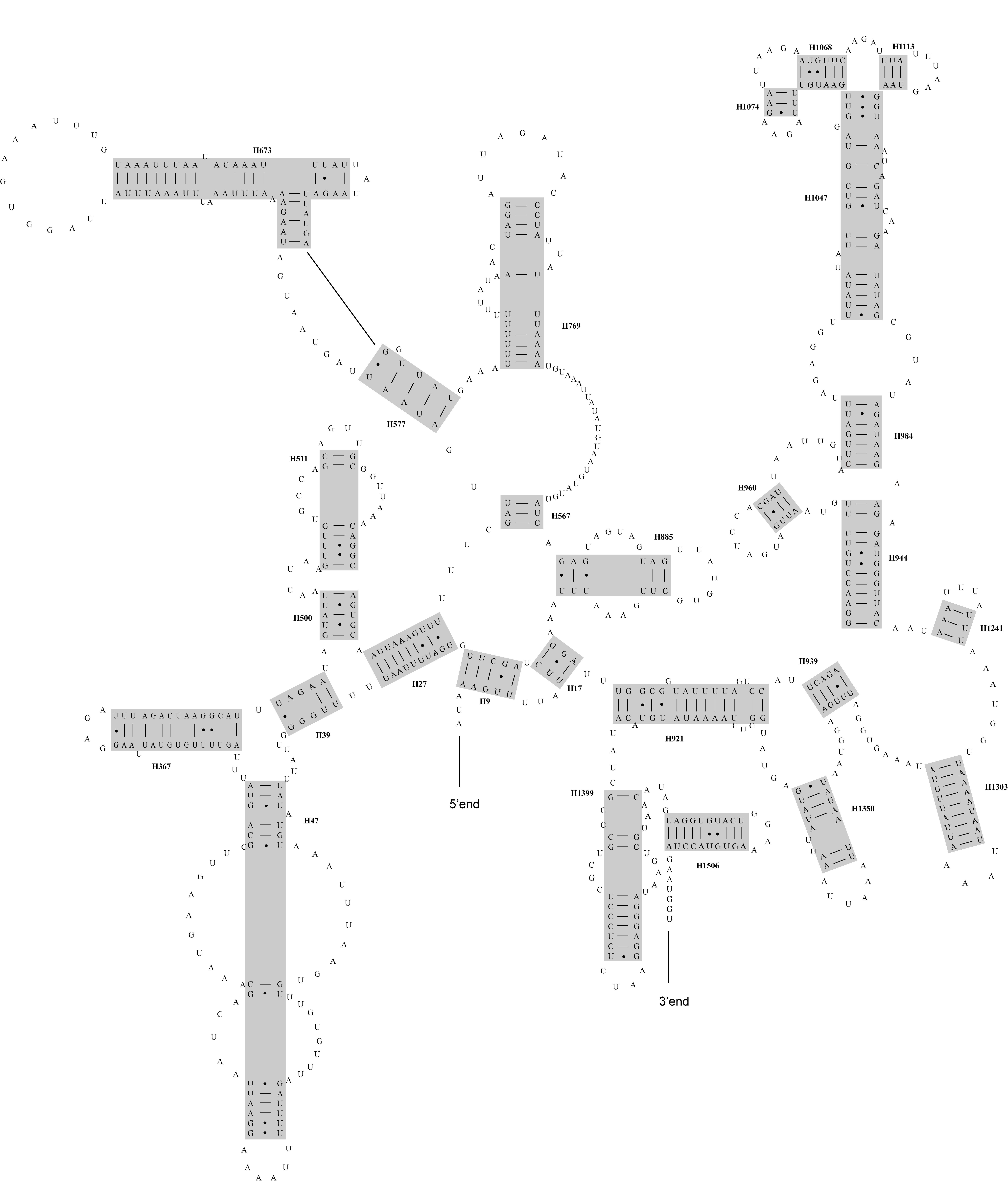
For the P. mutans and H. oblita, the complete mitochondrial rRNA genes were identified between trnL-CUN and trnV and between trnV and the control region, with the length of 1,270 or 1,289 bp for rrnL and 799 or 800 bp for rrnS. The secondary structures of mitochondrial rRNA genes were inferred (Fig. 1A and B for rrnL and rrnS of P. mutans, and Supp. Fig. S2A and B for rrnL and rrnS of H. oblita). Because the P. mutans and H. oblita have the basically identical secondary structure predictions for two mitochondrial rRNA genes, the following description will focus on those of P. mutans. The secondary structure of rrnL consisted of five canonical structural domains (I-II, IV-VI) and 44 helices (Fig. 1A and Supp. Fig. S2A). Domain III was absent, which was the same as the published insect mitochondrial rrnL secondary models (Cannone et al. 2002, Gillespie et al. 2006). When compared with other scarab beetles, domains Ⅳ-Ⅵ located in the 3′ end of the rrnL molecule were highly conserved. The three domains contained the helices with more than 75% identical positions across species (e.g., H1755, H1835 and H1906). The mitochondrial rrnS secondary model predicted for P. mutans consisted of 27 helices (Fig. 1B and Supp. Fig. S2B), which formed four typical domains (labeled I, II, III and IV). Across taxa analyzed, domains III and IV were more conserved than domains I and II.
