

Abstract
Stoichiometric proton-coupled electron transfer (PCET) reactions of the metal−organic framework (MOF) MIL-125, Ti8O8(OH)4(bdc)6 (bdc = terephthalate), are described. In the presence of UV light and 2-propanol, MIL-125 was photoreduced to a maximum of 2(e−/H+) per Ti8 node. This stoichiometry was shown by subsequent titration of the photoreduced material with the 2,4,6-tri-tert-butylphenoxyl radical. This reaction occurred by PCET to give the corresponding phenol and the original, oxidized MOF. The high level of charging, and the independence of charging amount with particle size of the MOF samples, shows that the MOF was photocharged throughout the bulk and not only at the surface. NMR studies showed that the product phenol is too large to fit in the pores, so the phenoxyl reaction must have occurred at the surface. Attempts to oxidize photoreduced MIL-125 with pure electron acceptors resulted in multiple products, underscoring the importance of removing e− and H+ together. Our results require that the e− and H+ stored within the MOF architecture must both be mobile to transfer to the surface for reaction. Analogous studies on the soluble cluster Ti8O8(OOCtBu)16 support the notion that reduction occurs at the Ti8 MOF nodes and furthermore that this reduction occurs via e−/H+ (H-atom) equivalents. The soluble cluster also suggests degradation pathways for the MOFs under extended irradiation. The methods described are a facile characterization technique to study redox-active materials and should be broadly applicable to, for example, porous materials like MOFs.
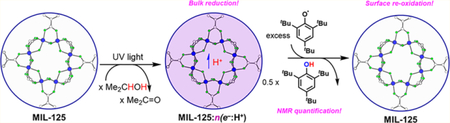
Graphical Abstract:
■INTRODUCTION
Metal−organic frameworks (MOFs) are emerging as promising materials for facilitating redox reactions, including multi-e−/multi-H+ transformations. Recent studies highlight the ability of the organic linkers or metal ions in or on the MOF nodes to undergo 1−e− oxidations,1–4 for the MOFs themselves to serve as conductive materials5–7 or semi-conductors,8,9 and for the MOFs to facilitate redox reactions that are pertinent to fuel cells and energy.10–12 In this latter context, the development of MOF photocatalysts that mimic the reactivity of bulk TiO2 is of timely interest.13–19
Common photocatalytic reactions such as water splitting or CO2 reduction are fundamentally proton-coupled electron transfer (PCET) processes. Therefore, tuning the PCET properties of the catalyst is of the utmost importance.20–22 In this context, several groups have developed MOFs that show PCET behavior resulting in good photo- and electro-chemical activities.22–24 PCET has been suggested to occur on the linker,24 the node,25 and the linker and node combined22,23 but has not been studied in well-known photoactive MOFs. In this work, we provide an in-depth analysis of the PCET behavior in the widely used MIL-125.
The Ti-based MOFs MIL-12516 (Ti8O8(OH)4(bdc)6) and NH2-MIL-12513 (Ti8O8(OH)4(bdc-NH2)6) (bdc-NH2 = 2- aminoterephthalate) have planar Ti8 nodes with oxide and hydroxide ligands, linked with terephthalate (bdc) groups. These materials have been shown to be photoactive; for instance, UV irradiation of a slurry of MIL-125 in benzyl alcohol formed benzaldehyde with a color change to blue.16 It was suggested that this process reduces each MOF node (Ti8 cluster) by a net H-atom (eq 1). Though the stoichiometry of the reduction was not established, the proposed eq 1 led us to hypothesize that the reduced MOF should be able to facilitate well-defined PCET reactions.
| (1) |
NH2-MIL-125 can be reduced by visible light in the presence of a sacrificial reductant, which is highly desirable for photocatalysts.13 In the presence of triethanolamine (TEOA), visible-light irradiation of NH2-MIL-125 results in the catalytic reduction of nitrobenzene to aniline,15 CO2 to formate,13 and protons (aq) to H2.14,15,26
Herein, we describe fundamental studies of MIL-125 that address basic mechanistic questions associated with MOF photocatalysis, such as whether the redox chemistry takes place only at the surface of the MOF particles or occurs throughout the pores. Specifically, we develop a solution NMR assay to determine the reduction stoichiometry, the number of e− + H+ (H-atoms) transferred per node. We show by judicious choice of substrates that MIL-125 can undergo well-defined PCET reactions. The results show that irradiation causes reduction throughout the MOF, but all of the (e−/H+) can be removed through PCET reactions that occur only at the surface. This work thus develops our understanding of the redox capabilities in MOFs, which should advance the promising area of using MOF materials to facilitate multi-e−/multi-H+ transformations.
