

Abstract
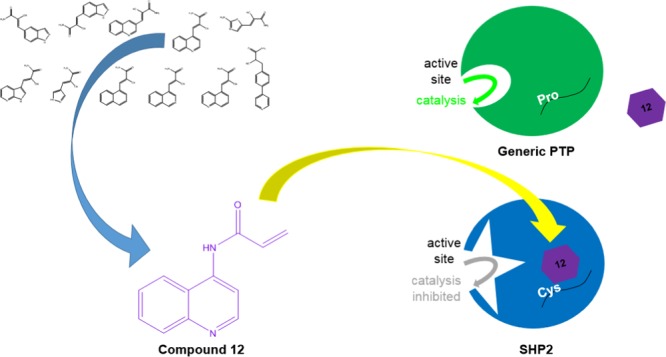
Difficulties in developing active-site-directed protein tyrosine phosphatase (PTP) inhibitors have led to the perception that PTPs are “undruggable”, highlighting the need for new means to target pharmaceutically important PTPs allosterically. Recently, we characterized an allosteric-inhibition site on the PTP domain of Src-homology-2-domain-containing PTP 2 (SHP2), a key anticancer drug target. The central feature of SHP2’s allosteric site is a nonconserved cysteine residue (C333) that can potentially be labeled with electrophilic compounds for selective SHP2 inhibition. Here, we describe the first directed discovery effort for C333-targeted allosteric SHP2 inhibitors. By screening a previously reported library of reversible, covalent inhibitors, we identified a lead compound, which was modified to yield an irreversible inhibitor (12), that inhibits SHP2 allosterically and selectively through interaction with C333. These findings provide a novel paradigm for allosteric-inhibitor discovery on SHP2, one that may help to circumvent the challenges inherent in targeting SHP2’s active site.
Introduction
Protein tyrosine phosphatases (PTPs) catalyze the dephosphorylation of phosphotyrosine, a critical control element in mammalian signal transduction,1 and misregulation of PTP activity has been implicated in a range of human pathologies, including cancer, diabetes, and autoimmune disorders.2−6 Src-homology-2-domain-containing PTP 2 (SHP2) provides a particularly strong example of the connection between improperly regulated PTP activity and human disease: SHP2 mutations cause Noonan and LEOPARD syndromes, and mutations that increase SHP2’s basal PTP activity are the most common cause of sporadic juvenile myelomonocytic leukemia.7 SHP2 activity also has been strongly implicated in oncogenesis even in instances in which the enzyme does not itself harbor mutations.8−11 Therefore, SHP2 has emerged as a bona fide anticancer target for inhibitor discovery, and compounds that can effectively target either (or both) the wild-type enzyme and its improperly activated variants hold significant therapeutic potential.
PTPs, including SHP2, however, have proven notoriously difficult to drug. Active-site-directed PTP inhibitors often display limited selectivity, owing to the high degree of sequence and structural homology in PTP active sites.12 Bioavailability is also a major hurdle, as the potencies of many active-site-directed compounds are dependent on negatively charged phosphotyrosine-mimetic pharmacophores that hinder cell permeability.13 The specific challenges inherent in identifying target-specific and bioactive SHP2 inhibitors have been further highlighted by a recent study, which showed that several previously reported compounds, some of which have been widely used in the SHP2-signaling literature, either (i) fail to inhibit SHP2 in a cellular context or (ii) induce non-SHP2-mediated signaling effects that are likely the result of off-target protein-kinase inhibition.14
Allosteric inhibition represents an attractive approach for circumventing the difficulties that have limited successful development of active-site-directed SHP2 inhibitors for two key reasons: allosteric sites are typically much less strongly conserved than PTP active sites and allosteric sites do not necessarily privilege charged pharmacophores.15,16 In principle, allosteric PTP inhibition could be achieved either through engagement with a site on the PTP domain or by targeting a regulatory site beyond the PTP domain.17 For SHP2, noteworthy success on the latter front has been realized through the development of compounds that stabilize an autoinhibitory interaction between SHP2’s SH2 domains and its PTP domain.18 Although small-molecule stabilizers of SHP2’s autoinhibition (e.g., SHP099) hold enormous promise for targeting cancers in which SHP2 activity is implicated,19,20 the potency of such compounds can be strongly attenuated by cancer-associated gain-of-function mutations that disrupt the integrity of the autoinhibitory interaction.21 Compounds that can inhibit PTP activity at an allosteric site directly on SHP2’s PTP domain would provide an alternate mode of allosteric SHP2 targeting that is likely to remain functional regardless of the strength of the SH2/PTP domain interaction in the particular SHP2 variant that is targeted therapeutically.
We have previously demonstrated that the SHP2 catalytic domain contains an allosteric-inhibition site that is not present in most other PTPs and that renders the enzyme sensitive to selective inhibition.22 This finding stemmed from an unexpected observation that SHP2 is inhibited by a biarsenical compound, FlAsH-EDT2,23,24 which does not inhibit other PTPs.25 It was further shown that the FlAsH-EDT2 sensitivity of SHP2’s allosteric site is dependent on the presence of a nonconserved cysteine residue (C333) that lies outside of the active site (Figure 1A,B).22 Importantly, SHP2’s unusual sensitivity to FlAsH-EDT2 is completely removed by mutation of C333 to proline,25 the amino acid that occupies the analogous position in most classical PTPs (Figure 1B).26 C333, therefore, represents the key selectivity determinant of a nonconserved inhibitory site that may provide a new means for targeting the SHP2 catalytic domain allosterically and with high selectivity.
Figure 1.