Fig. 1.


(A) The secondary structure of rrnL gene predicted for Popillia mutans. (B) The secondary structure of rrnS gene predicted for Popillia mutans.
The full control region located between rrnS and trnI was identified only in the mitogenome of P. mutans, with the length of 1,497 bp and A + T content of 82.6%. There were a series of poly-A or poly-T stretches and [T]n[A]n structures randomly scattered in this region. However, no obviously tandem repeat motifs were found.
Characteristics of Data Matrices
Saturation tests demonstrated that rRNA genes and the third codon positions of protein-coding genes were saturated when assuming an asymmetrical topology (Iss < Iss.cAsym, Supp. Table S4). Evolutionary rate analyses showed that the major lineages analyzed shared the similar dS values (Table 2). In comparison, several outgroup taxa (e.g., Glaresis sp.) and ingroup taxa (e.g., Polyphylla laticollis mandshurica and Cheirotonus jansoni) had the higher dN values, while the Rutelinae and Cetoniinae displayed the lower dN values. The dN/dS values showed the same trend as that of the dN values (P = 0.910). The statistical analyses revealed no significant differences of dS values among the species studied. However, there were significant differences for dN or dN/dS values (P < 0.05), with or without outgroup taxa.
Table 2.
Estimation of synonymous and nonsynonymous substitution rates by yn00 implemented in PAML
| Item | Subfamily | Species | Group | dN | dS | dN/dS |
|---|---|---|---|---|---|---|
| Ingroup | Aphodiinae | Aphodius sp. | 0 | 0.1553 | 4.3560 | 0.0356 |
| Scarabaeinae | Sarophorus sp. | 1 | 0.1304 | 4.2417 | 0.0307 | |
| Scarabaeinae | Scarabaeidae sp. BMNH 1274750 | 1 | 0.1386 | 4.2943 | 0.0323 | |
| Scarabaeinae | Scarabaeidae sp. BMNH 1274752 | 1 | 0.1294 | 4.2988 | 0.0301 | |
| Scarabaeinae | Scarabaeidae sp. BMNH 1274753 | 1 | 0.1423 | 4.2896 | 0.0332 | |
| Scarabaeinae | Xinidium sp. | 1 | 0.1390 | 4.2969 | 0.0324 | |
| Cetoniinae | Leucocelis sp. | 2 | 0.1146 | 4.3489 | 0.0264 | |
| Cetoniinae | Myodermum sp. | 2 | 0.1280 | 4.4122 | 0.0290 | |
| Cetoniinae | Osmoderma opicum | 2 | 0.1372 | 4.3791 | 0.0313 | |
| Cetoniinae | Protaetia brevitarsis | 2 | 0.1179 | 4.3100 | 0.0274 | |
| Dynastinae | Cyphonistes vallatus | 3 | 0.1463 | 4.5169 | 0.0324 | |
| Melolonthinae | Asthenopholis sp. | 4 | 0.1228 | 4.5031 | 0.0273 | |
| Melolonthinae | Cheirotonus jansoni | 4 | 0.1574 | 4.4242 | 0.0356 | |
| Melolonthinae | Holotrichia oblita | 4 | 0.1389 | 4.4084 | 0.0315 | |
| Melolonthinae | Holotrichia sp. | 4 | 0.1402 | 4.3853 | 0.0320 | |
| Melolonthinae | Polyphylla laticollis mandshurica | 4 | 0.1665 | 4.4430 | 0.0375 | |
| Melolonthinae | Rhopaea magnicornis | 4 | 0.1289 | 4.3641 | 0.0295 | |
| Melolonthinae | Schizonycha sp. | 4 | 0.1266 | 4.3786 | 0.0289 | |
| Rutelinae | Adoretus sp. | 5 | 0.1143 | 4.2931 | 0.0266 | |
| Rutelinae | Popillia mutans | 5 | 0.1226 | 4.3500 | 0.0282 | |
| Rutelinae | Popillia sp. | 5 | 0.1258 | 4.3227 | 0.0291 | |
| Sericinae | Pleophylla sp. | 6 | 0.1253 | 4.3275 | 0.0290 | |
| Sericinae | Serica orientalis | 6 | 0.1236 | 4.3431 | 0.0285 | |
| Sericinae | Serica sp. | 6 | 0.1257 | 4.3992 | 0.0286 | |
