

Abstract
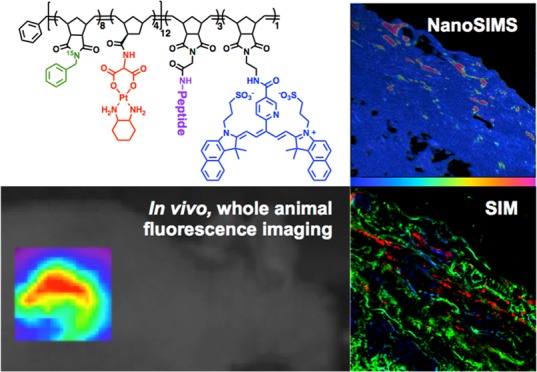
In nanomedicine, determining the spatial distribution of particles and drugs, together and apart, at high resolution within tissues, remains a major challenge because each must have a different label or detectable feature that can be observed with high sensitivity and resolution. We prepared nanoparticles capable of enzyme-directed assembly of particle therapeutics (EDAPT), containing an analogue of the Pt(II)-containing drug oxaliplatin, an 15N-labeled monomer in the hydrophobic block of the backbone of the polymer, the near-infrared dye Cy5.5, and a peptide that is a substrate for tumor metalloproteinases in the hydrophilic block. When these particles reach an environment rich in tumor associated proteases, the hydrophilic peptide substrate is cleaved, causing the particles to accumulate through a morphology transition, locking them in the tumor extracellular matrix. To evaluate the distribution of drug and EDAPT carrier in vivo, the localization of the isotopically labeled polymer backbone was compared to that of Pt by nanoscale secondary ion mass spectrometry (NanoSIMS). The correlation of NanoSIMS with super-resolution fluorescence microscopy revealed the release of the drug from the nanocarrier and colocalization with cellular DNA within tumor tissue. The results confirmed the dependence of particle accumulation and Pt(II) drug delivery on the presence of a Matrix Metalloproteinase (MMP) substrate and demonstrated antitumor activity. We conclude that these techniques are powerful for the elucidation of the localization of cargo and carrier, and enable a high-resolution assessment of their performance following in vivo delivery.
Short abstract
Multiscale imaging methods to track drug-loaded nanoparticles are needed. Correlated optical and isotopic nanoscopy were used to study Pt(II)-loaded enzyme responsive micelle distribution in tissues.
1. Introduction
We have developed a method, termed EDAPT for “enzyme-directed assembly of particle therapeutics”, for targeting nanoparticles that accumulate and become locked in malignant tissues when they encounter tumor proteases.1−3 This platform technology targets tumors via disease-associated enzymes acting on these responsive materials leading to a morphology transition from small spherical nanoparticles to microscale accumulated material within tumor tissue leading to retention and accumulation of fluorescent probes and therapeutics after intratumoral (IT) or intravenous (IV) administration (Figure 1).1,4 This approach, as with other targeted nanoparticle technologies,5,6 presents a challenge in terms of determining particle localization, drug release and distribution at high resolution within the targeted tissue.7 Meeting this challenge requires that carrier and drug are labeled, or are naturally predisposed for high resolution imaging. Several techniques, including magnetic resonance imaging (MRI), positron emission tomography (PET), and computed tomography (CT), are available and widely used for the study of chemical systems in vivo, especially for whole animal imaging.8,9 To obtain higher resolution, ex vivo techniques such as fluorescence microscopy, scanning transmission X-ray microscopy (STXM), Raman microspectral imaging, electron microscopy (EM), or nanoscale secondary ion mass spectrometry (NanoSIMS) have proven useful.10−12 However, these techniques vary in sensitivity, resolution, nanoparticle labeling requirements, cost, availability, and sample preparation needs.13 Further, to fully understand the relationship between nanomaterials and their biological behavior using these techniques individually is problematic.
Figure 1.

Polymer structure, assembly, and characterization of labeled micellar nanoparticles. Left: Pep-Pt-P chemical structure. Dialysis of the 15N, Pt, and Cy5.5 multilabeled polymer (Pep-Pt-P) from DMSO to water yields well-defined micellar nanoparticles of approximately 20 nm in diameter. l-Amino acid peptide sequence “GPLGLAGGERDG” for L-Pep-Pt-NP and d-amino acid sequence “gplglaggerdg” for D-Pep-Pt-NP. As previously shown, cleavage of the l-amino acid peptide sequence “GPLGLAGGERDG” occurs between the italicized G and L, promoting the release of the hydrophilic peptide sequence “LAGGERDG” which triggers the nano- to micrometer morphology change.1 Right: TEM image (dry state, negative uranyl acetate stain) of L-Pep-Pt-NP before (left) and after (right) exposure to MMP, demonstrating enzyme-induced morphology change. For TEM images of D-Pep-Pt-NP before and after MMP exposure, see Figure S3.
