

Abstract
Protein tyrosine phosphatases (PTPs) play essential roles in regulating signaling events in multiple cells by tyrosine dephosphorylation. One of them, PTPσ, appears important in regulating function of plasmacytoid dendritic cells (pDC). Here we report that PTPσ deletion in knockout mice and inhibition with a selective antagonist peptide exacerbated symptoms of experimental autoimmune encephalomyelitis (EAE) by enhancing axon and myelin damage in the spinal cord. PTPσ −/− mice displayed pro-inflammatory profiles in the spinal cord and lymphoid organs following MOG peptide immunization. PTPσ deletion promoted a pro-inflammatory phenotype in conventional DCs and directly regulated differentiation of CD4+ T cells. It also facilitated infiltration of T lymphocytes, activation of macrophages in the CNS and development of EAE. Therefore, PTPσ is a key negative regulator in EAE initiation and progression, which acts by regulating functions of DCs, T cells, and other immune cells. PTPσ may become an important molecular target for treating autoimmune disorders.
Keywords: Protein tyrosine phosphatase σ, multiple sclerosis, experimental autoimmune encephalomyelitis, immune cell, dendritic cell function
1. Introduction
Multiple sclerosis (MS) is an autoimmune disease characterized by myelin and axonal damage in the central nervous system (CNS), especially the spinal cord. Following activation of antigen-presenting dendritic cells, myelin-specific T cells are stimulated in peripheral lymphoid organs and enter CNS after penetrating the blood-brain barrier (Martino and Hartung, 1999; Merrill and Benveniste, 1996). T cells that have accessed the CNS initiate and coordinate immune attack against myelin sheaths by recruiting other inflammatory cells from the immune system, including activated macrophages and microglia. Transmigration of activated B lymphocytes and plasma cells contributes to subsequent damage progression by generating antibodies against myelin structures. The intensive attack of immune cells and generation of various cytokines induce myelin and axon damage in the spinal cord and brain (Friese et al., 2006; Hauser and Oksenberg, 2006). A feature of pathologic change in MS is formation of multiple demyelinated plaques dispersed in the CNS, predominantly in white matter areas (Friese et al., 2006; Hauser and Oksenberg, 2006). The accrued myelin and axon damage causes signal conduction failure along fiber tracts and consequent neurological deficits. In the early stage, most MS patients follow a relapsing-remitting course with partial or complete recovery, but intermittent inflammation over time usually results in severe structural damage in the CNS and persistent impairment of neurological functions. Experimental autoimmune encephalomyelitis (EAE) is widely employed as an animal model to study human CNS demyelinating diseases, including MS.
Protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) play essential roles in regulating signaling events in multiple cell types by tyrosine phosphorylation and dephosphorylation, respectively. PTPs include non-receptor PTPs and receptor PTPs (RPTPs), which comprise a family of vital cell surface proteins that have intracellular PTP activity and extracellular domains with sequence homology to cell adhesion molecules. The RPTP family includes eight sub-families based on their extracellular domain structures. In contrast to the well-studied PTKs, the properties of most RPTPs remain unclear, including their specific substrates, regulation, biological functions and possible roles in human disorders (Takahashi and Craig, 2013; Um and Ko, 2013). Type IIa RPTPs include three members in vertebrates, the leukocyte common antigen-related (LAR), PTPσ, and PTPδ. All of them contain typical cell adhesion Ig and fibronectin III domains, which contribute to their interactions with the extracellular matrix (ECM) molecules. Structurally, these three RPTPs have two cytoplasmic tandem regions, the membrane-proximal PTP domain (D1, catalytically active) and the membrane-distal PTP domain (D2, inactive).
During development, LAR and PTPσ are widely expressed by multiple tissues, including the nervous system, lung, kidney and thymus, while PTPδ is mainly present in the nervous system. In mammalian nervous system, presynaptic type-IIa RPTPs contribute to synaptic differentiation of neurons by binding cognate postsynaptic partners through their extracellular regions and forming trans-synaptic adhesions (Takahashi and Craig, 2013; Um and Ko, 2013). In immune system, LAR is expressed in immature thymocytes, but it is not required for T cell development and function (Kondo et al., 2010; Terszowski et al., 2001). The expression and significance of PTPσ in immune system are largely unknown. A recent study demonstrated that PTPσ is expressed in plasmacytoid dendritic cells (pDCs) and downregulated by pDC activation, and that PTPσ deletion induces generation of interferon α (IFNα) in pDCs (Bunin et al., 2015). Because DC-specific deletion of PTPσ on a LAR-deficient background enhances production of IFN by pDCs, leukocyte infiltration in the intestine and mild colitis, PTPσ appears essential for inhibiting spontaneous IFN production and immune-mediated intestinal inflammation. Whether PTPσ is expressed by other immune cells and whether it has a role in regulating autoimmune disorders in the nervous system are unknown. Here, we identify PTPσ as an important negative regulator of EAE development that acts predominantly by regulating functions of immune cells, including conventional DCs (cDCs), cluster of differentiation 4 (CD4) T lymphocytes, regulatory T (Treg) cells and others. Suppression of PTPσ exacerbated the progression of clinical symptoms and axonal damage in the spinal cord of mice with EAE. PTPσ deficiency stimulated a pro-inflammatory phenotype in conventional DCs, directly regulated differentiation of CD4+ T cells, reduced activity of Treg, and facilitated infiltration of T lymphocytes and activation of macrophages in the CNS. Our findings suggest that PTPσ offers a novel and crucial molecular target for treating MS.
2. Materials and Methods
2.1. Reagents
LPS (Escherichia coli O55:B5), phorbol myristate acetate (PMA), ionomycin, complete Freund’s adjuvant, Mayer’s Hematoxylin solution, Eosin Y solution and Eriochrome Cyanine R were purchased from Sigma-Aldrich (St. Louis, MO). MOG peptide fragment 35–55 (MOG35–55) was synthesized by CHI Scientific, Inc. (Maynard, MA). Pertussis toxin was purchased from List Biological Laboratories (Campbell, CA). Histo-Clear II was purchased from National Diagnostics (Atlanta, GA). Fast SYBR Master Mix and Trizol reagent were purchased from Life Technologies (Carlsbad, CA). The CD4+ CD62L+ T cell Isolation Kit II was purchased from Miltenyi Biotec (Bergish-Gladbach, Germany). Recombinant murine GM-CSF, IL-12p70, IL-6, and IFNγ were purchased from Peprotech, Inc. (Rocky Hill, NJ). FITC-conjugated anti-mouse CD80 (RRID: AB_10896321), CD86 (RRID: AB_10896136), CD40 (RRID: AB_10897019), MHCII (RRID: AB_10893593); PE-conjugated anti-mouse IL-17 (RRID: AB_10584331), recombinant mouse IL-10, recombinant mouse IL-23; capture and biotinylated anti-mouse IL-12 (RRIDs: AB_394097 & AB_395419), IL-10 (RRIDs: AB_394093 & AB_395382), IL-6 (RRIDs: AB_398549 & AB_395368), IFNγ (RRIDs: AB_394145 & AB_395374), TNFα (RRIDs: AB_398625 & AB_395378), GolgiPlug, Cytofix/Cytoperm fixation, permeabilization solution, Perm/Wash buffer, TMB Substrate Reagent Set and H37Ra Mycobacterium tuberculosis were purchased from BD (San Diego, CA). Capture and biotinylated anti-mouse IL17 (RRIDs: AB_2125017 & AB_356467), recombinant mouse IL17, recombinant TGFβ, capture and biotinylated anti-mouse IL-27 antibody (RRIDs: 355012 & AB_2231063), and recombinant mouse IL-27 were purchased from R&D Systems (Minneapolis, MN). APC-conjugated anti-mouse IFNγ (RRID: AB_469503), capture and biotinylated anti-mouse IL-23 antibody (RRIDs: AB_2637368 & AB_466928) were purchased from eBioscience (San Diego, CA).
2.2. PTPσ knockout (KO) mice, EAE induction, clinical score evaluation and sIg1 treatment
PTPσ−/− mice on BALB/c background were generated as described previously (Elchebly et al., 1999). C57BL6 mice were purchased from Jackson Laboratory. For EAE immunization, adult mice (7–10 weeks old) were induced by subcutaneous injection of 200 μl of emulsion containing 200 μg of 35-55 MOG peptide in complete Freund’s adjuvant with 200 μg of H37Ra Mycobacterium tuberculosis. Bordetella pertussis toxin (50 ng) was injected intraperitoneally on the same day and 48 hrs after MOG peptide injection. Following immunization, animals were evaluated for clinical EAE scores with the following criteria: 0, no detectable sign of EAE; 1, weakness of the tail; 2, definite tail paralysis and hind limb weakness; 3, partial paralysis of the hind limbs; 4, complete paralysis of the hind limbs; 5, complete paralysis of the hind limbs with incontinence and partial or complete paralysis of forelimbs. During the clinical score evaluations, the examiner was unaware of the drug treatment or genotypes of transgenic mice. For the experiments with peptide treatments, mice received subcutaneous injections (two times per day) of random peptide or sIg1 (143 μg/mouse/day) beginning 3 hrs after MOG peptide injections for 21 successive days.
2.3. Immunohistochemistry and axon and myelin analyses
Mice were perfused with 4% paraformaldehyde 4 weeks after EAE immunization, and the spinal cord was dissected out. Fixed spinal cord was immersed in the same fixative for 1 day at 4°C, transferred into 30% sucrose in PBS and incubated overnight. Blocks from the spinal cords at the L4 level were cut into sets of 30 μm thick transverse sections and placed on gelatin-coated glass slides. Following PBS washing, sections were stained with H&E or EC. For H&E staining, sections were incubated with hematoxylin solution for 5 min, differentiated in 70% ethanol containing 1% HCl for 5 seconds, incubated with eosin solution for 5 seconds, dehydrated through ascending ethyl alcohols, cleared in Histo-Clear II, and cover-slipped with Permount mounting medium. For EC staining, the sections were stained with EC solution (0.2% EC, 0.5% sulfuric acid and 0.4% ferric chloride) at room temperature for 20 min. After a gentle rinse in distilled water, slides were differentiated in 0.5% ferric ammonium sulfate at room temperature for 2 min and cover-slipped using VectaMount mounting medium. For immunohistochemistry staining for IBA-1 and CD3, transverse floating sections were blocked with 10% goat serum, 1% bovine serum albumin, and 0.3% Triton X-100 in TBS for 2 hrs at room temperature. Samples were then incubated with primary antibody diluted in TBS containing 5% goat serum, 0.1% bovine serum albumin, and 0.3% Triton X-100 overnight at 4°C. The following primary Wako, RRID: AB_2314667) antibodies were used: microglia-specific protein IBA-1 (1:1,000, rabbit polyclonal, and cluster of differentiation 3 (CD3, mouse monoclonal 1:50, Santa Cruz Biotechnology, RRID: AB_627014). After incubation with primary antibodies, sections were incubated with secondary antibodies conjugated with Alexa488 or Alexa594 (1:200; Invitrogen, RRIDs: AB_2534069 & AB_2534095).