| Outgroup | Geotrupidae | Anoplotrupes stercorosus | 7 | 0.1420 | 4.2712 | 0.0332 |
| Geotrupidae | Bolboceratex sp. | 7 | 0.1520 | 4.5252 | 0.0336 | |
| Glaphyridae | Glaphyrus comosus | 8 | 0.1411 | 4.3209 | 0.0326 | |
| Glaresidae | Glaresis sp. | 9 | 0.1598 | 4.4248 | 0.0361 | |
| Trogidae | Trox sp. | 10 | 0.1309 | 4.2923 | 0.0305 |
Phylogenetic Analysis
ML Analysis
Although results of the ML analyses based on various datasets were similar, the trees differed to a greater extent in the relationships of Melolonthinae and Rutelinae. The monophyly of Scarabaeidae was strongly supported (e.g., BP = 82 in PCGRNA-ML-tree, Fig. 2). The coprophagous scarab beetles comprising the clade Aphodiinae + Scarabaeinae were recovered as the sister group to the phytophagous lineages. The phytophagous clade contained five subfamilies, namely the Sericinae, Melolonthinae, Cetoniinae, Rutelinae, and Dynastinae. In all ML analyses but PCG_AA, the Sericinae was recovered outside of the assemblage of Melolonthinae, and as a sister lineage to all other phytophagous scarabs. In the ML tree from the PCG_AA dataset, the Sericinae was recovered as a sister group to a species from Melolonthinae (i.e., Holotrichia sp.), with weak node support value (BP = 74). The Melolonthinae as a non-monophyletic group was divided into two different non-sister clades.
Fig. 2.

Maximum likelihood tree inferred from the PCGRNA dataset using IQ-TREE under the partitions and best-fitting models selected by PartitionFinder. Branch support values (>70) are presented near each node. Scale bar represents substitutions/site. Asterisks designate the species newly sequenced in this study. The colored lines correspond to the subfamilies recovered.
The following phylogenetic results changed depending on the dataset. 1) For the relationship of melolonthine lineages, Holotrichia sp. formed a sister-group to Cheirotonus jansoni in the analyses from the PCG, PCGRNA, PCGDegen and PCGDegenRNA datasets (Fig. 2 and Fig. 3A, C, and D), whereas Holotrichia sp. was sister to the clade Sericinae in the analysis from the PCG_AA dataset (Fig. 3B); 2) The second major melolonthine clade containing the other five melolonthine species was sister to a large clade including Dynastinae, Rutelinae, and Cetoniinae, the relationships among melolonthine species were different across analyses: in the ML analyses using the PCG, PCGRNA and PCGDegenRNA datasets was Rhopaea magnicornis sister to Polyphylla laticollis mandshurica, and both together sister to Holotrichia oblita (Fig. 2 and Fig. 3A and D), in contrast, in other two analyses (i.e., PCG_AA and PCGDegen) was R. magnicornis sister to a clade P. laticollis mandshurica + H. oblita (Fig. 3B and C); 3) the monophyly of Rutelinae was supported by the PCG, PCG_AA, and PCGRNA datasets, whereas the degenerated datasets (i.e., PCGDegen and PCGDegenRNA) retrieving the C. vallatus (Dynastinae) as sister to Adoretus sp. led to a non-monophyletic Rutelinae (Fig. 3C and D).
Fig. 3.

Maximum likelihood trees inferred from the datasets of (A) PCG, (B) PCG_AA, (C) PCGDegen, and (D) PCGDegenRNA using IQ-TREE. Branch support values are presented near each node. Scale bar represents substitutions/site.
