

Abstract
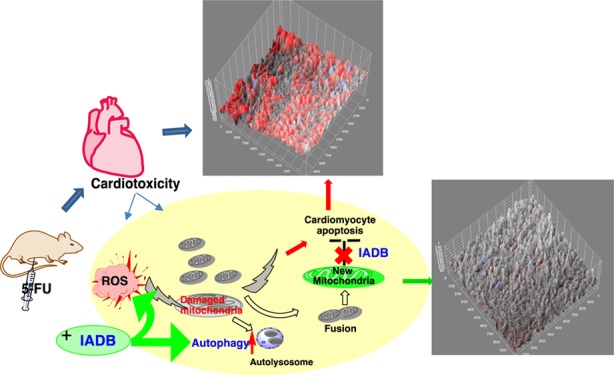
Clinically approved therapeutics that mitigate chemotherapy-induced cardiotoxicity, a serious adverse effect of chemotherapy, are lacking. The aim of this study was to determine the putative protective capacity of a novel indole alkaloid derivative B (IADB) against 5-fluorouracil (5-FU)-induced cardiotoxicity. To assess the free-radical scavenging activities of IADB, the acetylcholine-induced relaxation assay in rat thoracic aorta was used. Further, IADB was tested in normal and cancer cell lines with assays gauging autophagy induction. We further examined whether IADB could attenuate cardiotoxicity in 5-FU-treated male ICR mice. We found that IADB could serve as a novel bifunctional agent (displaying both antioxidant and autophagy-modulating activities). Further, we demonstrated that IADB induced production of cytosolic autophagy-associated structures in both cancer and normal cell lines. We observed that IADB cytotoxicity was much lower in normal versus cancer cell lines, suggesting an enhanced potency toward cancer cells. The cardiotoxicity induced by 5-FU was significantly relieved in animals pretreated with IADB. Taken together, IADB treatment, in combination with chemotherapy, may lead to reduced cardiotoxicity, as well as the reduction of anticancer drug dosages that may further improve chemotherapeutic efficacy with decreased off-target effects. Our data suggest that the use of IADB may be therapeutically beneficial in minimizing cardiotoxicity associated with high-dose chemotherapy. On the basis of the redox status difference between normal and tumor cells, IADB selectively induces autophagic cell death, mediated by reactive oxygen species overproduction, in cancer cells. This novel mechanism could reveal novel therapeutic targets in chemotherapy-induced cardiotoxicity.
Introduction
Chemotherapy-induced cardiotoxicity is a serious adverse outcome of several chemotherapeutics that severely impact therapeutic efficacy.1,2 For example, anthracycline intervention can lead to life-threatening cardiomyopathy.3 One of the most widely accepted mechanisms for chemotherapy-induced cardiotoxicity involves the overproduction of free radicals associated with oxidative stress and eventual apoptosis of cardiac cells.4,5 Unfortunately, many (but not all) chemotherapeutics employ reactive oxygen species (ROS)-dependent mechanisms to target cancer cells. These observations underscore the challenges in defining the approaches to alleviate the adverse outcome of cardiotoxicity from the intended mechanism of action (MOA) of drugs targeting cancerous cells.
5-Fluorouracil (5-FU), a cytotoxic thymidylate synthase inhibitor, is widely used for the treatment of multiple cancers; yet, its use is associated with cardiotoxicity, which can include myocardial ischemia, cardiac arrhythmias, hyper- and hypotension, left ventricular dysfunction, cardiac arrest, and sudden death.6,7 The heart is susceptible to ROS damage and oxidative stress, as this organ exhibits relatively low levels of antioxidant enzymes.8 For example, cardiac muscle contains 150 times less catalase (CAT) and 4 times less superoxide dismutase (SOD) than liver.9 5-FU has been shown to induce apoptosis of rat cardiac cells via generation of ROS.10 Additionally, in rabbits, the antioxidant probucol protected against 5-FU-induced endothelial damage.11 Accordingly, if ROS is the main instigator of 5-FU-induced cardiotoxicity, the compounds limiting free radical formation may have cardioprotective effects when combined with 5-FU, although studies thus far examining this approach have had only marginal success.12,13 Currently, there is no FDA-approved treatment that can ameliorate 5-FU-induced cardiotoxicity.
Cyclic nitroxides represent a diverse group of stable free-radical molecules with unique antioxidant properties.14 The antioxidant capacity of 4-hydroxyl-TEMPO (TEMPOL) is linked to its stable nitroxide radical structure.15 TEMPOL has a low molecular weight, permeates biological membranes, and scavenges both intra- and extracellular deleterious ROS. Moreover, TEMPOL’s antioxidant capacity is enhanced by its SOD-mimicking activity.16 TEMPOL can scavenge various ROS species, including carbon-centered, •OH, peroxyl, and thiyl radicals, as well as dinitrogen species .14 In addition to the reduction of ROS as a mechanism for reducing chemotherapeutic toxicity, another approach involves the alteration of autophagic mechanisms.17,18
Autophagy is a catabolic process in which long-lived proteins, damaged cell organelles, and other cellular particles are sequestered and degraded. Autophagy contributes to the maintenance of cellular energy homeostasis and survival in times of stress. Most anticancer drugs have been reported to induce autophagy in tumor cells.17 However, there are conflicting results from the ongoing clinical trials which target the inhibition of autophagy. Currently, there is no consensus on how to manipulate autophagy to improve the clinical course of cancer patients.
Alkaloids isolated from the plants used in Chinese herbal medicines are an important source for anticancer drug discovery, many of which work via autophagic mechanisms.19,20 For example, the alkaloid berberine produces its anticancer effects via the induction of autophagic cell death and mitochondrial apoptosis in liver carcinoma.21 Conversely, tetrandrine acts as an autophagy enhancer, inducing early G1 arrest in colon carcinoma cells.22 Additionally, camptothecin and vinblastine, the FDA-approved chemotherapeutics, also work via autophagic processes.19
To develop effective and safe therapeutic strategies to prevent the adverse effects of 5-FU on cardiac tissues without compromising its antitumor activity, we synthesized a series of novel compounds that conjoined indole alkaloid and nitroxide scaffolds (the latter based on our successful work with TEMPOL). Our objective was the development of new compounds whose cardioprotective actions would involve both ROS-scavenging and autophagic processes, with a minimal impact on the anticancer effect of 5-FU. Among these newly synthesized indole alkaloid derivatives (the detailed synthesis of the series compounds will be reported elsewhere), we discovered an indole alkaloid derivative B (IADB) that serves as a novel bifunctional agent (antioxidant and autophagy-modulating activity). The present study was designed to examine the hypothesis that cardiotoxicity induced by chemotherapy could be attenuated by IADB when administrated in conjunction with chemotherapeutical agents.
Results
Synthesis of IADB
The preparation of IADB followed the synthetic route outlined in Figure 1. l-Tryptophan was first reacted with formaldehyde which affords compound 1 via a Pictet–Spengler intramolecular cyclization. N-Boc-protected compound 2 was subjected to a coupling reaction with l-leucine benzyl ester to afford compound 3. To avoid the possible aminolysis, the deprotected compound 3 was then directly converted to compound 4 through the intramolecular cyclization with a ketone. After deprotection, compound 5 was subjected to a coupling reaction with l-theanine-OBzl·HCl to afford compound 6. Finally, compound 6 was readily converted to the target compound, IADB, after deprotection and coupling reactions with 4-amino-TEMPO. The chemical structure of IADB was confirmed by proton and carbon nuclear magnetic resonance studies and high-resolution mass spectrometry. The observed paramagnetic broadening was due to the existence of the nitroxide radical. IADB was further analyzed by electron paramagnetic resonance spectroscopy to confirm the intact nitroxide moiety. IADB was stable and was stored at room temperature in sealed bottles until use.
Figure 1.

Synthetic scheme of an IADB. Reagents and conditions: (i) H2SO4, HCHO; (ii) Boc2O; (iii) EDC, HOBt, L-Leu-OBzl·HCl; (iv) EtOAc·HCl (4N), Et3N; acetone; (v) Pd/C, H2; (vi) EDC, HOBt, l-theanine-OBzl·HCl, (vii) H2, Pd/C, and (viii) EDC, HOBt, 4-amino-TEMPO.
Acetylcholine-Induced Relaxation Could be Significantly Reversed by IADB in a Dose-Dependent Manner
The free-radical scavenging potential of IADB was evaluated in the acetylcholine (Ach)-induced relaxation of the rat thoracic aorta assay. In this assay, the isolated aortic rings were contracted with norepinephrine (NE). Upon stabilization, Ach was added to induce vasorelaxation. Exposure of the aortic rings to TEMPOL (10–6 mol/L) and IADB (10–6, 10–7, and 10–8 mol/L) was performed, and then the percentage inhibition of Ach-induced vasorelaxation was determined. As shown in Figure S1, the treatment of the aortic rings with the free-radical scavenger TEMPOL (10–6 mol/L) reduced the Ach-induced relaxation of the aortic rings to some degree (35.8 ± 4.2% inhibition). Compared to TEMPOL (10–6 mol/L), the treatment with IADB (10–6 mol/L) could substantially inhibit Ach-induced vasorelaxation (95.6 ± 5.7% inhibition, in comparison with TEMPOL, p < 0.001). Even at a lower concentration (10–7 mol/L), IADB still could reverse the Ach-induced vasorelaxation to a significant degree (inhibition percentage: 72.5 ± 3.8% inhibition, in comparison with TEMPOL, p < 0.01) (Figure S1).
IADB Induces GFP-LC3 Puncta Formation in Cancer and Normal Cells
During preliminary cellular assays, we observed that IADB intervention induced the presence of cytoplasmic structures, indicative of autophagy. Thus, to verify the potential role of IADB in autophagy induction, we evaluated IADB in both cancer and normal cell lines. Initially, HeLa cells were transiently transfected with a green fluorescent protein (GFP)-light chain 3 (LC3) followed by incubation with IADB for 24 h. Under nutrient-rich conditions, less than 5% of HeLa cells displayed GFP-LC3 puncta. Nutrient starvation conditions increased the percent of cells showing punctuation of GFP-LC3 to 35% in HeLa cells, and low numbers of disk-shaped small GFP-LC3 puncta in individual cells were detected (Figure S2). However, the addition of IADB dramatically increased the percent of cells containing fluorescent puncta and the number of GFP-LC3 puncta per cell. One GFP-LC3 fluorescent puncta was regarded as equivalent to one autophagosome. To examine whether IADB-mediated formation of autophagosome could occur in other cell types, several cancer cell lines (colon cancer HT-29, lung cancer A549, and breast cancer MDA-MB-231) and normal H9C2 rat cardiomyoblasts were also examined. A significant increase in fluorescent GFP-LC3 puncta per cell and the intensified green fluorescence (puncta) induced by IADB were observed in both the cancer and normal cells tested (Figure S2).