2.4. Isolation of mononuclear cells from brain and spinal cord
BALB/c wild type and PTPσ−/− mice were immunized as described before. Mice were anesthetized with 20 μl mixture of ketamine HCl and xylazine and perfused through the left cardiac ventricle with 30 ml of HBSS containing 2mM EDTA. The brain was dissected out and the spinal cord flushed with HBSS. CNS tissue was digested with 10 ml HBSS containing DNAse I (0.1 mg/ml for brain and 0.05 mg/ml for spinal cord) and Liberase (0.05 mg/ml for brain and 0.025 mg/ml for spinal cord) for 45 min at 37°C with shaking, followed by blocking solution (10% fetal calf serum, 10 mM EDTA in HBSS). The tissue was pelleted and re-suspended in 10 ml of 30% isotonic Percoll (diluted with 10× HBSS and distilled water), underlaid with 5 ml of 70% isotonic Percoll. Mononuclear cells were isolated from the 30/70 interphase after gradient centrifugation. Cells were washed with RPMI 1640 medium, stained with fluorescence conjugated anti CD45, anti CD11b, anti CD4, anti-IL-17 and anti IFNγ and analyzed by FACS to characterize mononuclear cells and detect IFNγ and IL17 producing CD4+ T cells. Fixation/permeabilization solution and Perm/Wash buffer were used in the experiments with Th1 and Th17 detection.
2.5. Generation of bone marrow derived dendritic cells
Bone marrow precursors from wild type and PTPσ−/− mice were cultured in the presence of 20 ng/ml rGM-CSF to generate cDCs as previously described (Kong et al., 2010). On day 7 non-adherent cells were collected and purified by anti-CD11c-coated magnetic beads using the autoMACS system according to the manufacturer’s instructions (Miltenyi Biotec). The purity of the purified cells was determined by FACS analysis (>96% for CD11c+ cells). To generate pDC (CD11c+B220+), we differentiated bone marrow precursors in medium containing Flt3L (7.5% supernatant from the Flt3L cell line, a kind gift from Dr. Stefania Gallucci Temple University Lewis Katz School of Medicine). After 9 days in culture, non-adherent cells were collected. The purity of CD11c+B220+ pDC determined by FACS was 38%.
2.6. RT2 Profiler PCR array and qRT-PCR assays
Three spinal cords were pooled together in each group at day 18 after immunization. Spinal cord tissue was homogenized, and the expression of cytokines, chemokines, chemokine receptors and other mediators was investigated using the autoimmune disease and inflammatory response RT2 Profiler PCR array (Qiagen, Valencia, CA) according to the manufacturer’s protocol. Cycle threshold values were determined by automated threshold analysis, and the results were standardized based on 5 housekeeping genes, including glyceraldehyde-3 phosphate dehydrogenase (Gapdh), β2 microglobulin (B2m), glucuronidase beta (Gusβ), heat-shock protein 90 alpha (Hsp90sb1), and β-actin.
Homogenized tissues or cells underwent mRNA extraction, reverse transcription and qRT-PCR to detect gene expression. The expression of IFNγ Il17a, Foxp3, Rorc, Il12b (p40), Il12a (p35), Il23a (p19), Il6, Ccl2, Ccl5, Ccr2, Ccr5, Ccr7, Cxcl10, Vcam1, Icam1, Tbx21, Il10, Il-27p28, IFNγ, and PTPσ was detected by SYBR Green-based qRT-PCR as previously described (Kong et al., 2010). Real-time PCR was performed using StepOnePlus Real-Time PCR System (AB Applied Biosystems). The following primers were used: IFNγ sense, 5′-AGCTCATCCGAGTGGTCCAC-3′ and antisense, 5′-GCTTCCTGAGGCTGGATACC-3′; Il17 sense, 5′-CCTGGACTCTCCACCGCAAT-3′ and antisense, 5′-ATGTGGTGGTCCAGCTTTCC-3′; Foxp3 sense, 5′-CAGCTGCCTACAGTGCCCCTA-3′, and antisense 5′-CATTTGCCAGCAGTGGGTAG-3′; RORC sense, 5′-GCGGAGCAGACACACTTACA-3′, and antisense, 5′-TCCACCACCACAGCTGAGAGG-3′; Il12b(p40) sense, 5′-GACCCTGCCGATTGAACTGGC-3′ and antisense, 5′-CAACGTTGCATCCTAGGATCG-3′; Il12a(p35) sense, 5′-GAGGACTTGAAGATGTACAG-3′ and antisense, 5′-TTCTATCTGTGTGAGGAGGGC-3′; Il23a(p19) sense, 5′-TGCTGGATTGCAGAGCAGTAA-3′ and antisense, 5′-ATGCAGAGATTCCGAGAGA-3′; Il6 sense, 5′-TCCTCTCTGCAAGAGACTTCCATCC and antisense, 5′-GGGAAGGCCGTGGTTGTCACC-3′; Ccl1 sense, 5′-AAGAGCATGCTTACGGTCTCC-3′ and antisense, 5′-GGCGCAGCTTTCTCTACCT-3′; Ccl2 sense, 5′-CACAGTTGCCGGCTGGAGCA-3′ and antisense, 5′-CAGCAGGTGAGTGGGGCGTT-3′; Ccl5 sense, 5′-GCTGCTTTGCCTACCTCTCC-3′ and antisense, 5′-TCGAGTGACAAAGACTGC-3′; Ccl8 sense, 5′-AGCTGTCCCTGTCAGCCCAGA-3′ and antisense, 5′-AGCAGCAGGTGACTGGAGCCT-3′; Ccl20 sense, 5′-CCTGATTTGTGTCCCAGTGGA-3′ and antisense, 5′-AATCCTTCCACTAAGCGCCC-3′; Cxcl10 sense, 5′-ATTCTTTAAGGGCTGGTCTGA-3′ and antisense, 5′-CACCTCCACATAGCTTACAGT-3′; Ccr2, sense, 5′-GCCTGCAAAGACCAGAAGAGGGC-3′ and antisense, 5′-GGTGTGGTGGCCCCTTCATCA-3′; Ccr5 sense, 5′-CATCGATTATGGTATGTCAGCACC-3′ and antisense, 5′-CAGAATGGTAGTGTGAGCAGGAA-3′; Ccr7 sense 5′-CCAGGAAAAACGTGCTGGTG-3′ and antisense 5′-GGCCAGGTTGAGCAGGTAGG-3′; Vcam1 sense, 5′-TGTGCGCTGTGACCTGTCTGC-3′, antisense 5′-TCTCCCATGCACAAGTGGCCC-3′; Icam1 sense, 5′-CTCCTGGCCCTGGTCACCGT-3′, antisense, ACCCACCCTGGGGCAGGAAG, Tbx21 sense, 5′-CGGTACCAGAGCGGCAAGT-3′, and antisense, 5′-CATGCTGCCTTCTGCCTTTC-3′; Il10 sense, 5′-CCTGGTAGAAGTGATGCCCC-3′ and antisense, 5′-TCCTTGATTTCTGGGCCATG-3′; Il27p28 sense, 5′-TCTGGTACAAGCTGGTTCCTGG-3′ and antisense, 5′-TAGCCCTGAACCTCAGAGAGCA-3′; IFNγ sense, 5′-CCCTATGGAGATGACGGAGA-3′ and antisense, 5′-ACCCAGTGCTGGAGAAATTG-3′; PTPσ sense, 5′-CATTTCCAGTTCACGGCATGGC-3′ and antisense, 5′-TAGTCAGAGCCCTCCACACCG-3′; β-actin sense, 5′-TCCACCACCACAGCTGAGAGG-3′ and antisense, 5′-CAGCTTCTCTTTGATGTCACG-3′. The expression level of each gene was indicated by the number of cycles needed for the cDNA amplification to reach a threshold. The amount of DNA was calculated from the number of cycles by using standard curves, and the results were normalized to β-actin.
2.7. CD4+ T cell differentiation
Naïve CD4+ T cells were purified from splenic cell suspension obtained from 6–8 weeks old WT and PTPσ−/− mice by using the CD4+CD62L+ T Cell Isolation Kit (Miltenyi Biotec). Naïve CD+ T cells were activated with plate bound anti-CD3 (3μg/ml) and soluble anti-CD28 (1μg/ml) in both non-polarizing (Th0) and polarizing (Th1 and Th17) conditions as follows: 20 ng/ml IL-12 (Th1) and 5 ng/ml TGFb1, 20ng/ml of IL-1b, 20ng/ml of IL-6 and 10 mg/ml of anti-IFNγ (Th17). After 72 h in culture, cytokine production was detected by both ELISA and flow cytometry. GolgiPlug, Fixation/permeabilization solution and Perm/Wash buffer were used in the experiments with CD4+ T cell differentiation in vitro.