Bayesian Analysis
Bayesian tree reconstructions were performed under the site-heterogeneous CAT-GTR or CAT-MTART model (Figs. 4 and 5). The monophyly of Scarabaeidae were recovered in all Bayesian analyses, except for that from the PCGDegenRNA dataset. The nested position of the outgroup Glaphyrus comosus resulted in a non-monophyletic Scarabaeidae in the PCGDegenRNA Bayes tree, but branch support measures indicated that this reconstruction was weakly supported (PP = 0.57). Monophyletic groupings of taxa corresponding to the subfamily level were congruent with the relationships found in the ML topologies. The Aphodiinae was well supported as a sister-group to Scarabaeinae in the coprophagous Scarabaeidae clade, with high posterior probabilities (PP ≥ 0.98). Among the phytophagous lineages, the Sericinae was consistently recovered as sister to the remaining phytophagous lineages. The non-monophyletic Melolonthinae branched next to the Sericinae. The terminal clades still consisted of the Cetoniinae, Rutelinae, and Dynastinae. The subfamily Rutelinae was rendered paraphyletic by an internal clade comprising C. vallatus + Adoretus sp. in all Bayesian analyses but for that from the PCG_AA dataset. The PCG_AA Bayes tree retrieved Dynastinae and Rutelinae to both be monophyletic.
Fig. 4.

Bayesian tree inferred from the PCGRNA dataset using PhyloBayes under the CAT-MTART model. Node numbers show poster probability values (>0.9). Scale bar represents substitutions/site. Asterisks designate the species newly sequenced in this study. The colored lines correspond to the subfamilies recovered.
Fig. 5.

Bayesian trees inferred from the datasets of (A) PCG, (B) PCG_AA, (C) PCGDegen, and (D) PCGDegenRNA using PhyloBayes. Node numbers show poster probability values. Scale bar represents substitutions/site.
Alternative Hypothesis Testing
The hypothesis that the basal division of Scarabaeidae into two clades (coprophagous and phytophagous) was strongly supported by the statistical testing with different datasets (Table 3). Moreover, hypothesis testing consistently supported Sericinae as an independent lineage and sister to all other phytophagous lineages. This hypothesis received the highest likelihood values in all analyses but for that from the PCG_AA dataset. Although the monophyly of Melolonthinae cannot be addressed unambiguously, the likelihood value for a tree constraining Melolonthinae as non-monophyletic was often higher than that constraining a monophyletic Melolonthinae. The paraphyly of Rutelinae was rejected by the PCG dataset (P < 0.05 in both SH and AU tests, Table 3), whereas the monophyly of Rutelinae was consistently supported by all testing. Based on five datasets and two inference methods, a total of 10 trees were produced (Figs. 2–5). The results of statistical testing of tree topology indicated that the topology resulting from the partitioned ML analyses of the PCGRNA and PCG datasets (Fig. 2 and 3A) was more likely to present the true tree, with respect to their highest likelihood values (Supp. Table S5). Whereas, some of the Bayesian trees were rejected (e.g., PCGRNA-Bayes-tree, P < 0.05 in all SH or AU tests).
Table 3.
Hypothesis testing: results of the SH and AU tests, with rejection at P < 0.05
| Hypothesis tested | PCG | PCG_AA | PCGDegen | PCGDegenRNA | PCGRNA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Log-likelihood | P values for | Log-likelihood | P values for | Log-likelihood | P values for | Log-likelihood | P values for | Log-likelihood | P values for | ||||||
| SH | AU | SH | AU | SH | AU | SH | AU | SH | AU | ||||||
| Coprophagous + Phytophagous | −163937.18 | 0.433 | 0.274 | −66075.52 | 0.907 | 0.731 | −54370.01 | 0.783 | 0.459 | −74014.86 | 0.836 | 0.358 | −194662.25 | 0.743 | 0.427 |
| Sericinae + all other phytophagous | −163907.95 | 0.915 | 0.786 | −66085.88 | 0.476 | 0.285 | −54365.57 | 0.721 | 0.611 | −74014.86 | 0.865 | 0.423 | −194656.99 | 0.979 | 0.796 |
| Melolonthinae as a nonmonophyletic group | −163988.42 | 0.040 | 0.010 | −66098.73 | 0.187 | 0.138 | −54389.19 | 0.181 | 0.040 | −74033.71 | 0.095 | 0.023 | −194664.16 | 0.658 | 0.468 |