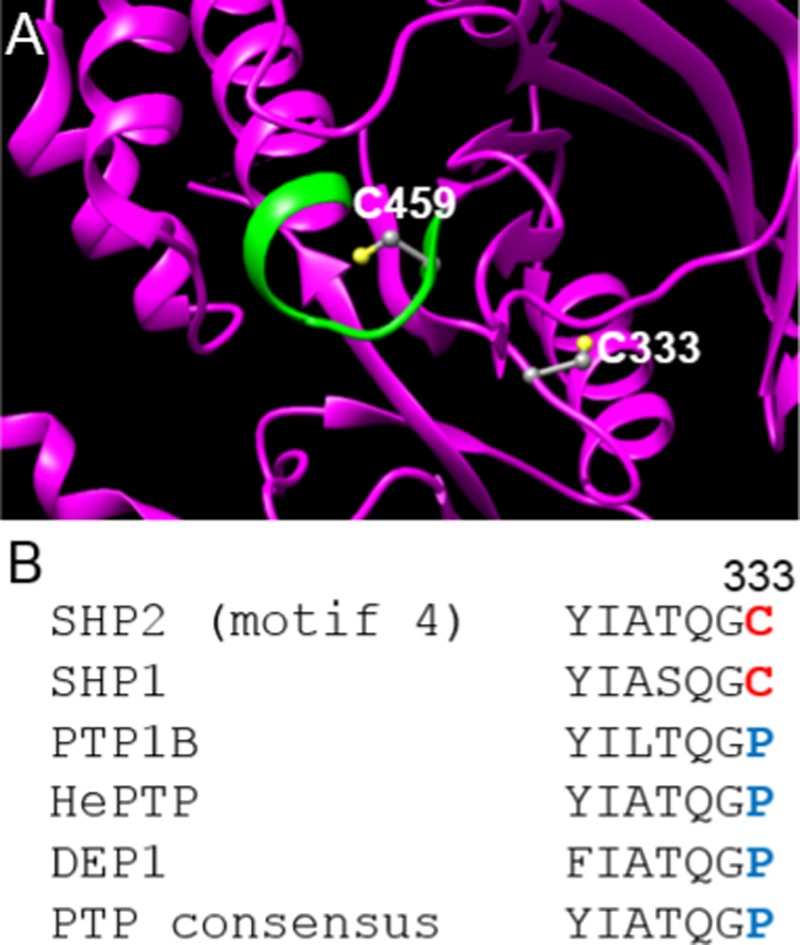
C333 is rare within classical PTP domains and lies outside of SHP2’s active site. (A) Three-dimensional structure of SHP2’s catalytic domain (PDB ID: 3B7O).27 SHP2 is shown as a magenta ribbon, with the conserved active-site motif highlighted in green. The side chains of C333 and the active-site cysteine, C459, are shown and colored by atom type. (B) Sequence alignment of motif 4 (as assigned by Andersen et al.26) of the PTPs discussed in this study. 34 of 37 classical human PTP domains contain proline at position 333, 1 PTP has serine (not shown), and only 2, SHP2 and SHP1, have cysteine.
Although it was previously shown that SHP2 can be selectively inhibited via its PTP-domain allosteric site, the biarsenical compound with which this discovery was made is, on its face, not viable as a lead for development of SHP2-directed therapeutics. FlAsH-EDT2 has been previously shown to associate with a significant number of cysteine-rich proteins nonspecifically28 and would be expected to present significant toxicity due to its arsenic atoms.29
In the current study, we identify the first druglike compounds that can bind to SHP2’s C333-containing allosteric site and inhibit the enzyme allosterically and selectively. To do so, we investigated compounds that contain two key elements: a “druglike” component that could potentially drive noncovalent binding interactions in the pocket surrounding C333 and an electrophilic component capable of reacting covalently with the nucleophilic C333, once the compound is noncovalently bound. The targeted discovery of compounds that covalently react with C333, and avoid reaction with conserved cysteines within the PTP domain, may represent an important new strategy for specific allosteric inhibition of SHP2.
Results and Discussion
To identify compounds that can allosterically inhibit SHP2 through a novel mechanism of action, we synthesized compound panel 1–8 (Figure 2A), a small library of cyanoacrylamides that was previously used to identify protein-kinase inhibitors that act via covalent interaction with a nonconserved cysteine residue in the kinase domain.30 We hypothesized that the compound panel could be remined with SHP2 (a member of a different enzyme family, the PTPs) to possibly yield lead compounds capable of inhibition through targeting the nonconserved cysteine C333. Because PTPs also contain a highly conserved cysteine residue that is necessary for catalytic activity (C459 in SHP2, one of two conserved cysteines in classical PTPs, vide infra), we screened compounds 1–8 both for inhibition of the wild-type SHP2 PTP domain and for selectivity against a SHP2 mutant, in which C333 is replaced with proline (C333P SHP2). A compound that selectively inhibits SHP2 over C333P SHP2 is likely to act at SHP2’s allosteric site, as the 333 position contains the only molecular difference between the two enzymes. Moreover, a compound that can target SHP2 over C333P SHP2 could be reasonably hypothesized to show selectivity over many other PTPs, as the vast majority of human classical PTPs (35 of 37) do not contain cysteine at the homologous position in their respective catalytic domains.26
Figure 2.

Screen for C333-directed allosteric SHP2 inhibitors. (A) Chemical structures of 1–11. Compounds 1–8 were reported previously by Miller et al.30 as putative protein-kinase inhibitors. (B) PTP activities of the SHP2 and C333P SHP2 catalytic domains (800 nM) were measured with para-nitrophenyl phosphate (pNPP, 4 mM) after preincubation with the indicated inhibitors (300 μM, 2 h, 23 °C). (C) PTP activities of the SHP2 and C333P SHP2 catalytic domains (800 nM) were measured with pNPP (4 mM) after preincubation with the indicated inhibitors (50 μM, 2 h, 37 °C).