■ RESULTS AND DISCUSSION
I. PCET Chemistry of the MOF MIL-125.
Irradiation of MIL-125 in neat iPrOH results in a color change from white to dark blue, indicating reduction of the MOF. The EPR spectrum of this material is similar to that reported for photoreduction with benzyl alcohol and exhibits overlapping broad EPR resonances at g = ca. 1.94 (see the SI).16 Quantification of the radical yield has not been attempted due to the nontrivial nature of the signal. The reduced MOF can be isolated and stored as a solid in the glovebox for days with little decay in the absence of oxygen. Resuspension of the reduced material in C6D6 and addition of 2,4,6-tri-tert-butylphenoxyl radical (tBu3ArO•) or the nitroxyl radical 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) resulted in a color change back to white over ∼30 min, indicative of oxidation of the reduced MOF. 1H NMR spectra of tBu3ArO• reactions showed exclusive formation of tBu3ArOH, indicative of a net PCET reaction (H-atom transfer reaction) (eq 2). No tBu3ArO− was observed, which would have suggested pure electron transfer (ET) from the reduced MOF. Thus, the photoreduced MIL-125 is capable of facilitating PCET.
| (2) |
To determine the stoichiometry and extent of reduction, a simple solution 1H NMR method was developed to quantify the e−/H+ equivalents transferred to MIL-125. This method uses 1H NMR quantification of a redox probe relative to an internal standard. After photoreduction, the MOF was isolated as a solid in the glovebox. A known mass was resuspended in C6D6, and a calibrated C6D6 solution of tBu3ArO• and di-p-tolyl-ether (Ar2O) was added. The suspension was stirred for at least 3.5 h to ensure complete oxidation of the MOF. 1H integration of tBu3ArOH vs the Ar2O standard provided quantification of the reducing equivalents transferred (Figure 1).
Figure 1.
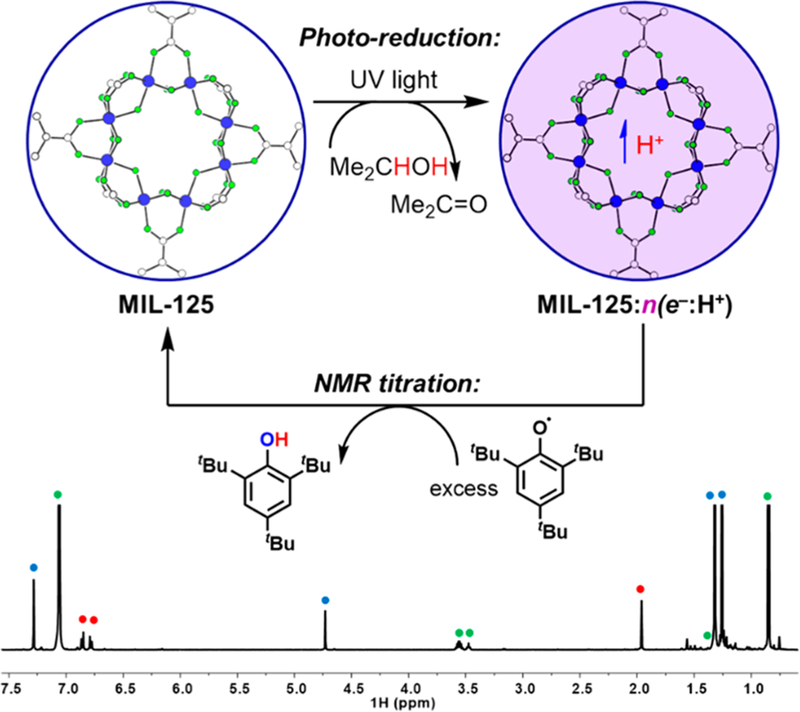
(Top) Scheme showing the photoreduction of MIL-125 and subsequent oxidation. For simplicity, only one Ti8 cluster is shown (the structure of MIL-125 is depicted within the blue circles). For maximum photoreduction, n ≅ 2. (Bottom) 1H NMR spectrum of the subsequent reaction with tBu3ArO• (C6D6). The colored dots identify tBu3ArOH (blue), Ar2O (internal standard, red), and C6D5H, THF, and iPrOH (green).