| Melolonthinae as a monophyletic group | −163968.76 | 0.112 | 0.027 | −66098.73 | 0.187 | 0.138 | −54393.38 | 0.156 | 0.038 | −74033.71 | 0.095 | 0.024 | −194668.23 | 0.578 | 0.179 |
| Cetoniinae + (monophyletic Rutelinae + Dynastinae) | −163926.75 | 0.601 | 0.349 | −66075.52 | 0.909 | 0.725 | −54371.56 | 0.632 | 0.412 | −74018.96 | 0.475 | 0.353 | −194680.67 | 0.292 | 0.065 |
| Cetoniinae + (paraphyletic Rutelinae + Dynastinae) | −163995.82 | 0.031 | 0.010 | −66087.21 | 0.378 | 0.132 | −54370.01 | 0.783 | 0.502 | −74014.86 | 0.948 | 0.758 | −194675.53 | 0.395 | 0.254 |
Discussion
The present study adds five new mitogenome sequences to the phytophagous scarab beetles, with the goal of investigating the higher-level relationships within Scarabaeidae. Our results demonstrate that NGS techniques adopted here can generate mitogenomes in a more efficient and cost-effective way, and that the mitogenomic data can provide insights into the subfamily relationships of Scarabaeidae. Although there have been numerous studies attempting to reconstruct the evolutionary history of scarab beetles (Howden 1982; Browne and Scholtz 1995, 1996, 1998; Ahrens 2005; Smith et al. 2006; Hunt et al. 2007; Ahrens et al. 2014; Bocak et al. 2014; Eberle et al. 2014), no authors utilized the mitogenomes alone to estimate the phylogeny of Scarabaeidae. This inspired us to perform this analysis to further assess the phylogenetic usefulness of mitogenomes. The backbone relationships recovered by the current mitogenomic data are concordant with previous morphological and molecular studies, namely that the Scarabaeidae comprises the traditionally recognized coprophagous and phytophagous lineages (Browne and Scholtz 1999, Smith et al. 2006, Ahrens et al. 2014, Bocak et al. 2014). The former clade includes the subfamilies Aphodiinae and Scarabaeinae, while the latter consists of Sericinae, Melolonthinae, Rutelinae, Dynastinae, and Cetoniinae. We acknowledge the effect of the sparse taxon sampling of several lineages on the recovery of relationships within this megadiverse insect group. There is an immediate need of an extensive sequencing program of scarab beetles. This article contributes to stimulate interest in phylogenomic study of Scarabaeidae by using whole mitogenome sequences.
The monophyly of Rutelinae and the placement of C. jansoni (Melolonthinae) were variable. Two ML analyses based on the degenerated datasets under the homogeneous model and four out of five Bayesian analyses under the site-heterogeneous model supported a paraphyletic Rutelinae. Recovery of the monophyly of Rutelinae depended on whether the single representative of Dynastinae (i.e., C. vallatus) clustered with one exemplar of Rutelinae (i.e., Adoretus sp.) or not. The majority of analyses retrieved a sister-group relationship of C. jansoni and Holotrichia sp., both of which were outside the main clade of Melolonthinae. Three species (i.e., C. jansoni, Holotrichia sp. and C. vallatus) leading to the unstable reconstructions exhibited the obviously long branch lengths, particularly in the Bayesian trees. In the analysis of substitution rates of protein-coding genes, we found the rate heterogeneity in the current data (Table 2). Moreover, three species motioned above had the greatly accelerated rates, compared with other scarab beetles. Long branches associated with rate heterogeneity may result in the incongruence between analyses.
With regard to the monophyly of the subfamily Melolonthinae, there has been no consensus opinion among coleopterists. Based on the nuclear gene datasets, Smith et al. (2006) did not provide definitive results on the monophyly of the Melolonthinae and suggested additional phylogenetically informative characters needed to tackle this problem. Machatschke (1959) proposed the monophyly of the subfamily Sericinae, which rendered the paraphyly of the Melolonthinae. This hypothesis was confirmed by another morphological study by Ahrens (2005). In this paper, the Sericinae was consistently recovered as an independent clade and placed as the most basal branching lineage within the phytophagous scarab beetles. Thus, our data supported the point of the elevated status of Sericini to be the subfamily rank (Sericinae). Meanwhile, a non-monophyletic Melolonthinae was supported in all analyses.
