

Abstract
While most disease-modifying drugs (DMDs) regulate multiple sclerosis (MS) by suppressing inflammation, they can potentially suppress antiviral immunity, causing progressive multifocal leukoencephalopathy (PML). The DMD glatiramer acetate (GA) has been used for MS patients who are at high risk of PML. We investigated whether GA is safe for use in viral infections by using a model of MS induced by infection with Theiler’s murine encephalomyelitis virus (TMEV). Treatment of TMEV-infected mice with GA neither enhanced viral loads nor suppressed antiviral immune responses, while it resulted in an increase in the Foxp3/Il17a ratio and IL-4/IL-10 production. This is the first study to suggest that GA could be safe for MS patients with a proven viral infection.
Introduction
Viral infections are considered to trigger multiple sclerosis (MS), which is an inflammatory demyelinating disease of the central nervous system (CNS). Clinically, several viruses, including human herpesvirus 6, have been isolated from MS patients, and higher antiviral immune responses have been reported in MS patients than in healthy controls [1]. Experimentally, viral infections have been shown to induce demyelination due to direct lytic viral infection of the CNS (viral pathology) and/or recruitment of inflammatory cells into the CNS (immunopathology) [2, 3].
Theiler’s murine encephalomyelitis virus (TMEV) is a non-enveloped, positive-sense, single-stranded RNA virus that belongs to the family Picornaviridae. TMEV is widely used as a viral model of MS, since TMEV infection induces chronic inflammatory demyelination in the CNS that resembles MS neuropathologically [4]. In the TMEV model, during the acute phase, around 1 week postinfection (p.i.), TMEV predominantly infects neurons in the brain and induces acute polioencephalomyelitis [5]. During the sub-clinical phase, 2–3 weeks p.i., although TMEV is largely cleared from the brain by antiviral T-cell and antibody responses, TMEV is axonally transported to the spinal cord [6]. Around 4 weeks p.i. (early chronic phase), mice begin to develop inflammatory demyelination in the spinal cord, where persistent viral infection in macrophages and glial cells and antiviral immunity contribute to pathogenesis [7, 8].
Immunomodulatory drugs, including natalizumab and fingolimod, have been used as disease-modifying drugs (DMDs) for MS patients, since DMDs reduce their disease activity and progression [9]. While DMDs are beneficial in MS by regulating immunopathology, they sometimes cause CNS viral reactivation syndrome (e.g., progressive multifocal leukoencephalopathy [PML]) by suppressing antiviral immunity [10]. The DMD glatiramer acetate (GA) has never been linked to PML, but it has anti-inflammatory effects, resulting in enhanced interleukin (IL)-4 and IL-10 production and increased induction of regulatory T cells (Tregs) expressing the forkhead box P3 (Foxp3) transcription factor.
In a study of TMEV-induced demyelinating disease (TMEV-IDD), Ure et al. [11] tested the effects of GA, focusing mainly on remyelination during the late chronic phase, 27–52 weeks p.i., since a lack of remyelination has been proposed to explain a lack of recovery from TMEV-IDD during the late chronic phase, but not during the early chronic phase. The authors demonstrated that passive transfer of anti-GA antibodies to infected mice significantly enhanced remyelination without alteration of demyelination, although GA treatment itself did not affect remyelination or demyelination despite the induction of anti-GA antibodies. Thus, large gaps in our knowledge exist as to whether and how GA could be beneficial in the treatment of virus-induced demyelinating diseases and could suppress pro-inflammatory responses without suppressing antiviral immune responses.
We first determined whether GA treatment could affect TMEV-IDD during the early chronic phase. SJL/J mice were infected with TMEV on day 0 and injected daily with a low dose (0.15 mg/mouse) of GA on days 0 to 27 (Whole group), days 0 to 6 (Early group), or days 21 to 27 (Late group) [12]. We monitored their clinical signs and body weight changes for 5 weeks (Supplementary Methods). During the acute phase, the infected mice, irrespective of GA treatment, had similar levels of impaired righting reflexes, indicating acute polioencephalomyelitis (Fig. 1A and Supplementary Fig. 1A–C). Around 3–4 weeks p.i., most of the mice in all groups recovered completely. During the chronic phase, the mice began to develop impaired righting reflexes, indicating the onset of TMEV-IDD. Mice from all GA-treated groups had significantly lower clinical scores than control mice without GA treatment (TMEV-alone group) (P < 0.05, Kruskal-Wallis test). We repeated the experiment and confirmed the effects of low-dose GA treatment on TMEV-IDD. In the second experiment, we again observed significant differences among the four groups in their clinical scores during the early chronic phase (e.g., mean clinical score ± standard error of the mean [SEM] on day 34: TMEV alone, 1.5 ± 0; Whole, 1.1 ± 0.3; Early, 0.8 ± 0.2; Late, 1.1 ± 0.2 [P < 0.05, Kruskal-Wallis test]), while there were no statistical differences in body weight changes among the groups (Supplementary Fig. 2).