For accurate NMR integration, it is essential that neither the internal standard nor the redox probe bind to or be taken up in the MOF, as this would diminish the observed concentration. Addition of MIL-125 or photoreduced MIL-125 to NMR samples containing tBu3ArOH and Ar2O does not change the relative integration of the two species, indicating that neither was adsorbed (see SI). Given the similarity in size and charge between tBu3ArO• and tBu3ArOH, it is very likely that the radical oxidant likewise is not taken up into the pores. By contrast, TEMPOH was shown to be taken up by MIL-125, precluding the use of TEMPO• as a redox probe (see the SI).
Several suspensions of MIL-125 in iPrOH were irradiated for various amounts of time and analyzed by titration with tBu3ArO•. A plot of H-atom equivalents transferred per Ti8 node versus photolysis time (Figure 2) shows that increasing the photolysis time increases the extent of reduction. Since EPR spectra indicate that reduction occurs at Ti,16 we report the number of reducing equivalents relative to the Ti8 nodes (the moles of Ti8 nodes was determined from the mass of the MOF and the moles of H-atoms transferred from the NMR method). The increased reduction with time was also evident by eye, as the blue color of the MOF suspension became darker with longer irradiation time. After 5 h, each Ti8 node is reduced by at least 0.4 H-atoms. At long irradiation times, MIL-125 can be reduced by up to 2 H-atom equivalents per Ti8 cluster.
Figure 2.
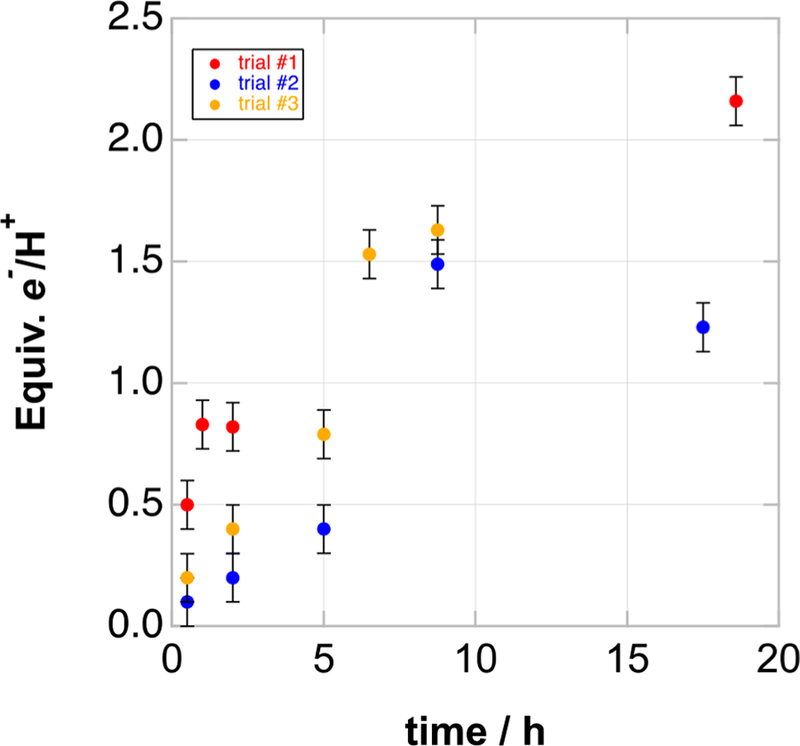
Number of reducing equivalents per Ti8 node vs time for three different photoreductions of MIL-125 using a 200 W Hg/Xe lamp. Errors for data points are estimated to be ≤±0.1 equiv of e−/ H+, based on the error of NMR analysis by integration also taking into account error propagation from the balance accuracy and the estimated maximal sample loss. The sample in trial #2 (blue circles) became black by 17.5 h of photolysis and the color persisted after addition of excess tBu3ArO•, indicating decomposition.
The variability in extent of reduction between the three experiments was attributed to differences in the irradiation conditions, such as distance from lamp, stirring rate, and nature of the suspension, solvent volume, type of quartz vessel, and beam focus (see the SI). In some instances, prolonged irradiation gave fewer H-atom equivalents than anticipated (Figure 2), which is attributed to MOF degradation (vide infra).
To test that the irradiation did not degrade the MOF, two types of experiments were done on various samples. In the first, powder XRD was collected on the photoreduced MOF material. The X-ray diffraction patterns showed no loss in crystallinity of the material upon photoreduction, and no new reflections were observed (see the SI). The second experiment used 1H NMR analysis of material formed after titration with tBu3ArOH. After removal of the C6D6 solvent and resuspension in d7-DMF, the 1H NMR spectrum only showed resonances attributed to solvent, Ar2O, and tBu3ArOH. No terephthalic acid or soluble terephthalate species were detected in the samples in which the titration restored the white color of the oxidized MOF. By contrast, terephthalic acid was present in the solution that corresponds to the blue point at 17.5 h in Figure 2, for which the dark color persisted after oxidation. These observations are indicative of MOF degradation and suggest that extended UV irradiation may degrade MIL-125 under certain conditions.