Our compound screen revealed, unsurprisingly, that most of the compounds in the small panel did not demonstrate selective inhibition of the wild-type SHP2 PTP domain over the C333P mutant (Figure 2B). However, two compounds (4 and 7) showed some selectivity for the wild-type enzyme, with 4 being the more active of the two. Interestingly, the relative placement of 4’s cyanoacrylamide moiety and the nitrogen atom in its quinoline ring system appears to be critical for activity, as isomer 3 showed very little activity on SHP2 (Figure 2A,B). To further test the importance of the presence and/or placement of 4’s quinoline nitrogen atom, we synthesized compounds 9–11 which either lack the nitrogen atom (9, Figure 2A) or have the nitrogen atom in a different location within the heteroaromatic ring system (10 and 11, Figure 2A). We found that the 4-substituted quinoline pharmacophore of compound 4 was necessary for activity, as 9–11 showed no selective inhibition of wild-type SHP2 under the screening conditions (Figure 2C).
The electrophilic cyanoacrylamide functional group of compounds 1–11 has previously been shown to react reversibly with cysteine residues.30,31 Although reversible covalent inhibitors have been fruitfully used to target nonconserved cysteine residues in the protein-kinase superfamily,32 we were interested in developing an irreversible analogue of 4, which would potentially enable the unambiguous mapping of the interaction between SHP2 and the 4-quinoline pharmacophore. To develop an irreversible inhibitor that retains the lead compound’s SHP2-binding properties, we explored derivatives of 4, in which the 4-quinoline pharmacophore was unchanged, but the compound’s cyanoacrylamide was replaced with electrophiles that could potentially react irreversibly with C333 of SHP2.
Notably, we found that compound 12 (Figure 3A), in which the cyanoacrylamide is replaced with an acrylamide, not only retained the inhibitory activity of compound 4 against SHP2, but it demonstrated increased potency (Figure 3B), whereas some measurable SHP2 activity remained after pre-incubation with 300 μM 4 (Figure 2B), essentially all SHP2 activity was abolished in the presence of 150 μM compound 12 (Figure 3B). Inhibition by 12 was shown to be dose-dependent, and we estimated a 50% inhibitory concentration (IC50) of approximately 35 μM with the wild-type SHP2 PTP domain. Importantly, compound 12 retains the specificity of 4 against C333P SHP2, showing little to no significant inhibition of the mutant enzyme, even at the highest compound concentration assayed (150 μM, Figure 3B). In addition, the IC50 of compound 12 on wild-type SHP2 is not affected by the addition of detergent, further suggesting that the compound does not act through nonspecific enzyme aggregation (Figure S1, Supporting Information).33
Figure 3.

Compound 12 is a C333-targeted, time-dependent SHP2 inhibitor. (A) Chemical structure of 12. (B) Dose dependence of inhibition by 12. PTP activities of the SHP2 and C333P SHP2 catalytic domains (250 nM) were measured with pNPP (5 mM) after preincubation (2 h, 37 °C) with vehicle or 12 at the indicated concentrations. (C) Time dependence of inhibition by 12. PTP activities of the SHP2 and C333P SHP2 catalytic domains (250 nM) were measured with pNPP (5 mM) after preincubation with vehicle or 12 (150 μM) for the indicated times at 37 °C. (D) Kinetic analysis of SHP2 inhibition. Time-dependent inhibition assays were carried out essentially as described for panel (C) to determine the half-life (t1/2) of PTP activity at various concentrations of 12. The linear relationship between t1/2 and the reciprocal of inhibitor concentration (1/[12]) was analyzed to estimate the indicated kinetic constants as previously described.34,35
Because compound 12 was designed as a cysteine-targeting covalent inhibitor, we next sought to investigate whether it acts in a time-dependent manner. We found that, indeed, the degree of SHP2 inhibition at a fixed concentration of 12 (150 μM) is highly dependent on the time of preincubation (Figure 3C). In agreement with the dose-dependence experiments (Figure 3B), C333P SHP2 was not significantly inhibited by 12 regardless of the duration of the preincubation (Figure 3C). Interestingly, a mutant of SHP2, in which C333 has been replaced with alanine (C333A SHP2), shows attenuated, yet still significant, time-dependent inhibition by 12 (Figure S2, Supporting Information), suggesting that covalent bond formation between C333 and 12’s acrylamide moiety is not strictly rate limiting for SHP2 inhibition. It may be that a conformational change that is necessary for inhibition of wild-type (or C333A) SHP2 is precluded in the rigidified C333P mutant.22 Regardless of the precise mechanism of inhibition, the time- and concentration-dependence of SHP2 inhibition were investigated more quantitatively to estimate the kinetics of inhibition, yielding an inactivation constant (kinact) of 0.021 min–1 and an inhibition constant (KI) of 279 μM (Figure 3D) for 12 on SHP2.
We next sought to directly assess which, if any, of SHP2’s catalytic-domain cysteine residues are labeled in the presence of compound 12. It is important to note that PTP domains contain two highly conserved cysteine residues (C367 and C459 in human SHP2 numbering),26 and that reagents that inhibit PTP activity through covalent reaction with these conserved cysteines have been developed previously.16,36,37 Compounds that label C367 and C459 (and the homologous cysteines in other PTPs), however, are generally not selective, as their mechanisms of action rely on conserved elements within the PTP domain.