Dose-Responsive and Time-Dependent Effects of IADB on Autophagy
In GFP-LC3 transiently transfected cells, LC3 overexpression may result in protein aggregation. It is important to distinguish protein aggregates from true autophagosomes. In a stably transfected system, the levels of GFP-LC3 could be controlled at an appropriate level to avoid artificial aggregation. With this in mind, we next analyzed the IADB-induced changes in the level and distribution of microtubule-associated protein 1 LC3 (an autophagosomal marker) in HeLa cells that had been transfected with LC3 fused to GFP. Under nutrient-rich conditions, small GFP-LC3 puncta (weakly fluorescent) were homogeneously distributed throughout the cytoplasm of the majority of transfected cells. Under nutrient starvation, the number of GFP-LC3 puncta per cell increased. Nutrient starvation again served as a positive control for the induction of autophagy. Under IADB treatment, the number of GFP-LC3 puncta per cell was significantly increased. As shown in Figure S3 (Supporting Information), IADB intervention resulted in significant increases in the number of GFP-LC3 puncta per cell in a dose-and time-dependent manner (Figure S3). With these findings, we next sought to verify that the increased number of GFP-LC3 puncta was not the result of large protein aggregation.
IADB Enhances Autophagic Flux in GFP-LC3 Stably Transfected HeLa Cells
As autophagy is a dynamic process, the accumulation of GFP-LC3 puncta may indicate enhanced autophagosome formation or impaired autophagic degradation. Monitoring autophagic flux is generally utilized as a measure of autophagic degradation activity.28 Previously, we developed a new assay for monitoring the autophagic flux. In this assay, we utilized (i) GFP-LC3 levels to monitor autophagosome formation (Figure 3B); (ii) LysoProbe26,27,37 as an estimate of changes in lysosomal activities (Figure 3C; LysoProbe stains acidic vesicular organelles, including lysosomes and autolysosomes); and (iii) colocalization of these two probes to estimate the autophagic flux via monitoring of autolysosome formation (i.e., the fusion of autophagosome and lysosome) and turnover/degradation (Figure 3D).
Figure 3.
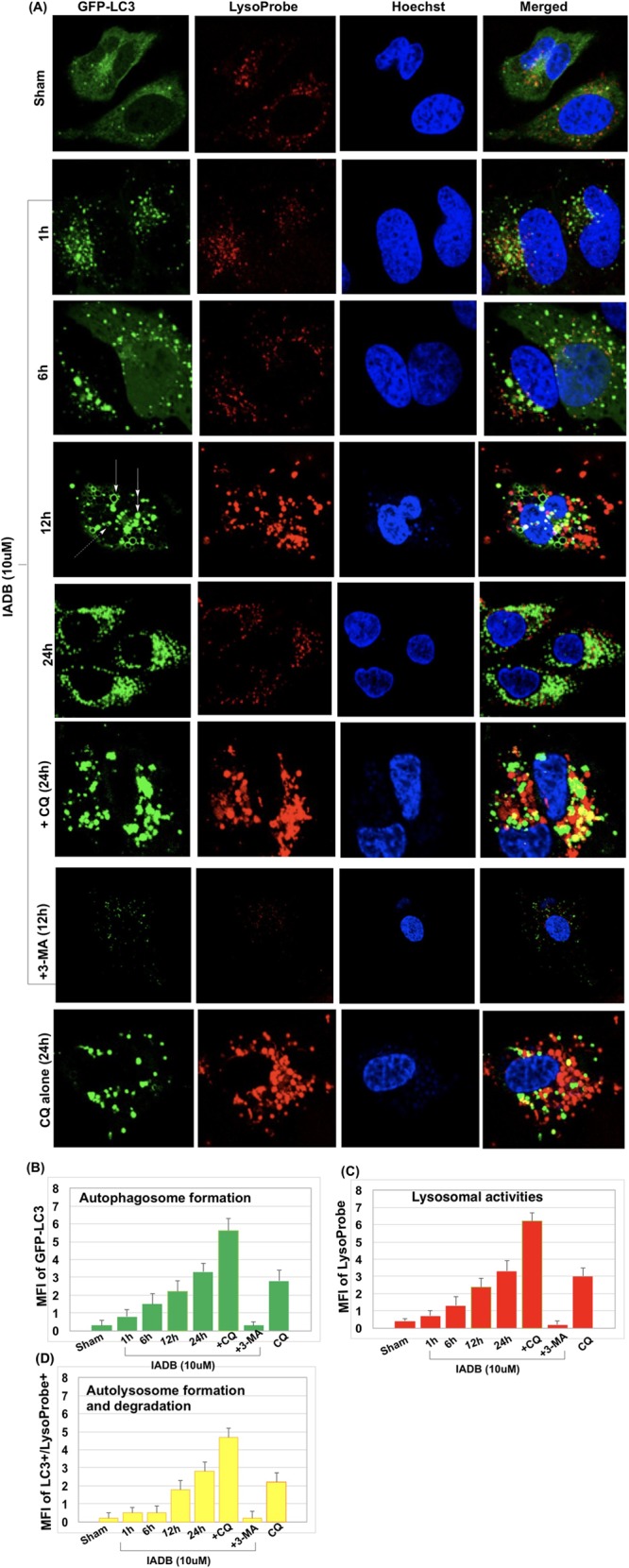
IADB enhances autophagic flux in GFP-LC3 stably transfected HeLa cells. (A) Representative confocal fluorescence images of HeLa cells subjected to different treatment protocols (first row: control cells; second row: IADB (10 μM) for 1 h; third row: IADB (10 μM) for 6 h; fourth row: IADB (10 μM) for 12 h; fifth row: IADB (10 μM) for 24 h; sixth row: IADB (10 μM) + CQ (80 μM) for 24 h; seventh row: IADB (10 μM) + 3-MA (2 mM) for 12 h; and eighth row: CQ (80 μM) alone for 24 h), counterstained with LysoProbe (red fluorescence), GFP-LC3 (green fluorescence), Hoechst 33342 (blue fluorescence). (B) By measuring the mean fluorescence intensity (MFI) of GFP-LC3 changes, the total autophagosome number was determined; (C) by measuring the MFI of LysoProbe changes, lysosomal formation was estimated; and (D) by measuring the MFI of GFP-LC3+/LysoProbe+ double positive, the colocalization index was produced, estimating the autophagic flux (N = 6 independent experiments).
As shown in Figure 3A, untreated sham control cells demonstrated a diffuse cytoplasmic staining of GFP-LC3 with very few punctate autophagosomes and lysosomes (first row, Figure 3A): GFP-LC3, green; LysoProbe, red; merged (autophagy flux), yellow. Induction of autophagosome formation was observed following IADB treatment (10 μM, 1 h) in complete medium (second row, Figure 3A). Selected GFP-LC3 patches appeared following 6 h of IADB treatment (third row, Figure 3A). Representative GFP-LC3 puncta appeared in numerous rings (solid arrow), disks (double arrows), and cup-shaped structures (dashed arrow) (Figure 2). GFP-labeled rings (arrow, Figure 2) and GFP-LC3-labeled cups (dashed arrow, Figure 2) may represent optical sections traversing the center of autophagosomes, whereas GFP-labeled solid disks (double-headed arrow, Figure 2) likely represent optical sections that include the edges of autophagosomes extending from lateral to lateral edge. We refer to these disks, cups, and ring-shaped structures as GFP puncta. After 12 h of treatment with IADB, the number of GFP-LC3 patches decreased, whereas GFP-labeled rings, GFP-labeled cups, and GFP-labeled solid disks increased with IADB (fourth row, Figure 3A). A prolonged treatment with IADB (24 h) resulted in an extensive production of GFP-LC3 puncta (fifth row, Figure 3A). To validate the enhanced autophagic flux, IADB-induced autophagic activity was validated using 3-methyladenine (3-MA). The class III PI3K inhibitor 3-MA is an inhibitor of the early steps of autophagy, which blocks autophagosome formation.28 The addition of 3-MA prior to IADB treatment abrogated IADB-mediated autophagy (decreased GFP-LC3 puncta; seventh row, Figure 3A). Next, we treated the HeLa cells stably expressing GFP-LC3 with IADB in the absence or presence of chloroquine (CQ) (80 μM). CQ, a known inhibitor of the later steps of autophagy, has the capacity to prevent the fusion of autophagosomes with lysosomes with a concomitant inhibition of the lysosomal degradation of proteins late in autophagy.28 We found that increases in the number of GFP-LC3 puncta per cell induced by IADB were further elevated in the presence of CQ. Combinatorial treatment (IADB + CQ) significantly increased the number of GFP-LC3 puncta and a higher colocalization index (monitoring autolysosomes formation and degradation) in comparison to either compound alone (eighth and sixth rows, Figure 3A), suggesting that the autophagic degradation was inhibited in the presence of CQ. These results further confirmed that the increased number of GFP-LC3 puncta was due to enhanced autophagosome formation rather than impaired autophagic degradation.
Figure 2.
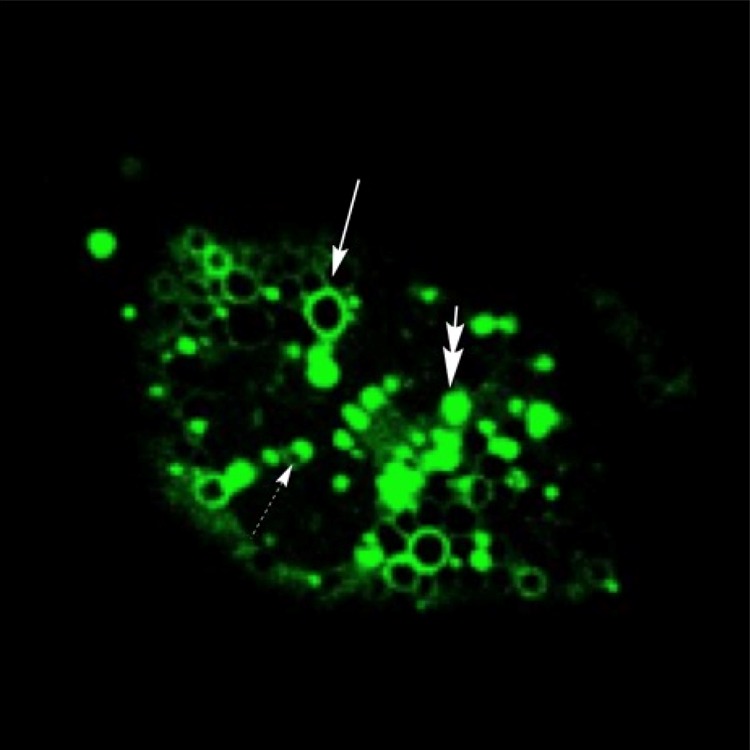
Representative autophagic structures induced by IADB incubation (10 μM) for 12 h. GFP-LC3 fluorescence appeared in numerous rings (solid arrow), disks (double arrows), and cup-shaped structures (dashed arrow).
