

Abstract
BACKGROUND
Despite the high rate of sudden death after myocardial infarction among patients with a low ejection fraction, implantable cardioverter-defibrillators are contraindi-cated until 40 to 90 days after myocardial infarction. Whether a wearable cardio- verter-defibrillator would reduce the incidence of sudden death during this high-risk period is unclear.
METHODS
We randomly assigned (in a 2:1 ratio) patients with acute myocardial infarction and an ejection fraction of 35% or less to receive a wearable cardioverter-defibrillator plus guideline-directed therapy (the device group) or to receive only guideline-directed therapy (the control group). The primary outcome was the composite of sudden death or death from ventricular tachyarrhythmia at 90 days (arrhythmic death). Secondary outcomes included death from any cause and nonarrhythmic death.
RESULTS
Of 2302 participants, 1524 were randomly assigned to the device group and 778 to the control group. Participants in the device group wore the device for a median of 18.0 hours per day (interquartile range, 3.8 to 22.7). Arrhythmic death occurred in 1.6% of the participants in the device group and in 2.4% of those in the control group (relative risk, 0.67; 95% confidence interval [CI], 0.37 to 1.21; P=0.18). Death from any cause occurred in 3.1% of the participants in the device group and in 4.9% of those in the control group (relative risk, 0.64; 95% CI, 0.43 to 0.98; uncorrected P=0.04), and nonarrhythmic death in 1.4% and 2.2%, respectively (relative risk, 0.63; 95% CI, 0.33 to 1.19; uncorrected P=0.15). Of the 48 participants in the device group who died, 12 were wearing the device at the time of death. A total of 20 participants in the device group (1.3%) received an appropriate shock, and 9 (0.6%) received an inappropriate shock.
CONCLUSIONS
Among patients with a recent myocardial infarction and an ejection fraction of 35% or less, the wearable cardioverter-defibrillator did not lead to a significantly lower rate of the primary outcome of arrhythmic death than control. (Funded by the National Institutes of Health and Zoll Medical; VEST ClinicalTrials.gov number, NCT01446965.)
THE INCIDENCE OF SUDDEN CARDIAC death is high during the early months after a myocardial infarction,1–3 particularly among patients with a low left ventricular ejection fraction.2–7 Implantable cardioverter- defibrillators (ICDs) reduce mortality among patients with a reduced ejection fraction when the devices are implanted months to years after myocardial infarction.8–10 However, two randomized trials did not show a long-term mortality benefit from ICDs that had been implanted immediately after myocardial infarction.11,12
The wearable cardioverter-defibrillator may protect against sudden death during the immediate period after myocardial infarction, before ICD implantation is indicated under current guidelines (beginning 40 days after myocardial infarction or 90 days if the patient has undergone revascularization).13,14 Registries and case series involving high-risk patients have shown that wearable cardioverter-defibrillators are effective in terminating ventricular tachyarrhythmias.15–19 We conducted the Vest Prevention of Early Sudden Death Trial (VEST) — a multicenter, randomized, controlled trial — to determine the efficacy of a wearable cardioverter- defibrillator during the period before ICDs are indicated in patients who have had a myocardial infarction and have a reduced ejection fraction.
METHODS
TRIAL DESIGN AND OVERSIGHT
The trial protocol (available with the full text of this article at NEJM.org) was designed by the investigators and originally included two components: the VEST randomized trial and the observational Prediction of ICD Treatment Study (PREDICTS)20; only the results of VEST are reported in this article. The protocol was approved by the institutional review boards of the University of California, San Francisco, and the other trial sites. Details of the history of the trial, the role of the sponsors, and the trial oversight are provided in Figure S1 and Tables S1, S2, and S3 in the Supplementary Appendix (available at NEJM.org).
The trial was initially funded by the National Institutes of Health, which appointed the members of the independent data and safety monitoring board, with additional support from Zoll Medical. After 2011, funding was provided exclusively by Zoll Medical. Zoll Medical had no role in the trial design, the selection or supervision of trial centers, the analysis or interpretation of the data, the preparation of the manuscript, or the decision to submit the manuscript for publication. Zoll Medical did participate in site monitoring. The authors vouch for the completeness and accuracy of the data and for the fidelity of the trial to the protocol.