The sister-group relationship between Rutelinae and Dynastinae was always recovered by previous studies (Howden 1982, Browne and Scholtz 1999, Hunt et al. 2007, Bocak et al. 2014). Smith et al. (2006) indicated that some taxa of the Dynastinae might be moved to the subfamily Rutelinae. In the PCGDegen and PCGDegenRNA ML analyses and in all Bayesian analyses but PCG_AA, the Rutelinae was recovered as a paraphyletic group with respect to the single representative of Dynastinae. Despite this, the preferred trees (i.e., PCG-ML-tree and PCGRNA-ML-tree, see results in Supp. Table S5) supported the monophyly of Rutelinae (BP = 95 in both trees). Future studies should focus on adding whole mitogenome sequences of other underrepresented taxa, especially Dynastinae, in order to advance understanding of the subfamily level relationships of the phytophagous clades. The sister group relationship between the clade comprising Rutelinae and Dynastinae and the subfamily Cetoniinae was strongly supported in all analyses (BP ≥ 88 and PP ≥ 0.98). This arrangement is in line with the result of Bocak et al. (2014), but conflict with Browne and Scholtz (1999) and Hunt et al. (2007). The latter two researches recovered a close affinity of the clade Rutelinae + Dynastinae and the Melolonthinae (Browne and Scholtz 1999, Hunt et al. 2007).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This research is supported by grants from the National Natural Science Foundation of China (31402002), Key Scientific Research Projects of Henan Province (14B210036, 16A210029, 18B210006), and Agricultural Science and Technology Innovation Project of Henan Province (30600309).
References Cited
- Ahrens D. 2005. The phylogeny of Sericini and their position within the Scarabaeidae based on morphological characters (Coleoptera: Scarabaeidae). Syst. Entomol. 31: 113–144. [Google Scholar]
- Ahrens D., J. Schwarzer, and Vogler A. P.. 2014. The evolution of scarab beetles tracks the sequential rise of angiosperms and mammals. Proc. Biol. Sci. 281: 20141470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: A quality control tool for high throughput sequence data. Bioinformatics Group, Babraham Institute, Cambridge, United Kingdom: (www.bioinformatics.babraham.ac.uk/projects/fastqc). [Google Scholar]
- Balthasar V. 1963. Monographie der Scarabaeidae und Aphodiidae der palaearktischen und orientalischen Region. Lamellicornia. Band 1. Der Tschechoslowakischen Akadademie der Wissenschaften. Verlag, Prague. [Google Scholar]
- Bernt M., A. Donath F. Jühling F. Externbrink C. Florentz G. Fritzsch J. Pütz M. Middendorf, and Stadler P. F.. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69: 313–319. [DOI] [PubMed] [Google Scholar]
- Bocak L., Barton C., Crampton-Platt A., Chesters D., Ahrens D., and Vogler A. P.. 2014. Building the Coleoptera tree‐of‐life for> 8000 species: composition of public DNA data and fit with Linnaean classification. Syst. Entomol. 39: 97–110. [Google Scholar]
- Breeschoten T., Doorenweerd C., Tarasov S., and Vogler A. P.. 2016. Phylogenetics and biogeography of the dung beetle genus Onthophagus inferred from mitochondrial genomes. Mol. Phylogenet. Evol. 105: 86–95. [DOI] [PubMed] [Google Scholar]
- Brown J., Scholtz C. H., Janeau J. L., Grellier S., and Podwojewski P.. 2010. Dung beetles (Coleoptera: Scarabaeidae) can improve soil hydrological properties. Appl. Soil Ecol. 46: 9–16. [Google Scholar]
- Browne J. and Scholtz C. H.. 1995. Phylogeny of the families of Scarabaeoidea (Coleoptera) based on characters of the hindwing articulation, hindwing base and wing venation. Syst. Entomol. 20: 145–173. [Google Scholar]
- Browne J. and Scholtz C. H.. 1996. Morphology of the hind wing articulation and wing base of the Scarabaeoidea (Coleoptera) with some phylogenetic implications. Bonn. Zool. Monogr. 40: 1–200. [Google Scholar]
- Browne J. and Scholtz C. H.. 1998. Evolution of the scarab hind wing articulation and wing base: a contribution toward the phylogeny of the Scarabaeidae (Scarabaeoidea: Coleoptera). Syst. Entomol. 23: 307–326. [Google Scholar]
- Browne J. and Scholtz C. H.. 1999. A phylogeny of the families of Scarabaeoidea (Coleoptera). Syst. Entomol. 24: 51–84. [Google Scholar]