The measured stoichiometries show that the photo-reductions must be reducing the Ti8 clusters throughout the MOF particles, not only at the surface. The fraction of Ti8 nodes that are at the surface was estimated using the roughly elliptical morphologies of the MIL-125 particles observed by SEM, with semiaxes that range from 300 (±100) to 850 (±50) nm (SI). Assuming perfect ellipsoids, the volumetric fraction of the outermost shell — one layer of Ti8 nodes and linker, 1.9 nm — contains only 1.3% of the Ti8 clusters. If reduction occurred exclusively in this outer shell, then each surface Ti8 cluster would have to be reduced by ∼88 H-atoms, or ∼11 H-atoms for each Ti atom, to achieve the observed 1 e−/H+ per Ti8 cluster throughout the MOF. Studies with different batches of MIL-125 that differ in size and morphology, ranging from spheres with 85 nm radii to truncated octahedra with radii of 2650 nm, all showed facile reduction to 1 e−/H+ per bulk Ti8 cluster. All of these results would require unrealistically high numbers of e−/H+ per surface Ti (see the SI). Thus, both the Ti8 nodes at the surface and within the MOF must have been reduced.
The above studies indicate photoreduced MIL-125 can transfer net H-atom equivalents (e−/H+) to tBu3ArO•. It is also possible to remove some of the electrons with the simple outer-sphere oxidant decamethylferrocenium hexafluorophos-phate ([FeCp*2+]PF6−). Treatment of reduced MIL-125 with [FeCp*2+]PF6− resulted in some lightening of the blue color, and 1H NMR analysis showed the presence of both FeCp*2+/ FeCp*2, indicative of oxidation of the MOF solid. (FeCp*2 does not fit in the pores of MIL-125 or photoreduced MIL-125; see the SI). As FeCp*2+ and FeCp*2 undergo fast electron exchange on the NMR time scale, and a single resonance for the two species is observed. The position of this resonance indicates the mole fractions (χ) of the two species (eq 3).27
| 3 |
Identical samples of reduced MIL-125 transferred fewer reducing equivalents to FeCp*2+ (electron transfer, ET) than to tBu3ArO• (PCET/H-atom transfer). In one comparison, FeCp*2+ removed 0.36 e− versus 0.83 H-atom equivalents per Ti8 node by tBu3ArO •. Further, the resulting solution NMR spectrum of MIL-125 that has been treated with FeCp*2 had several new, unidentifiable resonances, indicative of side reactions. Attempts to use other oxidants, including anilinium radicals, likewise did not give clean electron transfer chemistry. Thus, PCET seems to be more facile and reversible chemistry for the MOF than simple ET. The advantage of chemical reactions that move electrons and protons together is that charge balance is maintained. A principle of solid-state chemistry is that each unit cell must be electrically neutral, and that should apply to MOF materials as well. Removal of just electrons from the MOF leaves an unfavorable positive charge, which likely makes the remaining protons more acidic and leads to decay of the MOF.
The PCET reaction with tBu3ArO• must occur at the surface because tBu3ArOH is too large to enter the MOF pores (see above). Since complete reoxidation is observed, the e−/H+ equivalents must be able to migrate through the MOF to the surface. Thus, the material allows for e−/H+ hopping. Proton28–31 and electron 5–7 conductivity in MOFs have already been described; in this study, the two occur together here to maintain charge balance (see above). The proton migration likely occurs via trapped alcohol in the MOF pores and through mobility of the H+ from the clusters’ bridging hydroxides, a process very similar to that observed in Zr-based MOFs.32,33 The H+ ion accompanying the e− will likely bind to a bridging oxygen on the MIL-125 cluster to form a hydroxide. A similar mechanism has been shown for COK-69, another photoreducible Ti-MOF.25 Similarly, the PCET reduction of bulk TiO2 and other oxides is thought to form hydroxides.34 Broad EPR resonances at rt for reduced MIL-125 have previously been ascribed to e− hopping between different Ti centers.16 The e−/H+ hopping mechanism between different Ti8 nodes is not clear. The kinetics of this process are likely complicated and multiexponential due to the size dispersion of the MIL-125 crystals. Monitoring the time course of the formation of tBu3ArOH from reaction of photoreduced MIL- 125 with tBu3ArO• over time is consistent with multi-exponential kinetics and shows that the reaction is complete within 30−60 min (SI).