PARTICIPANTS
Patients who had been hospitalized with an acute myocardial infarction21 and who had an ejection fraction of 35% or less (assessed ≥8 hours after myocardial infarction) were enrolled within 7 days after hospital discharge. For patients who had undergone revascularization, the ejection fraction was assessed 8 or more hours after percutaneous coronary intervention (PCI) or 48 or more hours after coronary-artery bypass grafting. Patients were excluded if they had an ICD or unipolar pacemaker, had clinically significant valve disease, were undergoing long-term hemodialysis, or had a chest circumference that was too small or too large to accommodate the wearable cardioverter-defibrillator. Patients were also excluded if they were pregnant or had been discharged to a nursing facility with an anticipated stay of more than 7 days. The inclusion and exclusion criteria are provided in Table S4 in the Supplementary Appendix. All the participants provided written informed consent.
TRIAL PROCEDURES
Eligible participants were randomly assigned in a 2:1 ratio to receive a wearable cardioverter-defibrillator plus guideline-directed medical therapy (the device group)22–27 or to receive guideline- directed medical therapy alone (the control group) at hospital discharge. The Zoll LifeVest wearable cardioverter-defibrillator16–18,28,29 that was used in this trial was commercially available in the United States and Germany (Fig. S2 in the Supplementary Appendix). Participants in the device group were fitted with the device, trained in its use, and instructed to wear the device continuously for 3 months (except while bathing). Sites were alerted if a participant wore the device for less than 15 hours in a 24-hour period (monitored through the device itself). Arrhythmias that were detected by the device were not reported to treating physicians or the trial sites unless a shock was delivered or cardiac arrest occurred. Per protocol, crossovers from the control group to the wearable cardioverter-defibrillator were not allowed, and early ICD implantation (<3 months) was allowed only for guideline-based secondary prevention of sudden death.14,30,31
FOLLOW-UP AND OUTCOMES
Participants were followed at 1 month with a telephone call and at 3 months with an in-person visit. At the conclusion of the trial, the National Death Index was searched for U.S. participants for whom vital status was unknown.
Initially, the primary outcome of the trial was death from any cause at 60 days; however, slower- than-expected recruitment made the originally planned sample of 4506 patients infeasible. On January 29, 2010, after the first 244 participants had been enrolled, the data and safety monitoring board, the steering committee, and the institutional review boards approved a change in the primary outcome to the combined 90-day incidence of sudden death and nonsudden death due to ventricular tachyarrhythmia; we refer to this outcome as arrhythmic death. The cause of death was adjudicated by an independent panel of experts who were unaware of the group assignments (and therefore did not have any data from the wearable cardioverter-defibrillator). With the revised primary outcome, the sample-size target was changed to 1890 (see the Supplementary Appendix). In October 2015, on the basis of lower-than-expected device wear time and without the inspection of outcome differences according to trial group32,33 (as prespecified in the protocol), the data and safety monitoring board recommended increasing the sample to 2300 patients.
Secondary outcomes were death from any cause; nonarrhythmic death; hospitalization for myocardial infarction, atrial fibrillation, congestive heart failure, stroke, or sustained ventricular tachyarrhythmia; wearable cardioverter-defibrillator wear time (as monitored by the device); and adverse events (Table S5 in the Supplementary Appendix). Definitions for the adjudicated out-comes are provided in the Supplementary Appendix.
