

Metabolic engineering and synthetic biology are now the key enabling technologies for manipulating microorganisms to suit the practical outcomes desired by humankind. The introduction of exogenous DNA into cells is an indispensable step for this purpose. However, some microorganisms, including the important industrial workhorse Corynebacterium glutamicum, possess a complex cell wall structure to shield cells against exogenous DNA. Although genes responsible for cell wall synthesis in C. glutamicum are known, engineering of related genes to improve cell competency has not been explored yet. In this study, we demonstrate that mutations in cell wall synthesis genes can significantly improve the electrotransformation efficiency of C. glutamicum. Notably, the Y489C mutation in bifunctional peptidoglycan glycosyltransferase/peptidoglycan dd-transpeptidase PonA increased electrotransformation efficiency by 19.25-fold by affecting peptidoglycan synthesis.
KEYWORDS: Corynebacterium glutamicum, electrotransformation, peptidoglycan, ponA
ABSTRACT
Corynebacterium glutamicum is frequently engineered to serve as a versatile platform and model microorganism. However, due to its complex cell wall structure, transformation of C. glutamicum with exogenous DNA is inefficient. Although efforts have been devoted to improve the transformation efficiency by using cell wall-weakening agents, direct genetic engineering of cell wall synthesis for enhancing cell competency has not been explored thus far. Herein, we reported that engineering of peptidoglycan synthesis could significantly increase the transformation efficiency of C. glutamicum. Comparative analysis of C. glutamicum wild-type strain ATCC 13869 and a mutant with high electrotransformation efficiency revealed nine mutations in eight cell wall synthesis-related genes. Among them, the Y489C mutation in bifunctional peptidoglycan glycosyltransferase/peptidoglycan dd-transpeptidase PonA dramatically increased the electrotransformation of strain ATCC 13869 by 19.25-fold in the absence of cell wall-weakening agents, with no inhibition on growth. The Y489C mutation had no effect on the membrane localization of PonA but affected the peptidoglycan structure. Deletion of the ponA gene led to more dramatic changes to the peptidoglycan structure but only increased the electrotransformation by 4.89-fold, suggesting that appropriate inhibition of cell wall synthesis benefited electrotransformation more. Finally, we demonstrated that the PonAY489C mutation did not cause constitutive or enhanced glutamate excretion, making its permanent existence in C. glutamicum ATCC 13869 acceptable. This study demonstrates that genetic engineering of genes involved in cell wall synthesis, especially peptidoglycan synthesis, is a promising strategy to improve the electrotransformation efficiency of C. glutamicum.
IMPORTANCE Metabolic engineering and synthetic biology are now the key enabling technologies for manipulating microorganisms to suit the practical outcomes desired by humankind. The introduction of exogenous DNA into cells is an indispensable step for this purpose. However, some microorganisms, including the important industrial workhorse Corynebacterium glutamicum, possess a complex cell wall structure to shield cells against exogenous DNA. Although genes responsible for cell wall synthesis in C. glutamicum are known, engineering of related genes to improve cell competency has not been explored yet. In this study, we demonstrate that mutations in cell wall synthesis genes can significantly improve the electrotransformation efficiency of C. glutamicum. Notably, the Y489C mutation in bifunctional peptidoglycan glycosyltransferase/peptidoglycan dd-transpeptidase PonA increased electrotransformation efficiency by 19.25-fold by affecting peptidoglycan synthesis.
INTRODUCTION
Corynebacterium glutamicum, a Gram-positive and generally recognized as safe (GRAS) soil bacterium, is widely used for the industrial production of various amino acids, such as glutamate and lysine, the market sizes of which are approximately 3.1 million and 2.2 million tons per year, respectively (1, 2). With the availability of genome sequences (3, 4) and the development of systems metabolic engineering and synthetic biology, C. glutamicum has become an industrial platform organism for the production of amino acids, chemicals, materials, fuels, and recombinant proteins (5–8). High-efficiency introduction of exogenous DNA into cells is required for multiplex and automated strain modification (9–11). Currently, electrotransformation is the most frequently used method for C. glutamicum due to its simplicity and efficiency. However, compared with another widely used platform strain Escherichia coli (about 109 CFU μg−1 DNA), the electrotransformation efficiency of C. glutamicum has only been improved up to 107 CFU μg−1 DNA (for C. glutamicum strain ATCC 13032), even though a complicated competent cell treatment strategy was applied (12). CRISPR/Cas9 genome editing technologies recently reported in C. glutamicum also showed that low transformation efficiency is a main limitation for improving editing efficiency and performing multilocus editing (10, 11). Therefore, efforts are still indispensable to further improve the electrotransformation efficiency of C. glutamicum.
It has been demonstrated that the restriction-modification (RM) system is one of the key barriers for electrotransformation in C. glutamicum (13–15). The typical type II RM system cglMRR was experimentally verified to mainly restrict foreign DNA in C. glutamicum (15). The effect of the RM system on electrotransformation can be avoided by cglMRR deletion or employing E. coli expressing methyltransferase CglM as a xenogeneic plasmid donor host (16, 17). Alternatively, a 6-min heat shock treatment at 46°C immediately following electroporation has frequently been applied to deactivate the restriction system (14). The complex cell wall structure of C. glutamicum (18, 19) is another major barrier that significantly reduces transformation efficiency. The cell wall core of Gram-positive C. glutamicum consists of a thicker peptidoglycan layer than with E. coli, a mycolic acid layer that is unique to the Corynebacteriales order, and a heteropolysaccharide arabinogalactan layer (20). To decrease the cell wall barrier and consequently enhance the cell competency, different cell wall-weakening agents, such as glycine, isonicotinic acid hydrazide (INH), and Tween 80, were added to the medium used for preparing competent cells (21–24). Glycine is known to replace d-alanine in peptidoglycan precursors and loosen the cross-linking between peptidoglycan chains (13, 25, 26). INH is generally recognized to inhibit the synthesis of mycolic acids (22), and Tween 80 changes the carbon chain length and the degree of unsaturation of mycolic acids (27). After systematically optimizing their dosage, the contribution of cell wall-weakening agents to the improvement in electrotransformation efficiency has been fully explored (12). On the contrary, few efforts have been devoted toward identifying and manipulating the genetic factors involved in cell wall synthesis to improve cell competency. Jang and Britz isolated a cell surface mutant of C. glutamicum MLB133 that diminished the proportion of mycolic acids attached to the cell surface and the thickness of the cell wall (19). In combination with use of glycine and INH, the mutant showed increased electrotransformation efficiency. Unfortunately, the underlying genetic mutations were not identified. Therefore, uncovering the genetic basis and molecular mechanism for weakening the cell wall barrier is urgently demanded to further improve electrotransformation efficiency.
We previously isolated a glutamate-hyperproducing mutant of C. glutamicum ATCC 13869 (strain SL4) via physical and chemical mutagenesis and selection. In subsequent studies, it was unexpectedly discovered that the electrotransformation efficiency of strain SL4 was extremely high even in the absence of cell wall modifiers (1.94 × 106 ± 0.48 × 106 CFU μg−1 DNA) (10). Since strain ATCC 13869 lacks the cglMRR RM system, it is speculated that the enhanced transformation efficiency is caused by mutations in cell wall synthesis-related genes. In this study, nine mutations in cell wall synthesis-related genes were discovered by comparative genome analysis, and their effects on transformation efficiency were characterized by introducing them individually into the wild-type strain ATCC 13869. Among two contributing mutations, a Y489C mutation in the ponA gene encoding bifunctional peptidoglycan glycosyltransferase/peptidoglycan dd-transpeptidase significantly increased the electrotransformation efficiency of strain ATCC 13869 by 19.25-fold. Experimental evidence indicated that the Y489C mutation in ponA affected peptidoglycan synthesis. The molecular mechanism reported here will provide a new perspective to manipulate the cell wall synthesis of C. glutamicum and improve its electrotransformation efficiency.