One way to overcome the specific limitations of each technique is to image samples by multiple methods simultaneously. As an example of this approach, correlated optical and isotopic nanoscopy (abbreviated as COIN) has been introduced for multimodal imaging pertaining to the nanometer length scale.14,15 In one such application of this approach, NanoSIMS, exhibiting high spatial resolution, sensitivity, and mass resolution yielding some chemical information,11,16 is linked with super-resolution fluorescence microscopy techniques. Although methodologically complex, this combination of techniques has been applied to great effect in the study of biological processes.14,17
In this paper we describe the behavior of Pt(II) drug-loaded, matrix-metalloproteinase (MMP) responsive nanoparticles in vivo following IT administration. These EDAPT nanoparticles were generated through the assembly of amphiphilic block copolymers designed to undergo a change in size following exposure to MMPs that are highly overexpressed in tumor tissues.18−20 This was achieved through incorporation of peptide substrates for MMPs in the micellar nanoparticle shell. As previously shown, upon exposure to proteolytic enzymes, the cleavage of a specific hydrophilic peptide fragment on the shell of the small, spherical micellar nanoparticles occurs, changing the amphiphilic nature of the polymer. This trigger causes the polymers to pack differently, leading to a phase transition, or morphology change to a larger, microscale assembly, which locks the material within tumor tissue in vivo.(1−4,21−24) As a negative control for this process, analogous systems were generated using all d-amino acids in the peptide sequence as they are not recognized as substrates by MMPs or other proteases in the body. We employed NanoSIMS correlated with structured illumination microscopy (SIM) to study the distribution of a drug cargo and nanocarrier in tumor tissues. This allowed a quantification of drug release from the nanocarrier to tumor tissues and confirmed drug targets in vivo. This is an unprecedented study demonstrating the application of such techniques in the elucidation of specific interactions and distribution of drug loaded nanoparticles in tissues.
2. Results and Discussion
We designed Pt(II)-loaded nanoparticles (Pep-Pt-NPs) using amphiphilic block copolymers containing an oxaliplatin analogue and MMP-responsive peptides (Pep-Pt-P, Figure 1). These polymers were synthesized through ring opening metathesis polymerization (ROMP).25 The monomers were specifically designed to form polymers that would self-assemble into particles with three important capabilities: (1) retention in tumor tissue; (2) antitumor activity in a murine xenograft of a cancer type that overexpresses MMPs; and (3) contrast for optical (in vivo and ex vivo) and isotopic imaging (ex vivo). These polymers were synthesized using four different ROMP monomers: (1) an oxaliplatin analogue monomer containing a norbornene polymerizable moiety (Pt-Mon),26 known to be cytotoxic to a variety of cancer cells and useful as an isotopic label because of the lack of Pt in normal tissues; (2) a 15N-labeled phenyl monomer (15N-Mon) which serves as a isotopic label for the polymer backbone of the nanocarrier; (3) a peptide substrate monomer (Pep-Mon) as an MMP recognition sequence (l-amino acid sequence “GPLGLAGGERDG” for L-Pep-Mon and d-amino acid sequence “gplglaggerdg” for D-Pep-Mon); and (4) a cyanine 5.5 dye (Cy5.5) monomer (Cy-Mon) as a fluorescent polymer label for super-resolution fluorescence microscopy (Figure S1). Copolymerization of 15N-Mon with Pt-Mon in a 2:1 ratio to form the hydrophobic block, followed by Pep-Mon to form the hydrophilic block and Cy-Mon as a third block generated polymer Pep-Pt-P (Figure 1 and Figure S1). Nanoparticles were generated from Pep-Pt-P by dialyzing the polymers from DMSO into water as drug-containing, enzyme-responsive, isotopically, and fluorescently labeled L-Pep-Pt-NP (Figures 1 and S2). Nonresponsive, negative control nanoparticles were synthesized by incorporating d-amino acid peptides into the hydrophilic block of the polymers, yielding D-Pep-Pt-NP, Figure S3. Compared to other previously reported oxaliplatin loaded micelles,27−29 our synthetic approach generated functional nanoparticles with more than 35 wt % drug loading without the need of post-polymerization or postassembly conjugations.
After confirming the ability of L-Pep-Pt-NP to aggregate upon MMP exposure in vitro (Figures 1, S2 and S3), we examined the capability of the material to be retained in tumor tissue in vivo and explored their ability to inhibit tumor growth as a result. We utilized human HT-1080 fibrosarcoma subcutaneous xenografts for these studies as this cell line overexpresses MMPs.30,4 Mice bearing HT-1080 tumors were injected IT with either L-Pep-Pt-NP or D-Pep-Pt-NP at 2.5 mg/kg with respect to Pt content and monitored over the course of 12 days for retention and efficacy. A third cohort received saline alone as an additional negative control, with a fourth receiving oxaliplatin alone, which is the clinical analogue of the compound loaded in the particles.
Importantly, L-Pep-Pt-NP was able to inhibit tumor growth relative to both the saline and D-Pep-Pt-NP controls (Figure 2). Additionally, it performed as well as oxaliplatin, suggesting that packaging the Pt drug in a nanocarrier does not adversely affect its therapeutic potential. As previously demonstrated for other EDAPT systems, a key advantage of this platform is its ability to deliver its cargo specifically and selectively to tumor tissues, while limiting off-target toxicity (Figure S4).4
Figure 2.
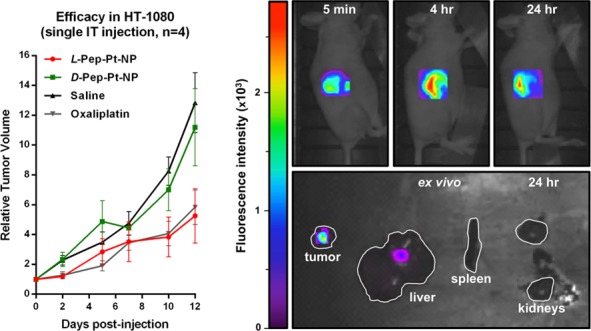
Efficacy and whole animal and ex vivo organ targeting data. Left: Comparison of L-Pep-Pt-NP to D-Pep-Pt-NP, oxaliplatin at 2.5 mg/kg dose with respect to Pt and saline following IT injection. L-Pep-Pt-NP effectively inhibited tumor growth up to 12 days postinjection with efficacy similar to oxaliplatin, whereas D-Pep-Pt-NP showed no observable effect. Right-top: Time course of live-animal fluorescence imaging following IT injection of L-Pep-Pt-NP. Right-bottom: Ex vivo tissue analysis. Fluorescence imaging of tumor, liver, spleen, and kidney excised at 24 h from animal administered L-Pep-Pt-NP. White line shows the outline of the organs. Highest signal was observed in tumor.