- Cameron S. L. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59: 95–117. [DOI] [PubMed] [Google Scholar]
- Cannone J. J., Subramanian S., Schnare M. N., Collett J. R., D’Souza L. M., Du Y., Feng B., Lin N., Madabusi L. V., Müller K. M.,. et al. 2002. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinf. 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherman M. and Morón M. A.. 2014. Validación de la familia Melolonthidae Leach, 1819 (Coleoptera: Scarabaeoidea). Acta Zoológica Mexicana (Nueva Serie). 30: 201–220. [Google Scholar]
- Cherman M. A., Moron M. A., and Almeida L. M.. 2016. Phylogenetic relationships within Diplotaxini Kirby (Coleoptera: Melolonthidae: Melolonthinae) with emphasis on Liogenys Guérin-Méneville. Syst. Entomol. 41: 744–770. [Google Scholar]
- Coates B. S. 2014. Assembly and annotation of full mitochondrial genomes for the corn rootworm species, Diabrotica virgifera virgifera and Diabrotica barberi (Insecta: Coleoptera: Chrysomelidae), using Next Generation Sequence data. Gene. 542: 190–197. [DOI] [PubMed] [Google Scholar]
- Crampton-Platt A., M. J. Timmermans M. L. Gimmel S. N. Kutty T. D. Cockerill C. Vun Khen, and Vogler A. P.. 2015. Soup to tree: the phylogeny of beetles inferred by Mitochondrial Metagenomics of a Bornean Rainforest sample. Mol. Biol. Evol. 32: 2302–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle J., R. Myburgh, and Ahrens D.. 2014. The evolution of morphospace in phytophagous scarab chafers: no competition–no divergence?PLoS One. 9: e98536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erichson W. F. 1847. Naturgeschichte der Insecten Deutschlands. Erste Abtheilung. Coleoptera. Nicolaischen Buchhandlung. 3: 481–640. [Google Scholar]
- Gillespie J. J., J. S. Johnston J. J. Cannone, and Gutell R. R.. 2006. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): structure, organization, and retrotransposable elements. Insect Mol. Biol. 15: 657–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillett C. P., A. Crampton-Platt M. J. Timmermans B. H. Jordal B. C. Emerson, and Vogler A. P.. 2014. Bulk de novo mitogenome assembly from pooled total DNA elucidates the phylogeny of weevils (Coleoptera: Curculionoidea). Mol. Biol. Evol. 31: 2223–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall T. A. 1999. BioEdit: a user–friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41: 95–98. [Google Scholar]
- Howden H. F. 1982. Larval and adult characters of Frickius Germain, its relationship to the Geotrupini, and a phylogeny of some major taxa in the Scarabaeoidea (Insecta: Coleoptera). Can. J. Zool. 60: 2713–2724. [Google Scholar]
- Hunt T., J., Bergsten Z., Levkanicova A., Papadopoulou O. S., John R., Wild P. M., Hammond D., Ahrens M., Balke M. S., Caterino, et al. 2007. A comprehensive phylogeny of beetles reveals the evolutionary origins of a superradiation. Science. 318: 1913–1916. [DOI] [PubMed] [Google Scholar]
- Katoh K. and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kück P. and Meusemann K.. 2010. FASconCAT: convenient handling of data matrices. Mol. Phylogenet. Evol. 56: 1115–1118. [DOI] [PubMed] [Google Scholar]
- Lanfear R., B. Calcott S. Y. Ho, and Guindon S.. 2012. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29: 1695–1701. [DOI] [PubMed] [Google Scholar]
- Lartillot N., T. Lepage, and Blanquart S.. 2009. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 25: 2286–2288. [DOI] [PubMed] [Google Scholar]
- Lartillot N. and Philippe H.. 2004. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 21: 1095–1109. [DOI] [PubMed] [Google Scholar]
- Machatschke J. W. 1959. Phylogenetische Untersuchungen über die Sericini (sensu Dalla Torre 1912) (Coleoptera: Lamellicornia, Melolonthidae). Beiträge zur Entomologie. 9: 730–746. [Google Scholar]
- Nguyen L. T., H. A. Schmidt A. von Haeseler, and Minh B. Q.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols E., Spector S., Louzada J., Larsen T., Amezquita S., Favila M. E., and Network T. S. R.. 2008. Ecological functions and ecosystem services provided by Scarabaeinae dung beetles. Biol. Conser. 141: 1461–1474. [Google Scholar]