RESULTS
Comparative genome analysis between strains SL4 and ATCC 13869.
The genome of C. glutamicum SL4 was sequenced, and its sequence was compared with that of ATCC 13869. The cell wall synthesis-related genes were identified according to COG annotation and the published literature. For these genes, only 9 single nucleotide polymorphisms (SNPs) located in 8 genes were discovered in the SL4 strain (Table 1), with two mutations in the murJ gene. Insertions, deletions, and rearrangements were not found. The products of the fli, ponA, ddl, and murJ genes are important enzymes responsible for peptidoglycan biosynthesis (28–30). The gene emb, encoding arabinose transferase, is crucial for arabinogalactan synthesis (31, 32). The genes ufaA, fadD4, and otsB are involved in mycolic acid biosynthesis (28, 33).
TABLE 1.
Mutations related to cell wall synthesis in strain SL4
| Gene name | Locus_taga | Functional annotationb | Nucleotide change | Amino acid change | Category |
|---|---|---|---|---|---|
| fli | BBD29_RS00175 | Flippase | GCC/GTC | A63V | Peptidoglycan synthesis |
| ponA | BBD29_RS01645 | Penicillin binding protein 1A | TAC/TGC | Y489C | |
| ddl | BBD29_RS07135 | d-Alanine-d-alanine ligase | GGC/AGC | G256S | |
| murJ | BBD29_RS15295 | Lipid II flippase MurJ/MviN | CCG/TCG | P373S | |
| ACC/ATC | T882I | ||||
| emb | BBD29_RS01155 | Arabinosyl transferase | GCC/ACC | A962T | Arabinogalactan synthesis |
| ufaA | BBD29_RS07910 | Cyclopropane fatty acid synthase | AGG/AAG | R90K | Mycolic acid synthesis |
| fadD4 | BBD29_RS06420 | Fatty-acid-CoA ligase | GGC/AGC | G32S | |
| otsB | BBD29_RS12855 | Trehalose-6-phosphatase | GTT/ATT | V69I |
Locus_tag is the identifier of each gene in strain ATCC 13869 genome sequence from NCBI database (accession number NZ_CP016335).
CoA, coenzyme A.
Identification of mutations responsible for improved electrotransformation efficiency.
To evaluate the effects of the mutations on electrotransformation efficiency, eight mutant strains harboring individual gene mutations were constructed from the wild-type strain C. glutamicum ATCC 13869 by using the pK18mobsacB system via homologous double-crossover recombination (34). Competent cells of the resulting mutants and the wild-type strain were prepared using LBG medium (LB supplemented with 0.5% [wt/vol] glucose) without cell wall modifiers, and the electrotransformation efficiency was then determined (Fig. 1). The ponAY489C mutant showed the highest efficiency (6.36 × 104 ± 1.00 × 104 CFU μg−1 DNA), 19.25-fold higher than that of the wild-type strain (3.14 × 103 ± 0.32 × 103 CFU μg−1 DNA). Another mutant, the ddlG256S mutant, also achieved an improved efficiency (1.72 × 104 ± 0.30 × 104 CFU μg−1 DNA), 4.48-fold higher than that of the wild-type strain. On the other hand, the mutants with ufaAR90K and fadD4G32S had 1.34-fold and 1.82-fold lower efficiencies than that of the wild-type strain, respectively. The rest of the mutants presented efficiencies similar to that of the wild-type strain. These results showed that mutations in genes ponA and ddl both improved the electrotransformation efficiency of C. glutamicum. To test the combinational effect of ponAY489C and ddlG256S mutations, the ponAY489C ddlG256S double-mutant strain was then constructed. However, combination of the two mutations did not further increase the efficiency (5.51 × 104 ± 0.24 × 104 CFU μg−1 DNA), which is only 16.55-fold higher than that of the wild-type strain. Since the ponAY489C mutant showed the highest transformation efficiency, it was further studied in the subsequent experiments.
FIG 1.
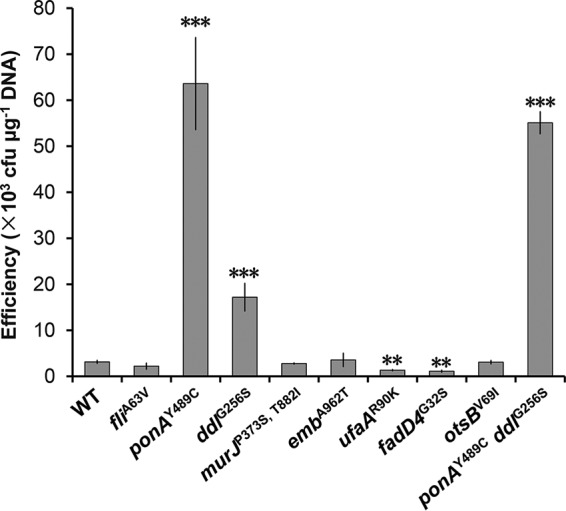
Electrotransformation efficiencies of the ATCC 13869 wild-type (WT) strain and mutants. Competent cells were prepared using LBG medium without cell wall-weakening agents. The number of cells used for each electrotransformation was approximately 108. Error bars indicate standard deviations from the results from three parallel experiments. Asterisks indicate significant changes in electrotransformation efficiency based on a comparison between the mutants and the wild-type strain. **, P ≤ 0.01; ***, P ≤ 0.001 (Student’s two-tailed t test).
Different effects of Y489C point mutation and deletion of ponA on electrotransformation efficiency.
PonA, encoded by ponA (gene synonym, pbp1a), is a bifunctional enzyme acting as both transpeptidase and transglycosylase in the peptidoglycan synthesis (Fig. 2A). To verify whether the Y489C mutation in ponA improved electrotransformation efficiency by deactivating PonA, a ponA-deleted mutant (ΔponA) was constructed. The transformation efficiency of the ΔponA mutant was compared with those of the wild-type strain and ponAY489C mutant. Similar to the result shown in Fig. 1, the efficiency of ponAY489C mutant (5.05 × 104 ± 0.78 × 104 CFU μg−1 DNA) was 17.04-fold higher than that of the wild-type strain (2.8 × 103 ± 0.46 × 103 CFU μg−1 DNA). Surprisingly, the efficiency of the ΔponA mutant (1.65 × 104 ± 0.46 × 104 CFU μg−1 DNA) was only 4.89-fold higher than that of the wild-type strain (Fig. 2B), which was much lower than that of the ponAY489C mutant. Since ponA plays an important dual role in cell wall synthesis, its deletion may make cells more susceptible to electroporation or affect cell growth, which consequently have negative effects on the electrotransformation efficiency. To investigate these possibilities, the cell viability after electroporation was first determined. However, no significant decrease in cell viability was observed in the case of ponA deletion (Fig. 2B). Then, the growth curves of the ATCC 13869, ponAY489C mutant, and ΔponA mutant strains were determined. The ponAY489C mutant had the same growth rate as the wild-type strain, and the ΔponA mutant exhibited a slight but nonsignificant decrease in growth, suggesting that the transformation efficiency changes were not caused by different growth characteristics (Fig. 2C). These results demonstrate that the site mutation Y489C does not disrupt the function of PonA, at least not completely.
FIG 2.