II. PCET Chemistry of the Soluble Cluster Ti8O8(OOCtBu)16.
Parallel studies using a known, well-defined molecular cluster, Ti8O8(OOCtBu)16 (Ti8),35 provide additional insight into the nature of the photoreduced state and possible degradation pathways. Cluster Ti8 serves as a model for the nodes of MIL-125, with a similar ring of eight Ti atoms that are connected via bridging oxo ligands in a relatively flat arrangement (Figure 3). The ligand arrangement differs from that in MIL-125 in that the MOF has four fewer bridging carboxylates decorating the outside of the ring and an additional four bridging hydroxo ligands in the interior of the ring. The empirical cluster formula in the MOF is Ti8O8(OH)4(OOCR)12 (Figure 1) vs the molecular Ti8O8(OOCtBu)16. We chose the Ti8 cluster with alkyl carboxylates rather than the related benzoate that is more analogous to MIL-125 because of the limited solubility and uninformative 1H NMR spectrum of Ti8O8(OOCPh)16. Ti8 is readily identified by its two sharp 1H NMR resonances at 1.28 and 1.37 ppm (C6D6) that correspond to tBu groups that are axial and equatorial with respect to the planar Ti8 ring.
Figure 3.
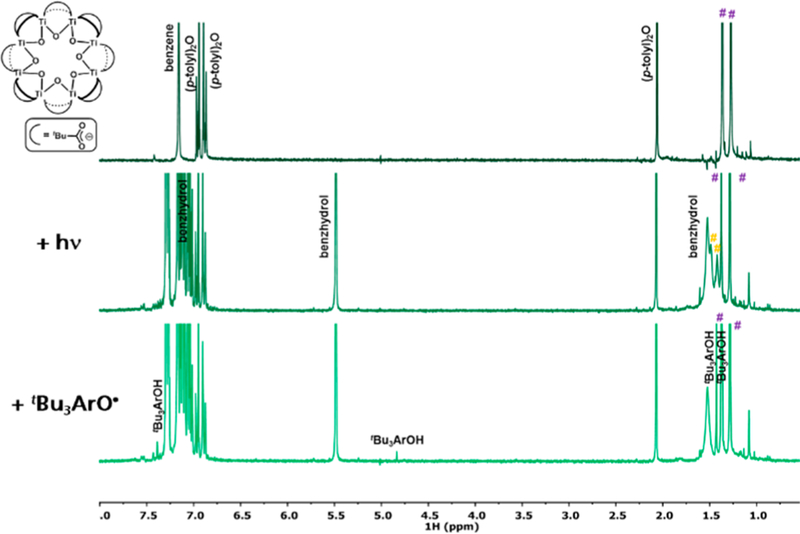
1H NMR spectra (C6D6, 500 MHz): (top) Ti8 with Ar2O standard; (middle): after 15 min of irradiation with excess benzhydrol; (bottom) after addition of tBu3ArO•. Peaks marked with purple # correspond to Ti8, while orange # correspond to the speculative H atom reduced cluster.
Irradiation of a C6D6 solution of Ti8 (0.95 mM), benzhydrol (Ph2CHOH, 45 mM), and Ar2O internal standard for 15 min resulted in darkening of the solution to blue. The 1H NMR spectrum of this reaction revealed the presence of two broad new resonances at 1.41 and 1.48 ppm, along with the presence of unreacted Ti8 (Figure 3). Integration showed 80% of starting Ti8 accounted for, and of this material, ∼43% has been converted to the new species. Treatment of this solution with 1 equiv of tBu3ArO• caused an immediate color change from blue to pale gray. The 1H NMR spectrum of this solution shows the single set of tBu resonances of Ti8 (85% mass balance from initial mass) and resonances ascribed to tBu3ArOH (64% yield by initial mass). These results suggest that the new resonances correspond to a photoreduced cluster. Its reactivity with tBu3ArO• suggests that the new cluster has been reduced by 1e− and 1H+.
The observation of only two tBu resonances for the reduced cluster indicates that this cluster retains the effective 8-fold symmetry of the ring on the NMR time scale. This suggests that the proton is moving around the Ti8 ring rapidly on the NMR time scale. Such rapid proton movement is consistent with the proton movement during oxidation of the reduced MIL-125.