IADB Exhibits Specific Cytotoxic Effects toward Cancer Cells
To explore whether IADB could act as an autophagy-promoting compound with anticancer activity, we examined a panel of cancer cell types (HeLa, HT29, A-549, and MDA-MB-231 cells), employing a cytotoxicity assay. Normal H9C2 rat cardiomyoblasts were employed as control. IADB demonstrated differential toxicity in cancer cells (mean IC50 of IADB in MDA-MB-231 cells, 30.4 ± 1.9–45.7 ± 2.8 μM; mean IC50 in HT-29 cells, 25.3 ± 1.5–37.5 ± 2.2 μM; and mean IC50 in HeLa cells, 46.4 ± 2.3–61.8 ± 1.7 μM). The cytotoxicity of IADB was considerably lower in H9C2 rat cardiomyoblasts (mean IC50 greater than 100 μM), suggesting enhanced potency in cancer cells.
Combined IADB/5-FU Treatment Induces Significant Mitochondrial Oxidative Damage in Colon Cancer HT-29 Cells, Not in Normal H9C2 Cardiac Cells
Previous studies suggested that ROS-mediated cardiotoxicity is induced by various chemotherapeutic agents, suggesting that a concomitant administration of antioxidants may provide some mitigation of cardiotoxicity.1 Accordingly, we examined the in vitro effects of IADB and 5-FU in a combinatorial paradigm. Initially, we evaluated the antioxidant effects of IADB, alone or in combination with 5-FU, in the control H9C2 rat cardiomyoblasts and the colon cancer cell line HT-29. We employed MitoProbe,32−36 a sensitive fluorogenic reporter, for real-time monitoring of mitochondrial ROS (mtROS) generation. The MFI of MitoProbe was quantified as a gauge of mtROS production.
The HT-29 sham control cells (untreated) contained primarily long tubular mitochondria, evenly distributed throughout the cell (first row, Figure 4A). The 5-FU exposure resulted in the formation of intermediate/fragmented mitochondria. Conversely, the 5-FU-treated cells displayed a more dispersed and irregular staining pattern associated with increased MitoProbe fluorescence (second row, Figure 4A). Pretreatment (12 h) of IADB to HT-29 cells resulted in a proportion of cells exhibiting a slight increase in MitoProbe fluorescence (Figure 4B) associated with a dispersed network of tubular structures surrounding the nucleus (third row, Figure 4A). Increasing the exposure time to IADB + 5-FU treatment led to progressive cellular changes in HT-29 cells: (a) initially, the mitochondria randomly aggregated in small groups throughout the cytoplasm, with a higher concentration near the nucleus (fourth row, Figure 4A); (b) this progressed to further aggregation of mitochondrial clusters around the nucleus in a concentric appearance (fifth row, Figure 4A); (c) subsequently, the aggregated mitochondria appeared as large round clumps 24 h after the combinatorial treatment (sixth row, Figure 4A), potentially associated with cell death; and (d) after 48 h of combinatorial exposure, cells with fragmented nuclei were obvious, indicating apoptosis (seventh row, Figure 4A). Furthermore, the combinatorial treatment enhanced mtROS levels (Figure 4B), indicative of oxidative stress and mitochondrial damage in HT-29 cells.
Figure 4.
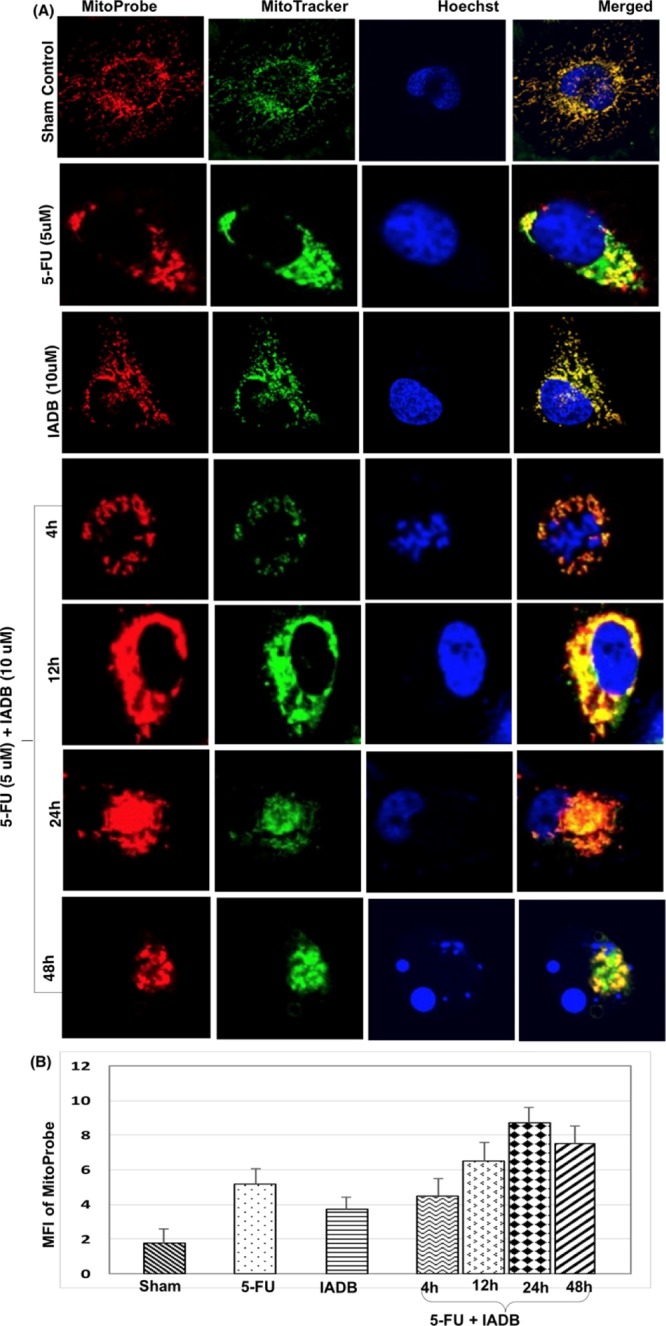
Combined IADB/5-FU treatment induces significant mitochondrial oxidative damage in colon cancer HT-29 cells; detection of mitochondrial oxidative damage in HT-29 cells before and after exposure to 5-FU (5 μM) without or with IADB (10 μM) treatment: (A) representative confocal fluorescence images of HT-29 cells subjected to different treatment protocols––counter-stained with MitoProbe (red fluorescence), MitoTracker (green fluorescence), and Hoechst 33342 (blue fluorescence); (B) MFI of MitoProbe changes, estimating total mtROS generation.
Untreated H9C2 cardiomyoblasts (first row, Figure 5A) presented normal cell morphology with well-defined, filamentous mitochondrial networks and low levels of mtROS. H9C2 cardiomyoblasts treated with 5-FU revealed more fragmented mitochondrial networks and extensive accumulation of mitochondria (second row, Figure 5A) associated with significantly increased levels of mtROS (48 h treatment; Figure 5B). As shown in Figure 5B, pretreatment of H9C2 cardiomyoblasts with IADB (10 μM) for 12 h, followed by combinatorial treatment [IADB (10 μM) + 5-FU (5 μM)] for 48 h, normalized mtROS at all time points measured (4, 12, 24 and 48 h).
Figure 5.
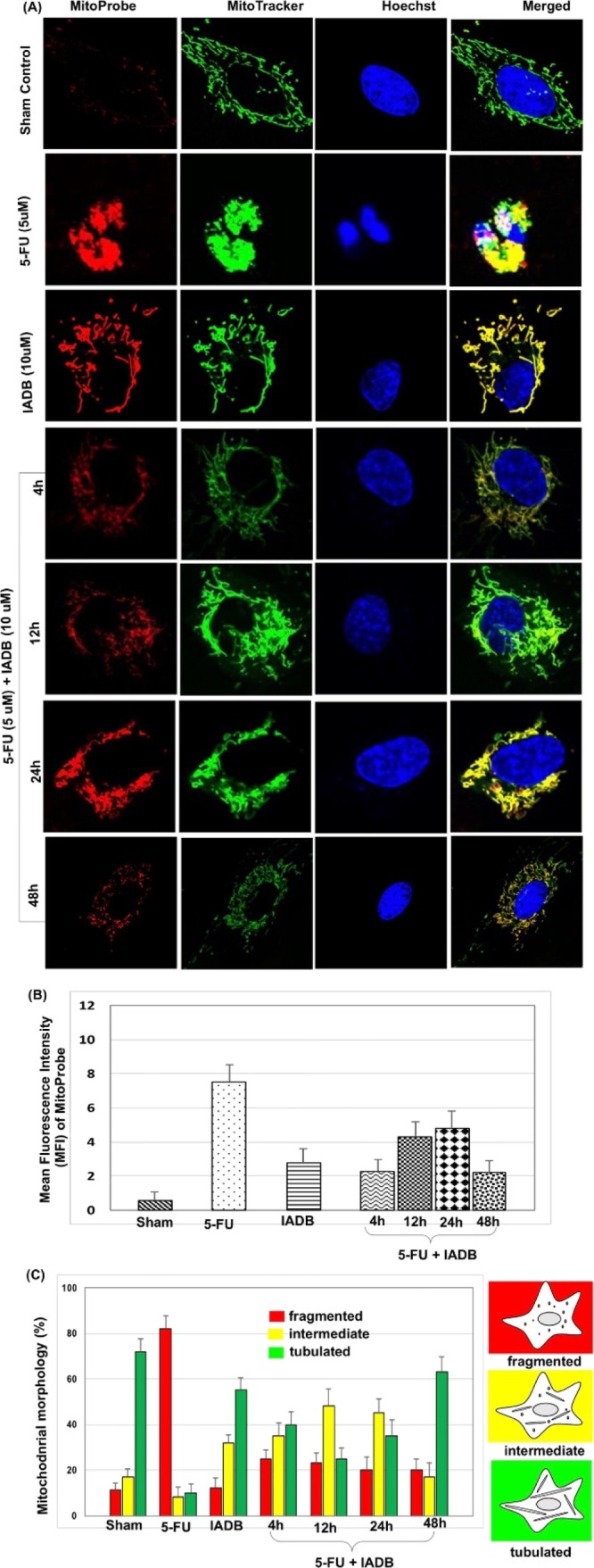
Combined IADB/5-FU treatment induces significant mitochondrial oxidative damage in colon cancer HT-29 cells, but not in normal H9C2 cardiac cells. Detection of mitochondrial oxidative stress in H9C2 cardiac cells pre-/post-5-FU (5 μM) exposure with/without IADB (10 μM) treatment: (A) representative confocal fluorescence images of H9C2 cardiomyoblasts subjected to different drug interventions––counterstained with MitoProbe (red fluorescence), MitoTracker (green fluorescence), and Hoechst 33342 (blue fluorescence); (B) total mtROS generation in H9C2 cardiomyoblasts as determined by MFI of MitoProbe; and (C) mitochondrial morphological changes during different treatment regimens (percent of cells with ≥100 cells per experiment). N = 6 independent experiments.
Interestingly, we observed that the combinatorial treatment [IADB (10 μM) + 5-FU (5 μM)] resulted in a return of mitochondrial morphology to the expected tubular structure in H9C2 cardiomyoblasts (fourth to seventh row, Figure 5A). The mitochondrial morphology was scored as follows: fragmented, mainly small and round; intermediate, mixture of round and shorter tabulated; and tabulated, long with higher interconnectivity. The method for quantification involved determining the percentage of cells with abnormal mitochondrial morphologies as a surrogate measure for the proportion of cells with fragmented mitochondria. The nontreated cells employed as the control contained predominantly long and evenly distributed tubular mitochondria throughout the cell, and the cells with intermediate or fragmented mitochondria were expressed as a percentage of the total cells counted (100 cells were counted per experiment, and the data were averaged over four independent experiments per treatment).