Effects of Y489C point mutation and deletion of ponA on electrotransformation efficiency. (A) Function of PonA in transglycosylation and transpeptidation of cell wall synthesis. (B) Electrotransformation efficiency and susceptibility to electroporation of the wild-type (WT), ponAY489C, and ΔponA strains in the absence of cell wall-weakening agents. Competent cells were prepared using LBG medium without cell wall-weakening agents. The cells used for each electrotransformation were approximately 108. After electroporation and recovery, cells were spread on selective LBG plates supplemented with 25 µg/ml kanamycin to determine the electrotransformation efficiency and LBG plates without kanamycin to determine the susceptibility to electroporation. (C) Growth curves of wild-type (WT), ponAY489C, and ΔponA strains. Cells were cultured in LBG medium with an initial OD600 value of 0.2. The cell density was determined every 3 hours. (D) Electrotransformation efficiency of wild-type (WT), ponAY489C, and ΔponA strains in the presence of cell wall-weakening agents. Competent cells were prepared using NCM medium supplemented with glycine, threonine, INH and Tween 80 according to the protocol described previously (12). Error bars indicate standard deviations from three parallel experiments. Asterisks indicate significant changes in electrotransformation efficiency and susceptibility to electroporation based on a comparison between the mutants and the wild-type strain. **, P ≤ 0.01; ***, P ≤ 0.001 (Student’s two-tailed t test). NS indicates nonsignificant change between the wild-type strain and the mutant based on Student’s two-tailed t test (P > 0.05).
Although the ponAY489C mutant shows enhanced electrotransformation efficiency compared with the wild-type ATCC 13869 strain, the efficiency is still lower than that of mutant SL4 (1.94 × 106 ± 0.48 × 106 CFU μg−1 DNA). Next, we tested whether the combinational use of the ponAY489C mutation and cell wall-weakening agents could further boost the electrotransformation efficiency. By using the cell wall-weakening agents described by Ruan et al. (12), the electrotransformation efficiencies of all the ATCC 13869 wild-type strain and its derivatives increased. Notably, the use of ponAY489C mutation along with cell wall-weakening agents boosts the electrotransformation efficiency to 2.36 × 106 ± 0.53 × 106 CFU μg−1 DNA, which was approximately 50-fold higher than the efficiency obtained without cell wall-weakening agents (Fig. 2D). These results suggest that the combined use of genetic modification and cell wall-weakening agents is a promising strategy to improve the electrotransformation efficiency of C. glutamicum. It should be noted that although the protocol described by Ruan et al. (12) was also useful for strain ATCC 13869 and its derivatives, the electrotransformation efficiencies obtained were not as high as that of strain ATCC 13032 (107 CFU μg−1 DNA). This might be because the protocol was optimized for strain ATCC 13032 rather than strain ATCC 13869. There should still be room for improvement in the electrotransformation efficiency of strain ATCC 13869.
Detecting membrane localization of the PonAY489C mutant.
In a previous study, Valbuena and coworkers constructed and overexpressed a green fluorescent protein (GFP)-PonA fusion protein in C. glutamicum to investigate the localization of PonA (enzyme synonym penicillin-binding protein 1a [PBP1a]). The results suggest that PonA is a membrane-bound enzyme with a transmembrane region (29). According to the motif analysis of PonA in C. glutamicum (KEGG; http://www.kegg.jp/ssdb-bin/ssdb_motif?kid=cgl:NCgl0274), the point mutation Y489C was located on the transpeptidase domain of PonA but not the conserved active sites (35). To determine whether the mutation would affect the membrane localization of PonA, we constructed gfp-ponA and gfp-ponAY489C fusion genes according to the design by Valbuena and coworkers, which was fusing gfp to the 5′ end of the ponA gene. The gfp-ponA and gfp-ponAY489C fusion genes were overexpressed by plasmids in the ponA-deleted C. glutamicum host, and cells in the exponential phase of growth were collected for observation. As shown in Fig. 3, there was no obvious difference in the localization between the GFP-PonA and GFP-PonAY489C fusion proteins. More fluorescence was localized at the cell division septa, which was consistent with previous reports (29). This result suggests that the Y489C mutation has no obvious effect on the membrane localization of PonA.
FIG 3.

Membrane localization of GFP-PonA and GFP-PonAY489C fusion proteins in the ΔponA mutant. Microscope images under visible light and fluorescence are shown. White arrows point out the typical membrane localization of GFP-PonA and GFP-PonAY489C fusion proteins. Bars represent 5 μm. Fluorescence was excited at 480 nm, and the emission was monitored at 527 nm.
Effects of Y489C point mutation and deletion of ponA on peptidoglycan structure.
To investigate the mechanism of ponA mutation/deletion-induced efficient electrotransformation, unincorporated peptidoglycan precursors of the wild-type strain and mutants were analyzed using vancomycin fluorescence (Van-FL) staining. Vancomycin can bind to the un-cross-linked d-Ala-d-Ala termini within peptidoglycan at the outer site of the cell surface. Therefore, the labeled vancomycin staining can be used to detect the distribution and amount of un-cross-linked d-Ala-d-Ala termini in the cell wall (36). Wild-type C. glutamicum cells were stained exclusively at the cell poles and later in the cell cycle at midcell (Fig. 4A), which was consistent with previous studies (29, 37). The ponAY489C and ΔponA mutants showed the same Van-FL staining patterns on the cell wall as the wild-type strain (Fig. 4A). However, the fluorescence intensities of vancomycin stained ponAY489C and ΔponA mutants were 38.9% and 82.0% stronger compared than the wild-type strain, respectively, suggesting that the mutation and deletion of ponA result in changes in peptidoglycan structure (Fig. 4B). Changes in the cell wall structure may affect the susceptibility of cells to beta-lactam antibiotics (38). Therefore, the susceptibility to penicillin, a beta-lactam antibiotic, was investigated for the wild-type strain and ponA mutants by determining the MIC. Tetracycline that inhibits protein synthesis was used as a control. The ponAY489C mutation or deletion of ponA made cells more susceptible to penicillin than was the wild-type strain, while the ponA-deleted mutant had the lowest MIC (Fig. 4C and D). No difference in the MIC of tetracycline was observed for the three strains (Fig. 4C). The results are consistent with the Van-FL data, suggesting that ponA deletion alters the cell wall structure more dramatically than does the ponAY489C mutation.
FIG 4.

Vancomycin fluorescence (Van-FL) staining and penicillin susceptibility of the WT, ponAY489C mutant, and ΔponA mutant strains. (A) Microscope images of Van-FL-stained cells under fluorescence. Bars represent 5 μm. (B) Fluorescence intensities of Van-FL stained cells. When the OD600 of the culture reached 1.0, Van-FL was added to the culture with a final concentration of 100 µg/ml. The mixture was incubated for another 30 min at 30°C. After washing 3 times with PBS buffer, the Van-FL-stained cells were used for fluorescence microscope observation (λ excitation = 480 nm, λ emission = 527 nm) and fluorescence intensity determination by using a fluorimeter (λ excitation = 490 nm, λ emission = 520 nm). Error bars indicate standard deviations from the results from three parallel experiments. Asterisks indicate significant changes in fluorescence intensity based on a comparison between the mutants and the wild-type strain. ***, P ≤ 0.001 (Student’s two-tailed t test). (C) MICs of penicillin and tetracycline for different strains. Three parallel experiments were conducted, and the same MIC was obtained. (D) Determination of penicillin MIC using M.I.C.Evaluator strips (256 to 0.015 μg/ml). One of the three parallel experiments is shown here.
Effects of ponAY489C mutation on glutamate production.