The discrepancies in mass balance in the Ti8 chemistry indicate that partial cluster degradation accompanies photo-reduction and that some of the NMR silent TiIII species can reduce tBu3ArO• (see the SI for UV−vis studies). Increasing the irradiation time gave rise to several additional resonances in the corresponding 1H NMR spectrum, indicative of multiple reduction products. Similar results are noted when pivalic acid is added as a sacrificial reductant. Resonances that correspond to pivalic acid, isobutane, and isobutene implicate the intermediacy of tBu•, which disproportionates to the hydro-carbon products. Thus, the carboxylate ligands of Ti8 can serve as a sacrificial reductant (photoreduction also occurs in the absence of an added alcohol). A similar process has been reported for pivalate on single-crystal TiO2 surfaces.36 The reduced Ti8 cluster reacts much more rapidly with tBu3ArO• than the MOF does (≤ seconds vs hours). This likely reflects the requirement that e− and H+ diffuse to the surface of the MOF to react with tBu3ArO•.
■ CONCLUSIONS
In summary, the proton-coupled electron transfer (PCET) chemistry of MIL-125 has been developed. Irradiation of MIL-125 with iPrOH as a sacrificial reductant gives a maximum of two (e−/H+) per Ti8 cluster node. Subsequent oxidation by tBu3ArO• occurs by PCET to give tBu3ArOH. The extent of reduction of the MOF shows that the reducing equivalents must be stored within the MOF and not only on the outer surface. Since tBu3ArO• is too large to enter the MOF, the PCET oxidation must, however, occur at the surface, with the e− and H+ diffusing from the bulk to the surface. The coupling of e− and H+ in the MOF likely results from the requirement for charge balance, that it is energetically unfavorable for charges to build up inside the MOF. This study thus presents an unusually detailed look at PCET involving a MOF material. Though there are currently very few well-defined examples, we believe that PCET will prove to be a very common type of redox reactivity in MOFs and other porous materials. This work may therefore be relevant to a number of emerging MOF applications, such as CO2 reduction and water splitting. Our results also show the utility of the simple solution NMR methods developed for quantification of e−/H+ equivalents transferred to the MOF. These methods should be widely applicable to other redox-active MOFs and heterogeneous materials, showing that they can store redox equivalents and that reactivity can be limited to the surface.
■ ACKNOWLEDGMENTS
C.T.S. acknowledges financial support from the U.S. National Institutes of Health through a postdoctoral fellowship (1F32GM099316). M.F.D. thanks the Swiss National Science Foundation (SNF) for financial support. J.M.M. acknowledges support from the U.S. National Science Foundation awards (CHE-1151726 and CHE-1609434). S.S. and D.E.D.V. gratefully acknowledge the FWO (Aspirant grant and project funding) and KUL Methusalem for funding.
Footnotes
Notes
The authors declare no competing financial interest.
■ REFERENCES
- (1).Brozek CK; Dinca M Ti3+-, V2+/3+-, Cr2+/3+-, Mn2+-, and Fe2+-Substituted MOF-5 and Redox Reactivity in Cr- and Fe-MOF-5. J. Am. Chem. Soc 2013, 135, 12886−12891. [DOI] [PubMed] [Google Scholar]
- (2).Cozzolino AF; Brozek CK; Palmer RD; Yano J; Li M; Dinca M Ligand Redox Non-Innocence in the Stoichiometric Oxidation of Mn2(2,5-Dioxidoterephthalate) (Mn-MOF-74). J. Am. Chem. Soc 2014, 136, 3334−3337. [DOI] [PubMed] [Google Scholar]
- (3).Smolders S; Lomachenko KA; Bueken B; Struyf A; Bugaev AL; Atzori C; Stock N; Lamberti C; Roeffaers MBJ; De Vos DE Unravelling the Redox-Catalytic Behavior of Ce4+ Metal− Organic Frameworks by X-Ray Absorption Spectroscopy. ChemPhy-sChem 2018, 19, 373−378. [DOI] [PubMed] [Google Scholar]