The mitochondria of H9C2 cardiac cells remained elongated with entangled tubules at 24 h (sixth row, Figure 5A) after combinatorial intervention (IADB + 5-FU), whereas the HT-29 colon cancer cells continued to manifest significant mitochondrial damage under identical treatment conditions. The combinatorial intervention [IADB (10 μM) + 5-FU (5 μM)] appeared to only transiently alter mitochondrial morphology. As shown in Figure 5A,C, the combinatorial therapy for 48 h resulted in a complete correction of mitochondrial structure in the majority of H9C2 myoblasts to their expected tubular structure. Interestingly, cell-specific recovery was observed only in the H9C2 cardiac cells (seventh row, Figure 5A) but not in the colon cancer HT-29 cells (seventh row, Figure 4A). Our results suggest that IADB provides greater protection to normal cardiac cells (H9C2) as opposed to tumor cells (HT-29). These findings implied that unbalanced mitochondrial fusion/fission, or possibly failure of autophagy in HT-29 cells, may have induced the accumulation of damaged mitochondria.
Combinatorial IADB/5-FU Intervention Induces Autophagic Cell Death in Colon Cancer HT-29 Cells
The combinatorial application of autophagy inhibitors coupled with chemotherapeutic agents is a common strategy for cancer cell sensitization;29−31 yet, it remains to be determined whether cancer chemotherapy would benefit from up- or downregulation of autophagy. Thus, we next evaluated the autophagic responses in HT-29 cells undergoing combinatorial intervention. The treatment of HT-29 cells with 5-FU (5 μM) alone (48 h) resulted in an increased GFP-LC3 fluorescence puncta formation (second row, Figure 6A, compared to control, first row, Figure 6A). The GFP-LC3 puncta formation was dramatically increased by IADB pretreatment (third row, Figure 6A) or combinatorial treatment (fourth to seventh row, Figure 6A) as compared to the 5-FU treatment alone. The combinatorially treated HT-29 cells revealed the presence of autophagic vesicles as early as 4 h (fourth row, Figure 6A), which progressively increased with time. The number of GFP-LC3 fluorescence puncta increased steadily over 24 h of combinatorial treatment, indicative of autophagosome accumulation (Figure 6B). The HT-29 cells showed similar results, with the appearance of LysoProbe-labeled organelles (red fluorescence images) (Figure 6C) that correlated with the appearance of GFP-labeled autophagosomes (green fluorescence images), the combined occurrence of which can be labeled as autophagy process-related vesicular organelles (AVOs). After 24 h of combinatorial intervention, the clustering of AVOs was obvious. Simultaneously, an extensive lysosomal accumulation was also observed (Figure 6C). The cumulative data indicate that IADB pretreatment activated autophagy, as the combinatorial treatment resulted in a continuous activation of autophagy and a concomitant imbalance between the rate of AVO formation and degradation (Figure 6D). The lysosomal findings support this, suggesting that “autophagic stress” occurred and preceded both apoptosis and necrosis. Normally, autophagy and apoptosis remain in balance and control the organelle numbers. However, our observation of continuous autophagy induced by the combinatorial treatment eventually activated apoptosis and cell death.
Figure 6.
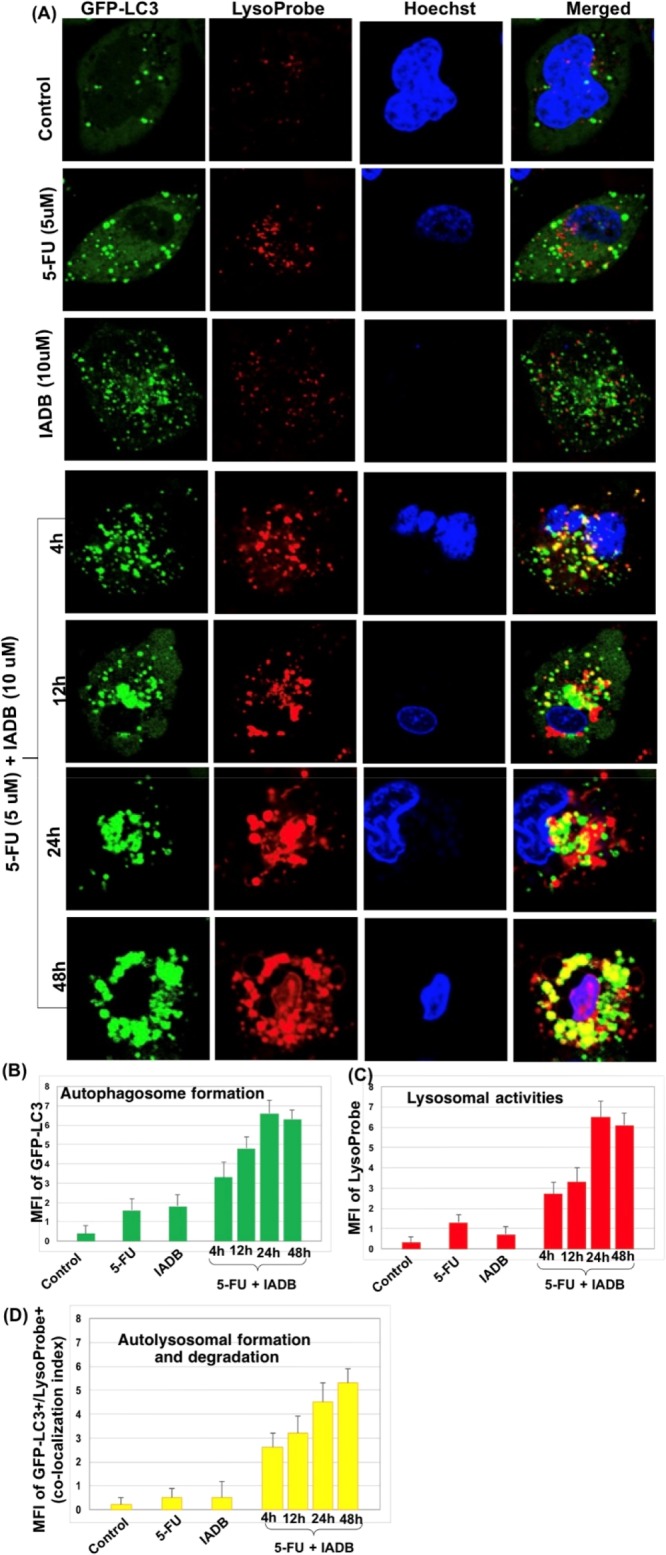
Confirmation of “autophagic stress” induced by combined IADB + 5-FU treatment using confocal laser scanning microscopy. (A) Representative confocal fluorescence images of HT-29 cells subjected to different treatment protocols––counterstained with LysoProbe (red fluorescence), GFP-LC3 (green fluorescence) transfected cells, and Hoechst 33342 (blue fluorescence) (control cells (first row); 5-FU (5 μM) alone for 48 h (second row); pretreatment IADB (10 μM) for 12 h (third row); IADB-pretreated cells at 4 h (fourth row), 12 h (fifth row), 24 h (sixth row), and 48 h (seventh row) after exposure to 5-FU, respectively. (B) Total autophagosome number in HT-29 cells determined by measuring the MFI of GFP-LC3 changes. (C) Lysosomal activities evaluated in HT-29 cells by quantitation of MFI of LysoProbe changes. (D) Colocalization index determined as a measure of autolysosome formation (i.e., the fusion of autophagosome and lysosome) and degradation events (autophagic flux) by measuring the MFI of GFP-LC3+/LysoProbe+ double-positive HT-29 cells. N = 6 independent experiments.
Combinatorial Treatment (IADB + 5-FU vs 5-FU Alone) Potentiated Growth Inhibition of Colon Cancer HT-29 Cells
The treatment with 5-FU (5 μM; 48 h) inhibited the growth of colon cancer HT-29 cells by one-third in comparison to the vehicle-treated cells. The pretreatment with IADB (10 μM; 12 h) prior to that with 5-FU doubled the inhibitory effect, as measured by trypan blue exclusion (Figure S4).
Confirmation of “Autophagic Stress” Induced by Combined IADB + 5-FU Treatment Using Transmission Electron Microscopy
The transmission electron microscopy (TEM) images of HT-29 cells treated with 5-FU (5 μM) for 24 h revealed minimal autophagic vesicles, with the mitochondria that were slightly swollen with a faint double-membrane structure (Figure 7A). Conversely, the IADB pretreatment followed by the IADB + 5-FU combined treatment for 24 h revealed the presence of numerous AVOs with cellular material that appeared degenerated (Figure 7B). The AVOs resembled autophagosomes and autolysosomes, and several of them showed entrapped intracellular organelles such as mitochondria or endoplasmic reticulum, as well as digested residual material. The overproduction of AVOs in combinatorially treated cells (Figure 7B) correlated with the accumulation of LysoProbe-/GFP-LC3-labeled AVOs (Figure 6). The data suggest “autophagic stress” associated with the dysregulation of autophagic responses, or perhaps a situation in which “excessive” autophagic demand goes unbalanced by the consumption of cellular reserves. Whichever the exact mechanism, it would seem reasonable that the overproduction or impaired clearance of AVO degradation eventually leads to cell death.
Figure 7.