According to previous studies, modifications in cell wall synthesis, such as treatment with penicillin, caused glutamate excretion (39). Since the ponAY489C mutation affected the peptidoglycan synthesis, its effects on glutamate production were investigated. In shake flasks, no glutamate production was observed under biotin-rich conditions for both the wild-type strain and the ponAY489C mutant, suggesting that the ponAY489C mutation did not cause constitutive glutamate secretion (Fig. 5A). Under biotin limitation, extracellular glutamate was accumulated by both the wild-type strain (10.7 ± 1.5 g/liter) and the ponAY489C mutant (9.3 ± 1.5 g/liter). No significant differences in glutamate titers, cell growth, or glucose consumption were observed for these two strains (Fig. 5A). Then, fermentations in bioreactors were conducted to further investigate the effects of ponAY489C mutation on glutamate production. After 38 h of fermentation, the wild-type strain produced 65.7 g/liter glutamate, with a conversion yield of 0.52 g/g. The final glutamate titer and conversion yield obtained by the ponAY489C mutant were 71.7 g/liter and 0.51 g/g, respectively (Fig. 5B). The slight advantage of the ponAY489C mutant in glutamate titer was due to its slightly higher biomass (optical density at 600 nm [OD600], 38.3) than that of the wild-type strain (OD600, 41.1). However, the amount of glutamate produced per OD600 was almost the same for these two strains (1.7 g glutamate/OD600). The results suggest that the ponAY489C mutation is unlikely to cause dramatic changes to metabolic flux, which is important for further metabolic engineering of C. glutamicum strains harboring this mutation.
FIG 5.

Glutamate fermentation by the WT strain and the ponAY489C mutant. (A) Glutamate fermentation in shake flasks. Strains were cultivated in biotin-rich or biotin-poor fermentation medium supplemented with 80 g/liter glucose at 30°C and 220 rpm. OD600, glucose consumption, and glutamate production were detected after 24 h of fermentation. Error bars indicate standard deviations from the results from three parallel experiments. NS indicates a nonsignificant change based on Student’s two-tailed t test (P > 0.05). (B) Glutamate fermentation in 5-liter bioreactors. Strains were cultivated in biotin-poor fermentation medium supplemented with 140 g/liter glucose. Samples were picked periodically, and the OD600, glucose consumption, and glutamate production were detected. The data shown are the average and standard deviations of the results from three parallel determinations.
DISCUSSION
Metabolic engineering and synthetic biology are the mainly used strategies for rational or semirational design of industrial strains (1, 2, 8). However, the low transformation efficiency mainly caused by cell wall barriers severely limits efficient genetic engineering of C. glutamicum, an important industrial workhorse (8). Although exploiting cell wall-weakening agents can weaken the barrier and improve the electrotransformation efficiency to a certain extent (13), it is very difficult for further improvement due to unclear molecular mechanisms. In this study, the ponA gene encoding bifunctional peptidoglycan glycosyltransferase/peptidoglycan dd-transpeptidase was identified as an effective target for increasing the cell competency of C. glutamicum by altering the cell wall structure. The point mutation (Y489C) and deletion of ponA improved the electrotransformation efficiency by 19.25-fold and 4.89-fold, respectively (Fig. 1 and 2B). The ponAY489C mutant can be used as an industrial chassis due to its higher electrotransformation efficiency and no obvious effects on growth and metabolic flux (Fig. 2C and 5). We demonstrate that genetic modification of genes involved in cell wall synthesis is a useful strategy to improve the cell competency of C. glutamicum.
The mechanism for ponA mutation-induced efficient electrotransformation is probably relevant to the changes in peptidoglycan structure. A Van-FL staining assay suggested that the point mutation (Y489C) and deletion of ponA showed increased fluorescence intensity (Fig. 4B). There are several possible interpretations for this phenomenon. Because Van-FL can bind to the un-cross-linked d-Ala-d-Ala termini within peptidoglycan (36), the obvious interpretation is that the amount of un-cross-linked d-Ala-d-Ala termini within peptidoglycan increases due to ponA mutations. That means that ponA mutations may directly affect the transpeptidation degree of peptidoglycan. Second, the cross-linking reaction may be decelerated by ponA mutations, resulting in more un-cross-linked d-Ala-d-Ala termini bound by Van-FL. Third, ponA mutations may allow more Van-FL to contact with d-Ala-d-Ala termini due to structure changes in peptidoglycan. Overall, ponA mutations affected the cell wall synthesis, specifically peptidoglycan synthesis, and improved the electrotransformation efficiency of C. glutamicum.
Besides the ponA gene, there are four paralogous genes of ponA in C. glutamicum, including the pbp1b gene encoding an enzyme with both transpeptidase and transglycosylase activities and other three genes (pbp2a, pbp2b, and pbp3) encoding enzymes with only transpeptidase activity (29). These genes are very important for polar cell wall synthesis and cell division. It has been proven that pbp3 is an essential gene for C. glutamicum (40). The deletion of pbp2b resulted in elongated cells with multiple septa and reduced polar growth. Although a single deletion of ponA, pbp1b, and pbp2a did not cause significant change in cell morphology, double deletion of any two of ponA, pbp1b, pbp2a, and pbp2b gave rise to noticeable changes in cell morphology and growth (29). Therefore, effects of the four paralogous genes on electrotransformation efficiency of C. glutamicum deserve to be deeply analyzed in future work. In addition to genes encoding transpeptidase and transglycosylase, a mutation in the ddl gene that encodes d-alanine-d-alanine ligase, another key enzyme involved in peptidoglycan synthesis, also achieved an improved electrotransformation efficiency (4.48-fold higher; Fig. 1). Therefore, there is reason to believe that genetic modification targeting genes involved in peptidoglycan synthesis is a promising strategy to improve the electrotransformation efficiency of C. glutamicum.
MATERIALS AND METHODS
Comparative genome analysis.
The complete genome sequence of C. glutamicum ATCC 13869 was obtained from the NCBI database (GenBank accession number NZ_CP016335). Insertions, deletions, and rearrangements in the genome sequence of strain SL4 were determined by aligning the genome sequences of strains SL4 and ATCC 13869 using progressiveMauve (41). The mutations, including indels and SNPs between strains SL4 and ATCC 13869, were analyzed by using the SAMtools package (42) with second-generation sequencing data of strain SL4 after quality filtering. COG annotation of the strain ATCC 13869 genome sequence was performed by using the COG functional classification system (https://www.ncbi.nlm.nih.gov/COG/). Genes belonging to the COG functional category “cell wall/membrane/envelope biogenesis” were extracted, and the cell wall synthesis-related genes were selected based on their specialized functions. The functions of other genes involved in the synthesis of mycolic acid and arabinogalactan were obtained from published literature (20, 28, 43).
Bacterial strains, cultivation conditions, plasmids, and general methods.
All bacterial strains used in this study are listed in Table 2. E. coli DH5α was used for general cloning and was cultivated aerobically in Luria-Bertani broth (LB) (10 g/liter tryptone, 5 g/liter yeast extract, 10 g/liter NaCl) at 37°C. Kanamycin (50 μg/ml) was added to LB broth as required. C. glutamicum ATCC 13869 and its derivatives were cultivated aerobically at 30°C in LBG (LB supplemented with 0.5% [wt/vol] glucose). Kanamycin (25 μg/ml) was added to the medium as required. All plasmids used in this study (Table 2) were constructed using the ClonExpress II one-step cloning kit or ClonExpress MultiS one-step cloning kit (Vazyme, Nanjing, China), which facilitated the ligation of two or more DNA fragments through 15- to 20-bp overlaps. The primers used for genetic manipulation are listed in Table 3.
TABLE 2.