To test if allosteric inhibitor 12 is capable of inactivating SHP2 without labeling its conserved PTP-domain cysteines, we performed liquid-chromatographic tandem-mass-spectrometric analysis (LC-MS/MS) on SHP2 after incubation with the compound (50 μM, 3 h). Consistent with the previous observation that C333P SHP2 (which contains both C367 and C459) is resistant to inhibition by 12, LC-MS/MS revealed that these conserved residues do not form covalent adducts with 12 to any appreciable degree (Figure 4 and Tables S1 and S2, Supporting Information). We observed either no or almost no detectable (<0.25%) labeling of SHP2’s conserved cysteines in both SHP2 and C333P SHP2. By contrast, we observed approximately 20% labeling of C333 in SHP2, even though this cysteine residue is buried in SHP2 crystal structures. These findings are consistent with a “cryptic-allosteric-site” hypothesis, which posits that a conformational change is necessary to expose the C333 sidechain to solvent for allosteric inhibition to occur.22
Figure 4.
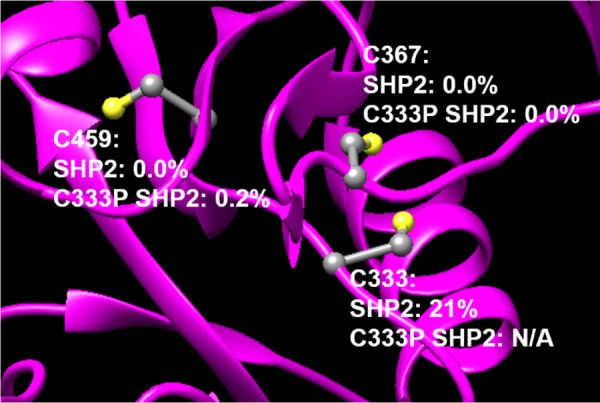
Compound 12 irreversibly labels C333, but not the conserved cysteines C367 and C459, in the SHP2 catalytic domain. After preincubation of SHP2 or C333P SHP2 with 12 (50 μM, 3 h), the degree of labeling at the indicated cysteine residues was determined by LC-MS/MS.
In addition to C333, 12 did also label other nonconserved cysteine residues (C259, C318, and C486) that are surface-exposed and/or positioned on loops (Tables S1 and S2). These labeling events presumably have no significant effects on SHP2 activity, as they occur in both the wild-type and C333P enzymes upon treatment with 12 (Tables S1 and S2). The off-target labeling caused by 12, coupled with its modest kinetics of inactivation (Figure 3D), underscore the need for further efforts focused toward the discovery of compounds that can target the SHP2 allosteric site with greater target potency, specificity, and rapidity. Nevertheless, compound 12’s demonstrated ability to engage C333 over SHP2’s conserved cysteines represents a key proof of principle for targeting SHP2’s allosteric site with covalent inhibitors.
The significance of the data presented above depends on a key assumption that selectivity for wild-type over C333P SHP2 is a useful proxy for selectivity against other PTP domains that contain proline at the position homologous to 333. To test the validity of this assumption directly, we expressed and purified the catalytic domains of four other PTPs, three that possess the consensus amino acid proline at position 333 (PTP1B, hematopoietic protein tyrosine phosphatase (HePTP), density-enhanced phosphatase 1 (DEP1)), as well as the one other PTP that has a cysteine at the equivalent position, SHP1 (Figure 1B). We then assayed the activities of all of the PTP domains after incubation with compound 12, under conditions that give strong inhibition of SHP2’s catalytic domain (Figure 5). We found, without exception, that PTP domains that contain proline at position 333 are resistant to compound 12, whereas the only two human PTPs that have cysteine at this position, SHP2 and SHP1, are sensitive to inhibition (Figure 5). These results confirm that the presence or absence of proline at position 333 is a strong determinant for the efficacy of compound 12. Moreover, because almost all human PTPs contain proline at this position, our data suggest that most PTPs will also be resistant to 12. It is also reasonable to infer that other yet-to-be-discovered compounds that can effectively engage C333 in the context of SHP2’s PTP-domain allosteric site will show similar levels of selectivity.
Figure 5.

Residue 333 is a strong determinant of sensitivity to 12 in multiple PTP domains. Activities of the indicated PTP domains (800 nM) were measured with pNPP (4 mM) in the absence (vehicle only) or presence of compound 12 (50 μM) after a 4 h incubation at 37 °C.
SHP2’s catalytic domain is widely used in inhibitor-discovery efforts, including the studies presented above.8 The isolated PTP domain is used largely for the purpose of experimental efficiency, as full-length SHP2 possesses strongly attenuated in vitro PTP activity due to the presence of the enzyme’s autoinhibitory SH2 domains.38,39 Nevertheless, for a small-molecule SHP2 inhibitor to be useful, it must of course be able to engage its target site in the context of the full-length protein.
To test whether compound 12 is capable of targeting SHP2’s allosteric site in the presence of the enzyme’s SH2 domains, we expressed a full-length construct (fl-SHP2, comprising residues 1–541, lacking only the disordered C-terminal tail) and assayed its sensitivity to inhibition by 12. In agreement with the results on the SHP2 catalytic domain, we found that 12 induces strong and dose-dependent inhibition of fl-SHP2 (Figure 6). Importantly, fl-SHP2’s sensitivity to 12 is eliminated by mutation of cysteine 333 to proline (fl-C333P SHP2). These results suggest that 12’s mechanisms of inhibition on the SHP2 PTP domain and fl-SHP2 are identical, i.e., that the compound inhibits allosterically through direct engagement of C333, either in the context of the PTP domain or the full-length enzyme.