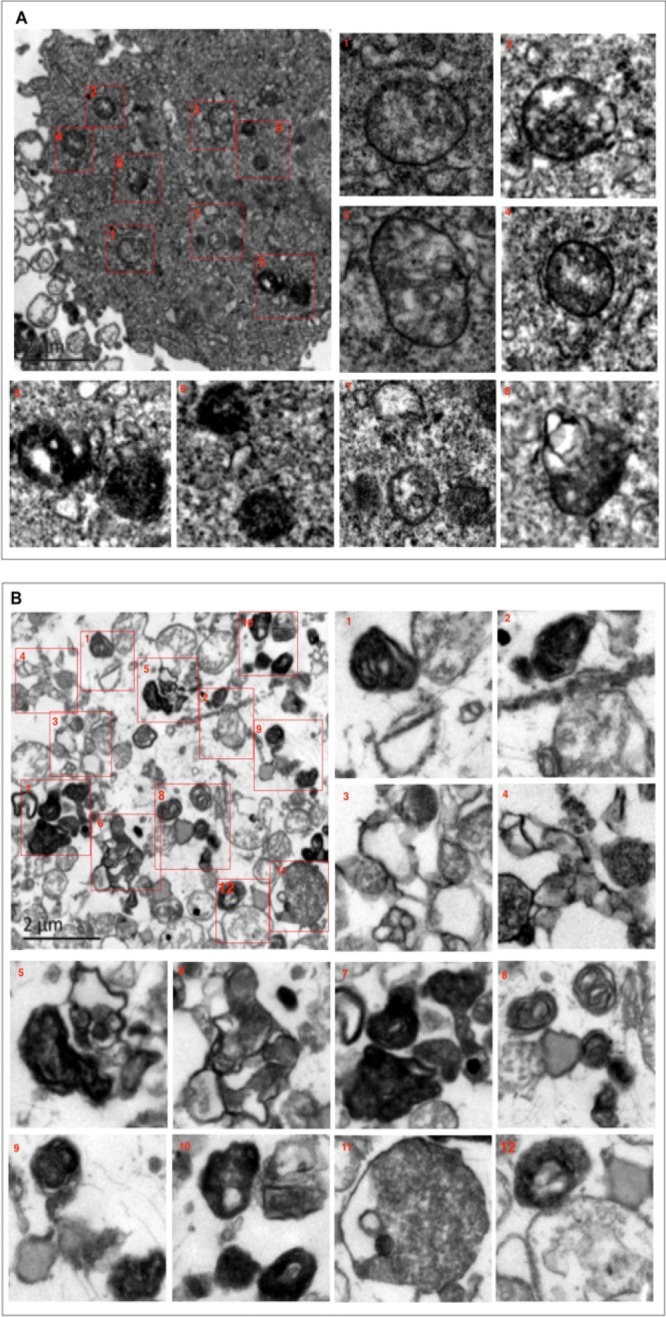
Confirmation of “autophagic stress” induced by combined IADB + 5-FU treatment using TEM: (A) TEM images of HT-29 cells treated with 5-FU (5 μM) for 24 h, revealing minimal autophagic vesicles accompanied by modestly swollen mitochondria with a relatively indistinct double-membrane structure. (B) TEM images of HT-29 cells co-treated with 5-FU (5 μM) + IADB (10 μM) for 24 h, revealing numerous autophagosomes and autolysosomes (electron-dense organelles engulfing double-membraned vesicles). These structures appeared to contain degraded cellular material, and the mitochondria were swollen with crista fragmentation.
Mitigation of 5-FU-Induced Cardiac Structural Damage by Combined IADB + 5-FU Treatment
We next investigated the potential of the IADB pretreatment to alleviate 5-FU-induced cardiac structural damage. The myocardial cells and the interstitium of IADB-treated animals did not reveal evidence of obvious pathological alterations, comparable to the vehicle-treated tissues. In contrast, major histopathological alterations (myocardial fiber disarrangement, extracellular edema, leukocyte infiltration, and enlarged intercellular spaces) were observed in 5-FU-treated tissues (Figure 8).
Figure 8.
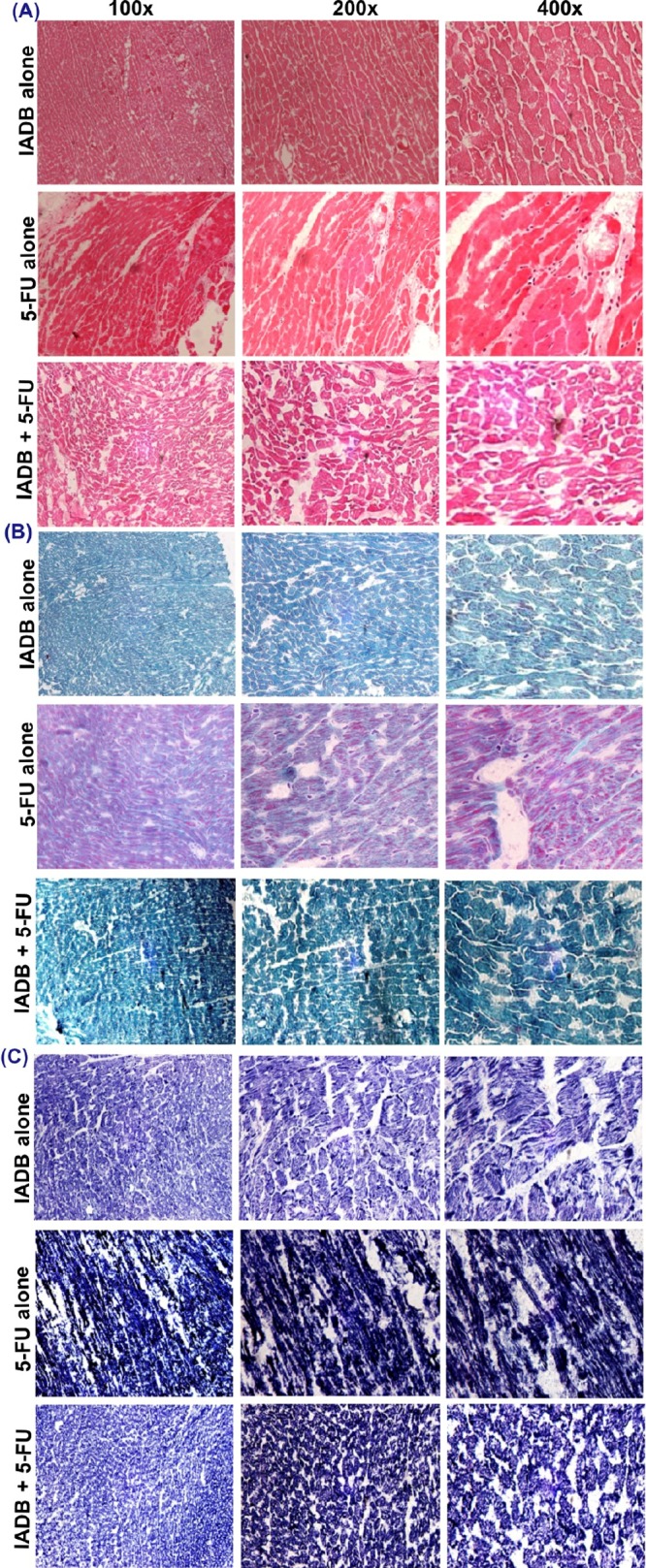
Mitigation of 5-FU-induced cardiac structural damage by combined IADB + 5-FU treatment. Representative photomicrographs of myocardial sections from IADB/5-FU alone and IADB + 5-FU combined treatments groups: (A) hematoxylin–eosin (H & E) staining; (B) modified trichrome staining; and (C) NADH staining (N = 6).
Further, modified Gomori trichrome staining of the 5-FU cohort revealed loose collagen fibers surrounding individual myocardial tracts and clear evidence of edema between the wavy muscle fibers. These features suggest the disruption of the collagen fibers in the interstitial region and the interstitial fluid separating the lateral myocyte arrangement. The myocardium samples taken from the combinatorially treated cohort revealed a significantly less disruption of myocardial fibers, although a mild degree of swelling around the nuclei and congestion of capillaries was still observed (Figure 8B).
The NADH-stained sections of myocardial samples derived from the 5-FU-treated cohort revealed an extensive interstitial edema between the damaged cardiomyocytes. Striated cardiac muscle disarray, local myocardial swelling, necrosis, and intercellular space expansion were frequently observed. Conversely, the tissues derived from the combinatorially treated animals revealed only a slight myocardial edema associated with the partially ruptured cardiac muscle fibers (Figure 8C).
Attenuation of 5-FU-Induced Cardiac Oxidative Damage by IADB + 5-FU Treatment Confirmed with the Oxidative Stress Biomarkers
To examine the potential protective effect of IADB on cardiomyocytes, lipid peroxidation (malondialdehyde, MDA) was estimated in heart extracts. Increased MDA was observed in 5-FU-treated cohorts. Conversely, no significant difference was observed between the sham controls and IADB alone or IADB + 5-FU groups (Figure S5), consistent with the protective capacity of IADB against lipid peroxidation. The SOD, CAT, and thiol glutathione (GSH) systems in heart detoxify ROS to nontoxic forms.30 Accordingly, we examined the effects of 5-FU and IADB on GSH, SOD, and CAT activities. 5-FU alone significantly decreased GSH; conversely, the IADB + 5-FU treatment essentially normalized the GSH levels, raising them significantly above the 5-FU treatment groups (Figure S5). 5-FU and 5-FU + IADB significantly increased SOD in comparison to control (Figure S5). For the IADB + 5-FU groups, CAT activities were significantly higher than that in the 5-FU-treated groups (Figure S5).
Alleviation of 5-FU-Induced Mitochondrial Oxidative Damage by IADB + 5-FU Treatment Confirmed by Direct Visualization of Tissue Oxidative Status
To assess tissue protection associated with reduced ROS production, mtROS production was determined in myocardial tissues using MitoProbe.36 The fluorescence intensity of MitoProbe was higher in the tissues of the 5-FU cohort (Figure 9A,C,D). Conversely, the fluorescence intensity in the combinatorially treated tissues was significantly reduced (Figure 9B–D), further underscoring the potential of IADB to alleviate mitochondrial oxidative stress.
Figure 9.

Alleviation of 5-FU-induced mitochondrial oxidative damage by IADB + 5-FU treatment confirmed by direct visualization of the tissue oxidative status: mitochondrial oxidative status was evaluated in myocardial tissues using mtROS fluorescent probe (MitoProbe). Representative fluorescent stained images of myocardium derived from the 5-FU-treated rat cohort (A) and IADB + 5-FU rat cohort (red: MitoProbe fluorescence; blue: Hoechst 33342 fluorescence) (B). (C) Interactive 3D surface plots of merged images and (D) Interactive 3D surface plots of merged images (fire LUT images).
Attenuation of 5-FU-Induced Cardiac Ultrastructural Damage by IADB + 5-FU Treatment Confirmed with TEM
We used TEM to further explore the structure/function correlates in myocardial organelles under various interventions. We found that the IADB-treated cohorts (Figure 10B) revealed the evidence of relatively normal mitochondria with intact cristae density (Figure 10A). On the other hand, we noted marked ultrastructural myocardial damage (myofibril disorganization, vacuolation, mitochondrial swelling, and cell lysis) in the samples of 5-FU-treated animals (Figure 10C). In addition, mitochondria became sparse at the remote regions of the nucleus (Figure 10D). Pretreatment with IADB, followed by 5-FU + IADB treatment, provided significant protection from these ultrastructural abnormalities (elongated and transverse cristae (indicated as “EM”); larger, rounded to ovular mitochondria with vacuolation (indicated as “M”); Figure 10E,F). At the perinuclear region, the majority of mitochondria showed relatively normal morphology, although mild edema was still noted (Figure 10F) (Figure S6). Moreover, we observed autophagosomes with double-membrane structures, and a number of these autophagic vacuoles contained identifiable organelles, whereas some late autophagosomes contained lamellar and vesicular structures [magnified autophagic vesicles are shown in the red box (Figure 10F)]. Cumulatively, more autophagosomes were observed with combinatorial intervention versus 5-FU alone (Figure S7). Taken together, our data suggest that under “mild” stress, the mitochondrial elongation response can be induced in cardiomyocytes [indicated as “EM” (elongated mitochondria) in Figure 10E]. Severely damaged cardiomyocyte mitochondria are unable to fuse into the mitochondrial network, making them susceptible to degradation. We therefore suggest that mitochondrial elongation can protect the mitochondria from mitophagy, coupled to autophagic degradation of cytosolic material (red box; Figure 10E), all of which will enhance energy production and the production of macromolecular precursors to promote cardiomyocyte survival during cellular stress.