Bacterial strains and plasmids used in this studya
| Strain or plasmid | Relevant genotype or description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| DH5α | F− supE44 ΔlacU169 (φ80lacZΔM15)hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | CWBiotech |
| C. glutamicum | ||
| ATCC 13869 | Wild-type C. glutamicum | ATCC |
| SL4 | Mutant strain derived from ATCC 13869 | Lab stock |
| fliA63V mutant | ATCC 13869 derivative with A63V mutation of fli | This work |
| ponAY489C mutant | ATCC 13869 derivative with Y489C mutation of ponA | This work |
| ddlG256S mutant | ATCC 13869 derivative with G256S mutation of ddl | This work |
| murJP373S-T882I mutant | ATCC 13869 derivative with P373S and T882I mutations of murJ | This work |
| embA962T mutant | ATCC 13869 derivative with A962T mutation of emb | This work |
| ufaAR90K mutant | ATCC 13869 derivative with R90K mutation of ufaA | This work |
| fadD4G32S mutant | ATCC 13869 derivative with G32S mutation of fadD4 | This work |
| otsBV69I mutant | ATCC 13869 derivative with V69I mutation of otsB | This work |
| ponAY489C ddlG256S mutant | ATCC 13869 derivative with both Y489C mutation of ponA and G256S mutation of ddl | This work |
| ΔponA mutant | ATCC 13869 with ponA deletion | This work |
| ΔponA(pEC-gfp-ponA) mutant | ΔponA derivative harboring pEC-gfp-ponA | This work |
| ΔponA(pEC-gfp-ponAY489C) mutant | ΔponA derivative harboring pEC-gfp-ponAY489C | This work |
| Plasmid | ||
| pK18mobsacB | Vector for allelic exchange in C. glutamicum, pK18 oriVE. coli sacB lacZα (Kanr) | 44 |
| pEC-XK99E | E. coli-C. glutamicum shuttle expression vector (Kanr) | 13 |
| pTRCmob-egfp | pTRCmob derivative carrying gfp | 39 |
| pK-fliA63V | pK18mobsacB harboring a 1.9-kb homologous fragment for fliA63V construction | This work |
| pK-ponAY489C | pK18mobsacB harboring a 2.3-kb homologous fragment for ponAY489C construction | This work |
| pK-ddlG256S | pK18mobsacB harboring a 2.0-kb homologous fragment for ddlG256S construction | This work |
| pK-murJP373S,T882I | pK18mobsacB harboring a 3.8-kb homologous fragment for murJP373,T882I construction | This work |
| pK-embA962T | pK18mobsacB harboring a 2.4-kb homologous fragment for embA962T construction | This work |
| pK-ufaAR90K | pK18mobsacB harboring a 2.4-kb homologous fragment for ufaAR90K construction | This work |
| pK-fadD4G32S | pK18mobsacB harboring a 2.1-kb homologous fragment for fadD4G32S construction | This work |
| pK-otsBV69I | pK18mobsacB harboring a 1.6-kb homologous fragment for otsBV69I construction | This work |
| pK-ΔponA | pK18mobsacB harboring a 2.1-kb homologous fragment for ponA deletion | This work |
| pEC-gfp-ponA | pEC-XK99E derivative containing gfp-ponA under Ptrc promoter | This work |
| pEC-gfp-ponAY489C | pEC-XK99E derivative containing gfp-ponAY489C under Ptrc promoter | This work |
ATCC, American Type Culture Collection. Kanr, kanamycin resistance.
TABLE 3.
Primers used in this study
| Primer | Sequence (5′ to 3′) |
|---|---|
| Hfli-F | GAGCTCGGTACCCGGGGATCCTTTTTGCTCGCTCTGACACATCTTC |
| Hfli-R | ACGACGGCCAGTGCCAAGCTTACGAGGTTGAGCACCACATTGAGTA |
| HponA-F | GAGCTCGGTACCCGGGGATCCGCGTACCAATGACACGATGCAGACC |
| HponA-R | ACGACGGCCAGTGCCAAGCTTTTGAGCAACTAGGTATTTCTAGCGG |
| Hddl-F | GAGCTCGGTACCCGGGGATCCACTGAAATCGGTGGTGCGTGTAAGA |
| Hddl-R | ACGACGGCCAGTGCCAAGCTTATGCACTTCGCTTTTCGACGCCCTC |
| HmurJ-F | GAGCTCGGTACCCGGGGATCCGCCACCAACCCACTAACGTA |
| HmurJ-R | ACGACGGCCAGTGCCAAGCTTTGGAAAATGCCCTGGAATCG |
| Hemb-F | GAGCTCGGTACCCGGGGATCCACCACGAATATGTGCGCTACCAAAC |
| Hemb-R | ACGACGGCCAGTGCCAAGCTTGGTTAGAAACCAGATAGCACAGATG |
| HufaA-F | GAGCTCGGTACCCGGGGATCCTCAGCTGGATCCTCGCAGATCTCAG |
| HufaA-R | ACGACGGCCAGTGCCAAGCTTGTGCAGCTATTGCACCGTTCATCGC |
| HfadD4-F | GAGCTCGGTACCCGGGGATCCCGGACTCCTTCGCTGCAACA |
| HfadD4-R | ACGACGGCCAGTGCCAAGCTTGCCCTGGGAGACTCGATAATTCCAC |
| HotsB-F | GAGCTCGGTACCCGGGGATCCGGCAGCTGTCCATGATTTGAAG |
| HotsB-R | ACGACGGCCAGTGCCAAGCTTGATCTCACCACCGTCATCCAAC |
| fliA63V-F | CAAGCCTGGGAAACTAGCACTTA |
| fliA63V-R | CCGGAATTTAGGATCCAACAACTTCG |
| ponAY489C-F | CGGTGTCCTCCCACGTTGATCC |
| ponAY489C-R | GCAGTTCCGCTGACCGTATCCTT |
| ddlG256S-F | AGCCTGTATGCCACCGCTGAT |
| ddlG256S-R | GCTGCACCAACACATCTAGCAAT |
| murJP373S,T882I-F1 | GCGTGGGCACCTGTTGTCAA |
| murJP373S,T882I-R1 | CAACAGGGACAGCACGACCTT |
| murJP373S,T882I-F2 | CAACGGTTGCCTCATTGTG |
| murJP373S,T882I-R2 | ATCTGTTCCTGCACCGTCATCC |
| embA962T-F | ACCATCAATCAGCCAGGACCAAG |
| embA962T-R | TTTCGTAGGCGTTGTTTACTGTTT |
| ufaAR90K-F | ATCGCCGGATTAAACCCACTTCG |
| ufaAR90K-R | CACTGCTGGGAAACTCCAACACA |
| fadD4G32S-F | CCCCTTCAGCAATTCTCAGTCACA |
| fadD4G32S-R | CGCTAGAGAAATCATTCGGTCCAA |
| otsBV69I-F | CTGCCTGGTTCTTTTGAAATCTC |
| otsBV69I-R | ATTCCTTCAGCCAGGTGCCCTT |
| LHΔponA-F | GAGCTCGGTACCCGGGGATCCGTTAGTAATGTGCGGCTTCT |
| LHΔponA-R | TCCAACAGCCTAGCGATAAC |
| RHΔponA-F | GGTTATCGCTAGGCTGTTGGAACAACCATTGAAGACGCCAT |
| RHΔponA-R | ACGACGGCCAGTGCCAAGCTTCCATCCGTGTTCGATGAAAG |
| ΔponA-F | GAGCCAAATATTCAGCCC |
| ΔponA-R | CTTTTGGTTGGCGTCTTG |
| gfp-F | ACAGGCCAAAGGAGTTGAGAATGAGTAAAGGAGAAGAACTTTTCA |
| gfp-R | ATGTTTGTATAGTTCATCCATGCCA |
| ponA-F | TGGATGAACTATACAAACATGTGTCCACCACGAATTCTCTGA |
| ponA-R | CCAAGCTTGCATGCCTGCAGCTAGCGGAAGAACTGGTTGATG |
| pEC-F | CTGCAGGCATGCAAGCTTGG |
| pEC-R | TCTCAACTCCTTTGGCCTGTGTGAAATTGTTATC |
Construction of C. glutamicum ATCC 13869 mutants.