Figure 6.

Compound 12 inhibits fl-SHP2, but not fl-C333P SHP2. PTP activities of the indicated enzymes (250 nM) were measured with 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) (50 μM) in the absence (vehicle only) or presence of the indicated concentrations of compound 12 after a 2 h incubation at 37 °C (the reduced activity of fl-SHP2, as compared to the SHP2 PTP domain, necessitated a switch of substrate to DiFMUP, a small-molecule PTP substrate that is more efficient than pNPP).
Conclusions
Collectively, our findings establish that a nonconserved allosteric site on the catalytic domain of the oncogenic PTP SHP2 can be targeted for selective inhibition with druglike small molecules. Moreover, our study validates the strategy of counter-screening putative SHP2 inhibitors against C333P SHP2 as an efficient means to identify allosteric SHP2 inhibitors. Despite assaying only a small panel of compounds, we identified a selective, C333-targeted inhibitor (12), using inactivity on C333P SHP2 as a key criterion for further investigation. It must be noted that significant challenges remain for future studies that target SHP2’s PTP-domain allosteric site. Compound 12’s modest potency and inhibition kinetics, as well its lack of selectivity over SHP1, are significant obstacles that will no doubt require larger screens of cysteine-reactive compound libraries to overcome. Given the demonstrated targetability of SHP2’s nonconserved allosteric site, as well as the importance of allosteric SHP2 inhibitors in anticancer pharmaceutical development, C333-directed compound screens may constitute an important new tool for PTP-targeted drug discovery.
Materials and Methods
General
“PTP activity (%)” is defined as the initial velocity of a PTP reaction in the presence of an inhibitor divided by the initial velocity of a vehicle-only control, which is defined as 100% activity. Dimethyl sulfoxide (DMSO) (5% v/v in the assay) was the vehicle in all inhibition experiments. Error bars and “±” values represent the standard deviations of three independent measurements.
Cloning and Mutagenesis of PTP-Encoding Genes
Plasmids encoding His6-tagged constructs of human SHP2 catalytic domain, human PTP1B catalytic domain, human DEP1 catalytic domain, and human full-length SHP2 (1–541) were purchased from VectorBuilder (Cyagen Biosciences Inc). The plasmid encoding the His6-tagged catalytic domain of HePTP has been described previously.25 Site-directed mutations were introduced using the Quikchange mutagenesis kit (Stratagene) according to the manufacturer’s instructions. Desired mutations were confirmed by sequencing.
Protein Expression and Purification
All PTP catalytic domains were expressed as His6-tagged constructs and purified using HisPur Ni-NTA resin (Thermo Scientific) according to the manufacturer’s instructions and as previously described.22,25 After purification, proteins were exchanged into storage buffer (50 mM 3,3-dimethyl glutarate at pH 7.0, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM tris(2-carboxyethyl)phosphine, concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C). Bradford assays were used to measure enzyme concentrations, and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to assess purity.
Phosphatase Activity Assay Using pNPP as Substrate
Quenched PTP assays using pNPP as substrate were carried out in a total volume of 200 μL, containing PTP buffer (50 mM 3,3-dimethyl glutarate at pH 7.0, 1 mM EDTA, 50 mM NaCl), enzyme (varying concentrations: see figure legends), 5% DMSO or inhibitor in DMSO solution (varying concentrations: see figure legends), and pNPP (added after varying preincubation times and at varying concentrations and temperatures: see figure legends). PTP reactions were quenched by the addition of 40 μL of 5 M NaOH, and the absorbances (405 nm) of 200 μL of the resulting solutions were measured.
Phosphatase Activity Assay Using DiFMUP as Substrate
After preincubations of varying times at 37 °C (see figures), PTP reactions were started by the addition of DiFMUP (50 μM) to solutions containing PTP buffer (see above), enzyme (250 nM), inhibitor (at varying concentrations) or DMSO (5%). The sample was excited (360 nm) and fluorescence (450 nm) of the resulting 200 μL solutions were measured continuously for 1 min. The slopes of the lines deriving from inhibitor-treated enzyme were compared to the corresponding DMSO controls.
Mass Spectrometry
Purified SHP2 or C333P SHP2 at 1 mg/mL was incubated with compound 12 (50 μM) for 3 h at 37 °C in PTP buffer. After the incubation, the 250 μL solution was added to 50 μL of HisPur Ni-NTA resin. The suspension was mixed well, and the beads were subsequently washed three times with 500 μL of cold PTP buffer. The His-tagged PTP was eluted with 50 μL of elution buffer (50 mM Tris pH 7.8, 500 mM NaCl, 250 mM imidazole). Eluent (18.75 μL) was purified by SDS-PAGE. The gel was then stained with Coomassie brilliant blue G-250, and the SHP2 bands were excised. Contents of the excised gel bands were identified by LC-MS/MS, as previously described.22,40
Compound Synthesis
Compounds 1–8 were synthesized essentially, as described previously.30 Spectral data were consistent with previously reported values.30
2-Cyano-3-(naphthalen-1-yl)prop-2-enamide (9)
To a solution of naphthalene-1-carboxaldehyde (0.087 mL, 0.64 mmol) in 2:1 tetrahydrofuran (THF)/isopropanol (6 mL) were added 2-cyanoacetamide (65.2 mg, 0.78 mmol) and piperidine (0.1 mL, 1.01 mmol). The reaction was stirred overnight at room temperature with production of a precipitate. The reaction was filtered, and the solids were washed with cold THF and dried in vacuo to afford 30.9 mg (22%) of 9 as a tan solid. Spectral data were consistent with previously reported values.41
2-Cyano-3-(isoquinolin-4-yl)prop-2-enamide (10)
To a solution of isoquinoline-4-carboxaldehyde (96.4 mg, 0.62 mmol) in 2:1 THF/isopropanol (6 mL) were added 2-cyanoacetamide (65.9 mg, 0.78 mmol) and piperidine (0.1 mL, 1.01 mmol). The reaction was stirred for 72 h at room temperature with production of a precipitate. The reaction was filtered, and the solids were washed with cold THF and dried in vacuo to afford 46.5 mg (35%) of 10 as a light yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 7.82 (td, 1H), 7.92 (br s, 1H), 7.96 (td, 2H), 8.20 (dd, 1H), 8.24 (br s, 1H), 8.28 (d, 1H), 8.79 (s, 1H), 8.94 (d, 1H), 9.48 (s, 1H). 13C NMR (400 MHz, DMSO-d6) δ 113.09, 116.32, 123.54, 124.37, 128.02, 128.98, 129.07, 132.54, 133.38, 143.05, 146.30, 155.84, 162.76. HRMS-EI (m/z): calculated for C13H9N3O: 223.0746; found: 223.0753.