2.8. FACS Analysis
To study the effect of PTPσ on cDC maturation, wild type and PTPσ−/− cDC were treated with LPS for 24 h. Then cells were collected, washed with PBS, and incubated for 30 min at 4°C with anti-CD80 FITC, anti-CD86 FITC, anti-CD40 FITC, anti-MHCⅡ FITC, and appropriate isotype-matched controls. Data were collected for 10,000 cells on FACSCalibur (BD Biosciences) and analyzed using Cellquest software from BD Biosciences (San Jose, CA). Cell profiles of thymus and spleen were characterized by surface staining. Thymus cell suspension was stained with anti-CD4 FITC (RRID: AB_394970), and anti-CD8 PE (RRID: AB_394571). Splenic cell suspension was stained with anti-CD4 FITC, anti-CD8 PE, anti-CD19 PE (RRID: AB_395050), anti-B220 APC (RRID: AB_398531), anti-CD11c FITC (RRID: AB_395060) and anti-CD11b PE (RRID: AB_394775). Flow cytometry was also used to detect intracellular cytokine production in CD4+ T cells. Naïve CD4+ T cells from wild type and PTPσ−/− mice were cultured in different conditions. Cells were collected after stimulation with PMA (50 ng/ml) and ionomycin (500 ng/ml) in the presence of Golgiplug (1 ml/ml) for 4 h. Then cells were fixed, permeabilized, and stained with anti-INFγ and anti-IL17.
2.9. Chemotaxis and cytokine ELISA assays
cDC were treated with 1 μg/ml LPS for 24 h, and migration in response to the chemokine CCL5 (100 ng/ml) was analyzed. The lower chambers of Transwell plates (8.0 μm pore size; Corning, Acton, MA) were loaded with 600 μl serum-free medium with or without CCL5. DC (1×105 cells in 0.1 ml) resuspended in serum-free medium were deposited in the upper chambers of the Transwell plates and allowed to migrate for 24 h at 37°C in 5% CO2. The numbers of migrated DC harvested from the lower chambers were counted by FACS (60-second counts). Cytokine production was determined by sandwich ELISA. Supernatants from cell cultures were harvested and subjected to ELISA. The detection limits were: 15 pg/ml for IL17, IL6 and IL10; 30 pg/ml for IL23, IL12p70 and IFNγ; and 4.7 pg/ml for IL27.
2.10. Conventional dendritic cell adoptive transfer of EAE
BALB/c mice (n=5 in each group) were immunized with MOG emulsified in CFA on day 0 and 7 with pertussis toxin on day 0 and 2. cDC generated from WT and PTPσ−/− mice were treated with LPS and pulsed with MOG for 24 hrs. After extensive washing with PBS, 2 × 106 cDC were injected intravenously on day 4 and day 8 post immunization. Mice were followed daily using the same scoring system as described above.
2.11. Statistical analysis
The behavioral data evaluated at multiple time points were analyzed with repeated-measures ANOVA; p values are provided in Figure 1. The other data collected at one time point were analyzed with one-way ANOVA followed by Bonferroni post-hoc tests or with Student’s t-test. The data in the graphs are means ± SEM (Figures. 1, 2 and 4) and mean ± SD (the other figures). The differences with p<0.05 were considered significant between different groups (*<0.05; **<0.01).
Figure 1. PTPσ transgenic deletion or inhibition with a sequence-targeting peptide aggravates the clinical symptoms of EAE mice.
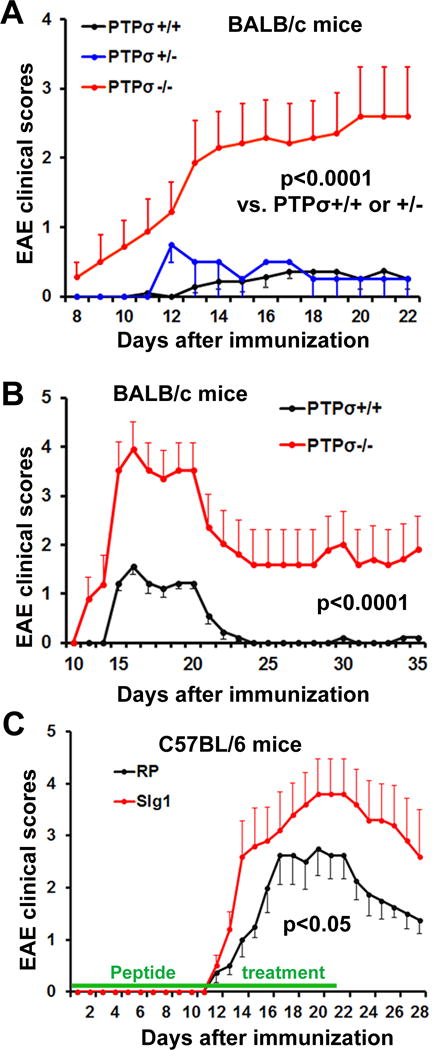
Graphs indicate clinical EAE scores as a function of time after EAE onset. (A, B) PTPσ+/+, PTPσ+/− and PTPσ−/− mice with BALB/c background were immunized with MOG peptide. PTPσ−/− mice exhibit more severe EAE symptoms than two groups of controls (PTPσ+/+ and PTPσ+/−). In the second set of experiments (B), animals were sacrificed for histology 35 days after immunization, instead of 22 days as in the first set. (C) C57BL/6 mice were immunized with MOG peptide and then received sIg1 peptides subcutaneously for 3 weeks. Inhibition of PTPσ with peptide also enhances EAE scores and mimics the effect of transgenic deletion in A, B. n= 5–9 mice per group.
Figure 2. PTPσ deficiency attenuates axon number and myelination in the lumbar spinal cord 25 days after EAE onset.
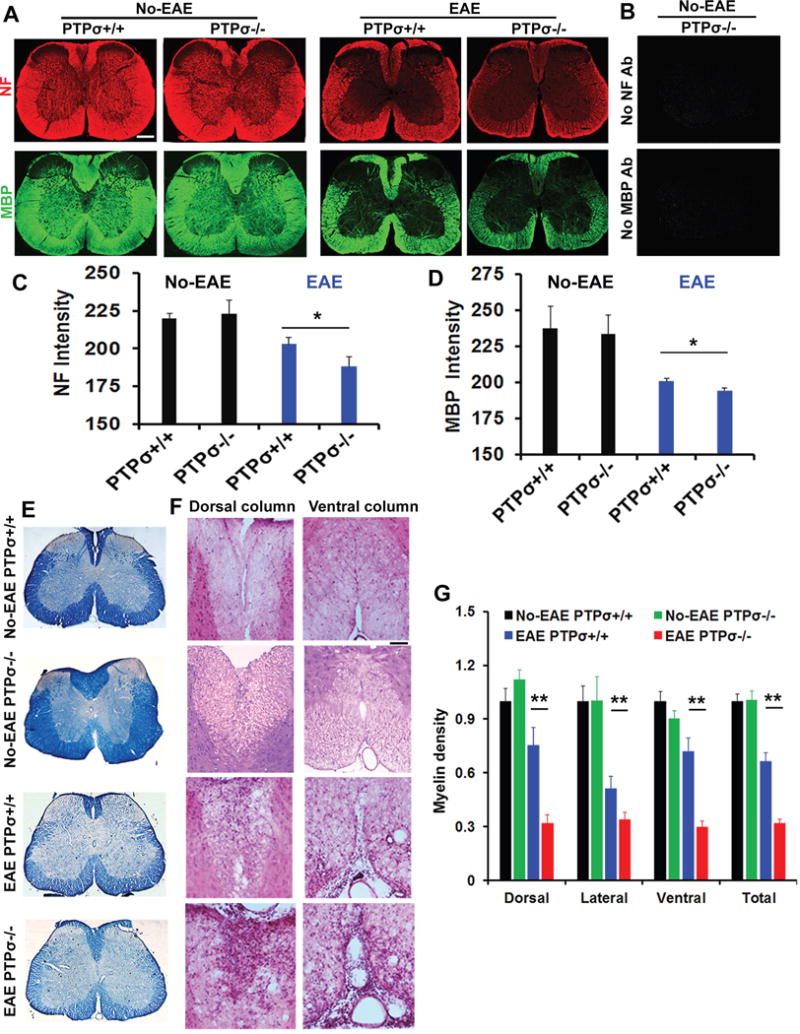
(A) Representative images of transverse sections of spinal cord from PTPσ+/+ and −/− mice immunostained for NF and MBP. (B) Images of transverse sections of spinal cord from PTPσ−/− animals show lack of specific immunostaining signals when the primary antibodies for NF and MBP were omitted. (C, D) The intensity of signals from stained axonal cylinders (NF) and myelin (MBP) were quantified in different white matter areas of transverse sections in a blind manner. (E) Representative transverse sections of stained spinal cord illustrate reduced myelin in PTP −/− mice. (F) Myelin signals stained by EC were quantified in different locations. (G) Representative images of transverse sections of spinal cord stained by H&E indicate more severe damage of white matter tracts in PTPσ−/− animals than controls. In all the sections, dorsal is up. Scale: 250 (low power) or 100 (high power) μm. n = 5–9 mice in each group.
Figure 4. PTPσ deletion increases infiltration of IBA-1+ macrophages and CD3+ T cells into spinal cord of EAE mice.
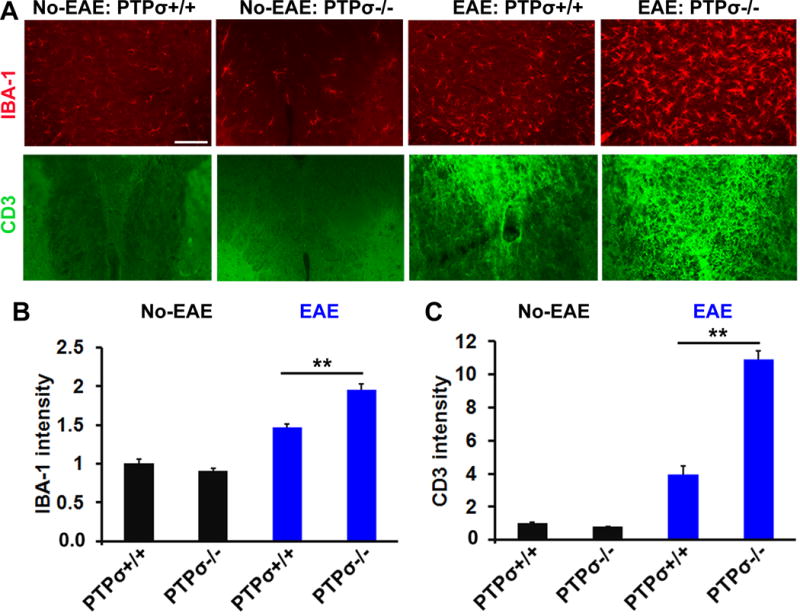
(A) Representative transverse spinal cord sections stained for IBA-1 (red) and CD3 (green) display greater numbers of IBA-1+ macrophages and CD3+ T cells in PTPσ−/− EAE mice. (B, C) Graphs indicate quantification of IBA-1 and CD3+ immunostaining signals from the spinal cord of no-EAE and EAE mice. Scale: 50 μm. n = 3–5 mice/group.