To construct the fliA63V, ponAY489C, ddlG256S, embA962T, ufaAR90K, fadD4G32S, and otsBV69I single-point mutants by using the sacB-based system (34), plasmids pK-fliA63V, pK-ponAY489C, pK-ddlG256S, pK-emb962T, pK-ufaAR90K, pK-fadD4G32S, and pK-otsBV69I were constructed, respectively. The homologous sequences containing single-nucleotide substitutions in the fli, ponA, ddl, emb, ufaA, fadD4, and otsB genes were amplified by PCR from the genomic DNA of C. glutamicum SL4 using the specific primer pairs Hfli-F/Hfli-R, HponA-F/HponA-R, Hddl-F/Hddl-R, Hemb-F/Hemb-R, HufaA-F/HufaA-R, HfadD4-F/HfadD4-R, and HotsB-F/HotsB-R, respectively. The homologous segments were subcloned into the HindIII and BamHI sites of pK18mobsacB, respectively. The recombinant plasmids were individually introduced into C. glutamicum ATCC 13869 through electrotransformation, according to the previously described procedure (12). Mutants were finally confirmed by sequencing with the fliA63V-F/fliA63V-R, ponAY489C-F/ponAY489C-R, ddlG256S-F/ddlG256S-R, embA962T-F/embA962T-R, ufaAR90K-F/ufaAR90K-R, fadD4G32S-F/fadD4G32S-R, and otsBV69I-F/otsBV69I-R primer pairs, respectively.
To construct the murJP373S-T882I dinucleotide mutant, plasmid pK-murJP373S-T882I was constructed. The homologous fragment containing dinucleotide substitution of gene murJ was amplified by PCR from the genomic DNA of C. glutamicum SL4 using the specific primer pair HmurJ-F/HmurJ-R and inserted into the HindIII and BamHI sites of pK18mobsacB. Plasmid pK-murJP373S-T882I was introduced into C. glutamicum ATCC 13869, and the murJP373S single-nucleotide mutant was first obtained. Then, plasmid pK-murJP373S-T882I was further introduced into the murJP373S mutant to construct the murJP373S-T882I dinucleotide mutant. The murJP373S mutant and murJP373S-T882I dinucleotide mutant were confirmed by sequencing with the murJP373S-T882I-F1/murJP373S-T882I-R1 and murJP373S-T882I-F2/murJP373S-T882I-R2 primer pairs, respectively. To construct the ponAY489C ddlG256S dinucleotide mutant, pK-ddlG256S was introduced into the ponAY489C mutant, and the ponAY489C ddlG256S mutant was verified using the same set of primers for ddlG256S.
To construct the ΔponA mutant, plasmid pK-ΔponA was constructed. The left and right homologous fragments for deleting ponA were amplified from the genomic DNA of C. glutamicum ATCC 13869 by PCR using the LHΔponA-F/LHΔponA-R and RHΔponA-F/RHΔponA-R primer pairs, respectively. The left and right homologous fragments were then subcloned into the HindIII and BamHI sites of pK18mobsacB. Then, plasmid pK-ΔponA was introduced into strain ATCC 13869 to construct the ΔponA mutant, which was confirmed by colony PCR with the ΔponA-F/ΔponA-R primer pair.
Testing electrotransformation efficiency of C. glutamicum.
C. glutamicum ATCC 13869 and its derivatives cultivated overnight in LBG medium were inoculated into 100 ml fresh LBG medium to an optical density at 600 nm (OD600) of 0.2. When the OD600 of the culture reached ∼1.0, cells were chilled on ice for 15 min and harvested by centrifugation at 4°C and 5,000 × g for 5 min. After washing with ice-cold deionized distilled water twice and then with 10% (vol/vol) glycerol twice, cells were resuspended in 0.8 ml of 10% (vol/vol) glycerol, and 100-µl aliquots (approximately 108 cells) of competent cells were obtained. Plasmid DNA (100 ng, ∼2 μl) was added to the competent cells and transferred to a precooled 2-mm electroporation cuvette (Bio-Rad). Electroporation was performed using the Eppendorf Electroporator 2510, with parameters set at 2,500 V and 5 ms. After electroporation, 1 ml LBG medium was added immediately, and the suspension was quickly incubated at 46°C for 6 min. Cells were incubated for 1 h at 30°C and spread on selective LBG medium supplemented with 25 µg/ml kanamycin. The number of colonies was counted from selective plate after 36 h of culture at 30°C.
To investigate the synergy between the ponAY489C mutation and cell wall-weakening agents on electrotransformation efficiency, competent cells of C. glutamicum ATCC 13869 and its derivatives were prepared using non-colony type monolayer culture (NCM) medium supplemented with glycine, threonine, INH, and Tween 80, according to the protocol described previously (12). After electroporation and recovery, cells were spread on selective LBHIS (5 g/liter tryptone, 5 g/liter NaCl, 2.5 g/liter yeast extract, 18.5 g brain heart infusion [BHI], 91 g/liter sorbitol, 18 g/liter agar, pH 7.2) plates supplemented with 25 µg/ml kanamycin (12). The number of colonies was counted from the selective plates after 36 h culture at 30°C.
Membrane localization analysis of PonA.
To analyze the membrane localization of PonA and PonAY489C, the gfp-ponA and gfp-ponAY489C fusion genes were constructed by fusing gfp to the 5′ ends of ponA and ponAY489C, respectively. First, gfp was amplified from pTRCmob-egfp with the gfp-F/gfp-R primer pair. The stop codon of gfp was removed, and a 20-bp overlap with the pEC-XK99E backbone was added to the 5′ end of the PCR product. Second, the wild-type and mutant ponA were PCR amplified from the genomic DNAs of C. glutamicum strains ATCC 13869 and SL4, respectively, using the ΔponA-F/ΔponA-R primer pair. Meanwhile, 20-bp overlaps with the gfp gene and the pEC-XK99E backbone were added to the 5′ end and the 3′ end of the PCR product, respectively. Third, the pEC-XK99E backbone was amplified from the circular plasmid using the pEC-F/pEC-R primer pair. Finally, the three fragments were assembled using the ClonExpress MultiS one-step cloning kit (Vazyme, Nanjing, China). The resulting plasmids, pEC-gfp-ponA and pEC-gfp-ponAY489C, were separately introduced into the ΔponA mutant through electrotransformation. The transformants were cultivated in LBG medium containing 25 μg/ml kanamycin and 0.1 mM isopropyl thio-β-d-galactopyranoside (IPTG). Cells of the exponential-growth phase were observed with a Leica DM5000B fluorescence microscope equipped with an HCX PL APO ×100/1.40-0.70 oil objective. Images were taken with a DN100 Nikon digital camera and assembled using the Corel Draw software. Fluorescence was excited at 480 nm, and the emission was monitored at 527 nm.
Van-FL staining and fluorescence measurement.
Vancomycin (Sigma, USA) and 5(6)-carboxyfluorescein-N-hydroxysuccinimide ester (FLUOS; Thermo Fisher Scientific, USA) were used to prepare Van-FL (vancomycin-FLUOS) solution according to the procedure described by Daniel and Errington (36). Cells were grown in 20 ml LBG medium with an initial OD600 of 0.3. When the OD600 of the culture reached 1.0, Van-FL was added to a final concentration of 100 µg/ml. The culture was then incubated at 30°C for another 30 min to allow absorption of Van-FL. Then, the Van-FL stained cells were washed with phosphate-buffered saline (PBS) buffer for three times and observed by using a Leica DM5000B fluorescence microscope equipped with an HCX PL APO ×100/1.40-0.70 oil objective. Images were taken with a DN100 Nikon digital camera and assembled using the Corel Draw software. Fluorescence was excited at 480 nm, and the emission was monitored at 527 nm. The fluorescence intensity of the Van-FL-stained cells was determined by using a FluoroMax-3 fluorimeter (Horiba, Japan) (λ excitation = 490 nm, λ emission = 520 nm).