2-Cyano-3-(quinolin-5-yl)prop-2-enamide (11)
To a solution of quinoline-5-carboxaldehyde (99.3 mg, 0.64 mmol) in 2:1 THF/isopropanol (6 mL) were added 2-cyanoacetamide (69.3 mg, 0.82 mmol) and piperidine (0.1 mL, 1.01 mmol). The reaction was stirred for 72 h at room temperature with production of a precipitate. The reaction was filtered, and the solids were washed with cold THF and dried in vacuo to afford 61.1 mg (43%) of 2-cyano-3-(quinolin-5-yl)prop-2-enamide as a white solid. 1H NMR (400 MHz, DMSO-d6): δ 7.69 (dd, 1H), 7.89 (overlap t at 7.93, br s, 1H), 7.93 (t, 1H), 8.16 (d, 1H), 8.20–8.25 (br s, 1H), 8.22 (d, 1H), 8.66 (d, 1H), 8.84 (s, 1H), 9.03 (dd, 1H). 13C NMR (400 MHz, DMSO-d6) δ 112.53, 116.42, 122.83, 126.73, 127.94, 129.58, 130.51, 132.85, 133.07, 147.67, 147.97, 151.79, 163.01. HRMS-EI (m/z): calculated for C13H9N3O: 223.0746; found: 223.0739.
N-(Quinolin-4-yl)prop-2-enamide (12)
To a solution of 4-aminoquinoline (0.2986 g, 2.06 mmol) in anhydrous THF (25 mL) was added acryloyl chloride (0.3343 g, 3.69 mmol) dissolved in anhydrous THF (5 mL). The reaction mixture was stirred under nitrogen at 0 °C for 3 h, quenched with aqueous sodium bicarbonate, and extracted with ethyl acetate three times. The organic layers were combined and concentrated, and the product was purified by column chromatography on silica gel (4:1 ethyl acetate/hexanes) to afford 22.9 mg (8.5%) of 12 as a white solid. 1H NMR (400 MHz, DMSO-d6): δ (5.91 dd, 1H), 6.40 (dd, 1H), 6.85 (dd, 1H), 7.66 (td, 1H), 7.79 (td, 1H), 8.02, 8.21 (d, 1H), 8.38 (dd, 1H), 8.82 (d, 1H), 10.38 (br s, 1H). 13C NMR (400 MHz, DMSO-d6) δ 112.47, 121.11, 122.71, 126.45, 129.03, 129.97, 131.80, 141.72, 149.10, 151.30, 164.83. HRMS-EI (m/z): calculated for C12H10N2O: 198.0793; found: 198.0790.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R15GM071388 to A.C.B. LC-MS/MS experiments were carried out by the Mass Spectrometry Facility at the University of Massachusetts Medical School. Small-molecule mass-spectral data were obtained at the University of Massachusetts Amherst Mass Spectrometry Center.
Glossary
Abbreviations
- DEP1
density-enhanced phosphatase 1
- DiFMUP
6,8-difluoro-4-methylumbelliferyl phosphate
- HePTP
hematopoietic protein tyrosine phosphatase
- IC50
50% inhibitory concentration
- KI
inhibition constant
- kinact
inactivation constant
- pNPP
para-nitrophenyl phosphate
- PTP
protein tyrosine phosphatase
- PTP1B
protein tyrosine phosphatase 1B
- SHP1
Src-homology-2-domain containing protein tyrosine phosphatase 1
- SHP2
Src-homology-2-domain containing protein tyrosine phosphatase 2
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02200.
Figure S1, S2 and Tables S1, S2 (PDF)
Author Present Address
† Brigham and Women’s Hospital, Harvard Medical School, Boston, Massachusetts 02115, United States (J.M.F.).
Author Present Address
‡ University of Florida, College of Medicine, Gainesville, Florida 32610, United States (S.K.).