Further, live animal fluorescence microscopy (Figure 2) was used to monitor the retention of both L-Pep-Pt-NP and D-Pep-Pt-NP postinjection by tracking the NIR fluorophore on the polymer backbone of both systems (λex = 635 nm and λem = 693 nm). Fluorescence was observable up to 5 days following IT injection of L-Pep-Pt-NP, suggesting that these materials were retained over a long time scale. Importantly, fluorescence was only observed for the first 5 h following IT injection of D-Pep-Pt-NP, indicative of rapid clearance of the nonresponsive material which had an efficacy comparable to that observed for saline (Figure S5). Furthermore, ex vivo tissue analysis of tumor-bearing mice injected IT with either L-Pep-Pt-NP, D-Pep-Pt-NP, or saline solution and sacrificed 24 h postinjection shows the highest fluorescence signal intensity in the excised tumors, with fluorescence observed to a lesser extent in the liver, spleen, and kidneys (Figures 2 and S6).
Tumor tissue was additionally examined ex vivo utilizing a multimodal super-resolution imaging approach that combines optical (SIM) with isotopic (NanoSIMS) nanoscopy. Both techniques have been proven useful for imaging biological samples.15,16,31,32 SIM, a light microscopy technique capable of subdiffraction limit imaging, has been widely used in the field of neuroscience, cell biology, and microbiology to observe cellular and subcellular processes that occur at the nanometer scale. NanoSIMS, on the other hand, has been used in the field of paleobiology, microbial ecology, and cellular biology to collect isotopic information and thus quantify the sample’s elemental composition.16 In our study, the combination of these techniques is necessary to provide a complete distribution map of the nanomaterial in the tumor tissue, with nanometric resolution. While NanoSIMS allows determination of the distribution of both the nanocarrier and the drug within the tumor independently, SIM provides three-dimensional structural information such as a specific extracellular or intracellular nanocarrier localization.
Once the nanomaterial aggregates in tumor tissues and the polymer hydrophobic block are exposed to the tissue milieu, nucleophiles such as chloride ions and water are expected to promote oxaliplatin drug release from the nanocarrier by ligand exchange reactions on the Pt(II) to displace the labile carboxylate ligands.33 Ultimately, oxaliplatin binding to nuclear DNA is expected to trigger cytotoxic effects in tumor cells.34,35 To validate this pathway, we performed super-resolution imaging of tumor tissue and tracked the nanocarrier (the polymer NIR dye label with SIM and the 15N label with NanoSIMS) and the Pt(II)-drug (NanoSIMS) separately. Similar to the procedure used for efficacy studies, tumor-bearing mice were injected IT with either L-Pep-Pt-NP, D-Pep-Pt-NP, or saline solution and sacrificed 24 h later. The tumors were harvested and cryosectioned into 5 μm sections and placed on indium-tin-oxide (ITO) coverslips. Immunostaining was performed on the sections with an anti-α-actinin antibody to label cytoplasmic dense structures (α-actinin, AF 488). Samples were subsequently stained with DAPI as a cell nucleus indicator and then dehydrated for imaging (see Experimental Section).
Fluorescent images were acquired at different magnifications as part of preliminary investigations. Low magnification images showed fluorescent signals from both the L-Pep-Pt-NP and D-Pep-Pt-NP polymeric nanomaterials distributed mainly at the periphery of the tumor tissue (Figure S7). As previously observed in time-course studies of fluorescence in live animals, only a minimal amount of Cy5.5 signal was detected for the D-Pep-Pt-NP sample because of its reduced retention in tumor tissues. Increased magnification revealed the polymeric probes preferentially accumulating in the extracellular space, as the fluorescent signal associated with the polymer backbone (Cy5.5) was only poorly correlated with intracellular fluorescently labeled, green actin filaments (Figures 3 and S8).
Figure 3.
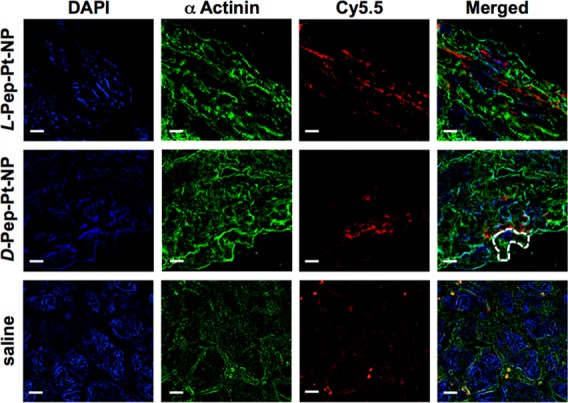
SIM analysis of 5 μm thick tumor tissue sections of animals pretreated with L-Pep-Pt-NP (top), D-Pep-Pt-NP (middle), or saline (bottom). The samples were examined using specific fluorescent probes. The polymers used to obtain the nanostructures contain a Cy5.5 NIR dye chemically bound to the polymer backbone used to track the nanomaterials (red). Actin filaments were visualized by staining with a mouse anti-α-actinin antibody followed by AF 488 goat antimouse antibody (green). Nucleus was stained with DAPI (blue). Although DAPI is known to bind and stain DNA, some nonspecific binding to other subcellular structures was observed. From these fluorescent images, it is clear that the signal for both the responsive and the nonresponsive nanoparticles are not correlated with nuclear and actin staining, suggesting an extracellular localization of the nanomaterials. Interestingly, samples from animals treated with saline solution showed fluorescent signals in the far red channel, attributed to tissue autofluorescence. The dashed line outlines a cell boundary. Scale bar represents 5 μm.