Determination of MIC of antibiotics.
M.I.C.Evaluator strips (Oxoid; penicillin, 256 to 0.015 μg/ml, product no. MA0101D; tetracycline, 256 to 0.015 μg/ml, product no. MA0105D) were used for determining the MICs of penicillin and tetracycline. C. glutamicum strains cultivated in LBG medium were used as seed cultures to inoculate shake flasks containing fresh LBG medium with an initial OD600 of 0.2. The cells were cultivated at 30°C and with shaking at 220 rpm. When the OD600 of the culture reached approximately 1.0, 100 μl culture was spread on LBG plates with the MIC strip placed in the center. After 10 to 14 h of cultivation, the MIC was determined.
Glutamate fermentation in shake flasks.
Seed cultures of C. glutamicum strains were prepared by transferring the overnight cultures prepared in LBG medium into 500-ml shake flasks containing 50 ml seed medium at an initial OD600 of 0.2. Cells at the exponential-growth phase were transferred into 500-ml shake flasks containing 20 ml fermentation medium with an inoculum size of 5% (vol/vol). The cells were cultivated at 30°C and with shaking at 220 rpm. The pH of the medium was adjusted by supplementation with 20% urea solution. The seed medium contains the following, per liter: 30 g glucose, 30 g corn steep liquor, 5.6 g urea, and 1.6 g K2HPO4·3H2O. The pH of the medium was 6.7. The fermentation medium contains the following, per liter: 80 g glucose, 0.8 g MgSO4·7H2O, 0.7 g H3PO4, 1.2 g KCl, 40 g 3-(N-morpholino) propanesulfonic acid (MOPS), and 2 ml of 1% phenol red solution. The pH of the medium was 6.7. When biotin-rich fermentation medium was used, 200 μg/liter biotin was added.
Glutamate fermentation in bioreactors.
Seed cultures of C. glutamicum strains were prepared by transferring the overnight cultures prepared in LBG medium into 500-ml shake flasks containing 50 ml seed medium at an initial OD600 of 0.2. Cells at the exponential-growth phase were transferred into 5-liter bioreactors containing 2 liters of fermentation medium, with an inoculum size of 0.05% (vol/vol). The temperature of bioreactors was set at 34°C for 0 to 16 h, 35°C for 17 to 22 h, 36°C for 23 to 28 h, and 37°C for 29 to 38 h. The dissolved oxygen was set at 30% by adjusting the aeration rate and agitation speed. The pH of the medium was adjusted by supplementation with ammonium hydroxide. The seed medium contains the following, per liter: 30 g glucose, 30 g corn steep liquor, 5.6 g urea, and 1.6 g K2HPO4·3H2O. The pH of the medium was 6.7. The fermentation medium contains the following, per liter: 140 g glucose, 3 g corn steep liquor, 1 g molasses, 0.87 g MgSO4·7H2O, 0.7 g H3PO4, 1.45 g KCl, 2.4 mg MnSO4·H2O, and 0.15 mg thiamine hydrochloride. The pH of the medium was 7.0.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grants 31800038, 31870044, and 31700044), the Key Research Program of Chinese Academy of Sciences (grant ZDRW-ZS-2016-2), the International Partnership Program of Chinese Academy of Sciences (grant 153D31KYSB20170121), and the first Special Support Plan for Talents Development and High-level Innovation and Entrepreneurship Team of the Tianjin Municipal City.
J.L., Y.W., Y.L, P.Z., and J.S. conceived and designed the experiments. J.L., Y.W., Y.L., X.G., J.Z., and J.C. performed the experiments. J.L., Y.W., X.N., and T.D.-O. analyzed the data. P.Z., J.S., and Y.M. contributed reagents and analytic tools. All authors contributed to the writing of the manuscript.
REFERENCES
- 1.Pfeifer E, Gatgens C, Polen T, Frunzke J. 2017. Adaptive laboratory evolution of Corynebacterium glutamicum towards higher growth rates on glucose minimal medium. Sci Rep 7:16780. doi: 10.1038/s41598-017-17014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woo HM, Park JB. 2014. Recent progress in development of synthetic biology platforms and metabolic engineering of Corynebacterium glutamicum. J Biotechnol 180:43–51. doi: 10.1016/j.jbiotec.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda M, Nakagawa S. 2003. The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62:99–109. doi: 10.1007/s00253-003-1328-1. [DOI] [PubMed] [Google Scholar]
- 4.Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Kramer R, Linke B, McHardy AC, Meyer F, Mockel B, Pfefferle W, Puhler A, Rey DA, Ruckert C, Rupp O, Sahm H, Wendisch VF, Wiegrabe I, Tauch A. 2003. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of l-aspartate-derived amino acids and vitamins. J Biotechnol 104:5–25. doi: 10.1016/S0168-1656(03)00154-8. [DOI] [PubMed] [Google Scholar]
- 5.Wendisch VF, Jorge JMP, Perez-Garcia F, Sgobba E. 2016. Updates on industrial production of amino acids using Corynebacterium glutamicum. World J Microbiol Biotechnol 32:105. doi: 10.1007/s11274-016-2060-1. [DOI] [PubMed] [Google Scholar]
- 6.Becker J, Wittmann C. 2012. Bio-based production of chemicals, materials and fuels–Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotechnol 23:631–640. doi: 10.1016/j.copbio.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Yim SS, Choi JW, Lee RJ, Lee YJ, Lee SH, Kim SY, Jeong KJ. 2016. Development of a new platform for secretory production of recombinant proteins in Corynebacterium glutamicum. Biotechnol Bioeng 113:163–172. doi: 10.1002/bit.25692. [DOI] [PubMed] [Google Scholar]
- 8.Lee JY, Na YA, Kim E, Lee HS, Kim P. 2016. The actinobacterium Corynebacterium glutamicum, an industrial workhorse. J Microbiol Biotechnol 26:807–822. doi: 10.4014/jmb.1601.01053. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Liu Y, Liu J, Guo Y, Fan L, Ni X, Zheng X, Wang M, Zheng P, Sun J, Ma Y. 2018. MACBETH: multiplex automated Corynebacterium glutamicum base editing method. Metab Eng 47:200–210. doi: 10.1016/j.ymben.2018.02.016. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Wang Y, Lu YJ, Zheng P, Sun JB, Ma YH. 2017. Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb Cell Fact 16:205. doi: 10.1186/s12934-017-0815-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho JS, Choi KR, Prabowo CPS, Shin JH, Yang D, Jang J, Lee SY. 2017. CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng 42:157–167. doi: 10.1016/j.ymben.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Ruan Y, Zhu L, Li Q. 2015. Improving the electro-transformation efficiency of Corynebacterium glutamicum by weakening its cell wall and increasing the cytoplasmic membrane fluidity. Biotechnol Lett 37:2445–2452. doi: 10.1007/s10529-015-1934-x. [DOI] [PubMed] [Google Scholar]
- 13.Kirchner O, Tauch A. 2003. Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104:287–299. doi: 10.1016/S0168-1656(03)00148-2. [DOI] [PubMed] [Google Scholar]
- 14.van der Rest ME, Lange C, Molenaar D. 1999. A heat shock following electroporation induces highly efficient transformation of Corynebacterium glutamicum with xenogeneic plasmid DNA. Appl Microbiol Biotechnol 52:541–545. doi: 10.1007/s002530051557. [DOI] [PubMed] [Google Scholar]