Figure 10.

Attenuation of 5-FU-induced cardiac ultrastructural damage by IADB + 5-FU treatment confirmed by TEM. Representative TEM of myocardium derived from the sham control and different drug-treated animal groups: (A) sham cardiomyocytes revealed typical myofibrillar arrangements and intact mitochondria (M); (B) IADB-treated cardiomyocytes were comparable to sham treatment, but with a small number of autophagosomes; (C,D) 5-FU-treated cardiomyocytes revealed lesions with myofibrillar disorganization and swollen mitochondria with lower density structures; (E,F) combinatorially treated myocardium revealed typical mitochondrial structures combined with a small number of autophagosomes surrounded by double membranes (red box), in addition to autophagic vacuoles containing identifiable organellar material, as well as late autophagosomes containing lamellar and vesicular structures.
Discussion
Conventional anticancer drugs induce oxidative stress in cancer patients being treated with chemotherapy.9−13 Previous studies suggest that the MOA of some of these chemotherapeutic agents for triggering apoptosis in cancer cells involves ROS-dependent pathways.17,18 The present study was designed to answer one question: “is it possible to induce apoptosis only in cancer cells while sparing normal cells/tissues?”
Most anticancer drugs activate autophagy in tumor cells. Therefore, a common strategy in several clinical trials is to inhibit the autophagic activity to enhance the potency of conventional chemotherapy.29,30 However, some of these clinical trials have been terminated because the research findings are contradictory.29,30 So far, no conclusive results have been published. The contrasting results for autophagy in cancer cells remain difficult to understand because the process itself can be either protective or detrimental during tumorigenesis and treatment.17 Whether the role of autophagy is positive or negative for the growth of the tumor depends upon the tissue, the stage and type of tumor, and the extent of active autophagic processes. Additionally, autophagy can be affected by cellular stress such as ROS levels.17 Many anticancer drugs have been reported to activate ROS-induced autophagy, which can either lead to the induction of apoptosis or the development of drug resistance, or both.17−19 Currently, there is no consensus as to whether inhibition or promotion of autophagy is beneficial in cancer treatment. Because of the complex interplay between ROS signaling and autophagy, identification of the optimal combination of autophagy modulators and antioxidants that can be combined with anticancer drugs to improve the efficacy of treatment remains a challenge. Our data indicate that IADB, a novel agent that simultaneously serves as an antioxidant and autophagy-modulating bifunctional agent, may well represent a novel breakthrough in the approach to cancer therapy.
As antioxidant activity plays a role in the protection against 5-FU-induced cardiotoxicity, we first evaluated IADB for the antioxidant activity, employing the Ach-induced relaxation of the rat thoracic aorta assay.23 In the Ach-induced relaxation assay, injury of the arterial wall triggers the production of ROS and alterations in the vascular redox equilibrium.24 The increase in the vascular ROS levels and oxidative stress induces endothelial cell layer damage. NO• is the primary driver for the vasorelaxation of a smooth muscle.24 Endothelial cells in the proximity to a smooth muscle also generate O2•– during vascular oxidative damage, and the production of one free radical can result in a cascade of further radical production (e.g., O2•– overproduction, coupled with NO•, can lead to ONOO– generation, the latter a physiologically active toxic metabolite of NO• that induces vascular and myocardial dysfunction).24,25 As shown in Figure S1 (Supporting Information), we observed that Ach-induced relaxation could be significantly reversed by IADB in a dose-dependent manner.
As autophagy is a complex process, we felt it important to accurately quantify the autophagic effects of IADB. Although immunoblotting for endogenous LC3 is commonly used to monitor autophagosome formation or degradation, we found the results from Western blotting inconsistent (data not shown) because of the high background, precluding the accurate measurement of small changes in LC3 protein levels. Moreover, Western blotting does not measure the autophagic flux which represents the rate of autophagic degradation. Therefore, we employed fluorescent imaging technology and TEM to accurately determine the impact of IADB treatment on autophagy. Compared to fluorescence microscopy, the resolution of TEM is superior. TEM remains one of the most accurate methods for the detection of autophagy. However, TEM does not facilitate the quantitative measurement of autophagic flux in real time, nor is it particularly suited for routine visualization in all steps involved in the entire autophagic process. A careful and rigorous evaluation of autophagy with a translational focus is critical if we are to improve the clinical outcomes in cancer patients, and thus we next pursued a series of dynamic imaging studies to confirm the autophagy-modulating capacity of IADB.
Earlier reports indicated that autophagy induced by chemotherapy (i.e. 5-FU) could limit the effects of drug efficacy, and thus inhibition of autophagy may augment efficiency. However, our data indicated that IADB pretreatment, followed by combinatorial intervention (IADB + 5-FU), induced continuous autophagy activation in colon cancer HT-29 cells. We speculate that “excessive” autophagy culminates in cancer cell death, and in this scenario, autophagy inhibition would result in depressed therapeutic efficacy and enhanced tumor progression. Additionally, cytotoxic stimuli could activate autophagic cell death in tumor cells that are resistant to apoptosis.20−22 Our data suggested that autophagic cell death could be induced by the IADB + 5-FU combined treatment, and this may spur further investigation for sensitizing apoptosis-resistant cancer cells to cell death.
Pretreatment of colon cancer HT-29 cells with IADB prior to 5-FU exposure activated autophagy, which leads to alterations in ROS generation; however, upon exposure to 5-FU + IADB, significant levels of ROS were produced in tumor cells. We suspect that the overproduction of ROS, coupled to continuous stimulation of autophagy, eventually would lead to “excessive” autophagic activation, and finally apoptosis, as shown with confocal imaging analysis and TEM. As shown in Figure 7B, it appears that the accumulation of autophagic vacuoles directly promotes cancer cell death. Surprisingly, the IADB-pretreated H9C2 cardiac cells subsequently exposed to IADB + 5-FU showed evidence of only minimal oxidative stress (Figure 5B). Our data suggested that the anticancer effect of IADB may be mediated through autophagy induction. However, the anticancer properties and molecular mechanisms of IADB seem to be cell-specific, and thus its beneficial effects in different cell types remain to be further investigated.
Autophagic cell death is characterized by extensive sequestration of cytoplasm leading to cell death and the formation of autophagosomes or autolysosomes. Autophagic cell death is autophagy-dependent cell death, which can be triggered by starvation or autophagy-promoting compounds that may result in excessive autophagy induction and cell death.31 Here, we demonstrated that an “exhausted” autophagic response induced by combined treatment (IADB and 5-FU) could eventually destroy cancer cells and lead to autophagic cell death. However, the exact mechanism of autophagic cell death induced by the combined treatment is still not mechanistically clear.
On the other hand, 5-FU treatment alone was associated with an increased mtROS level in myocardium (Figure 9A). The elevated mtROS levels induced by chemotherapy could severely compromise the cellular nucleus of energy production, for example, mitochondrial oxidative phosphorylation.1 Here, the histological analysis of myocardial sections revealed 5-FU-induced cardiotoxicity, as expected, and this was alleviated with IADB pretreatment. Subcellularly, 5-FU-driven mtROS overproduction correlated with mitochondrial oxidative damage. Surprisingly, the combinatorial intervention (IADB + 5-FU) could significantly prevent mtROS overproduction (Figure 9B). In support of these findings, TEM revealed evidence of mitochondrial damage (swollen mitochondria accompanied with lower density structures) with 5-FU treatment (Figure 10C,D). In contrast to this, the combinatorial treatment (IADB + 5-FU) significantly attenuated mitochondrial swelling with the maintenance of normal cristae (Figure 10E,F).
Although TEMPOL can scavenge O2•–, its t1/2 in blood is only 15 s,15 which is likely associated with the rapid reduction of nitroxide radical. Unlike TEMPOL, the antioxidant activity of IADB may not only result from the nitroxide moiety but also from amino acid residues of IADB, which would coordinately enhance the radical-scavenging capacity of nitroxide. The indole ring of Trp as a hydrogen donor and the hydrophobic microenvironments induced by Trp and theanine residues, in addition to the hydrophilic nature of theanine, are likely to contribute to the enhanced overall antioxidant-scavenging property of IADB. From the predicted lowest energy conformation of IADB (Figure 11A), we speculate that a nitroxide radical surrounded by hydrophobic and resonance-stabilized amino acid side chains may result in enhanced bioavailability that correlates to improved scavenging of O2•– or NO• and associated cardioprotection.
Figure 11.

Proposed mechanism(s) of action of IADB: (A) predicted lowest energy conformation of TEMPOL and IADB; (B) schematic representation of the redox transformations of nitroxide, hydroxylamine, and the oxoammonium cation of IADB; (C) distinct action mechanisms of IADB involved in normal and tumor cells. In oxygen-rich normal cells (i.e., cardiomyocytes), the nitroxide moiety of IADB remains in the oxidized form (IADB-NO•). In hypoxic tumor cells, the nitroxide moiety of IADB is rapidly reduced to the corresponding hydroxylamine form (IADB-NOH). (D) HOMO/LUMO plots for nitroxide IADB (IADB-NO•) and its hydroxylamine (IADB-NOH), in which ΔE determines the chemical reactivity. The higher EHOMO combined with the lower ΔE of the nitroxide form of IADB (IADB-NO•) confers a substantively higher radical-scavenging potential than the corresponding hydroxylamine form (IADB-NOH). These data provide a rationale and explanation as to why IADB confers greater protection to normal (i.e., heart) vs tumor tissues. (E) Schematic diagram of our conclusion that cardioprotection with IADB during chemotherapy is likely associated with the upregulation of autophagy and its ensuing antioxidant activity.
In biological systems, nitroxide metabolism occurs
predominantly
via the reduction of the nitroxide to a hydroxylamine. IADB-NO•, having a single unpaired electron, can undergo
redox transformations between the one-electron oxidized state, the
oxoammonium cation, and the one-electron reduced IADB-NOH. When IADB-NO• is incubated with
cells or administered in vivo, a dynamic equilibrium is established
between the three forms. The equilibrium levels of these forms are
dependent on tissue oxygenation and the levels of reducing equivalents
or the tissue “redox status”. The reduction rates of IADB-NO• in tumor and normal tissues are
different because the tissue redox status is quantitatively different.
In the tumor microenvironment, the reduction rate of IADB-NO• can be accelerated because of high oxidative
activity. Compared to normal cells (i.e. cardiomyocytes), the oxidative
activity of cancer cells is higher, which is due to the overproduction
of hydroxyl radical and superoxide generated under hypoxic conditions.