The fact that tumor regression occurred (Figure 2) means that Pt-containing oxaliplatin analogue must permeate into cells over this time frame. To directly visualize this, tissue sections previously imaged by SIM (Figure 3) were further analyzed with NanoSIMS (Figures 4, S9, S10, and S11). A secondary electron (SE) image was collected together with four masses of interest to create elemental maps for each sample: 31P as a nuclear indicator because of the phosphorus-rich DNA; 12C14N as an indicator for organic matter; 12C15N as a polymer backbone indicator; and 195Pt as a drug label. A hue-saturation-intensity (HSI) representation of the 12C15N/12C14N ratio map as a color scaled image was constructed in order to specifically identify 15N-rich areas (Figures 4 and S9). In addition to the tumor tissue, a sample of yeast was imaged daily and used to calibrate the 12C15N/12C14N measurements relative to air (Supporting Information). 195Pt counts obtained from images of samples of animals pretreated with saline solution were used to set baseline counts and the 15N distribution was in accordance with 15N natural abundance (Figure 5 and Supporting Information). It is important to note that fluorescence images from these saline samples (Figure 3, bottom panels) show signals in the far red channel (Cy5) attributed to autofluorescence, since no specific 195Pt or elevated 15N signals were observed by NanoSIMS (Figure 4, bottom panels). This highlights the need for, and importance of, multimodal imaging when studying labeled materials in complex biological systems.
Figure 4.

NanoSIMS imaging of tissue slices. Secondary electron images (SE) and NanoSIMS ion maps of dehydrated 5 μm tumor tissue sections of mice treated with L-Pep-Pt-NP (top), D-Pep-Pt-NP (middle), or saline (bottom). 31P–, 12C14N–, 12C15N–, and 195Pt– ion maps were obtained simultaneously, and intensities are shown in a fire scale, except for the 195Pt– ion map which is shown in white/black. HSI images represent the 12C15N/12C14N ratio and highlight specifically enriched areas as can be seen for L-Pep-Pt-NP and D-Pep-Pt-NP samples. The scales on the HSI images were adjusted depending on the relative intensities for each sample. Thus, the scale for L-Pep-Pt-NP is 0.0037–0.02, for D-Pep-Pt-NP is 0.0037–0.11, and for the saline solution sample 0.0037–0.15. Images, in all cases, represent an area of 48 μm × 48 μm.
Figure 5.

Quantitative analysis of 15N and 195Pt enrichment inside or outside of 15N- and 31P-rich ROIs. A: ROIs were defined according to 15N accumulation, as high 15N/14N (ROI number 1–16) and low 15N enrichment (ROI number 17–32). A.1: 15N ROIs on the L-Pep-Pt-NP HSI image. A.2: 15N ROIs on the D-Pep-Pt-NP HSI image. A.3: 15N/14N inside and outside of 15N-rich ROIs. A.4: 195Pt enrichment inside and outside of 15N-rich ROIs. B: ROIs were defined according to 31P signal, as high 31P (ROI number A1–10) and low 31P (ROI number B1–10). B.1: 31P ROIs on the L-Pep-Pt-NP31P ion map image. B.2: 31P ROIs on the D-Pep-Pt-NP31P ion map image. B.3: 15N/14N inside or outside of 31P-rich ROIs. B.4: 195Pt enrichment inside or outside of 31P-rich ROIs. Note that Pt– was collected as 196Pt– for the saline sample. Pt counts on saline ROIs were normalized to 195Pt by multiplying by 1.34, according to their isotopic abundance (33.8/25.2). Enrichment values were obtained from at least two independent images of each sample. For additional images, see Figure S12. A summary of these results and its statistical analysis can be found in Tables S1–S3. Minimum significant difference was defined as a p-value < 0.05.
As seen in Figure 4, samples exposed to D- and L-Pep-Pt-NP show 195Pt localized in areas enriched in 15N. Some of these hotspots correlated with Cy5.5 fluorescent signals on the SIM images (Figure S10). This suggests that, in certain areas of the tumor, the three labels incorporated in the polymer are still associated with the aggregated nanomaterials. To better understand the distribution of drug and nanocarrier in the tissue, regions of interest (ROIs) were defined. These ROIs were used to quantify the accumulation of the nanocarrier and drug labels within or outside of the ROIs. Thus, two types of ROIs were analyzed: within or outside highly 15N-rich areas defined from the corresponding HSI images and within or outside of 31P-rich areas defined from the corresponding 31P– images, to analyze accumulation of the labels on the aggregated nanomaterial (Figure 5 top panels) and on the nucleus of cells (Figure 5 bottom panels), respectively.
By averaging the values for ROIs inside or outside of 15N-rich areas (Figure 5A), it became clear that 15N enrichment was significantly greater in 15N-rich ROIs of L-Pep-Pt-NP than for either D-Pep-Pt-NP or the saline control, which should have an 15N enrichment at natural abundance (Figure 5A.3, dashed line at 0.0037). Interestingly, 15N enrichment in ROIs selected outside the main areas of 15N accumulations was larger for the Pep-Pt-NPs than for the saline sample, and again larger for the responsive L-Pep-Pt-NP compared to nonresponsive D-Pep-Pt-NP (Table S3). This indicates that some of the material becomes delocalized from the main 15N-rich areas. Although a similar distribution was observed for 195Pt, with a higher concentration of Pt in 15N-rich ROIs of L-Pep-Pt-NP samples than of D-Pep-Pt-NP and the saline control, no significant difference was observed between the Pt enrichment of ROIs outside the 15N-rich regions (Figure 5A.4, Table S3). This suggests a differential distribution of Pt and 15N in ROIs outside the main 15N enriched areas between samples treated with responsive and nonresponsive nanoparticles. For both labels (15N and Pt), the higher concentration of counts in L-Pep-Pt-NP samples than D-Pep-Pt-NP samples is in agreement with the greater retention in tumor tissues of the responsive material previously observed by fluorescence spectroscopy.