- (4).Su J; Yuan S; Wang H-Y; Huang L; Ge J-Y; Joseph E; Qin J; Cagin T; Zuo J-L; Zhou H-C Redox-Switchable Breathing Behavior in Tetrathiafulvalene-Based Metal−organic Frameworks. Nat. Commun 2017, 8, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sun L; Campbell MG; Dinca M Electrically Conductive Porous Metal−Organic Frameworks. Angew. Chem., Int. Ed 2016, 55, 3566−3579. [DOI] [PubMed] [Google Scholar]
- (6).Sheberla D; Bachman JC; Elias JS; Sun C-J; Shao-Horn Y; Dinca M Conductive MOF Electrodes for Stable Supercapacitors with High Areal Capacitance. Nat. Mater 2017, 16, 220−224. [DOI] [PubMed] [Google Scholar]
- (7).Feng D; Lei T; Lukatskaya MR; Park J; Huang Z; Lee M; Shaw L; Chen S; Yakovenko AA; Kulkarni A; Xiao J; Fredrickson K; Tok JB; Zou X; Cui Y; Bao Z Robust and Conductive Two-Dimensional Metal−organic Frameworks with Exceptionally High Volumetric and Areal Capacitance. Nat. Energy 2018, 3, 30−36. [Google Scholar]
- (8).Alvaro M; Carbonell E; Ferrer B; Llabreśi Xamena FX; Garcia H Semiconductor Behavior of a Metal-Organic Framework (MOF). Chem. - Eur. J 2007, 13, 5106−5112. [DOI] [PubMed] [Google Scholar]
- (9).Stassen I; Burtch N; Talin A; Falcaro P; Allendorf M; Ameloot R An Updated Roadmap for the Integration of Metal− organic Frameworks with Electronic Devices and Chemical Sensors. Chem. Soc. Rev 2017, 46, 3185−3241. [DOI] [PubMed] [Google Scholar]
- (10).Wu HB; Lou XWD Metal-Organic Frameworks and Their Derived Materials for Electrochemical Energy Storage and Con-version: Promises and Challenges. Sci. Adv 2017, 3, No. eaap9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhou J; Wang B Emerging Crystalline Porous Materials as a Multifunctional Platform for Electrochemical Energy Storage. Chem. Soc. Rev 2017, 46, 6927−6945. [DOI] [PubMed] [Google Scholar]
- (12).Mahmood A; Guo W; Tabassum H; Zou R Metal-Organic Framework-Based Nanomaterials for Electrocatalysis. Adv. Energy Mater 2016, 6, 1600423. [Google Scholar]
- (13).Fu Y; Sun D; Chen Y; Huang R; Ding Z; Fu X; Li Z An Amine-Functionalized Titanium Metal-Organic Framework Photo-catalyst with Visible-Light-Induced Activity for CO2 Reduction. Angew. Chem., Int. Ed 2012, 51, 3364−3367. [DOI] [PubMed] [Google Scholar]
- (14).Horiuchi Y; Toyao T; Saito M; Mochizuki K; Iwata M; Higashimura H; Anpo M; Matsuoka M Visible-Light-Promoted Photocatalytic Hydrogen Production by Using an Amino-Function-alized Ti(IV) Metal−Organic Framework. J. Phys. Chem. C 2012, 116, 20848−20853. [Google Scholar]
- (15).Toyao T; Saito M; Horiuchi Y; Mochizuki K; Iwata M; Higashimura H; Matsuoka M Efficient Hydrogen Production and Photocatalytic Reduction of Nitrobenzene over a Visible-Light-Responsive Metal−organic Framework Photocatalyst. Catal. Sci. Technol 2013, 3, 2092−2097. [Google Scholar]
- (16).Dan-Hardi M; Serre C; Frot T; Rozes L; Maurin G; Sanchez C; Ferey G; Maurín G A New Photoactive Highly Porous Titanium (IV) Dicarboxylate. J. Am. Chem. Soc 2009, 131, 10857− 10859. [DOI] [PubMed] [Google Scholar]
- (17).Nguyen HL; Gandara F; Furukawa H; Doan TLH; Cordova KE; Yaghi OM A Titanium-Organic Framework as an Exemplar of Combining the Chemistry of Metal- and Covalent-Organic Frameworks. J. Am. Chem. Soc 2016, 138, 4330−4333. [DOI] [PubMed] [Google Scholar]
- (18).Yuan S; Liu T-F; Feng D; Tian J; Wang K; Qin J; Zhang Q; Chen Y-P; Bosch M; Zou L; Teat SJ; Dalgarno SJ; Zhou H-C A Single Crystalline Porphyrinic Titanium Metal− organic Framework. Chem. Sci 2015, 6, 3926−3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yuan S; Qin J-S; Xu H-Q; Su J; Rossi D; Chen Y; Zhang L; Lollar C; Wang Q; Jiang H-L; Son DH; Xu H; Huang Z; Zou X; Zhou H-C [Ti8Zr2O12(COO)16] Cluster: An Ideal Inorganic Building Unit for Photoactive Metal−Organic Frameworks. ACS Cent. Sci 2018, 4, 105−111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chang X; Wang T; Gong J CO2 Photo-Reduction: Insights into CO2 Activation and Reaction on Surfaces of Photocatalysts. Energy Environ. Sci 2016, 9, 2177−2196. [Google Scholar]