Author Contributions
§ B.M.-A., J.M.F., and S.K. and contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Tonks N. K. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- Jiang Z. X.; Zhang Z. Y. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008, 27, 263–272. 10.1007/s10555-008-9113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Zhang Z. Y. PTP1B as a drug target: recent developments in PTP1B inhibitor discovery. Drug Discovery Today 2007, 12, 373–381. 10.1016/j.drudis.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Y.; Lee S. Y. PTP1B inhibitors as potential therapeutics in the treatment of type 2 diabetes and obesity. Expert Opin. Invest. Drugs 2003, 12, 223–233. 10.1517/13543784.12.2.223. [DOI] [PubMed] [Google Scholar]
- Easty D.; Gallagher W.; Bennett D. C. Protein tyrosine phosphatases, new targets for cancer therapy. Curr. Cancer Drug Targets 2006, 6, 519–532. 10.2174/156800906778194603. [DOI] [PubMed] [Google Scholar]
- Bialy L.; Waldmann H. Inhibitors of protein tyrosine phosphatases: next-generation drugs?. Angew. Chem., Int. Ed. 2005, 44, 3814–3839. 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- Tartaglia M.; Niemeyer C. M.; Fragale A.; Song X. L.; Buechner J.; Jung A.; Hahlen K.; Hasle H.; Licht J. D.; Gelb B. D. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat. Genet. 2003, 34, 148–150. 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- Butterworth S.; Overduin M.; Barr A. J. Targeting protein tyrosine phosphatase SHP2 for therapeutic intervention. Future Med. Chem. 2014, 6, 1423–1437. 10.4155/fmc.14.88. [DOI] [PubMed] [Google Scholar]
- Aceto N.; Sausgruber N.; Brinkhaus H.; Gaidatzis D.; Martiny-Baron G.; Mazzarol G.; Confalonieri S.; Quarto M.; Hu G.; Balwierz P. J.; Pachkov M.; Elledge S. J.; van Nimwegen E.; Stadler M. B.; Bentires-Alj M. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat. Med. 2012, 18, 529–537. 10.1038/nm.2645. [DOI] [PubMed] [Google Scholar]
- Sausgruber N.; Coissieux M. M.; Britschgi A.; Wyckoff J.; Aceto N.; Leroy C.; Stadler M. B.; Voshol H.; Bonenfant D.; Bentires-Alj M. Tyrosine phosphatase SHP2 increases cell motility in triple-negative breast cancer through the activation of SRC-family kinases. Oncogene 2015, 34, 2272–2278. 10.1038/onc.2014.170. [DOI] [PubMed] [Google Scholar]
- Wang H.-C.; Chiang W.-F.; Huang H.-H.; Shen Y.-Y.; Chiang H.-C. Src-homology 2 domain-containing tyrosine phosphatase 2 promotes oral cancer invasion and metastasis. BMC Cancer 2014, 14, 442 10.1186/1471-2407-14-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich M. A. Drug discovery and protein tyrosine phosphatases. Curr. Med. Chem. 2009, 16, 2095–2176. 10.2174/092986709788612693. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Y. Drugging the Undruggable: Therapeutic Potential of Targeting Protein Tyrosine Phosphatases. Acc. Chem. Res. 2017, 50, 122–129. 10.1021/acs.accounts.6b00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi R.; Ran H.; Neel B. G. Off-target inhibition by active site-targeting SHP2 inhibitors. FEBS Open Bio 2018, 1405. 10.1002/2211-5463.12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmann C.; Barr K. J.; Kung J.; Zhu J.; Erlanson D. A.; Shen W.; Fahr B. J.; Zhong M.; Taylor L.; Randal M.; McDowell R. S.; Hansen S. K. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 2004, 11, 730–737. 10.1038/nsmb803. [DOI] [PubMed] [Google Scholar]
- Hansen S. K.; Cancilla M. T.; Shiau T. P.; Kung J.; Chen T.; Erlanson D. A. Allosteric inhibition of PTP1B activity by selective modification of a non-active site cysteine residue. Biochemistry 2005, 44, 7704–7712. 10.1021/bi047417s. [DOI] [PubMed] [Google Scholar]
- Krishnan N.; Koveal D.; Miller D. H.; Xue B.; Akshinthala S. D.; Kragelj J.; Jensen M. R.; Gauss C.-M.; Page R.; Blackledge M.; Muthuswamy S. K.; Peti W.; Tonks N. K. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. 10.1038/nchembio.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. N. P.; LaMarche M. J.; Chan H. M.; Fekkes P.; Garcia-Fortanet J.; Acker M. G.; Antonakos B.; Chen C. H. T.; Chen Z. L.; Cooke V. G.; Dobson J. R.; Deng Z.; Fei F.; Firestone B.; Fodor M.; Fridrich C.; Gao H.; Grunenfelder D.; Hao H. X.; Jacob J.; Ho S.; Hsiao K.; Kang Z. B.; Karki R.; Kato M.; Larrow J.; La Bonte L. R.; Lenoir F.; Liu G.; Liu S. M.; Majumdar D.; Meyer M. J.; Palermo M.; Perez L.; Pu M. Y.; Price E.; Quinn C.; Shakya S.; Shultz M. D.; Slisz J.; Venkatesan K.; Wang P.; Warmuth M.; Williams S.; Yang G. Z.; Yuan J.; Zhang J. H.; Zhu P.; Ramsey T.; Keen N. J.; Sellers W. R.; Stams T.; Fortin P. D. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. 10.1038/nature18621. [DOI] [PubMed] [Google Scholar]