In an effort to study the differential accumulation of the nanocarrier (15N) and the Pt complex on the presumed drug target (DNA), a second set of ROIs was defined within or outside of 31P-rich areas found in the 31P– ion map (Figure 5B.1). As expected, the 15N enrichment inside and outside 31P-rich ROIs is much lower than for the 15N ROIs (Figure 5A.3 and 5B.3). As previously observed, 15N and Pt showed a different distribution, and in all cases the accumulation accounted for by the labels was significantly higher than in the saline control sample. Although no significant difference was observed for 15N enrichment inside or outside 31P-rich ROIs, differences between samples and between the local sample distribution was observed for Pt, with a higher concentration of Pt in the nucleus and for the samples exposed to the responsive system L-Pep-Pt-NP (Figure 5B.4).
In summary, these results show a different enrichment of the nanocarrier and the drug inside and outside 15N-rich ROIs, and, at the same time, differences were observed between their respective concentrations suggesting spatial dissociation of the species. The larger concentration of 15N and Pt in the tumor sections exposed to the responsive L-Pep-Pt-NP is in agreement with an extended retention of this material relative to the nonresponsive D-Pep-Pt-NP. As a consequence of this extended retention, inhibition of tumor growth was observed for the L-Pep-Pt-NP system but not for the d-amino acid control (D-Pep-Pt-NP), indicating that the Pt-drug is being released from the nanocarrier and binding to intracellular targets. A higher concentration of Pt was observed in 31P-rich ROIs and with respect to 15N enrichment for samples treated with the responsive nanoparticles, suggesting a specific association of Pt with 31P-rich structures such as DNA.
3. Conclusion
This study serves as a demonstration of the potential of COIN for the study of nanomaterials in complex biological systems in general and specifically for EDAPT nanoparticles. Fluorescence microscopy allowed for the differentiation of cellular structures and compartments with respect to the nanoparticle utilizing specific fluorescent labels. NanoSIMS imaging produced data consistent with the retention of the material in the extracellular space of tumor tissues, revealing the dissociation of the drug from the nanocarrier. This resulted in greater drug delivery for enzyme-responsive EDAPT particles than for a nonresponsive control nanoparticles which were poorly retained. By comparing the specific localization of the nanocarrier and the drug at the nanometer scale, we could track the specific association of Pt with the nuclear DNA of tumor cells from the in vivo sample. This work establishes COIN as a powerful methodology for tracking nanomaterials as delivery vehicles, where elucidation of the location of cargo and carrier at high-resolution is desirable for optimizing and assessing in vivo delivery.
4. Experimental Section
Polymer Synthesis
To a stirred solution of 15N-Mon was added a solution of the catalyst ((IMesH2)(C5H5N2)(Cl)2Ru=CHPh) in dry DMF and a solution of Pt-Mon in dry DMF. The reaction was allowed to stir under N2 for 2 h, after which an aliquot was removed and quenched with ethyl vinyl ether for SLS analysis. The remaining solution of 15N-Mon + Pt-Mon + catalyst was split into two separate reaction vessels. To one reaction vessel was added a solution of L-Pep-Mon in dry DMF (to ultimately afford L-Pep-Pt-P). To the second vessel was added a solution of D-Pep-Mon in dry DMF (to ultimately afford D-Pep-Pt-P). After three additional hours, a small aliquot was removed from each reaction vessel and terminated with ethyl vinyl ether for SLS analysis. Then, to each of the polymer solutions was added Cy-Mon and reaction was allowed to stir for an additional 2 h, before the polymer solutions were fully quenched with ethyl vinyl ether. The fully terminated polymers were precipitated with a cold 1:1 ether/methanol solution to afford the block copolymers as dark yellow solids (L-Pep-Pt-P, D-Pep-Pt-P).
Nanoparticle Preparation. (L-Pep-Pt-NP and D-Pep-Pt-NP)
Polymers (L-Pep-Pt-P or D-Pep-Pt-P) were dissolved in DMSO, and DPBS (Dulbecco’s phosphate buffered saline, no Ca, no Mg) was added over the course of 2 h. These solutions were transferred to 3500 MWCO snakeskin dialysis tubing and dialyzed against DPBS at pH 7.4 over 2 days with two buffer changes.
Intratumoral Efficacy
Tumor-bearing nu/nu female mice were randomly sorted into four groups (four mice per cohort) and treated with L-Pep-Pt-NP, D-Pep-Pt-NP, oxaliplatin, or saline at the dosage equivalent of 2.5 mg/kg of Pt as a single IT injection. Mouse weight and tumor volume were recorded once daily over the course of the 12-day study. Animals were imaged at 0, 4, 24, 48, and 72 h postinjection via live-animal optical imaging. To assess efficacy, relative tumor volume was calculated for each data point. The average relative tumor volume of each cohort at each time point was then calculated, along with standard deviation and standard error of mean. Animals were sacrificed at 12 days postinjection. The tumor was excised from each animal and treated as in the above protocol.