3. Results
3.1. PTPσ deletion in knockout (KO) mice and inhibition with a selective antagonist peptide enhance EAE symptoms
PTPσ is expressed in various types of cells, including DCs and CNS neurons, and regulates their functions during development (Bunin et al., 2015; Johnson and Van Vactor, 2003; Takahashi and Craig, 2013). Because of the importance of PTPs in regulating inflammatory and immune reactions under diverse pathologic conditions (Bunin et al., 2015; Harroch et al., 2002; Jacobsen et al., 2000), we evaluated the roles of PTPσ in regulating the immune response in the CNS of KO mice (with BALB/c genetic background) following immunization with myelin oligodendrocyte glycoprotein (MOG) peptide. The PTPσ KO mice were generated by inserting a selectable neomycin resistance gene into the D1 phosphatase (catalytic) domain, as reported previously (Elchebly et al., 1999). Consistent with an earlier study using BALB/c mice (Maatta et al., 1998), a portion of wild-type (WT) and heterozygous mice developed mild EAE symptoms following immunization with MOG peptide (5–9 mice group). Most of the mice with EAE symptoms had reduced tail tone (Fig. 1A). In contrast, all PTPσ−/− mice subjected to the same immunization procedures developed a typical course of neurological disability of EAE starting 9 14 days after immunization. The symptoms persisted and became more severe during the 3-week period of monitoring. To confirm this novel finding, we performed a second set of experiments with PTPσ +/+ and −/− mice and monitored EAE development and progression for 5 weeks. We consistently detected more severe EAE symptoms in the PTPσ deficient mice (Fig. 1B), especially during the 2–3 weeks after immunization.
To confirm the role of PTPσ in EAE development, we used a pharmacological method in a different strain of mice. We immunized EAE-susceptible C57BL/6 mice with MOG35–55 peptide and treated them with a sequence-targeting peptide against a functional domain of PTPσ for 3 weeks. This peptide was designed by our laboratory and contains 17 amino acids (KPRVTWNKKGKKVNSQR) that selectively target the first Ig domain of PTPσ (called sIg1), the region essential for ligand binding (Shen et al., 2009). We employed a random sequence peptide (RP) as the control; it lacks biological activity due to its nonsense sequence. Immunization of C57BL/6 EAE mice by subcutaneous injections of an emulsion containing autoantigen MOG peptide in complete Freund’s adjuvant induced a typical course of EAE (starting 12 14 days post-immunization); symptoms persisted for at least 5 weeks (Stromnes and Goverman, 2006). Importantly, systemic treatments with sIg1 for 3 weeks efficiently mimicked the effect of PTPσ deletion in KO mice (Fig. 1C), although the difference in EAE symptoms between these two groups was smaller than with PTPσ deletion in transgenic mice. Together, these findings show that PTPσ deletion or inhibition significantly facilitates the initiation and progression of EAE.
3.2. PTPσ deletion decreases axonal number and myelination in the spinal cord of EAE mice
Suppression of PTPσ by transgenic deletion or a pharmacological approach aggravated the clinical symptoms of EAE, suggesting that repressed function of PTPσ increases damage to CNS tissues. We therefore determined whether PTPσ−/− mice had reduced axon number and myelination in the spinal cord several weeks after EAE onset. We measured immunostained neurofilament (NF, a marker for axon cylinders) and myelin basic protein (MBP, a marker for myelin), and Eriochrome cyanine (EC) stained myelin sheath. Compared to non-EAE controls, BALB/c PTPσ +/+ mice immunized with MOG peptide exhibited reduced axonal and myelin structures in the spinal cord, especially at the lumbar level (Fig. 2A–E). Numerous groups have consistently reported obvious myelin and axonal loss along the white matter tracts in EAE mice (McGavern et al., 2000; Mi et al., 2007). The MOG-immunized littermate PTPσ−/− mice had even more greatly attenuated staining signals for axons and myelin structures in different white matter areas of the spinal cord. Further analyses with hemotoxylin and eosin (H&E) staining also demonstrated more severe damage of the white matter areas in PTPσ−/− EAE mice than in PTPσ+/+ controls (Fig. 2F, G). Notably, we did not detect obvious difference in the white matter structures of spinal cord between PTPσ+/+ and PTPσ−/− groups in no-EAE mice. Therefore, PTPσ deficiency exacerbated demyelination and axonal loss in mice with EAE.
3.3. PTPσ deletion enhances infiltration of CD4+ T cells and activation of macrophages in the CNS
Infiltration and activation of inflammatory cells in the CNS play a critical role in development of EAE following MOG peptide immunization (Goverman, 2009). To determine whether deletion of PTPσ facilitates infiltration of immune cells into the CNS, we collected fresh brain and spinal cord tissue 18 days after immunization and isolated and analyzed mononuclear cells by FACS based on expression of immune cell markers, including CD4, CD11b and CD45. Importantly, the numbers of infiltrating CD4+ T cells, CD11b+CD45Hi macrophages and CD11b+CD45Int microglia were significantly increased in both the brain and spinal cord of PTPσ−/− mice compared with PTPσ+/+ controls (Fig. 3A–C). Because IFNγ and interleukin 17 (IL17) generated by Th1 and Th17 cells are important for regulating pathology of EAE mice (Murphy et al., 2010), we determined the numbers of IFNγ and IL17 producing T cells in the brain and spinal cord by FACS 18 days after immunization. We consistently detected increased numbers of these subtypes of T cells in the PTPσ−/− group. Moreover, we confirmed the increased immune responses in the CNS by immunostaining fixed spinal cord with antibodies against IBA-1 and CD3, two widely used markers for activated microglia/macrophages and T cells (Fig. 4). Of note, we did not observe altered numbers of T cells and activated microglia in PTPσ−/− mice without immunization.
Figure 3. PTPσ deletion facilitates infiltration of CD4+ T cells and activation of macrophages in spinal cord and brain.
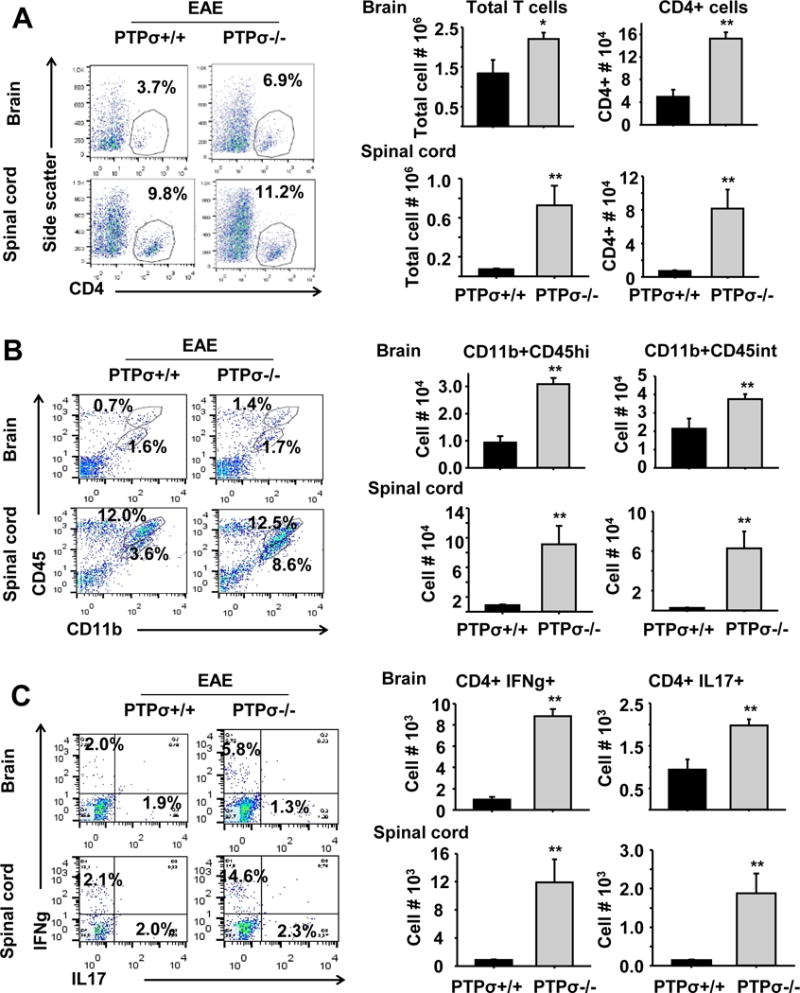
(A–C) Markers for immune cells, including CD4, CD11b and CD45, were used to isolate subsets of T cells, macrophages and microglia by FACS from fresh brain and spinal cord 18 days after immunization. PTPσ−/− mice exhibit increased numbers of IFNγ+ and IL17+ CD4+ T cells, CD11b+CD45Hi macrophages and CD11b+CD45Int microglia monocytes. n = 3–5 mice/group.
3.4. PTPσ−/− mice display pro-inflammatory profiles in the spinal cord and lymphoid organs following MOG peptide immunization
Because EAE is an autoimmune disease primarily localized to the spinal cord, we performed additional experiments on the expression profile of multiple genes related to autoimmunity and inflammatory responses in the spinal cord of PTPσ+/+ or −/− mice 18 days following MOG immunization. qRT-PCR assay revealed significant upregulation of a number of pro-inflammatory related genes in PTPσ−/− EAE mice and Microarrays confirmed upregulation of various specific cytokines, chemokines, and master transcriptional factors (Fig. 5). Because CD4+ T cells, especially the Th1 and Th17 subtypes, play an essential role in EAE development, we next focused on the signature gene expression in Th1 and Th17 cells. We confirmed the high levels of expression of Ifng (IFNγ), Il17a (IL17) and the corresponding transcription factors, including Tbx21 (Tbet) and Rorc (Rorγt), in the spinal cord of PTPσ−/− mice. Expression of Il12a, Il12b, and Il23a genes that encode for IL-12p70 and IL-23, two cytokines associated with Th1 and Th17 differentiation, was consistently increased in PTPσ−/− mice. In contrast, expression of Foxp3, a frequently-used marker for Treg activities (Fontenot et al., 2005; Ziegler, 2006), was significantly reduced in PTPσ−/− mice, indicating that PTPσ deletion lowered the numbers of Treg cells.