In hypoxic tumor cells, the nitroxide form of IADB (IADB-NO•) is rapidly reduced to the corresponding
hydroxylamine form (IADB-NOH) (Figure 11C). However, the hydroxylamine (IADB-NOH) and nitroxide forms (IADB-NO•) do not constitute an effective redox couple and therefore are incapable
of supporting cellular processes involving oxidation/reduction. In
oxygen-rich normal cells (i.e. cardiomyocytes), the nitroxide moiety
of IADB remains in the oxidized form (IADB-NO•) (Figure 11C). The nitroxide (IADB-NO•) and oxoammonium pair 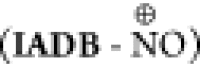 act as an efficient redox couple, which
can balance the cellular redox processes via reversible one-electron
transfer reactions. Thus, under “mild” oxidative stress,
the mitochondria may undergo constant damage repair, whereas severely
damaged mitochondria/organelles will be degraded and recycled by IADB-triggered autophagy. This may serve to explain why IADB would confer greater protection to normal tissue than
tumor tissue. To evaluate our hypothesis, we further examined the
ionization potential of the two forms of IADB. HOMO and
LUMO orbital energy levels for the two forms of IADB are
listed in Figure 11. From a calculation perspective, an electronic system with a larger
HOMO–LUMO gap is predicted to be less reactive than the one
having a smaller difference. As well, a small differential in the
HOMO and LUMO energy gap would shed light on the intramolecular charge
transfers.23 As predicted, the small energy
gap for IADB-NO• suggests that this
molecule possesses low ionization energies, revealing that it readily
donates electron, underscoring the favorable free-radical scavenging
activity. Thus, it is reasonable to assume that the higher EHOMO-combined coupling with the lower ΔE of the nitroxide form of IADB (IADB-NO•) would confer a considerably higher radical-scavenging
potential than the corresponding hydroxylamine form (IADB-NOH) (Figure 11D).
The computational analysis further supported our hypothesis that the
nitroxide radical of IADB (IADB-NO•) serves as an effective cellular antioxidant, whereas its corresponding
hydroxylamine form (IADB-NOH) only yields minimal antioxidant
effects. Taken together, the combined effects of our novel antioxidant
and autophagy-inducing agent, IADB, on 5-FU-induced cardiotoxicity
in vivo may form the platform for new approaches in cancer treatment
that combine antioxidants with autophagy modulators to manipulate
ROS-induced autophagy during chemotherapy.
act as an efficient redox couple, which
can balance the cellular redox processes via reversible one-electron
transfer reactions. Thus, under “mild” oxidative stress,
the mitochondria may undergo constant damage repair, whereas severely
damaged mitochondria/organelles will be degraded and recycled by IADB-triggered autophagy. This may serve to explain why IADB would confer greater protection to normal tissue than
tumor tissue. To evaluate our hypothesis, we further examined the
ionization potential of the two forms of IADB. HOMO and
LUMO orbital energy levels for the two forms of IADB are
listed in Figure 11. From a calculation perspective, an electronic system with a larger
HOMO–LUMO gap is predicted to be less reactive than the one
having a smaller difference. As well, a small differential in the
HOMO and LUMO energy gap would shed light on the intramolecular charge
transfers.23 As predicted, the small energy
gap for IADB-NO• suggests that this
molecule possesses low ionization energies, revealing that it readily
donates electron, underscoring the favorable free-radical scavenging
activity. Thus, it is reasonable to assume that the higher EHOMO-combined coupling with the lower ΔE of the nitroxide form of IADB (IADB-NO•) would confer a considerably higher radical-scavenging
potential than the corresponding hydroxylamine form (IADB-NOH) (Figure 11D).
The computational analysis further supported our hypothesis that the
nitroxide radical of IADB (IADB-NO•) serves as an effective cellular antioxidant, whereas its corresponding
hydroxylamine form (IADB-NOH) only yields minimal antioxidant
effects. Taken together, the combined effects of our novel antioxidant
and autophagy-inducing agent, IADB, on 5-FU-induced cardiotoxicity
in vivo may form the platform for new approaches in cancer treatment
that combine antioxidants with autophagy modulators to manipulate
ROS-induced autophagy during chemotherapy.
Conclusions
Taken
together, our data present the first evidence that IADB is not toxic to cardiac cells and demonstrates toxicity
toward only tumor cells. This unique feature is likely achieved by
its dual function (a combined modulation of autophagy and ROS production).
Because of the redox status difference between normal and tumor cells,
it selectively induces autophagic cell death, mediated by ROS overproduction,
in cancer cells. Furthermore, in vivo studies provide evidence that
cardioprotection associated with IADB may correlate with
its unique antioxidant activity, which originates from the unique
kinetic equilibrium profiles of its three forms (IADB-NO•, 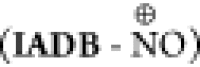 , and IADB-NOH), based on the
differences in the tumor redox status versus that in normal tissues.
Therefore, the IADB treatment in combination with chemotherapy
may lead to reduced cardiotoxicity, as well as the reduction of anticancer
drug dosages that may further improve chemotherapeutic efficacy with
decreased off-target effects. Overall, our data suggest that the use
of IADB may be of benefit in minimizing cardiotoxicity
associated with high-dose chemotherapy. These cellular studies pave
the way for future animal studies with IADB, the studies
currently underway in our laboratories.
, and IADB-NOH), based on the
differences in the tumor redox status versus that in normal tissues.
Therefore, the IADB treatment in combination with chemotherapy
may lead to reduced cardiotoxicity, as well as the reduction of anticancer
drug dosages that may further improve chemotherapeutic efficacy with
decreased off-target effects. Overall, our data suggest that the use
of IADB may be of benefit in minimizing cardiotoxicity
associated with high-dose chemotherapy. These cellular studies pave
the way for future animal studies with IADB, the studies
currently underway in our laboratories.
Materials and Methods
Cell Culture
All cell lines were obtained from American Type Cell Culture collection (ATCC). HeLa cells were grown in Eagle’s minimal essential medium and 10% fetal bovine serum (FBS; Sigma-Aldrich, heat inactivated). Human colonic adenocarcinoma HT-29, lung cancer A549, and breast cancer cell lines MDA-MB231 were routinely maintained in Dulbecco’s modified Eagle medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% FBS (Gibco). H9C2 cardiac myoblasts were obtained from ATCC and maintained in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin. All cells were grown at 37 °C in a humidified 5% CO2 atmosphere.
Rat Aortic Strip Assay To Measure Free-Radical Scavenging Capacity
The free-radical scavenging activity of IADB and TEMPOL in the rat aortic strip was determined as described.23 Briefly, after sacrifice, rat aortic strips were obtained and then immediately placed into a perfusion solution (5 mL oxygenated (95% O2, 5% CO2) Krebs solution, 37 °C). The aortic strips were then mounted to tension transducers, and an NE (final concentration 10–9 mol/L) solution was added to induce contraction. The relaxation contraction curves were recorded. When the hypertonic contraction reached a maximum, NE was flushed, and the vessel strips were stabilized for 30 min. Subsequently, the NE (final concentration 10–9 mol/L) solution was reintroduced. When the hypertonic contraction value of the aortic strips reached the previously defined maximum, 15 μL of NE or a solution of the test compounds in 15 μL of water were added, respectively. Upon stabilization, 1.5 μL of Ach (final concentration of 10–6 mol/L) was added, and the percent inhibition of Ach-induced vasorelaxation was determined.
Drug Treatment in HT-29 Cells
HT-29 cells were grown until they reached ∼50% subconfluence, and then the cells were subjected to drug treatment. To assess the effects of the co-treatment with 5-FU and IADB, the cells were pretreated with IADB (10 μM) for 12 h followed by treatment with 5-FU (5 μM) for another 48 h.
Cell Proliferation Assay
The drug-treated HT-29 cells and untreated control cells were cultured in 96-well plates at a density of 5 × 103 cells per cell in a 100 μL complete medium. The CellTiter96 cell proliferation assay kit was used following the manufacturer’s protocol, and the cell growth was evaluated using trypan blue exclusion. All experiments were performed in triplicate, and the proliferation of HT-29 cells was calculated as the ratio of each experimental condition to that of untreated control cells.
Expression of GFP LC3
The GFP human LC3 fusion protein expressing plasmid pEGFP LC3 was purchased from Life Technologies. The cells (5 × 104 per well) were seeded in six-well plates the day before transfection, and the cells were transfected with FuGENER HD Transfection Reagent (Roche, 04709691001). The GFP fusion proteins were observed under a laser scanning microscope system. The percentage of GFP LC3-positive cells with GFP LC3 punctate patterning was determined from three independent experiments and the means derived.
Live Cell Imaging
The cells were grown in 35 mm glass-bottom dishes for 24 h. The media were removed and the cells were washed three times with 1× Dulbecco’s phosphate-buffered saline (DPBS) without Ca2+ or Mg2+ (Hyclone, Fisher Sci.). The fluorescent probes were incubated with cells in the media without FBS. After each step, the cells were washed with DPBS buffer. The cells were imaged using an Olympus confocal laser scanning microscope.
Animal Test
All animal tests were performed in compliance with the “Guide for the Care and Use of Laboratory Animals” published by the US National Institutes of Health. The male ICR mice (20 ± 2 g) were housed in a 12/12 light/dark cycle at 21 ± 2 °C for 24 h before use. The mice were randomly divided into four groups: (i) Sham control group: the mice were intraperitoneally injected with saline for 14 consecutive days; (ii) 5-FU alone group: the mice were intraperitoneally injected with 5-FU (10 mg/kg) once over 3 consecutive days; (iii) IADBalone group: the mice were intraperitoneally injected with IADB (30 mg/kg) for 14 consecutive days; (iv) IADB+ 5-FU group: the mice were intraperitoneally injected with IADB (30 mg/kg) for 11 consecutive days, and then intraperitoneally administered 5-FU (10 mg/kg) + IADB (30 mg/kg) once over 3 consecutive days. Upon the conclusion of experiments, the mice were euthanized via sodium pentobarbital overdose. The heart tissue samples were immediately separated, and blood was rapidly obtained from the ascending aorta. The tissue samples were divided into two sections, one for the determination of lipid peroxidation and the other used for histological examination (Leica CM 1850 UV clinical cryostat) at −30 °C.
Histological Analysis
The tissues were mounted in gum tragacanth in an appropriate orientation and snap-frozen in isopentane chilled in liquid nitrogen. The frozen tissue sections (4–5 μm) were cut as previously described and stained with H & E, modified trichrome, or NADH followed by light microscopic examination.
Statistical Analysis
A two-way analysis of variance followed by Scheffé’s test was employed using the Origin program. If differences were observed, the values were then analyzed using Student’s t test for paired data. All values were expressed as mean ± SE, and significance was set at the 95th centile.
Acknowledgments
The authors acknowledge the support of NIH (GM088795-01), NSF (no. 1048655), NASA (# MSGC R85197), MIIE (#1204010), VPR-SI fund (#R01386), and the Research Excellence Fund (#R01121 and #R01323), Portage Health Foundation (#R01552), National Science Foundation of China (no. 81370419), Natural Science Foundation of HeBei, China (grant 062761266), Administration of Traditional Chinese Medicine of HeBei Province (grant 2007134).
Glossary
Abbreviations
- IADB
indole alkaloid derivative B
- 5-FU
5-fluorouracil
- ROS
reactive oxygen species
- TEMPOL
4-hydroxyl-TEMPO
- SOD
superoxide dismutase
- NE
norepinephrine
- Ach
acetylcholine
- LC3
microtubule-associated protein 1 light chain 3
- mtROS
mitochondrial reactive oxygen species
- MFI
mean fluorescence intensity
- 3-MA
3-methyladenine
- AVOs
autophagy process-related vesicular organelles
- MDA
malondialdehyde
- CAT
catalase
- GSH
thiol glutathione
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02139.