Fluorescent IHC Staining of Tissue Sections
Tissue sections on a 18 mm2 ITO coverslip were fixed with acetone at room temperature and washed three times with PBSt (0.05% Tween in PBS). Sections were incubated with blocking solution. The primary antibody mouse anti-α-actinin was added in blocking buffer and incubated at 4 °C overnight. Tissues were washed three times with PBSt, and the secondary antibody goat antimouse AlexaFluor 488 was added in blocking buffer and incubated for 30 min. Tissue sections were washed three times with PBSt and were incubated for 10 min with DAPI. The tissue sections were finally washed three times with PBSt and then subjected to a series of dehydration washes with 30%, 50%, 70%, 80% ethanol solutions and three times with 100% ethanol (30 min each).
Structural Illumination Microscopy Imaging
SIM imaging was performed on the Elyra S1 inverted fluorescence microscope (Zeiss). An objective with 10× and 40× magnification and 1.4 numerical aperture and an oil immersion objective with 100× magnification and 1.4 numerical aperture were used in this study. Three rotations and five phases were taken for each SIM image. The pixel size in the raw images was 80 nm per pixel, and in the resulting super resolution images was 40 nm per pixel. The camera exposure time was 100 ms per image. For every sample, three tracks were recorded sequentially for (1) image the NPs (Cy5.5) using 642 nm wavelength at 7 mW power laser excitation and emission wavelength longer than 655 nm; (2) image the actin filaments (α-actinin) using 488 nm at 2 mW laser excitation and emission band 495 to 550 nm; and (3) image the nucleus (DAPI) using 405 nm 2 mW laser excitation and emission band 420–480 nm. These tracks were later processed to obtain super resolution images using the ZEN software (Zeiss).
NanoSIMS Imaging
For the NanoSIMS analyses, the samples were coated with 10 nm of Au prior to analysis to minimize sample charging. Samples were presputtered with about 2 × 1016 ions cm–2 after which, images sized 48 μm × 48 μm containing 256 pixel × 256 pixel were acquired with a 16 keV ∼1.5 pA Cs+ primary ion beam (diameter ∼115 nm) using magnetic peak switching, where in the first two planes, 12C14N–, 31P–, and 195Pt– were collected (13.5 ms/pixel). After the first two planes, the detector collecting 12C14N was moved to collect 12C15N– , and in the second analysis, two consecutive planes collecting 12C15N–, 31P–, and 195Pt– were acquired. Image data were processed using OpenMIMS (National Resource for Imaging Mass Spectrometry, Harvard University, Cambridge), which is an ImageJ plugin (U.S. National Institutes of Health, Bethesda, Maryland) in which pixel by pixel deadtime and QSA corrections were applied. Data from ROIs were further processed in a spreadsheet. 15N data were normalized with respect to air by analyzing a yeast standard daily (see Supporting Information). There appeared to be a minor interference associated with 195Pt as evidenced by a small background; however, Pt treated cells had hotspots that were significantly higher in Pt than background.
SIM and NanoSIMS Correlation
NanoSIMS images were transformed using Matlab software to correlate with SIM images acquired on identical areas. Thus, all three signals observed on SIM images can be correlated with the eight acquired NanoSIMS images representing different ion maps.
NanoSIMS Data Statistical Analysis
The first step of NanoSIMS data processing involved correcting the counts obtained for 15N and 14N with the yeast standard, which was measured each day before any of the samples. The yeast standard has a known δ15N value of ∼0.34 ‰ relative to air. That daily correction factor was applied to all the 15N enrichment data obtained for the tissue samples. Those were called “corrected 15N enrichment values”. ROIs counts (as a ratio counts/area) were averaged for the different groups, leaving out of the average of outliers (values higher or lower than IQR*1.5). Additional statistical analyses were performed with Graphpad software and these values were used to compare bars on graphs in Figure 5 in the main text. The results of unpaired t-tests are shown in Table S3.
Acknowledgments
M.T.P. thanks the UCSD CRIN for a postdoctoral fellowship and the mentorship of Dr. A. Kummel within that program. C.E.C. thanks the UCSD CRIN program, as well as the Inamori Foundation, for graduate student fellowships. We thank ARO for a DURIP grant (W911NF-13-1-0321) to purchase a PerkinElmer plate reader used in these studies. We acknowledge use of the UCSD Cryo-EM Facility, which is supported by NIH Grant R37 GM-03350 to Dr. T. S. Baker and a gift from the Agouron Institute to UCSD. M.T.P. thanks the UCSD Neuroscience Microscopy Shared Facility which is supported via the P30 NS047101 grant. M.T.P. also thanks Dr. P. R. Castillo and C. MacIsaac from Scripps Institution of Oceanography, UCSD for their assistance with the ICP-OES experiments. M.T.P thanks the UCSD Chemistry and Biochemistry Molecular MS facility for the MS analysis. M.T.P. thanks Dr. Crystal Zuñiga, Valeria Reyes, Dr. Maria Bagur and Dr. Christopher Forman for their assistance with the preparation of this manuscript. Part of this research was performed using EMSL, a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and located at PNNL.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00444.