Figure 5. Gene expression profile of spinal cord from PTPσ deficient mice differs from WT.
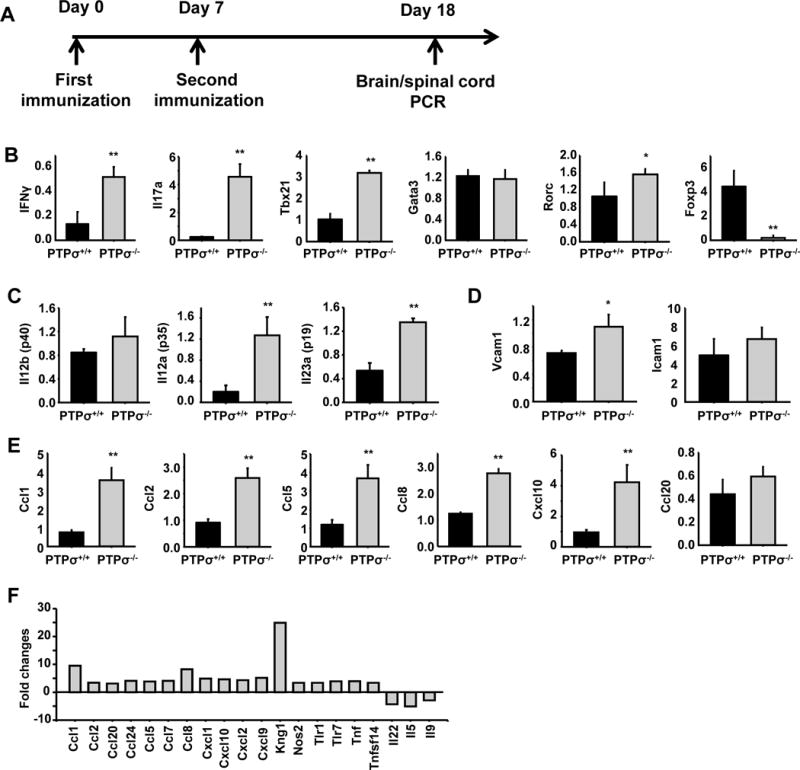
(A) Schematic drawing shows the time points for the experimental procedures. (B–E) qRT-PCR assay shows gene expression profile in spinal cord of PTPσ+/+ and −/− mice 18 days after immunization with MOG emulsified in CFA. (F) Microarray analyses show fold changes of different genes in the spinal cord 18 days after immunization. *p< 0.05, ** p<0.01 compared with WT mice; n = 3 mice/group.
To evaluate the role of PTPσ in regulating development of Th1 and Th17 cells in lymphoid organs, we isolated splenocytes from PTPσ+/+ or −/− mice 8 days post-immunization with MOG peptide and determined the cytokine recall response by qRT-PCR and ELISA. Similar to the T cells isolated from spinal cord, splenocytes derived from PTPσ−/− mice upregulated IFNγ, IL-17, cytokine-encoded genes Il12a, Il12b, Il23a, and the transcription factors of Tbx21 and Rorc (Fig. 6). Because immature thymocytes have been reported to express PTPσ (Ogata et al., 1994), we analyzed both thymocytes and splenic T cell subpopulations derived from PTPσ +/+ or −/− mice. In thymus of PTPσ−/− mice, double negative (CD4-CD8−) and single positive CD4 and CD8 thymocytes were decreased, and double positive thymocytes (CD4+CD8+) were slightly increased (sFig. 1A). In spleen, although PTPσ deletion significantly reduced the number of CD8 T cells, the percentages of CD4+ T, B, CD11b+ and CD11c+ cells were similar in the two groups (sFig. 1B), suggesting that PTPσ deficiency does not significantly affect the development of CD4+ T, B and CD11b+ cells.
Figure 6. PTPσ deletion alters expression levels of cytokines and transcription factors in splenocytes 8 days after immunization.
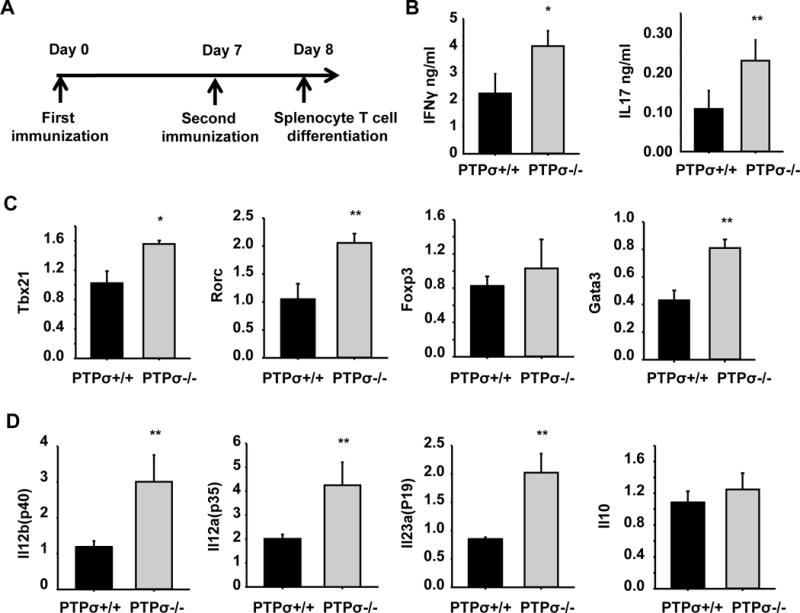
(A) Schematic drawing shows the time points for experimental procedures. (B) Splenocytes were collected from WT and PTPσ−/− mice 8 days after MOG immunization (before disease onset) and re-stimulated with MOG in vitro for 3 days. Cytokine production was then evaluated by ELISA. (C, D) Splenocytes collected as in A were re-stimulated with MOG for 48 h, then qRT-PCR was used to measure levels of transcription factors that regulate CD4+ T cell differentiation and production of cytokine-encoded genes. * p<0.05, ** p<0.01 compared with WT mice. n = 3 mice/group.
3.5. PTPσ deletion promotes a pro-inflammatory phenotype in cDCs and development of EAE
cDCs play a central role in inducing EAE through antigen presentation (Quintana et al., 2015; Weir et al., 2002). We generated both pDCs and cDCs from bone marrow cells of PTPσ +/+ and −/− mice and compared the expression levels of PTPσ in pDC, cDC, and naïve splenic CD4+ T cells by qRT-PCR. Both pDCs and cDCs expressed high levels of PTPσ, although the level was greater in pDCs. Naïve CD4+ T cells also expressed PTPσ, but at a lower level than DCs (Fig. 7A). PTPσ has recently been reported to be expressed in pDCs and to regulate their function by suppressing the inflammatory response (Bunin et al., 2015). The significance of PTPσ expression in cDC is unknown. We found that, similar to pDC (Bunin et al., 2015), lipopolysaccharide (LPS) stimulation temporarily attenuated PTPσ expression of cDC with a return to normal levels 24 hrs after LPS treatment (Fig. 7B). Although PTPσ deletion did not alter expression levels of MHCII, CD40, CD80 or CD86 or maturation status of cDCs 24 hrs after LPS stimulation (sFig. 2), PTPσ−/− cDCs expressed and secreted greater amounts of the cytokines that contribute to Th1 differentiation (IL-12p70 and IL-27) and pathogenic Th17 differentiation (IL-6 and IL-23, Fig. 7C). PTPσ−/− cDCs also upregulated IFN-β and iNOS, and downregulated IL-10 (Fig. 7D), suggesting that PTPσ deletion enhances the pro-inflammatory properties of cDCs. Because cDCs are also the major producers of chemokines, we examined expression of a number of chemokines and detected higher levels of CCL2, CCL5 and CXCL10, and chemokine receptors CCR2, CCR5 and CCR7 in PTPσ−/− cDCs (Fig. 7E). Importantly, functional experiments demonstrated enhanced migration of PTPσ−/− cDCs in response to CCL5 (Fig. 8A). In contrast to cDCs, similar experiments with PTPσ−/− bone marrow-derived macrophages showed statistically significant increases only in Il23a, Il6, Ccr5 and Ccr2 (sFig. 3). The lack of upregulation of Arg1 and Cd206 is evidence that PTPσ−/− macrophages do not present an M2 phenotype.
Figure 7. Dendritic cells and CD4+ naïve T cells express different levels of PTPσ; PTPσ deletion alters the cytokine production and gene expression of cDCs.
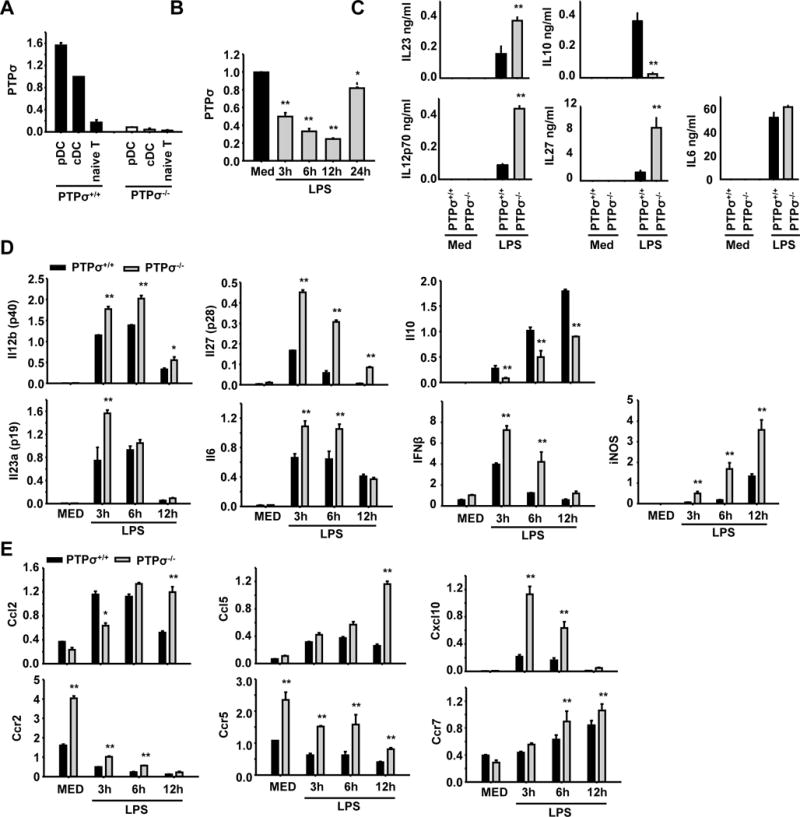
(A) Plasmacytoid dendritic cells (pDC) and conventional dendritic cells (cDC) were generated from bone marrow, and naïve CD4+ T cells were isolated from spleens of PTPσ+/+ and −/− mice. Expression levels of PTPσ were measured by RT-PCR. (B) Expression levels of PTPσ in cDCs were determined by RT-PCR following LPS treatment at the different time points indicated. (C) Cytokine levels were measured by ELISA in cDCs treated with medium (MED, as a negative control) or LPS for 12 (for IL23) or 24 hours (for others). (D, E) Levels of cytokines, chemokines and chemokine receptors were measured by RT-PCR in cDCs treated with MED or LPS for 3, 6 or 12 hours. * p<0.05, ** p<0.01 compared with WT group. n = 3 independent experiments.