STATISTICAL ANALYSIS
The primary analysis was performed according to the intention-to-treat principle. In the primary analysis, participants who had an indeterminate cause of death were assumed not to have had arrhythmic death but were counted in the out-come of death from any cause, and all the participants with missing vital status were assumed to be alive. The primary outcome as well as death from any cause, nonarrhythmic death, and rehospitalization were compared with the use of unadjusted log-binomial models (with relative risks reported), with P values assessed by Pearson chi-square tests. Time-to-event analyses were conducted with the use of Cox models and are reported as Kaplan-Meier plots with hazard ratios. Rare events (indeterminate cause of death and other clinically significant arrhythmias — nonatrial fibrillation and nonventricular tachyarrhythmias) were analyzed with the use of exact logistic regression. The risk of having an alarm indicating arrhythmia was estimated with the use of random-effects logistic models to account for within-person clustering. P values are reported without correction for multiple comparisons, except where noted. Additional analyses, including sensitivity analyses to account for missing data, survival analyses (performed with the Kaplan-Meier method), P value corrections for multiple comparisons, and as-treated analyses are described in the Supplementary Appendix.
RESULTS
CHARACTERISTICS OF THE PARTICIPANTS
From July 2008 through April 2017, we enrolled 2348 participants at 76 sites in the United States, at 24 in Poland, at 6 in Germany, and at 2 in Hungary (Table S6 in the Supplementary Appendix). One U.S. site was dismissed on June 24, 2014, and the 46 participants at that site were excluded from the analyses, owing to irregularities found by the institutional review board at that site.
Therefore, a total of 2302 participants were included in the analyses (1524 participants in the device group and 778 in the control group) (Fig. S3 in the Supplementary Appendix). The two groups were balanced with regard to the participants’ demographic characteristics, medical history, and characteristics of the index hospitalization for myocardial infarction (Table 1). The mean ejection fraction was 28%, and 83.6% of the participants underwent PCI during the index hospitalization. Table S7 in the Supplementary Appendix shows the baseline characteristics of the participants who were enrolled before versus after the protocol was amended to change the primary outcome.
Table 1.
Characteristics of the Participants.*
| Characteristic | Device Group (N = 1524) |
Control Group (N=778) |
|---|---|---|
| Age — yr | 60.9±12.6 | 61.4±12.3 |
| Male sex — no./total no. (%) | 1108/1521 (72.8) | 577/772 (74.7) |
| Body-mass index† | 28.4±5.5 | 28.4±5.5 |
| Race or ethnic group — no./total no. (%)‡ | ||
| White | 1279/1491 (85.8) | 636/751 (84.7) |
| Black | 143/1491 (9.6) | 75/751 (10.0) |
| Asian | 23/1491 (1.5) | 14/751 (1.9) |
| Native American or Alaskan | 25/1491 (1.7) | 12/751 (1.6) |
| Pacific Islander or Hawaiian | 1/1491 (0.1) | 0/751 |
| Multiple | 20/1491 (1.3) | 14/751 (1.9) |
| Hispanic | 85/1521 (5.6) | 34/772 (4.4) |
| Baseline condition before index hospitalization — no./total no. (%) | ||
| Current smoking | 561/1520 (36.9) | 273/770 (35.5) |
| Diabetes mellitus | 497/1521 (32.7) | 246/776 (31.7) |
| Hypertension | 994/1521 (65.4) | 501/776 (64.6) |
| Previous myocardial infarction | 380/1518 (25.0) | 193/775 (24.9) |
| Previous CABG | 133/1521 (8.7) | 70/776 (9.0) |
| Previous PCI | 374/1520 (24.6) | 202/776 (26.0) |
| Previous congestive heart failure | 247/1518 (16.3) | 146/774 (18.9) |
| NYHA functional class | ||
| I | 691/1520 (45.5) | 326/775 (42.1) |
| II | 529/1520 (34.8) | 286/775 (36.9) |
| III | 211/1520 (13.9) | 116/775 (15.0) |
| IV | 46/1520 (3.0) | 18/775 (2.3) |