Details on general methods, polymer synthesis, nanoparticle preparation and characterization, in vitro nanoparticle degradation via MMP-12, intratumoral efficacy studies, fluorescent IHC staining of tissue sections, fluorescence microscopy imaging and SIM Imaging, NanoSIMS imaging, SIM and NanoSIMS correlated images and statistical analysis of NanoSIMS data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chien M.-P.; Thompson M. P.; Barback C. V.; Ku T.-H.; Hall D. J.; Gianneschi N. C. Enzyme-Directed Assembly of a Nanoparticle Probe in Tumor Tissue. Adv. Mater. 2013, 25, 3599–3604. 10.1002/adma.201300823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M. M.; Carlini A. S.; Chien M. P.; Sonnenberg S.; Luo C. L.; Braden R. L.; Osborn K. G.; Li Y. W.; Gianneschi N. C.; Christman K. L. Enzyme-Responsive Nanoparticles for Targeted Accumulation and Prolonged Retention in Heart Tissue after Myocardial Infarction. Adv. Mater. 2015, 27, 5547–5552. 10.1002/adma.201502003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerleider J. L.; Kammeyer J. K.; Braden R. L.; Christman K. L.; Gianneschi N. C. Enzyme-Targeted Nanoparticles for Delivery to Ischemic Skeletal Muscle. Polym. Chem. 2017, 8, 5212–5219. 10.1039/C7PY00568G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callmann C. E.; Barback C. V.; Thompson M. P.; Hall D. J.; Mattrey R. F.; Gianneschi N. C. Therapeutic Enzyme-Responsive Nanoparticles for Targeted Delivery and Accumulation in Tumors. Adv. Mater. 2015, 27, 4611–4615. 10.1002/adma.201501803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalafatovic D.; Nobis M.; Son J. Y.; Anderson K. I.; Ulijn R. V. MMP-9 triggered Self-Assembly of Doxorubicin Nanofiber Depots halts Tumor Growth. Biomaterials 2016, 98, 192–202. 10.1016/j.biomaterials.2016.04.039. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Wang T.; Perche F.; Taigind A.; Torchilin V. P. Enhanced Anticancer Activity of Nanopreparation containing an MMP2-Sensitive PEG-Drug Conjugate and Cell-Penetrating Moiety. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17047–17052. 10.1073/pnas.1304987110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivask A.; Mitchell A. J.; Malysheva A.; Voelcker N. H.; Lombi E. Methodologies and Approaches for the Analysis of Cell-Nanoparticle Interactions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10, e1486 10.1002/wnan.1486. [DOI] [PubMed] [Google Scholar]
- Chen Z. Y.; Wang Y. X.; Lin Y.; Zhang J. S.; Yang F.; Zhou Q. L.; Liao Y. Y. Advance of Molecular Imaging Technology and Targeted Imaging Agent in Imaging and Therapy. BioMed Res. Int. 2014, 2014, 1–12. 10.1155/2014/819324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James M. L.; Gambhir S. S. A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications. Physiol. Rev. 2012, 92, 897–965. 10.1152/physrev.00049.2010. [DOI] [PubMed] [Google Scholar]
- Ostrowski A.; Nordmeyer D.; Boreham A.; Holzhausen C.; Mundhenk L.; Graf C.; Meinke M. C.; Vogt A.; Hadam S.; Lademann J.; Ruhl E.; Alexiev U.; Gruber A. D. Overview about the Localization of Nanoparticles in Tissue and Cellular Context by Different Imaging Techniques. Beilstein J. Nanotechnol. 2015, 6, 263–280. 10.3762/bjnano.6.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R. F. S.; Theiner S.; Meibom A.; Koellensperger G.; Keppler B. K.; Dyson P. J. Application of Imaging Mass Spectrometry Approaches to Facilitate Metal-Based Anticancer Drug Research. Metallomics 2017, 9, 365–381. 10.1039/C6MT00231E. [DOI] [PubMed] [Google Scholar]
- Battistella C.; Klok H. A. Controlling and Monitoring Intracellular Delivery of Anticancer Polymer Nanomedicines. Macromol. Biosci. 2017, 17, 1700022. 10.1002/mabi.201700022. [DOI] [PubMed] [Google Scholar]
- Le Trequesser Q.; Seznec H.; Delville M. H. Functionalized Nanomaterials: their Use as Contrast Agents in Bioimaging: Mono- and Multimodal Approaches. Nanotechnol. Rev. 2013, 2, 125–169. 10.1515/ntrev-2012-0080. [DOI] [Google Scholar]
- Saka S. K.; Vogts A.; Kröhnert K.; Hillion F.; Rizzoli S. O.; Wessels J. T. Correlated Optical and Isotopic Nanoscopy. Nat. Commun. 2014, 5, 1–8. 10.1038/ncomms4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetto M. T.; Anderton C. R.; Hu D.; Szymanski C. J.; Zhu Z.; Patterson J. P.; Kammeyer J. K.; Nilewski L. G.; Rush A. M.; Bell N. C.; Evans J. E.; Orr G.; Howell S. B.; Gianneschi N. C. Cellular Delivery of Nanoparticles Revealed with Combined Optical and Isotopic Nanoscopy. ACS Nano 2016, 10, 4046–4054. 10.1021/acsnano.5b06477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez J.; Renslow R.; Cliff J. B. 3rd; Anderton C. R. NanoSIMS for Biological Applications: Current Practices and Analyses. Biointerphases 2018, 13, 03B301 10.1116/1.4993628. [DOI] [PubMed] [Google Scholar]