Figure 8. PTPσ−/− cDCs show increased migration to CCL5 and aggravate clinical symptoms of cDC adoptive transfer EAE mice.
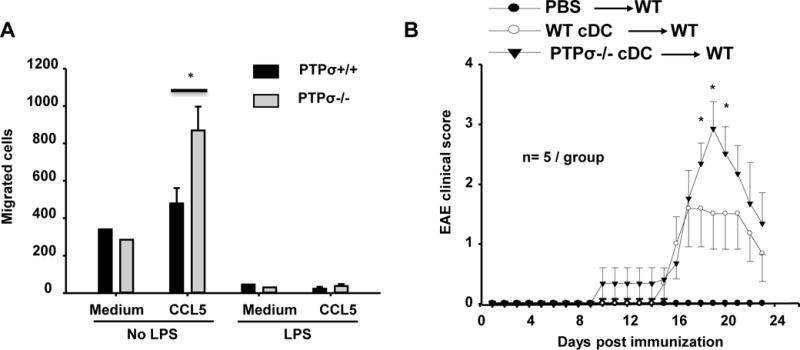
(A) cDCs collected from PTPσ+/+ or −/− mice were treated with or without LPS for 24 h and the migration of cDCs (1×105 cDCs) toward 100 ng/ml CCL5 was determined with a 24-h Transwell chemotaxis assay. n = 3 independent experiments. (B) cDCs were prepared from PTPσ+/+ or −/− mice, and then treated with LPS and pulsed with MOG for 24 hrs. On day 4 and 8 after immunization with MOG emulsified in CFA, adult BALB/c mice received intravenous injections of PTPσ+/+ or −/− cDCs (2 × 106 in 200 μl of PBS). A separate group of mice received injections of the same volume of PBS (200 μl) as negative controls. The daily EAE clinical scores were evaluated for 23 days. * p<0.05, compared with PTPσ+/+ cDC adoptive transfer EAE. n=5 mice/group.
To determine whether PTPσ−/− cDCs have a pro-inflammatory role in vivo, we adoptively transferred PTPσ+/+ and PTPσ−/− cDCs treated with LPS and pulsed with MOG35–55 to wild type mice immunized with CFA/MOG on day 4 and 7 after immunization. The mice receiving PTPσ−/− cDCs developed significantly higher clinical EAE scores than those that received PTPσ+/+ cDCs (Fig. 8B). Therefore, the findings of our in vitro and in vivo experiments provide evidence that PTPσ deficiency facilitates the pro-inflammatory activities of cDCs and development of EAE.
3.6. PTPσ directly regulates differentiation of CD4+ T cells
Upon antigen presentation by cDCs and depending on the cytokine milieu, CD4+ T cells differentiate into various subsets of effector T cells which play essential roles in EAE initiation and progression. To determine whether PTPσ directly affects T cell differentiation, we differentiated naïve CD4+ T cells isolated from spleens of PTPσ +/+ and −/− mice in non-polarizing conditions (Th0) and Th1 and Th17 polarizing conditions. PTPσ expression was induced following T cell differentiation, especially in Th17 polarizing conditions (Fig. 9A). Although both WT and PTPσ−/− T cells proliferated in a similar manner (Fig. 9B), PTPσ deletion led to upregulation of Tbet expression in Th1 cells, and of Rorγt expression in Th17 cells (Fig. 9C). WT Th0 and Th1 cells produced large amounts of IFNγ; Th17 cells produced much lower levels of IFNγ. Compared to WT, the amounts of IFNγ were significantly increased in both PTPσ−/− Th1 and Th17 cells (Fig. 9E). A statistically significant increase in IL-17 production was evident in PTPσ−/− Th17 cells. FACS analysis showed similar alterations in the percentages of IFNγ and IL-17 positive cells (Fig. 9D). Together, these results demonstrate that PTPσ expression in activated CD4+ T cells has an inhibitory effect on the differentiation of Th1 and especially of Th17 cells.
Figure 9. PTPσ deficiency promotes differentiation of CD4+ T cells into Th1 and Th17 cells.
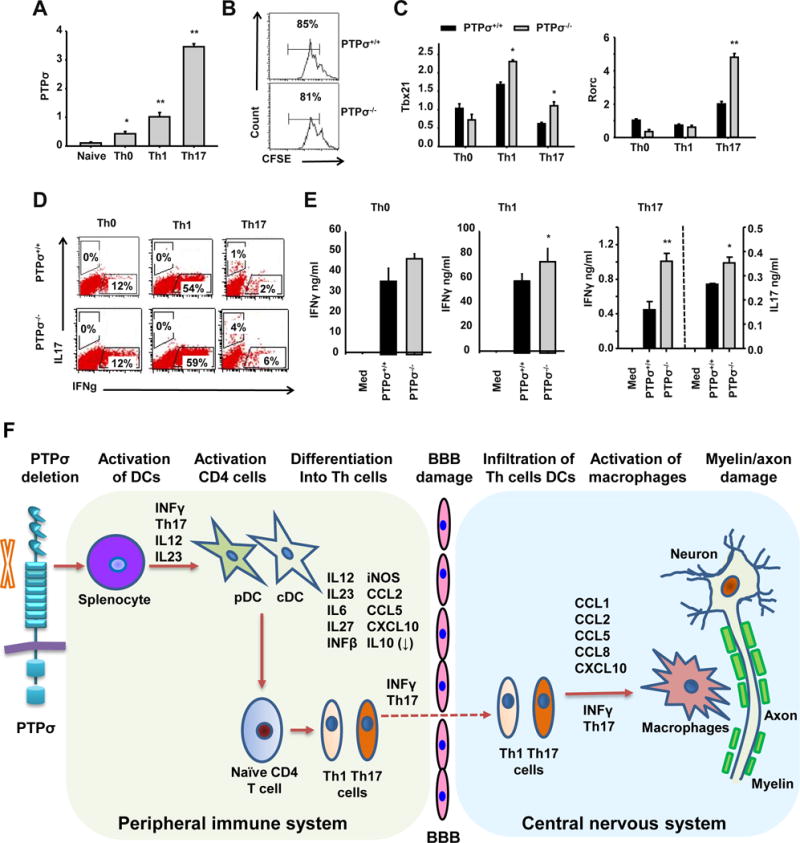
(A, C) Naïve CD4+ T cells purified from spleens of PTPσ +/+ and −/− mice were activated with anti-CD3 (3μg/ml) and anti-CD28 (1μg/ml) in Th0 (no cytokines were added), Th1 (+IL12 20 ng/ml) and Th17 (TGFb 3 ng/ml, IL23 50 ng/ml, IL6 20 ng/ml) conditions for 2 days. The expression levels of PTPσ (A), Tbx21 and Rorc (C) were measured by qRT-PCR. (B) Naïve CD4+ T cells purified from spleens of PTPσ +/+ and −/− mice were activated with anti-CD3 (3μg/ml) and anti-CD28 for 3 days and their proliferation then measured by carboxyfluorescein succinimidyl ester dilution. (D, E) Naïve CD4+ T cells purified from spleens of PTPσ +/+ and −/− mice were activated with anti-CD3 (3μg/ml) and anti-CD28 (1μg/ml) in Th0 (no cytokines), Th1 (+IL12 20 ng/ml) and Th17 (TGFb 3 ng/ml, IL23 50 ng/ml, IL6 20 ng/ml) conditions for 3 days. The levels of cytokines were measured by flow cytometry and ELISA. * p<0.05, ** p<0.01 compared with PTPσ +/+ group. n = 3 independent experiments. (F) Based on our results, we propose a model for the roles of PTPσ in regulating functions of immune cells and development of EAE.
4. Discussion
RPTPs are a family of cell surface phosphatases that have intracellular enzymatic activity and extracellular domains to interact with surrounding cell adhesion molecules due to sequence homologs. Some RPTPs are fundamental for controlling proliferation, differentiation, communication and adhesion in multiple cell types, and their disruption contributes to diverse human disorders, including cancer, diabetes, autoimmune diseases, and neurological deficits (Um and Ko, 2013; Xu and Fisher, 2012). Although most PTKs have been well studied, the function, substrate specificity, controlling mechanisms, and relevance to human diseases of most RPTPs are largely unknown. In this study, we demonstrated that transgenic deletion and pharmacological inhibition of PTPσ, one of 3 RPTPs in the LAR subfamily, significantly aggravated clinical symptoms and axon and myelin damage in the spinal cord of EAE mice. Following immunization with MOG35–55 peptide, PTPσ knockout mice exhibited pro-inflammatory profiles in the spinal cord and lymphoid organs by altering the expression levels of a number of inflammatory related genes, including specific cytokines (IFNγ and IL-17), chemokines (CCL2, CCL5 and CXCL10), and master transcriptional factors (such as Tbet and RORγt). Importantly, PTPσ deficiency stimulated a pro-inflammatory phenotype in cDCs, affected differentiation of CD4+ T lymphocytes directly, reduced the number of Treg cells, and facilitated infiltration of T cells and activation of macrophages/microglia in the CNS. These findings illustrate the novel role of PTPσ as an essential negative regulator for development of autoimmune CNS disease (Fig. 9F). Identification of the critical roles of PTPσ in mediating immune cell functions and developing EAE helps us better understand the molecular control of immune functions, and is potentially important for designing effective strategies to treat autoimmune disorders.