| Index hospitalization for myocardial infarction | ||
| Left ventricular ejection fraction§ | ||
| Mean | 28.2±6.1 | 28.2±5.8 |
| Distribution — no./total no. (%) | ||
| <25% | 301/1519 (19.8) | 148/777 (19.0) |
| 25 to 35% | 1217/1519 (80.1) | 627/777 (80.7) |
| >35% | 1/1519 (0.1) | 2/777 (0.3) |
| PCI during index hospitalization — no./total no. (%) | 1275/1513 (84.3) | 650/773 (84.1) |
| Thrombolytic agent during index hospitalization — no./total no. (%) | 118/1513 (7.8) | 71/773 (9.2) |
| CABG during index hospitalization — no. (%) | 14 (0.9) | 12 (1.5) |
| Cardiac arrest or ventricular fibrillation — no./total no. (%) | 169/1513 (11.2) | 70/773 (9.1) |
| Pulmonary edema leading to intubation — no./total no. (%) | 162/1513 (10.7) | 88/773 (11.4) |
| Intraaortic balloon pump — no./total no. (%) | 173/1513 (11.4) | 93/773 (12.0) |
| Cardiogenic shock — no./total no. (%) | 136/1513 (9.0) | 79/773 (10.2) |
| Atrial fibrillation during hospitalization — no./total no. (%) | 156/1513 (10.3) | 91/773 (11.8) |
| Median maximum creatinine level (IQR) — mg/dl¶ | 1.1 (0.9–1.3) | 1.1 (0.9–1.4) |
| Median no. of days from admission to randomization (IQR) | 5.0 (3.0–8.0) | 5.0 (3.0–7.0) |
Plus–minus values are means ±SD. There were no significant differences (P<0.05) between the trial groups. Percentages may not total 100 because of rounding. CABG denotes coronary-artery bypass grafting, IQR interquartile range, NYHA New York Heart Association, and PCI percutaneous coronary intervention.
The body-mass index is the weight in kilograms divided by the square of the height in meters.
Race and ethnic group were reported by the participant.
An ejection fraction of 35% or less was an inclusion criterion for the study. An ejection fraction of more than 35% represents a protocol violation.
To convert values for creatinine to micromoles per liter, multiply by 88.4.
INTERVENTION AND TREATMENT
The majority of the participants in each group received guideline-directed medical therapy for myocardial infarction and heart failure (Table 2). In the device group, 43 participants (2.8%) never wore the device after randomization; in the control group, 20 participants (2.6%) received the device outside the protocol. Including person-days in which the wearable cardioverter-defibrillator was not worn at all, participants in the device group wore the device for a median of 18.0 hours per day (interquartile range, 3.8 to 22.7) and for a mean (±SD) of 14.0±9.3 hours per day (Table 2), with decreasing use over time. Details are provided in Figures S3 and S4 in the Supplementary Appendix. There was no significant between- group difference in the rate of ICD implantation during the follow-up period, nor was there a significant between-group difference in the timing of or reason for implantation (Table 2, and Table S8 in the Supplementary Appendix).
Table 2.
Treatment Received during the Trial Period.*
| Treatment | Device Group (N = 1524) |
Control Group (N = 778) |
P Value |
|---|---|---|---|
| Wearable cardioverter-defibrillator | |||
| Patients with device — no. (%)† | 1481 (97.2) | 20 (2.6) | <0.001 |
| Hours per day that device was worn during follow-up‡ | |||
| Median (IQR) | 18.0 (3.8–22.7) | 0.0 (0.0–0.0) | <0.001 |
| Mean | 14.0±9.3 | 0.4±2.7 | <0.001 |
| Implantable cardioverter-defibrillator | |||
| Patients with device — no. (%)§ | 67 (4.4) | 44 (5.7) | 0.18 |
| Median no. of days from randomization to implantation (IQR) | 62 (24–81) | 58 (25–77) | 0.33 |
| Medication use — no. (%)¶ | |||
| Aspirin | 1329 (87.2) | 678 (87.1) | 0.97 |
| Other antiplatelet agent | 1379 (90.5) | 680 (87.4) | 0.02 |
| Statin | 1386 (90.9) | 696 (89.5) | 0.25 |
| Beta-blocker, including carvedilol | 1411 (92.6) | 720 (92.5) | 0.97 |
| ACE inhibitor or ARB | 1334 (87.5) | 667 (85.7) | 0.23 |