- Legin A. A.; Schintlmeister A.; Jakupec M. A.; Galanski M.; Lichtscheidl I.; Wagner M.; Keppler B. K. NanoSIMS Combined with Fluorescence Microscopy as a Tool for Subcellular Imaging of Isotopically Labeled Platinum-Based Anticancer Drugs. Chem. Sci. 2014, 5, 3135–3143. 10.1039/c3sc53426j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K.; Plaks V.; Werb Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E. S.; Jiang T.; Aguilera T. A.; Nguyen Q. T.; Ellies L. G.; Scadeng M.; Tsien R. Y. Activatable Cell Penetrating Peptides Linked to Nanoparticles as Dual Probes for in Vivo Fluorescence and MR Imaging of Proteases. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4311–4316. 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer C.; Bredow S.; Mahmood U.; Weissleder R.; Tung C. H. Optical Imaging of Matrix Metalloproteinase-2 Activity in Tumors: Feasibility Study in a Mouse Model. Radiology 2001, 221, 523–529. 10.1148/radiol.2212010368. [DOI] [PubMed] [Google Scholar]
- Chien M. P.; Carlini A. S.; Hu D. H.; Barback C. V.; Rush A. M.; Hall D. J.; Orr G.; Gianneschi N. C. Enzyme-Directed Assembly of Nanoparticles in Tumors Monitored by in Vivo Whole Animal Imaging and ex Vivo Super-Resolution Fluorescence Imaging. J. Am. Chem. Soc. 2013, 135, 18710–18713. 10.1021/ja408182p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Xu H.; Zhang X. Tuning the Amphiphilicity of Building Blocks: Controlled Self-Assembly and Disassembly for Functional Supramolecular Materials. Adv. Mater. 2009, 21, 2849–2864. 10.1002/adma.200803276. [DOI] [Google Scholar]
- Chien M. P.; Thompson M. P.; Lin E. C.; Gianneschi N. C. Fluorogenic Enzyme-Responsive Micellar Nanoparticles. Chem. Sci. 2012, 3, 2690–2694. 10.1039/c2sc20165h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel K. B.; Callmann C. E.; Gianneschi N. C.; Cohen S. M. Dual-Responsive Nanoparticles release Cargo upon Exposure to Matrix Metalloproteinase and Reactive Oxygen Species. Chem. Commun. 2016, 52, 2126–2128. 10.1039/C5CC09164K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y.; Olsen B. D.; Kornfield J. A.; Grubbs R. H. Efficient Synthesis of Narrowly Dispersed Brush Copolymers and Study of Their Assemblies: The Importance of Side Chain Arrangement. J. Am. Chem. Soc. 2009, 131, 18525–18532. 10.1021/ja908379q. [DOI] [PubMed] [Google Scholar]
- Proetto M. T.; Rush A. M.; Chien M.-P.; Abellan Baeza P.; Patterson J. P.; Thompson M. P.; Olson N. H.; Moore C. E.; Rheingold A. L.; Andolina C.; Millstone J.; Howell S. B.; Browning N. D.; Evans J. E.; Gianneschi N. C. Dynamics of Soft Nanomaterials Captured by Transmission Electron Microscopy in Liquid Water. J. Am. Chem. Soc. 2014, 136, 1162–1165. 10.1021/ja408513m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshayes S.; Cabral H.; Ishii T.; Miura Y.; Kobayashi S.; Yamashita T.; Matsumoto A.; Miyahara Y.; Nishiyama N.; Kataoka K. Phenylboronic Acid-Installed Polymeric Micelles for Targeting Sialylated Epitopes in Solid Tumors. J. Am. Chem. Soc. 2013, 135, 15501–15507. 10.1021/ja406406h. [DOI] [PubMed] [Google Scholar]
- Miura Y.; Takenaka T.; Toh K.; Wu S. R.; Nishihara H.; Kano M. R.; Ino Y.; Nomoto T.; Matsumoto Y.; Koyama H.; Cabral H.; Nishiyama N.; Kataoka K. Cyclic RGD-Linked Polymeric Micelles for Targeted Delivery of Platinum Anticancer Drugs to Glioblastoma through the Blood-Brain Tumor Barrier. ACS Nano 2013, 7, 8583–8592. 10.1021/nn402662d. [DOI] [PubMed] [Google Scholar]
- Tummala S.; Gowthamarajan K.; Kumar M. N. S.; Wadhwani A. Oxaliplatin Immuno Hybrid Nanoparticles for Active Targeting: an Approach for Enhanced Apoptotic Activity and Drug Delivery to Colorectal Tumors. Drug Delivery 2016, 23, 1773–1787. 10.3109/10717544.2015.1084400. [DOI] [PubMed] [Google Scholar]
- Yoon S. O.; Kim M. M.; Chung A. S. Inhibitory Effect of Selenite on Invasion of HT1080 Tumor Cells. J. Biol. Chem. 2001, 276, 20085–20092. 10.1074/jbc.M101143200. [DOI] [PubMed] [Google Scholar]
- Long B. R.; Robinson D. C.; Zhong H. N. Subdiffractive Microscopy: Techniques, Applications, and Challenges. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014, 6, 151–168. 10.1002/wsbm.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhorst M. F.; Schaffer J.; Goetze B. Structure brings Clarity: Structured Illumination Microscopy in Cell Biology. Biotechnol. J. 2009, 4, 858–65. 10.1002/biot.200900025. [DOI] [PubMed] [Google Scholar]
- Gibson D. The Mechanism of Action of Platinum Anticancer Agents-What do we Really know About it?. Dalton Trans. 2009, 0, 10681–10689. 10.1039/b918871c. [DOI] [PubMed] [Google Scholar]
- Wheate N. J.; Walker S.; Craig G. E.; Oun R. The Status of Platinum Anticancer Drugs in the Clinic and in Clinical Trials. Dalton Trans. 2010, 39, 8113–8127. 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- Wedlock L. E.; Kilburn M. R.; Liu R.; Shaw J. A.; Berners-Price S. J.; Farrell N. P. NanoSIMS Multi-Element Imaging Reveals Internalisation and Nucleolar Targeting for a Highly-Charged Polynuclear Platinum Compound. Chem. Commun. 2013, 49, 6944–6946. 10.1039/c3cc42098a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.