Fig. 1.
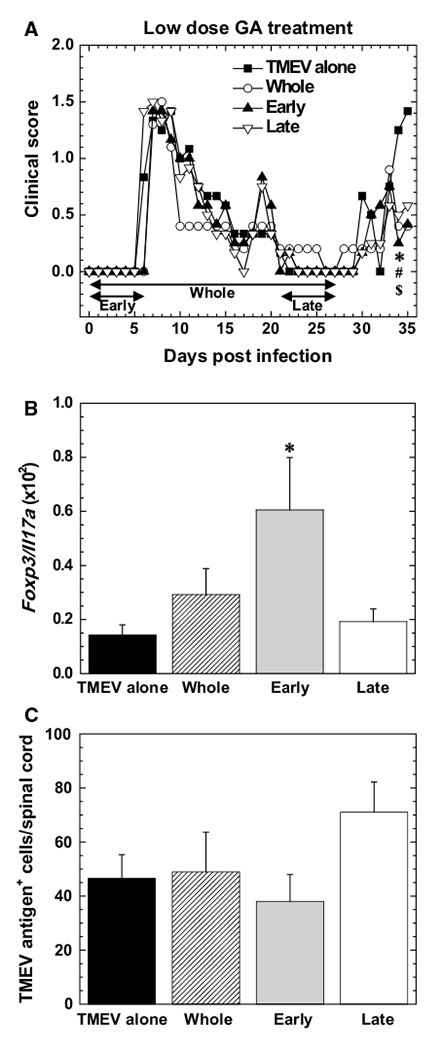
Safety of glatiramer acetate (GA) treatment for Theiler’s murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD). A Clinical scores of TMEV-IDD. Mice were infected with TMEV on day 0 and treated daily with a low dose of GA for 4 weeks (days 0 to 27, Whole, unfilled circles), during the acute phase (days 0 to 6, Early, filled triangles), or during the chronic phase (days 21 to 27, Late, open inversed triangle) of TMEV infection. Control mice had TMEV infection without GA treatment (TMEV alone, filled squares). Clinical scores were evaluated by impaired righting reflex scores (Supplementary Methods). P < 0.05, TMEV alone versus *Whole, #Early, and $Late, Kruskal-Wallis test. Results are representative of two independent experiments and expressed as mean clinical scores. Each experiment included five to six mice per group. B Ratios of Foxp3 to Il17a levels in the spinal cord 5 to 6 weeks postinfection (p.i.). Levels of Foxp3 and Il17a were semi-quantified by real-time PCR. *, P < 0.05, ANOVA. Results are the mean ratios + standard error of the mean (SEM). Each group was composed of three to four mice. C Numbers of viral antigen-positive cells visualized by immunohistochemistry in the spinal cord at 5 to 6 weeks p.i. Results are the averages of two independent experiments expressed as the mean + SEM. Each experiment included four to six mice per group
Interestingly, treatment with a high dose of GA (2 mg/mouse) did not alter clinical signs significantly (Supplementary Fig. 1D). This suggests that the beneficial effects of GA treatment on TMEV-IDD depend on the dosage of the treatment. Although the precise mechanism is unclear, this could be due to the high-dose treatment being outside of the therapeutic window (note: the two standard ranges of doses that have been tested in mice, 0.15 mg and 2 mg per mouse, are much higher than the currently approved 20-mg daily dose used in humans [e.g., the 0.15 mg/20 g mouse dosage corresponds to 450 mg/60 kg in humans]) [13, 14]. Alternatively, some unknown adverse effects could counter the beneficial effects of GA. Thus, in subsequent studies, we treated mice with the low dose of GA to further evaluate the effects of GA treatment on TMEV-IDD.
In an autoimmune model of MS, experimental autoimmune encephalomyelitis (EAE), GA treatment has been shown to increase anti-inflammatory Foxp3+ Tregs and decrease pro-inflammatory IL-17-producing T helper (Th) 17 cells [15], resulting in amelioration of EAE. To determine whether GA treatment could also alter the ratio of Tregs to Th17 cells in TMEV-IDD, we performed a semiquantitative analysis of Foxp3 and Il17a levels in the CNS by real-time PCR (Supplementary Methods). We found that the ratios of Foxp3 to Il17a levels were higher in all GA-treated groups, particularly the Early group (P < 0.05, ANOVA), compared with the control group (Fig. 1B), while there was no statistical difference in the expression level of either mRNA among the groups (Supplementary Fig. 3). This suggests that GA treatment may regulate the clinical signs of TMEV-IDD by shifting T-cell responses from the pro-inflammatory to the anti-inflammatory pathway.