A recent study reported important role of PTPσ in regulating function of pDCs and infiltration of leukocytes in immune-mediated intestinal inflammation (Bunin et al., 2015). This group demonstrates that human pDCs express PTPσ and their activation rapidly downregulates levels of surface PTPσ. Accordingly, PTPσ deletion increases generation of IFN-α and its crosslinking with an antibody suppresses activation of mouse pDCs. Moreover, mouse pDCs express both PTPσ and LAR (another member in the LAR subfamily) and DC-specific deletion of PTPσ on LAR-deficient background enhanced the IFN response of pDCs. Our results are generally consistent with the previous findings, but we provide further insight into the immune functions mediated by PTPσ in several aspects. We, for the first time, identified the critical role of PTPσ in regulating development of EAE, the most frequently used model of MS. Inhibiting PTPσ remarkably aggravated progression of clinical symptoms and axon damage in EAE mice. We also demonstrate that PTPσ is not only a crucial regulator of pDCs, it also plays significant roles in mediating functions of multiple immune cells, especially cDCs and T lymphocytes. Accordingly, PTPσ mediates immune responses by altering production of multiple cytokines and chemokines, in addition to IFN reported in previous study (Bunin et al., 2015).
Increased activation of diverse immune cells and their subsequent penetration into the CNS are presumably responsible for the susceptibility of PTPσ−/− mice to EAE. We observed increased numbers of CD4+ T cells, including IFNγ and IL-17 producing T cells, and of CD11b+CD45Hi macrophages and CD11b+CD45Int microglia in the brain and spinal cord of PTPσ−/− mice following immunization. We also detected upregulation of specific cytokines, chemokines, and master transcriptional factors that contribute to EAE pathogenesis. In agreement with our results, PTPσ has been shown to suppress immune-mediated intestinal inflammation by reducing spontaneous IFN production (Bunin et al., 2015). While we demonstrate the critical role of PTPσ in mediating immune function and EAE development, we cannot exclude the possibility that PTPσ also regulates demyelination and axonal damage in EAE mice by targeting neural cells directly. PTPσ is expressed in multiple tissues, including the CNS, and is a presynaptic receptor that regulates synaptic adhesion, organization and plasticity by interacting with multiple postsynaptic partners, such as cell adhesion molecules (Takahashi and Craig, 2013). PTPσ deletion alters hippocampal long-term potentiation, synaptic plasticity and brain functions. PTPσ deficient mice exhibit severe growth retardation, high neonatal mortality, and various neurological deficiencies, including motor dysfunction, defective proprioception, hippocampal dysgenesis, abnormal pituitary development, and thinning of the corpus callosum and cerebral cortex (Meathrel et al., 2002; Uetani et al., 2006). PTPσ is also important for controlling neuronal growth by functioning as a receptor for chondroitin sulfate proteoglycan (CSPG) axon growth inhibitors (Shen et al., 2009) and for heparan sulphate proteoglycan (HSPG) growth-promoting molecules (Coles et al., 2011). It will also be interesting to determine whether PTPσ is expressed in oligodendrocytes and directly regulates their functions. PTPRZ, another RPTP mainly expressed in the developing CNS, could inhibit oligodendrocyte differentiation during early development and remyelination following CNS demyelination by increasing apoptosis of mature oligodendrocytes (Harroch et al., 2002; Kuboyama et al., 2012).
Some PTPs are essential for regulating the functions of immune cells by reversing tyrosine phosphorylation of their target proteins (Dolton et al., 2006; Doody et al., 2009; Rhee and Veillette, 2012). For example, PTPN22, a non-receptor PTP, could regulate the development and activation of lymphocytes, establishment of tolerance, and host defense mediated by innate immune cells (Rieck et al., 2007; Stanford and Bottini, 2014). A recent study demonstrated expression of PTPσ in pDCs and its significant role in suppressing pDC function (Bunin et al., 2015). In this study, we found that cDCs and splenic CD4+ T cells also expressed PTPσ, although at relatively reduced levels compared with pDCs. PTPσ deletion augmented the pro-inflammatory properties of cDCs by increasing secretion of cytokines responsible for Th1 and Th17 differentiation, upregulating IFNβ and iNOS, downregulating IL-10, and promoting cDC migration following CCL5 stimulation. Importantly, adoptive transfer to wild type mice of PTPσ-deficient cDCs stimulated with LPS and pulsed with MOG peptide significantly increased the clinical EAE symptoms following MOG immunization. Moreover, differentiation of naïve CD4+ T cells upregulated PTPσ (particularly in Th17 polarizing conditions) and PTPσ deletion enhanced the generation of molecules critical for mediating immune responses, including Tbet in Th1 cells and Rorγt in Th17 cells. Interestingly, LAR, also a member of the LAR subfamily, is expressed in immature thymocytes and regulates their differentiation and expansion in mice (Kondo et al., 2010). Moreover, PTPσ deletion reduced expression levels of Foxp3, a lineage-specific marker for Treg (Fontenot et al., 2005; Ziegler, 2006). Foxp3 is a transcription factor in the forkhead-winged helix family and plays critical role in regulating suppressive properties of Treg cells. Consistently, Treg cells have been reported to prevent development (Hori et al., 2002; Olivares-Villagomez et al., 1998) and delay onset of spontaneous EAE (Lowther et al., 2013). Therefore, we have, for the first time, determined that PTPσ is an important negative regulator for pro-inflammatory properties of cDC, differentiation of CD4+ T cells into specific Th1 and Th17 cells, and development of EAE, a CNS autoimmune disease.
PTPσ deletion increases the levels of various cytokines, such as IL-12p70, IL-27, IL-23, IL-6, IFN and iNOS, suggesting that tyrosine dephosphorylation by PTPσ suppresses production of pro-inflammatory related cytokines. Tyrosine phosphorylation has consistently appeared to be vital for controlling cytokine generation in DCs (Fujita et al., 2013). However, the upstream signals that modulate PTPσ activity in immune cells and other cell types are not clear. As transmembrane receptors for multiple ECM molecules in diverse types of cells, PTPσ may attenuate tyrosine phosphorylation of target proteins upon interactions with its surrounding ligands. The known PTPσ ligands include a number of proteoglycans, especially the ECM proteins HSPGs and CSPGs (Coles et al., 2011; Ohtake and Li, 2015; Shen et al., 2009). HSPGs have been reported to inhibit production of cytokines by immune cells (Gordts et al., 2014). Syndecan-1, a HSPG used as a marker of plasma cells, may suppress various inflammatory responses and development of EAE in mice. Deletion of syndecan-1 increased disease severity and impaired recovery in mice with EAE (Zhang et al., 2013). PTPσ-dependent proteoglycan switch have been shown to regulate function of fibroblast-like synoviocytes (the joint-lining cells), including their invasiveness and migration (Doody et al., 2015). Incubating these cells with a proteoglycan-binding PTPσ decoy protein suppresses their invasiveness and attachment to cartilage by disrupting a constitutive interaction between PTPσ and HSPG syndecan-4. On the other hand, CSPGs are upregulated around the edge of active MS lesions and influence immune responses in several neurological disorders (Haylock-Jacobs et al., 2011; Sobel and Ahmed, 2001). Some cytokines, such as TGFβ1 and IFNγ, may alter expression levels of CSPGs (Fujiyoshi et al., 2010; Smith and Strunz, 2005). It will therefore be interesting to determine whether PTPσ regulates the functions of immune cells, such as DCs and T cells, by interacting with any specific ECM proteoglycans, such as syndecan-1 and 4.
Conclusion
Initiation and progression of EAE increase in PTPσ deficient mice, and deletion of this transmembrane phosphatase affects activation of cDCs, differentiation of T lymphocytes into Th1 and Th17 effectors, and activity of Treg cells. Together with a recent study (Bunin et al., 2015), our findings provide evidence that PTPσ is important for suppressing immune and autoimmune responses mediated by pDCs, cDCs and CD4+ T cells. Because suppression of PTPσ activity in immune cells contributes to EAE pathogenesis and progression, selective activation of this signaling protein may become an effective strategy for treating MS related disorders.
Supplementary Material
Highlights.
PTPσ inhibition exacerbates symptoms of experimental autoimmune encephalomyelitis
PTPσ deletion promotes a pro-inflammatory phenotype in immune cells
PTPσ is a key negative regulator in EAE initiation and progression
Acknowledgments
This work was supported by research grants to SL from NIH (1R01NS079432, 1R21NS066114 and 1R01EY024575) and Shriners Research Foundation (SHC-86300-PHI, SHC-86200-PHI-16, and SHC-446007).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions: YO, WK, DG and SL contributed to experimental designs, data analyses, figure making and paper writing. RH and MH contributed to the experiments. MLT contributed to experimental design and paper writing.
Conflict of Interest. The authors have no potential conflicts of interest to disclose.