Detailed synthetic procedure of preparation of IADB; cell culture; cell proliferation assay; expression of GFP LC3; percent inhibition of Ach-induced vasorelaxation by TEMPOL and IADB; IADB-induced GFP-LC3 puncta formation in cancer and rat normal H9C2 cardiac cells; IADB-induced autophagic vesicle accumulation and the appearance of punctate GFP-LC3 staining in a time- and dose-dependent manner; and effect of IADB on proliferation assay of HT-29 colon cancer cells (PDF)
Author Contributions
L.B., W.B., K.M.G., and J.J. designed the study. L.B., W.B., Y.B., P.L., and S.H. performed the synthesis, characterization, and cell assays. Y.B., P.L., and Y.Z. did the animal tests. L.B., W.B., C.H., K.M.G., J.J., and C.B., performed data analysis, wrote, and edited the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Angsutararux P.; Luanpitpong S.; Issaragrisil S. Chemotherapy-Induced Cardiotoxicity: Overview of the Roles of Oxidative Stress. Oxid. Med. Cell Longev. 2015, 2015, 1–13. 10.1155/2015/795602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudgil R.; Haddad H. Chemotherapy-related cardiac dysfunction: grey area in type I and type II classification. Curr. Opin. Cardiol. 2017, 32, 181–188. 10.1097/HCO.0000000000000361. [DOI] [PubMed] [Google Scholar]
- McGowan J. V.; Chung R.; Maulik A.; Piotrowska I.; Walker J. M.; Yellon D. M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. 10.1007/s10557-016-6711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deidda M.; Madonna R.; Mango R.; Pagliaro P.; Bassareo P. P.; Cugusi L.; Romano S.; Penco M.; Romeo F.; Mercuro G. Novel insights in pathophysiology of antiblastic drugs-induced cardiotoxicity and cardioprotection. J. Cardiovasc. Med. 2016, 17, e76–e83. 10.2459/jcm.0000000000000373. [DOI] [PubMed] [Google Scholar]
- Cadeddu C.; Mercurio V.; Spallarossa P.; Nodari S.; Triggiani M.; Monte I.; Piras R.; Madonna R.; Pagliaro P.; Tocchetti C. G.; Mercuro G. Preventing antiblastic drug-related cardiomyopathy. J. Cardiovasc. Med. 2016, 17, e64–e75. 10.2459/jcm.0000000000000382. [DOI] [PubMed] [Google Scholar]
- Layoun M. E.; Wickramasinghe C. D.; Peralta M. V.; Yang E. H. Fluoropyrimidine-Induced Cardiotoxicity: Manifestations, Mechanisms, and Management. Curr. Oncol. Rep. 2016, 18, 35. 10.1007/s11912-016-0521-1. [DOI] [PubMed] [Google Scholar]
- Papanastasopoulos P.; Stebbing J. Molecular basis of 5-fluorouracil-related toxicity: lessons from clinical practice. Anticancer Res. 2014, 34, 1531–1535. [PubMed] [Google Scholar]
- Paneni F.; Diaz Cañestro C.; Libby P.; Lüscher T. F.; Camici G. G.; Camici G. G. The Aging Cardiovascular System. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. 10.1016/j.jacc.2017.01.064. [DOI] [PubMed] [Google Scholar]
- Doroshow J. H.; Locker G. Y.; Myers C. E. Enzymatic Defenses of the Mouse Heart Against Reactive Oxygen Metabolites. J. Clin. Invest. 1980, 65, 128–135. 10.1172/jci109642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberti M.; Porto S.; Marra M.; Zappavigna S.; Grimaldi A.; Feola D.; Spina A.; Naviglio S.; Sannolo N.; Caraglia M.; Pesce D. 5-Fluorouracil induces apoptosis in rat cardiocytes through intracellular oxidative stress. J. Exp. Clin. Cancer Res. 2012, 31, 60. 10.1186/1756-9966-31-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinhult S.; Albertsson M.; Eskilsson J.; Cwikiel M. Effects of probucol on endothelial damage by 5-fluorouracil. Acta Oncol. 2003, 42, 304–308. 10.1080/02841860310004409. [DOI] [PubMed] [Google Scholar]
- Vincent D. T.; Ibrahim Y. F.; Espey M. G.; Suzuki Y. J. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharmacol. 2013, 72, 1157–1168. 10.1007/s00280-013-2260-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters K. A.; Kremer L. C. M.; Miller T. L.; Herman E. H.; Lipshultz S. E. Protecting against anthracycline-induced myocardial damage: a review of the most promising strategies. Br. J. Haematol. 2005, 131, 561–578. 10.1111/j.1365-2141.2005.05759.x. [DOI] [PubMed] [Google Scholar]
- Soule B. P.; Hyodo F.; Matsumoto K.; Simone N. L.; Cook J. A.; Krishna M. C.; Mitchell J. B. The chemistry and biology of nitroxide compounds. Free Radic. Biol. Med. 2007, 42, 1632–1650. 10.1016/j.freeradbiomed.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott C.; Bottle S. E. Biological Relevance of Free Radicals and Nitroxides. Cell Biochem. Biophys. 2016, 75, 227–240. 10.1007/s12013-016-0759-0. [DOI] [PubMed] [Google Scholar]
- Wilcox C. S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton E. L. The dual role of ROS, antioxidants and autophagy in cancer. Biomed J. 2016, 39, 89–92. 10.1016/j.bj.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Qu T.; Wang P.; Li X.; Qiang J.; Xia Z.; Duan H.; Huang J.; Zhu L. Unravelling the relationship between macroautophagy and mitochondrial ROS in cancer therapy. Apoptosis 2016, 21, 517–531. 10.1007/s10495-016-1236-3. [DOI] [PubMed] [Google Scholar]
- Hua F.; Shang S.; Hu Z.-W. Seeking new anti-cancer agents from autophagy-regulating natural products. J. Asian Nat. Prod. Res. 2017, 19, 305–313. 10.1080/10286020.2017.1304385. [DOI] [PubMed] [Google Scholar]
- Law B. Y.; Chan W. K.; Xu S. W.; Wang J. R.; Bai L. P.; Liu L.; Wong V. K. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci. Rep. 2014, 4, 5510. 10.1038/srep05510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillhon M.; Guamán Ortiz L. M.; Lombardi P.; Scovassi A. I. Berberine: new perspectives for old remedies. Biochem. Pharmacol. 2012, 84, 1260–1267. 10.1016/j.bcp.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Liu T.; Liu X.; Li W. Tetrandrine, a Chinese plant-derived alkaloid, is a potential candidate for cancer chemotherapy. Oncotarget 2016, 7, 40800–40815. 10.18632/oncotarget.8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi W.; Li X.; Bi Y.; Xue P.; Zhang Y.; Gao X.; Wang Z.; Li M.; Itagaki Y.; Bi L. Novel TEMPO-PEG-RGDs conjugates remediate tissue damage induced by acute limb ischemia/reperfusion. J. Med. Chem. 2012, 55, 4501–4505. 10.1021/jm201381w. [DOI] [PubMed] [Google Scholar]
- Guzik T. J.; West N. E. J.; Pillai R.; Taggart D. P.; Channon K. M. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension 2002, 39, 1088–1094. 10.1161/01.hyp.0000018041.48432.b5. [DOI] [PubMed] [Google Scholar]
- Dröge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- Chen X.; Bi Y.; Wang T.; Li P.; Yan X.; Hou S.; Bammert C. E.; Ju J.; Gibson K. M.; Pavan W. J.; Bi L. Lysosomal targeting with stable and sensitive fluorescent probes (Superior LysoProbes): applications for lysosome labeling and tracking during apoptosis. Sci. Rep. 2015, 5, 9004. 10.1038/srep09004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yapici N. B.; Bi Y.; Li P.; Chen X.; Yan X.; Mandalapu S. R.; Faucett M.; Jockusch S.; Ju J.; Gibson K. M.; Pavan W. J.; Bi L. Highly stable and sensitive fluorescent probes (LysoProbes) for lysosomal labeling and tracking. Sci. Rep. 2015, 5, 8576. 10.1038/srep08576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D. J.; Abdelmohsen K.; Abe A.; Abedin M. J.; Abeliovich H.; Acevedo Arozena A.; Adachi H.; Adams C. M. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T.-T.; Yu X.-X.; Yan L.-J.; Xiao H.-T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol 2017, 79, 287–294. 10.1007/s00280-016-3197-1. [DOI] [PubMed] [Google Scholar]
- Gewirtz D. A. The Challenge of Developing Autophagy Inhibition as a Therapeutic Strategy. Cancer Res. 2016, 76, 5610–5614. 10.1158/0008-5472.can-16-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L.; Bravo-San Pedro J. M.; Demaria S.; Formenti S. C.; Kroemer G. Activating autophagy to potentiate immunogenic chemotherapy and radiation therapy. Nat. Rev. Clin. Oncol. 2017, 14, 247–258. 10.1038/nrclinonc.2016.183. [DOI] [PubMed] [Google Scholar]
- Yapici N. B.; Mandalapu S.; Michael Gibson K.; Bi L. Targeted fluorescent probes for detection of oxidative stress in the mitochondria. Bioorg. Med. Chem. Lett. 2015, 25, 3476–3480. 10.1016/j.bmcl.2015.07.011. [DOI] [PubMed] [Google Scholar]
- Bi W.; Bi Y.; Gao X.; Li P.; Hou S.; Zhang Y.; Bammert C.; Jockusch S.; Legalley T. D.; Michael Gibson K.; Bi L. Indole-TEMPO conjugates alleviate ischemia-reperfusion injury via attenuation of oxidative stress and preservation of mitochondrial function. Bioorg. Med. Chem. 2017, 25, 2545–2568. 10.1016/j.bmc.2017.03.033. [DOI] [PubMed] [Google Scholar]
- Bi W.; Bi Y.; Gao X.; Yan X.; Zhang Y.; Harris J.; Legalley T. D.; Gibson K. M.; Bi L. Pharmacological protection of mitochondrial function mitigates acute limb ischemia/reperfusion injury. Bioorg. Med. Chem. Lett. 2016, 26, 4042–4051. 10.1016/j.bmcl.2016.06.079. [DOI] [PubMed] [Google Scholar]
- Bi W.; Bi Y.; Gao X.; Yan X.; Zhang Y.; Xue P.; Bammert C. E.; Legalley T. D.; Michael Gibson K.; Bi L.; Wang J.-X. Anti-inflammatory, analgesic and antioxidant activities of novel kyotorphin-nitroxide hybrid molecules. Bioorg. Med. Chem. Lett. 2016, 26, 2005–2013. 10.1016/j.bmcl.2016.02.086. [DOI] [PubMed] [Google Scholar]
- Bi L.Novel probes and targeting compounds for mitochondria. Patent PCT/US2013/065649 (2013), WO2014/063033A2 (2014).
- Bi L.Lysosomal targeting probes. Patent PCT/US2013/055578 (2013), WO2014/058535A1 (2014).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.