- 15.Schäfer A, Tauch A, Droste N, Puhler A, Kalinowski J. 1997. The Corynebacterium glutamicum cglIM gene encoding a 5-cytosine methyltransferase enzyme confers a specific DNA methylation pattern in an McrBC-deficient Escherichia coli strain. Gene 203:95–101. doi: 10.1016/S0378-1119(97)00519-2. [DOI] [PubMed] [Google Scholar]
- 16.Baumgart M, Unthan S, Ruckert C, Sivalingam J, Grunberger A, Kalinowski J, Bott M, Noack S, Frunzke J. 2013. Construction of a prophage-free variant of Corynebacterium glutamicum ATCC 13032 for use as a platform strain for basic research and industrial biotechnology. Appl Environ Microbiol 79:6006–6015. doi: 10.1128/AEM.01634-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li HD, Zhang LR, Guo W, Xu DQ. 2016. Development of a genetically engineered Escherichia coli strain for plasmid transformation in Corynebacterium glutamicum. J Microbiol Methods 131:156–160. doi: 10.1016/j.mimet.2016.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Jang KH, Chambers PJ, Chun UH, Britz ML. 2001. Characterization of the cell-surface barriers to plasmid transformation in Corynebacterium glutamicum. J Microbiol Biotechnol 11:294–301. [Google Scholar]
- 19.Jang KH, Britz ML. 2000. Improved electrotransformation frequencies of Corynebacterium glutamicum using cell-surface mutants. Biotechnol Lett 22:539–545. doi: 10.1023/A:1005629224109. [DOI] [Google Scholar]
- 20.Laneelle MA, Tropis M, Daffe M. 2013. Current knowledge on mycolic acids in Corynebacterium glutamicum and their relevance for biotechnological processes. Appl Microbiol Biotechnol 97:9923–9930. doi: 10.1007/s00253-013-5265-3. [DOI] [PubMed] [Google Scholar]
- 21.Kirchner O, Gartemann KH, Zellermann EM, Eichenlaub R, Burger A. 2001. A highly efficient transposon mutagenesis system for the tomato pathogen Clavibacter michiganensis subsp. michiganensis. Mol Plant Microbe Interact 14:1312–1318. doi: 10.1094/MPMI.2001.14.11.1312. [DOI] [PubMed] [Google Scholar]
- 22.Tomiyasu I, Yano I. 1984. Isonicotinic acid hydrazide induced changes and inhibition in mycolic acid synthesis in Nocardia and related taxa. Arch Microbiol 137:316–323. doi: 10.1007/BF00410728. [DOI] [PubMed] [Google Scholar]
- 23.Haynes JA, Britz ML. 1989. Electrotransformation of Brevibacterium lactofermentum and Corynebacterium glutamicum: growth in Tween 80 increases transformation frequencies. FEMS Microbiol Lett 61:329–334. doi: 10.1111/j.1574-6968.1989.tb03646.x. [DOI] [Google Scholar]
- 24.Haynes JA, Britz ML. 1990. The effect of growth conditions of Corynebacterium glutamicum on the transformation frequency obtained by electroporation. J Gen Microbiol 136:255–263. doi: 10.1099/00221287-136-2-255. [DOI] [Google Scholar]
- 25.Hammes W, Schleifer KH, Kandler O. 1973. Mode of action of glycine on biosynthesis of peptidoglycan. J Bacteriol 116:1029–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hopwood DA, Wright HM, Bibb MJ. 1977. Genetic recombination through protoplast fusion in Streptomyces. Nature 268:171–174. doi: 10.1038/268171a0. [DOI] [PubMed] [Google Scholar]
- 27.Chevalier J, Pommier MT, Cremieux A, Michel G. 1988. Influence of Tween 80 on the mycolic acid composition of three cutaneous corynebacteria. J Gen Microbiol 134:2457–2461. doi: 10.1099/00221287-134-9-2457. [DOI] [PubMed] [Google Scholar]
- 28.Jankute M, Cox JAG, Harrison J, Besra GS. 2015. Assembly of the mycobacterial cell wall. Annu Rev Microbiol 69:405–423. doi: 10.1146/annurev-micro-091014-104121. [DOI] [PubMed] [Google Scholar]
- 29.Valbuena N, Letek M, Ordonez E, Ayala J, Daniel RA, Gil JA, Mateos LM. 2007. Characterization of HMW-PBPs from the rod-shaped actinomycete Corynebacterium glutamicum: peptidoglycan synthesis in cells lacking actin-like cytoskeletal structures. Mol Microbiol 66:643–657. doi: 10.1111/j.1365-2958.2007.05943.x. [DOI] [PubMed] [Google Scholar]
- 30.Trunkfield AE, Gurcha SS, Besra GS, Bugg TDH. 2010. Inhibition of Escherichia coli glycosyltransferase MurG and Mycobacterium tuberculosis Gal transferase by uridine-linked transition state mimics. Bioorg Med Chem 18:2651–2663. doi: 10.1016/j.bmc.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alderwick LJ, Radmacher E, Seidel M, Gande R, Hitchen PG, Morris HR, Dell A, Sahm H, Eggeling L, Besra GS. 2005. Deletion of Cg-emb in Corynebacterianeae leads to a novel truncated cell wall arabinogalactan, whereas inactivation of Cg-ubiA results in an arabinan-deficient mutant with a cell wall galactan core. J Biol Chem 280:32362–32371. doi: 10.1074/jbc.M506339200. [DOI] [PubMed] [Google Scholar]
- 32.Escuyer VE, Lety MA, Torrelles JB, Khoo KH, Tang JB, Rithner CD, Frehel C, McNeil MR, Brennan PJ, Chatterjee D. 2001. The role of the embA and embB gene products in the biosynthesis of the terminal hexaarabinofuranosyl motif of Mycobacterium smegmatis arabinogalactan. J Biol Chem 276:48854–48862. doi: 10.1074/jbc.M102272200. [DOI] [PubMed] [Google Scholar]
- 33.Meena LS, Kolattukudy PE. 2013. Expression and characterization of Rv0447c product, potentially the methyltransferase involved in tuberculostearic acid biosynthesis in Mycobacterium tuberculosis. Biotechnol Appl Biochem 60:412–416. doi: 10.1002/bab.1112. [DOI] [PubMed] [Google Scholar]
- 34.Tan YZ, Xu DQ, Li Y, Wang XY. 2012. Construction of a novel sacB-based system for marker-free gene deletion in Corynebacterium glutamicum. Plasmid 67:44–52. doi: 10.1016/j.plasmid.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 36.Daniel RA, Errington J. 2003. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell 113:767–776. doi: 10.1016/S0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]
- 37.Sieger B, Schubert K, Donovan C, Bramkamp M. 2013. The lipid II flippase RodA determines morphology and growth in Corynebacterium glutamicum. Mol Microbiol 90:966–982. doi: 10.1111/mmi.12411. [DOI] [PubMed] [Google Scholar]
- 38.Łeski TA, Tomasz A. 2005. Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A. J Bacteriol 187:1815–1824. doi: 10.1128/JB.187.5.1815-1824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Cao G, Xu D, Fan L, Wu X, Ni X, Zhao S, Zheng P, Sun J, Ma Y. 2018. A novel Corynebacterium glutamicum l-glutamate exporter. Appl Environ Microbiol 84:e02691-17. doi: 10.1128/AEM.02691-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morlot C, Noirclerc-Savoye M, Zapun A, Dideberg O, Vernet T. 2004. The d,d-carboxypeptidase PBP3 organizes the division process of Streptococcus pneumoniae. Mol Microbiol 51:1641–1648. doi: 10.1046/j.1365-2958.2003.03953.x. [DOI] [PubMed] [Google Scholar]
- 41.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seidel M, Alderwick LJ, Birch HL, Sahm H, Eggeling L, Besra GS. 2007. Identification of a novel arabinofuranosyltransferase AftB involved in a terminal step of cell wall arabinan biosynthesis in Corynebacterianeae, such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J Biol Chem 282:14729–14740. doi: 10.1074/jbc.M700271200. [DOI] [PubMed] [Google Scholar]
- 44.Schäfer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. doi: 10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]