References
- Bunin A, Sisirak V, Ghosh HS, Grajkowska LT, Hou ZE, Miron M, Yang C, Ceribelli M, Uetani N, Chaperot L, Plumas J, Hendriks W, Tremblay ML, Hacker H, Staudt LM, Green PH, Bhagat G, Reizis B. Protein Tyrosine Phosphatase PTPRS Is an Inhibitory Receptor on Human and Murine Plasmacytoid Dendritic Cells. Immunity. 2015;43:277–288. doi: 10.1016/j.immuni.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, Gallagher JT, Jones EY, Flanagan JG, Aricescu AR. Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science (New York, NY) 2011;332:484–488. doi: 10.1126/science.1200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolton GM, Sathish JG, Matthews RJ. Protein tyrosine phosphatases as negative regulators of the immune response. Biochem Soc Trans. 2006;34:1041–1045. doi: 10.1042/BST0341041. [DOI] [PubMed] [Google Scholar]
- Doody KM, Bourdeau A, Tremblay ML. T-cell protein tyrosine phosphatase is a key regulator in immune cell signaling: lessons from the knockout mouse model and implications in human disease. Immunol Rev. 2009;228:325–341. doi: 10.1111/j.1600-065X.2008.00743.x. [DOI] [PubMed] [Google Scholar]
- Doody KM, Stanford SM, Sacchetti C, Svensson MN, Coles CH, Mitakidis N, Kiosses WB, Bartok B, Fos C, Cory E, Sah RL, Liu-Bryan R, Boyle DL, Arnett HA, Mustelin T, Corr M, Esko JD, Tremblay ML, Firestein GS, Aricescu AR, Bottini N. Targeting phosphatase-dependent proteoglycan switch for rheumatoid arthritis therapy. Sci Transl Med. 2015;7:288ra276. doi: 10.1126/scitranslmed.aaa4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elchebly M, Wagner J, Kennedy TE, Lanctot C, Michaliszyn E, Itie A, Drouin J, Tremblay ML. Neuroendocrine dysplasia in mice lacking protein tyrosine phosphatase sigma. Nat Genet. 1999;21:330–333. doi: 10.1038/6859. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Friese MA, Montalban X, Willcox N, Bell JI, Martin R, Fugger L. The value of animal models for drug development in multiple sclerosis. Brain. 2006;129:1940–1952. doi: 10.1093/brain/awl083. [DOI] [PubMed] [Google Scholar]
- Fujita H, Kitawaki T, Sato T, Maeda T, Kamihira S, Takaori-Kondo A, Kadowaki N. The tyrosine kinase inhibitor dasatinib suppresses cytokine production by plasmacytoid dendritic cells by targeting endosomal transport of CpG DNA. Eur J Immunol. 2013;43:93–103. doi: 10.1002/eji.201242699. [DOI] [PubMed] [Google Scholar]
- Fujiyoshi T, Kubo T, Chan CC, Koda M, Okawa A, Takahashi K, Yamazaki M. Interferon-gamma decreases chondroitin sulfate proteoglycan expression and enhances hindlimb function after spinal cord injury in mice. J Neurotrauma. 2010;27:2283–2294. doi: 10.1089/neu.2009.1144. [DOI] [PubMed] [Google Scholar]
- Gordts PL, Foley EM, Lawrence R, Sinha R, Lameda-Diaz C, Deng L, Nock R, Glass CK, Erbilgin A, Lusis AJ, Witztum JL, Esko JD. Reducing macrophage proteoglycan sulfation increases atherosclerosis and obesity through enhanced type I interferon signaling. Cell metabolism. 2014;20:813–826. doi: 10.1016/j.cmet.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goverman J. Autoimmune T cell responses in the central nervous system. Nature reviews. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harroch S, Furtado GC, Brueck W, Rosenbluth J, Lafaille J, Chao M, Buxbaum JD, Schlessinger J. A critical role for the protein tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nat Genet. 2002;32:411–414. doi: 10.1038/ng1004. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron. 2006;52:61–76. doi: 10.1016/j.neuron.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Haylock-Jacobs S, Keough MB, Lau L, Yong VW. Chondroitin sulphate proteoglycans: extracellular matrix proteins that regulate immunity of the central nervous system. Autoimmun Rev. 2011;10:766–772. doi: 10.1016/j.autrev.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8213–8218. doi: 10.1073/pnas.122224799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen M, Schweer D, Ziegler A, Gaber R, Schock S, Schwinzer R, Wonigeit K, Lindert RB, Kantarci O, Schaefer-Klein J, Schipper HI, Oertel WH, Heidenreich F, Weinshenker BG, Sommer N, Hemmer B. A point mutation in PTPRC is associated with the development of multiple sclerosis. Nat Genet. 2000;26:495–499. doi: 10.1038/82659. [DOI] [PubMed] [Google Scholar]
- Johnson KG, Van Vactor D. Receptor protein tyrosine phosphatases in nervous system development. Physiol Rev. 2003;83:1–24. doi: 10.1152/physrev.00016.2002. [DOI] [PubMed] [Google Scholar]
- Kondo S, Kishi H, Muraguchi A. Regulatory role of leukocyte-common-antigen-related molecule (LAR) in thymocyte differentiation. Eur J Immunol. 2010;40:1296–1302. doi: 10.1002/eji.200939743. [DOI] [PubMed] [Google Scholar]
- Kong W, Yen JH, Vassiliou E, Adhikary S, Toscano MG, Ganea D. Docosahexaenoic acid prevents dendritic cell maturation and in vitro and in vivo expression of the IL-12 cytokine family. Lipids Health Dis. 2010;9:12. doi: 10.1186/1476-511X-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboyama K, Fujikawa A, Masumura M, Suzuki R, Matsumoto M, Noda M. Protein tyrosine phosphatase receptor type z negatively regulates oligodendrocyte differentiation and myelination. PLoS One. 2012;7:e48797. doi: 10.1371/journal.pone.0048797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther DE, Chong DL, Ascough S, Ettorre A, Ingram RJ, Boyton RJ, Altmann DM. Th1 not Th17 cells drive spontaneous MS-like disease despite a functional regulatory T cell response. Acta Neuropathol. 2013;126:501–515. doi: 10.1007/s00401-013-1159-9. [DOI] [PubMed] [Google Scholar]
- Maatta JA, Kaldman MS, Sakoda S, Salmi AA, Hinkkanen AE. Encephalitogenicity of myelin-associated oligodendrocytic basic protein and 2′,3′-cyclic nucleotide 3′-phosphodiesterase for BALB/c and SJL mice. Immunology. 1998;95:383–388. doi: 10.1046/j.1365-2567.1998.00605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino G, Hartung HP. Immunopathogenesis of multiple sclerosis: the role of T cells. Curr Opin Neurol. 1999;12:309–321. doi: 10.1097/00019052-199906000-00010. [DOI] [PubMed] [Google Scholar]
- McGavern DB, Murray PD, Rivera-Quinones C, Schmelzer JD, Low PA, Rodriguez M. Axonal loss results in spinal cord atrophy, electrophysiological abnormalities and neurological deficits following demyelination in a chronic inflammatory model of multiple sclerosis. Brain 123 Pt. 2000;3:519–531. doi: 10.1093/brain/123.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meathrel K, Adamek T, Batt J, Rotin D, Doering LC. Protein tyrosine phosphatase sigma-deficient mice show aberrant cytoarchitecture and structural abnormalities in the central nervous system. Journal of neuroscience research. 2002;70:24–35. doi: 10.1002/jnr.10382. [DOI] [PubMed] [Google Scholar]
- Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu TH, Guo J, Zhang W, So KF, Pepinsky B, Shao Z, Graff C, Garber E, Jung V, Wu EX, Wu W. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nature medicine. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain, behavior, and immunity. 2010;24:641–651. doi: 10.1016/j.bbi.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Ogata M, Sawada M, Kosugi A, Hamaoka T. Developmentally regulated expression of a murine receptor-type protein tyrosine phosphatase in the thymus. J Immunol. 1994;153:4478–4487. [PubMed] [Google Scholar]
- Ohtake Y, Li S. Molecular mechanisms of scar-sourced axon growth inhibitors. Brain research. 2015;1619:22–35. doi: 10.1016/j.brainres.2014.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares-Villagomez D, Wang Y, Lafaille JJ. Regulatory CD4(+) T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. The Journal of experimental medicine. 1998;188:1883–1894. doi: 10.1084/jem.188.10.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Yeste A, Mascanfroni ID. Role and therapeutic value of dendritic cells in central nervous system autoimmunity. Cell death and differentiation. 2015;22:215–224. doi: 10.1038/cdd.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol. 2012;13:439–447. doi: 10.1038/ni.2246. [DOI] [PubMed] [Google Scholar]
- Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol. 2007;179:4704–4710. doi: 10.4049/jimmunol.179.7.4704. [DOI] [PubMed] [Google Scholar]
- Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, He Z, Silver J, Flanagan JG. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science (New York, NY) 2009;326:592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GM, Strunz C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia. 2005;52:209–218. doi: 10.1002/glia.20236. [DOI] [PubMed] [Google Scholar]
- Sobel RA, Ahmed AS. White matter extracellular matrix chondroitin sulfate/dermatan sulfate proteoglycans in multiple sclerosis. J Neuropathol Exp Neurol. 2001;60:1198–1207. doi: 10.1093/jnen/60.12.1198. [DOI] [PubMed] [Google Scholar]
- Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol. 2014;10:602–611. doi: 10.1038/nrrheum.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nature protocols. 2006;1:1810–1819. doi: 10.1038/nprot.2006.285. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Craig AM. Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. 2013;36:522–534. doi: 10.1016/j.tins.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terszowski G, Jankowski A, Hendriks WJ, Rolink AG, Kisielow P. Within the hemopoietic system, LAR phosphatase is a T cell lineage-specific adhesion receptor-like protein whose phosphatase activity appears dispensable for T cell development, repertoire selection and function. Eur J Immunol. 2001;31:832–840. doi: 10.1002/1521-4141(200103)31:3<832::aid-immu832>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Uetani N, Chagnon MJ, Kennedy TE, Iwakura Y, Tremblay ML. Mammalian motoneuron axon targeting requires receptor protein tyrosine phosphatases sigma and delta. J Neurosci. 2006;26:5872–5880. doi: 10.1523/JNEUROSCI.0386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Ko J. LAR-RPTPs: synaptic adhesion molecules that shape synapse development. Trends in cell biology. 2013;23:465–475. doi: 10.1016/j.tcb.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Weir CR, Nicolson K, Backstrom BT. Experimental autoimmune encephalomyelitis induction in naive mice by dendritic cells presenting a self-peptide. Immunology and cell biology. 2002;80:14–20. doi: 10.1046/j.1440-1711.2002.01056.x. [DOI] [PubMed] [Google Scholar]
- Xu Y, Fisher GJ. Receptor type protein tyrosine phosphatases (RPTPs) - roles in signal transduction and human disease. Journal of cell communication and signaling. 2012;6:125–138. doi: 10.1007/s12079-012-0171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wu C, Song J, Gotte M, Sorokin L. Syndecan-1, a cell surface proteoglycan, negatively regulates initial leukocyte recruitment to the brain across the choroid plexus in murine experimental autoimmune encephalomyelitis. J Immunol. 2013;191:4551–4561. doi: 10.4049/jimmunol.1300931. [DOI] [PubMed] [Google Scholar]
- Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.