On the other hand, we demonstrated previously that Tregs can be a double-edged sword in TMEV infection [16], enhancing CNS viral loads while decreasing CNS inflammation due to the anti-inflammatory responses. To determine whether GA treatment could affect viral loads after TMEV infection, we counted viral-antigen-positive cells in the CNS by immunohistochemistry with hyperimmune serum against TMEV (Supplementary Methods). As reported previously [17], control mice from the TMEV-alone group had viral-antigen-positive cells in the white matter of the spinal cord, particularly in the ventral and lateral funiculi, during the early chronic phase (Supplementary Fig. 4A). The number and location of viral-antigen-positive cells in all GA-treated groups were similar to those in the control group (Fig. 1C and Supplementary Fig. 4B). We also compared the levels of meningitis, parenchymal inflammation (perivascular cuffing), and demyelination in the CNS among the four groups and found no significant differences in CNS pathology (Supplementary Fig. 5). The discrepancies between CNS inflammation and clinical signs could be explained by increased migration of anti-inflammatory cells, including Tregs, which is consistent with the higher Foxp3 expression in the CNS in all GA-treated groups than in the control group.
We next compared the levels of TMEV-specific lymphoproliferation among the groups by [3H]thymidine incorporation assays (Supplementary Methods) and found that all four groups underwent substantial lymphoproliferative responses to TMEV without statistical differences (Fig. 2). We also examined serum anti-TMEV antibody titers by enzyme-linked immunosorbent assays (ELISAs) for the IgG1 and IgG2c isotypes (Supplementary Methods) that are produced by SJL/J mice (SJL/J mice lack the IgG2a isotype). Mice from all four groups had high anti-TMEV IgG1 and IgG2c titers without statistical differences (Fig. 2). Thus, GA treatment neither increased viral loads nor suppressed antiviral immunity.
Fig. 2.

GA-treatment-induced immune response to GA without alteration of the immune responses to TMEV. A, B Cellular immune responses to TMEV (A) and GA (B) 5 to 6 weeks p.i. Splenic mononuclear cells (MNCs) were stimulated with irradiated TMEV- or mock-infected antigen-presenting cells (APCs) or GA in the presence or absence of anti-CD4 or anti-CD8 antibody. Lymphoproliferative responses were quantified by [3H]thymidine incorporation assays and expressed as Δcpm (experimental cpm in TMEV-APCs or GA stimulation–control cpm in mock-APCs or no stimulation). Results are the average of two independent experiments expressed as the mean Δcpm + SEM of two to three spleen pools. Each spleen pool was composed of two to three spleens from four to six mice per group. All cultures were performed in triplicate. N.D.; not detectable. C–F Humoral immune responses to TMEV (C, E) and GA (D, F) 5 to 6 weeks p.i. TMEV alone, filled squares; Whole, unfilled circles; Early, filled triangles; Late, unfilled inverse triangles. Serum IgG1 and IgG2c titers for TMEV and GA were quantified by ELISA. *, P < 0.05; **, P < 0.01, ANOVA. Results are the average of two independent experiments expressed as the mean absorbance at 492 nm ± SEM. Each experiment included five to eight mice per group
Clinically, GA treatment has been shown to induce cellular and humoral immune responses to GA in the periphery [18]. We quantified lymphoproliferative responses to GA by [3H]thymidine incorporation assays (Supplementary Methods) and detected high levels of GA-specific lymphoproliferation in all GA-treated groups, but not in the TMEV-alone group (Fig. 2). The lymphoproliferative responses to GA in all GA-treated groups were inhibited in the presence of anti-CD4 antibody, but not in the presence of anti-CD8 antibody. This was consistent with previous findings that GA binds efficiently to major histocompatibility (MHC) class II molecules, inducing GA-specific CD4+ T cells [14]. We also determined serum anti-GA antibody titers by ELISAs (Supplementary Methods) and found that all mice from the three GA-treated groups had significantly higher titers of anti-GA IgG1 than control mice (P < 0.01, ANOVA, Fig. 2). Interestingly, although all mice from the Whole group had substantial titers of anti-GA IgG2c (P < 0.01, ANOVA), only 6 of 14 mice (43%) in the Early group and 9 of 14 mice (64%) in the Late group had low anti-GA IgG2c titers. Thus, GA treatment induced the anti-GA IgG1 isotype (Th2 associated) more effectively than anti-GA IgG2c isotype (Th1 associated). Similarly, in GA-treated MS patients, anti-GA IgG1 titers were two- to threefold higher than anti-GA IgG2 titers [18].