- Dardaei L.; Wang H. Q.; Singh M.; Fordjour P.; Shaw K. X.; Yoda S.; Kerr G.; Yu K.; Liang J. S.; Cao Y. C.; Chen Y.; Lawrence M. S.; Langenbucher A.; Gainor J. F.; Friboulet L.; Dagogo-Jack I.; Myers D. T.; Labrot E.; Ruddy D.; Parks M.; Lee D.; DiCecca R. H.; Moody S.; Hao H. X.; Mohseni M.; LaMarche M.; Williams J.; Hoffmaster K.; Caponigro G.; Shaw A. T.; Hata A. N.; Benes C. H.; Li F.; Engelman J. A. SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med. 2018, 24, 512–517. 10.1038/nm.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele C.; Ran H.; Diskin B.; Wei W.; Jen J.; Geer M. J.; Araki K.; Ozerdem U.; Simeone D. M.; Miller G.; Neel B. G.; Tang K. H. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discovery 2018, 1237–1249. 10.1158/2159-8290.CD-18-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Ren Y.; Gunawan S.; Teng P.; Chen Z.; Lawrence H. R.; Cai J.; Lawrence N. J.; Wu J. Selective inhibition of leukemia-associated SHP2(E69K) mutant by the allosteric SHP2 inhibitor SHP099. Leukemia 2018, 32, 1246–1249. 10.1038/s41375-018-0020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio C. M.; Lim C. S.; Bishop A. C. Targeting a cryptic allosteric site for selective inhibition of the oncogenic protein tyrosine phosphatase Shp2. Biochemistry 2015, 54, 497–504. 10.1021/bi5013595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin B. A.; Adams S. R.; Tsien R. Y. Specific covalent labeling of recombinant protein molecules inside live cells. Science 1998, 281, 269–272. 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- Pomorski A.; Krezel A. Exploration of biarsenical chemistry-Challenges in protein research. ChemBioChem 2011, 12, 1152–1167. 10.1002/cbic.201100114. [DOI] [PubMed] [Google Scholar]
- Zhang X. Y.; Chen V. L.; Rosen M. S.; Blair E. R.; Lone A. M.; Bishop A. C. Allele-specific inhibition of divergent protein tyrosine phosphatases with a single small molecule. Bioorg. Med. Chem. 2008, 16, 8090–8097. 10.1016/j.bmc.2008.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. N.; Mortensen O. H.; Peters G. H.; Drake P. G.; Iversen L. F.; Olsen O. H.; Jansen P. G.; Andersen H. S.; Tonks N. K.; Moller N. P. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 2001, 21, 7117–7136. 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr A. J.; Ugochukwu E.; Lee W. H.; King O. N. F.; Filippakopoulos P.; Alfano I.; Savitsky P.; Burgess-Brown N. A.; Muller S.; Knapp S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroffekova K.; Proenza C.; Beam K. G. The protein-labeling reagent FLASH-EDT2 binds not only to CCXXCC motifs but also non-specifically to endogenous cysteine-rich proteins. Pflügers Arch. 2001, 442, 859–866. 10.1007/s004240100619. [DOI] [PubMed] [Google Scholar]
- Shen S.; Li X. F.; Cullen W. R.; Weinfeld M.; Le X. C. Arsenic binding to proteins. Chem. Rev. 2013, 113, 7769–7792. 10.1021/cr300015c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. M.; Paavilainen V. O.; Krishnan S.; Serafimova I. M.; Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. J. Am. Chem. Soc. 2013, 135, 5298–5301. 10.1021/ja401221b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafimova I. M.; Pufall M. A.; Krishnan S.; Duda K.; Cohen M. S.; Maglathlin R. L.; McFarland J. M.; Miller R. M.; Frodin M.; Taunton J. Reversible targeting of noncatalytic cysteines with chemically tuned electrophiles. Nat. Chem. Biol. 2012, 8, 471–476. 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. M.; Taunton J. Targeting protein kinases with selective and semipromiscuous covalent inhibitors. Methods Enzymol. 2014, 548, 93–116. 10.1016/B978-0-12-397918-6.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B. Y.; Shoichet B. K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons Z. D.; Gates K. S. Redox regulation of protein tyrosine phosphatases: methods for kinetic analysis of covalent enzyme inactivation. Methods Enzymol. 2013, 528, 129–154. 10.1016/B978-0-12-405881-1.00008-2. [DOI] [PubMed] [Google Scholar]
- Seiner D. R.; LaButti J. N.; Gates K. S. Kinetics and mechanism of protein tyrosine phosphatase 1B inactivation by acrolein. Chem. Res. Toxicol. 2007, 20, 1315–1320. 10.1021/tx700213s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddraraju K. V.; Zhang Z. Y. Covalent inhibition of protein tyrosine phosphatases. Mol. BioSyst. 2017, 13, 1257–1279. 10.1039/C7MB00151G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punthasee P.; Laciak A. R.; Cummings A. H.; Ruddraraju K. V.; Lewis S. M.; Hillebrand R.; Singh H.; Tanner J. J.; Gates K. S. Covalent allosteric inactivation of protein tyrosine phosphatase 1B (PTP1B) by an inhibitor-electrophile conjugate. Biochemistry 2017, 56, 2051–2060. 10.1021/acs.biochem.7b00151. [DOI] [PubMed] [Google Scholar]
- Barford D.; Neel B. G. Revealing mechanisms for SH2 domain mediated regulation of the protein tyrosine phosphatase SHP-2. Structure 1998, 6, 249–254. 10.1016/S0969-2126(98)00027-6. [DOI] [PubMed] [Google Scholar]
- Neel B. G.; Gu H. H.; Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- Chio C. M.; Yu X.; Bishop A. C. Rational design of allosteric-inhibition sites in classical protein tyrosine phosphatases. Bioorg. Med. Chem. 2015, 23, 2828–2838. 10.1016/j.bmc.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamble S. B.; Rode C. V. Cascade synthesis of 2-cyanoacrylamides through deacetalization and/or Knoevenagel condensation followed by selective monohydration of zcetals and aldehydes over solid acid ferrites. ChemCatChem 2016, 8, 2678–2687. 10.1002/cctc.201600426. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.