- Nie R. E., Breeschoten T., Timmermans M. J., Nadein K., Xue H. J., Bai M., Huang Y., Yang X., and Vogler A. P.. 2018. The phylogeny of Galerucinae (Coleoptera: Chrysomelidae) and the performance of mitochondrial genomes in phylogenetic inference compared to nuclear rRNA genes. Cladistics. 34: 113–130. [DOI] [PubMed] [Google Scholar]
- Ouvrard D., B. C. Campbell T. Bourgoin, and Chan K. L.. 2000. 18S rRNA secondary structure and phylogenetic position of Peloridiidae (Insecta: Hemiptera). Mol. Phylogenet. Evol. 16: 403–417. [DOI] [PubMed] [Google Scholar]
- Patel R. K. and Jain M.. 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 7: e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., H. C. Leung S. M. Yiu, and Chin F. Y.. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 28: 1420–1428. [DOI] [PubMed] [Google Scholar]
- Péringuey L. 1904. Descriptive catalogue of the Coleoptera of South Africa. Fam. Lucanidae. Fam. Scarabaeidae. Sub-families: Sericinae, Melolonthinae, Trichiinae, Valginae, Cetoniinae. T. S. Afr. Philos. Soc. 13: 1–288. [Google Scholar]
- Regier J. C., J. W. Shultz A. Zwick A. Hussey B. Ball R. Wetzer J. W. Martin, and Cunningham C. W.. 2010. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 463: 1079–1083. [DOI] [PubMed] [Google Scholar]
- Ritcher P. O. 1958. Biology of Scarabaeidae. Annu. Rev. Entomol. 3: 311–334. [Google Scholar]
- Sanmartín I. and Mmartín-Piera F.. 2003. First phylogenetic analysis of the subfamily Pachydeminae (Coleoptera, Scarabaeoidea, Melolonthidae): the Palearctic Pachydeminae. J. Zool. Syst. Evol. Res. 41: 2–46. [Google Scholar]
- Schmidt H. A., K. Strimmer M. Vingron, and von Haeseler A.. 2002. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 18: 502–504. [DOI] [PubMed] [Google Scholar]
- Shimodaira H. 2002. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51: 492–508. [DOI] [PubMed] [Google Scholar]
- Shimodaira H. and Hasegawa M.. 1999. Multiple comparisons of loglikelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16: 114–116.10331256 [Google Scholar]
- Shimodaira H. and Hasegawa M.. 2001. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 17: 1246–1247. [DOI] [PubMed] [Google Scholar]
- Smith A. B. T. 2006. A review of the family-group names for the superfamily Scarabaeoidea (Coleoptera) with corrections to nomenclature and a current classification. Coleopt. Soc. Monogr. 5: 144–204. [Google Scholar]
- Smith A. B., Hawks D. C., and Heraty J. M.. 2006. An overview of the classification and evolution of the major scarab beetle clades (Coleoptera: Scarabaeoidea) based on preliminary molecular analyses. Coleopts. Bull. 60: 35–46. [Google Scholar]
- Song N., H. Li F. Song, and Cai W.. 2016. Molecular phylogeny of Polyneoptera (Insecta) inferred from expanded mitogenomic data. Sci. Rep. 6: 36175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2015. Using RAxML to Infer Phylogenies. Curr. Protoc. Bioinformatics. 51: 6.14.1–6.1414. [DOI] [PubMed] [Google Scholar]
- Tamura K., G. Stecher D. Peterson A. Filipski, and Kumar S.. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30: 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans M. J., S. Dodsworth C. L. Culverwell L. Bocak D. Ahrens D. T. Littlewood J. Pons, and Vogler A. P.. 2010. Why barcode? High-throughput multiplex sequencing of mitochondrial genomes for molecular systematics. Nucleic Acids Res. 38: e197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans M. J., Barton C., Haran J., Ahrens D., Culverwell C. L., Ollikainen A., Dodsworth S., Foster P. G., Bocak L., and Vogler A. P.. 2016. Family-level sampling of mitochondrial genomes in Coleoptera: compositional heterogeneity and phylogenetics. Genome Biol. Evol. 8: 161–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. 2013. DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 30: 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang Z. and Nielsen R.. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17: 32–43. [DOI] [PubMed] [Google Scholar]
- Zwick A., J. C. Regier, and Zwickl D. J.. 2012. Resolving discrepancy between nucleotides and amino acids in deep-level arthropod phylogenomics: differentiating serine codons in 21-amino-acid models. PLoS One. 7: e47450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.