Lastly, we determined whether GA treatment could alter the cytokine profile following TMEV infection. Splenic mononuclear cells (MNCs) were stimulated in vitro with a mitogen (concanavalin A [ConA]) or GA. The amounts of anti-inflammatory IL-10/IL-4 and pro-inflammatory interferon (IFN)-γ/IL-17 in the culture supernatants were quantified by ELISAs (Supplementary Methods). In ConA stimulation, the amounts of IL-10 were significantly higher in all GA-treated groups (Whole, P < 0.01; Early and Late, P < 0.05, ANOVA) than in the TMEV-alone group (Fig. 3). The amounts of IL-4 were also higher in the Whole and Late groups (Late, P < 0.05, ANOVA), but not in the Early group, when compared with the TMEV-alone group. All GA-treated groups had lower amounts of IFN-γ than the TMEV-alone group, without statistical differences. The amounts of IL-17 were lower in the Whole and Early groups and higher in the Late group than in the TMEV-alone group, without statistical differences. In GA stimulation, while the GA-treated groups showed substantial IL-10, IL-4, and IFN-γ production, but not IL-17 production, these four cytokines were not detectable in the TMEV-alone group. These results suggest that GA-specific IL-10/IL-4 production can affect the clinical signs of TMEV-IDD and serum anti-GA IgG1 titers.
Fig. 3.

Enhancement of anti-inflammatory cytokine production by GA treatment. A–D Cytokine profiles of splenic MNCs 5 to 6 weeks p.i. Splenic MNCs were stimulated with concanavalin A (ConA) or GA. Interleukin (IL)-10, IL-4, interferon (IFN)-γ, and IL-17 in the culture supernatant were quantified by ELISA. *, P < 0.05; **, P < 0.01; ANOVA. Results are the average expressed as the mean concentration + SEM of two pools of spleens from two to three mice. Each spleen pool was composed of two to three spleens from four to six mice per group. N.D.; not detectable
In summary, MS has been suggested to be an immune-mediated disease of the CNS associated with environmental factors, particularly viral infections [1]. Although the current FDA-approved DMDs have been effective in MS [9], most DMDs can potentially suppress not only antimyelin but also antiviral immune responses; suppression of the latter has been reported to cause latent viral reactivation. For example, natalizumab treatment triggers PML, particularly in patients who are positive for antibody against JC virus [10]. Among DMDs, GA is one of the most widely prescribed first-line drugs for MS [19]. In this study using the TMEV model, in which viral persistence and antiviral immunity contribute to pathogenesis, we demonstrated that GA treatment neither increased viral loads nor decreased antiviral immunity. Furthermore, GA treatment tended to be beneficial in the TMEV model by inducing anti-inflammatory immune responses. Thus, our findings suggest that GA treatment could be safe and effective for MS patients who are at risk of developing PML. However, we also found that Foxp3 levels in the CNS tended to be higher in all GA-treated groups. Since Tregs play a pathogenic role in some viral infections by affecting antiviral immunity [16, 20], GA treatment might carry the risk of increasing viral replication by inducing Tregs. Thus, Treg levels, together with viral loads and antiviral immunity, will be worth monitoring in a therapeutic application of GA in patients with proven viral infections.
Supplementary Material
Acknowledgements
We thank Dr. Viromi Fernando and Dr. Eiichiro Kawai for their helpful discussions, and Ms. Sadie Faith Pearson and Ms. Elaine Cliburn Stewart for their excellent technical assistance.
Funding This work was funded by the Teva Neuroscience Investigator-Initiated Program (TNSCOP0049), IDeA from the National Institute of General Medical Science of the NIH (5P30GM110703), KAKENHI from the Japan Society for the Promotion of Science (JP17K15628 and JP16H07356), Science Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan, and Kindai University Research Enhancement Grant.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00705-018-3729-6) contains supplementary material, which is available to authorized users.
Conflict of interest The decision to publish this article was solely the responsibility of the authors. All statements, opinions, and content presented in this article are those of the authors and do not represent the opinions of Teva. Teva provided a medical accuracy review of the article.
Compliance with ethical standards
Ethical approval All experimental procedures involving the use of animals were conducted according to the criteria outlined by the NIH and were approved by the IACUC of Louisiana State University.