- (21).Zhou T; Du Y; Borgna A; Hong J; Wang Y; Han J; Zhang W; Xu R Post-Synthesis Modification of a Metal-Organic Framework to Construct a Bifunctional Photocatalyst for Hydrogen Production. Energy Environ. Sci 2013, 6, 3229−3234. [Google Scholar]
- (22).Usov PM; Ahrenholtz SR; Maza WA; Stratakes B; Epley CC; Kessinger MC; Zhu J; Morris AJ Cooperative Electrochemical Water Oxidation by Zr Nodes and Ni-Porphyrin Linkers of a PCN-224 MOF Thin Film. J. Mater. Chem. A 2016, 4, 16818−16823. [Google Scholar]
- (23).Johnson BA; Bhunia A; Fei H; Cohen SM; Ott S Development of a UiO-Type Thin Film Electrocatalysis Platform with Redox-Active Linkers. J. Am. Chem. Soc 2018, 140, 2985−2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Celis-Salazar PJ; Epley CC; Ahrenholtz SR; Maza WA; Usov PM; Morris AJ Proton-Coupled Electron Transport in Anthraquinone-Based Zirconium Metal-Organic Frameworks. Inorg. Chem 2017, 56, 13741−13747. [DOI] [PubMed] [Google Scholar]
- (25).Bueken B; Vermoortele F; Vanpoucke DEP; Reinsch H; Tsou C-C; Valvekens P; De Baerdemaeker T; Ameloot R; Kirschhock CEA; Van Speybroeck V; Mayer JM; De Vos D A Flexible Photoactive Titanium Metal-Organic Framework Based on a [TiIV3(μ3-O)(O)2(COO)6] Cluster. Angew. Chem., Int. Ed 2015, 54, 13912−13917. [DOI] [PubMed] [Google Scholar]
- (26).Meyer K; Bashir S; Llorca J; Idriss H; Ranocchiari M; van Bokhoven JA Photocatalyzed Hydrogen Evolution from Water by a Composite Catalyst of NH2-MIL-125(Ti) and Surface Nickel(II) Species. Chem. - Eur. J 2016, 22, 13894−13899. [DOI] [PubMed] [Google Scholar]
- (27).Sandström J Dynamic NMR Spectroscopy; Academic Press, 1982. [Google Scholar]
- (28).Sadakiyo M; Yamada T; Kitagawa H Proton Conductivity Control by Ion Substitution in a Highly Proton-Conductive Metal− Organic Framework. J. Am. Chem. Soc 2014, 136, 13166−13169. [DOI] [PubMed] [Google Scholar]
- (29).Zhang F-M; Dong L-Z; Qin J-S; Guan W; Liu J; Li SL; Lu M; Lan Y-Q; Su Z-M; Zhou H-C Effect of Imidazole Arrangements on Proton-Conductivity in Metal−Organic Frameworks. J. Am. Chem. Soc 2017, 139, 6183−6189. [DOI] [PubMed] [Google Scholar]
- (30).Lin S; Usov PM; Morris AJ The Role of Redox Hopping in Metal−organic Framework Electrocatalysis. Chem. Commun 2018, 54, 6965−6974. [DOI] [PubMed] [Google Scholar]
- (31).Li A-L; Gao Q; Xu J; Bu X-H Proton-Conductive Metal- Organic Frameworks: Recent Advances and Perspectives. Coord. Chem. Rev 2017, 344, 54−82. [Google Scholar]
- (32).Ling S; Slater B Dynamic Acidity in Defective UiO-66. Chem. Sci 2016, 7, 4706−4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hajek J; Caratelli C; Demuynck R; De Wispelaere K; Vanduyfhuys L; Waroquier M; Van Speybroeck V On the Intrinsic Dynamic Nature of the Rigid UiO-66 Metal−organic Framework. Chem. Sci 2018, 9, 2723−2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Norby T; Widerøe M; Glöckner R; Larring Y Hydrogen in Oxides. Dalt. Trans 2004, 19, 3012−3018. [DOI] [PubMed] [Google Scholar]
- (35).Frot T; Cochet S; Laurent G; Sassoye C; Popall M; Sanchez C; Rozes L Ti8O8(OOCR)16, a New Family of Titanium- Oxo Clusters: Potential NBUs for Reticular Chemistry. Eur. J. Inorg. Chem 2010, 2010, 5650−5659. [Google Scholar]
- (36).Uetsuka H; Onishi H; Henderson MA; White JM Photoinduced Redox Reaction Coupled with Limited Electron Mobility at Metal Oxide Surface. J. Phys. Chem. B 2004, 108, 10621−10624. [Google Scholar]