References
- 1.Johnson RT (1998) Chronic inflammatory and demyelinating diseases In: Johnson RT (ed) Viral infections of the nervous system, 2nd edn. Lippincott-Raven, Philadelphia, pp 227–263 [Google Scholar]
- 2.Wu GF, Dandekar AA, Pewe L, Perlman S (2000) CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J Immunol 165:2278–2286 [DOI] [PubMed] [Google Scholar]
- 3.Tsunoda I, Kurtz CIB, Fujinami RS (1997) Apoptosis in acute and chronic central nervous system disease induced by Theiler’s murine encephalomyelitis virus. Virology 228:388–393 [DOI] [PubMed] [Google Scholar]
- 4.Sato F, Omura S, Kawai E, Martinez NE, Acharya MM, Reddy PC, Chaitanya GV, Alexander JS, Tsunoda I (2014) Distinct kinetics of viral replication, T cell infiltration, and fibrosis in three phases of myocarditis following Theiler’s virus infection. Cell Immunol 292:85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asakura K, Murayama H, Himeda T, Ohara Y (2002) Epitope-tagged L* protein of Theiler’s murine encephalomyelitis virus is expressed in the central nervous system in the acute phase of infection. J Virol 76:13049–13054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsunoda I, Fujinami RS (2002) Inside-out versus outside-in models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin Immunopathol 24:105–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murray PD, Pavelko KD, Leibowitz J, Lin X, Rodriguez M (1998) CD4+ and CD8+ T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J Virol 72:7320–7329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lipton HL, Twaddle G, Jelachich ML (1995) The predominant virus antigen burden is present in macrophages in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J Virol 69:2525–2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ransohoff RM, Hafler DA, Lucchinetti CF (2015) Multiple sclerosis—a quiet revolution. Nat Rev Neurol 11:134–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plavina T, Subramanyam M, Bloomgren G, Richman S, Pace A, Lee S, Schlain B, Campagnolo D, Belachew S, Ticho B (2014) Anti-JC virus antibody levels in serum or plasma further define risk of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol 76:802–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ure DR, Rodriguez M (2002) Polyreactive antibodies to glatiramer acetate promote myelin repair in murine model of demyelinating disease. FASEB J 16:1260–1262 [DOI] [PubMed] [Google Scholar]
- 12.Sato F, Martinez NE, Shahid M, Rose JW, Carlson NG, Tsunoda I (2013) Resveratrol exacerbates both autoimmune and viral models of multiple sclerosis. Am J Pathol 183:1390–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aharoni R, Sasson E, Blumenfeld-Katzir T, Eilam R, Sela M, Assaf Y, Arnon R (2013) Magnetic resonance imaging characterization of different experimental autoimmune encephalomyelitis models and the therapeutic effect of glatiramer acetate. Exp Neurol 240:130–144 [DOI] [PubMed] [Google Scholar]
- 14.Aharoni R (2014) Immunomodulation neuroprotection and remyelination—the fundamental therapeutic effects of glatiramer acetate: a critical review. J Autoimmun 54:81–92 [DOI] [PubMed] [Google Scholar]
- 15.Aharoni R, Eilam R, Stock A, Vainshtein A, Shezen E, Gal H, Friedman N, Arnon R (2010) Glatiramer acetate reduces Th-17 inflammation and induces regulatory T-cells in the CNS of mice with relapsing-remitting or chronic EAE. J Neuroimmunol 225:100–111 [DOI] [PubMed] [Google Scholar]
- 16.Martinez NE, Karlsson F, Sato F, Kawai E, Omura S, Minagar A, Grisham MB, Tsunoda I (2014) Protective and detrimental roles for regulatory T cells in a viral model for multiple sclerosis. Brain Pathol 24:436–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsunoda I, Tanaka T, Saijoh Y, Fujinami RS (2007) Targeting inflammatory demyelinating lesions to sites of Wallerian degeneration. Am J Pathol 171:1563–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenner T, Arnon R, Sela M, Abramsky O, Meiner Z, Riven-Kreitman R, Tarcik N, Teitelbaum D (2001) Humoral and cellular immune responses to Copolymer 1 in multiple sclerosis patients treated with Copaxone®. J Neuroimmunol 115:152–160 [DOI] [PubMed] [Google Scholar]
- 19.Arnon R, Aharoni R (2009) Neuroprotection and neurogeneration in MS and its animal model EAE effected by glatiramer acetate. J Neural Transm 116:1443–1449 [DOI] [PubMed] [Google Scholar]
- 20.Zelinskyy G, Dietze K, Sparwasser T, Dittmer U (2009) Regulatory T cells suppress antiviral immune responses and increase viral loads during acute infection with a lymphotropic retrovirus. PLoS Pathog 5:e1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.