| Eplerenone or spironolactone | 662 (43.4) | 343 (44.1) | 0.77 |
| Other diuretic agent | 736 (48.3) | 385 (49.5) | 0.59 |
| Amiodarone | 106 (7.0) | 55 (7.1) | 0.92 |
| Other antiarrhythmic agent of class IA, IC, or III | 5 (0.3) | 4 (0.5) | 0.50 |
| Digoxin | 86 (5.6) | 44 (5.7) | 0.99 |
P values were not corrected for multiple comparisons. ACE denotes angiotensin-converting enzyme, and ARB angio- tensin-receptor blocker
The use of any wearable cardioverter-defibrillator by participants in the control group was a protocol violation
The number of hours per day that the wearable cardioverter-defibrillator was worn included follow-up days after discharge from the hospital and before death or implantable cardioverter-defibrillator (ICD) implantation and also included participants who did not wear it at all (0 hours per day on those days) in the two groups in order to describe the difference in the device coverage according to group. A total of 11 participants (4 in the device group and 7 in the control group) who died before discharge from the hospital were excluded from this analysis
The implantation of an ICD in a participant in either group before 90 days of follow-up, unless for acceptable clinical indications for secondary prevention (e.g., cardiac arrest or sustained ventricular tachycardia during follow-up), was a protocol violation (Table S6 in the Supplementary Appendix)
Participants provided details regarding medication use at follow-up visits
FOLLOW-UP AND OUTCOMES
The mean follow-up was 84.3±15.6 days. A total of 10 participants (0.7%) in the device group and 12 (1.5%) in the control group were lost to follow-up, and their vital status at 90 days was unknown. An additional 2 participants in each group had insufficient data to determine whether the cause of death was arrhythmic or nonar- rhythmic; therefore, they were considered to have had an indeterminate cause of death.
There was no significant difference between the two groups in the primary outcome of arrhythmic death (1.6% in the device group and 2.4% in the control group; relative risk, 0.67; 95% confidence interval [CI], 0.37 to 1.21; P = 0.18) (Table 3 and Fig. 1). The total mortality was 3.1% in the device group, as compared with 4.9% in the control group (relative risk, 0.64; 95% CI, 0.43 to 0.98; uncorrected P = 0.04). The rate of nonarrhythmic death was 1.4% in the device group and 2.2% in the control group (relative risk, 0.63; 95% CI, 0.33 to 1.19; uncorrected P=0.15). With most approaches to correction for multiple testing, the P value for the analysis of total mortality was not significant (Table S9 in the Supplementary Appendix).
Table 3.
Primary, Secondary, and Other Outcomes.*
| Event | Device Group (N = 1524) |
Control Group (N = 778) |
Relative Risk (95% CI) |
P Value |
|---|---|---|---|---|
| Arrhythmic death | ||||
| No. of patients (%)† | 25 (1.6) | 19 (2.4) | 0.67 (0.37–1.21) | 0.18 |
| Device worn at time of death or event leading to death —no. | 9 | 0 | NA | |
| Nonarrhythmic death | ||||
| No. of patients (%)‡ | 21 (1.4) | 17 (2.2) | 0.63 (0.33–1.19) | 0.15 |
| Device worn at time of death or event leading to death —no. | 2 | 0 | NA | |
| Indeterminate death | ||||
| No. of patients (%)§ | 2 (0.1) | 2 (0.3) | 0.51 (0.04–7.05) | 0.83 |
| Device worn at time of death or event leading to death —no. | 1 | 0 | NA | |
| Death from any cause | ||||
| No. of patients (%) | 48 (3.1) | 38 (4.9) | 0.64 (0.43–0.98) | 0.04 |
| Device worn at time of death or event leading to death —no. | 12 | 0 | NA | |
| Rehospitalization, by cause — no. (%)¶ | ||||
| Any | 475 (31.2) | 253 (32.5) | 0.96 (0.85–1.09) | 0.51 |
| Cardiovascular or trial-related cause | 335 (22.0) | 174 (22.4) | 0.98 (0.84–1.16) | 0.83 |
| Recurrent myocardial infarction | 53 (3.5) | 32 (4.1) | 0.85 (0.55–1.30) | 0.44 |
| Stroke | 14 (0.9) | 8 (1.0) | 0.89 (0.38–2.12) | 0.80 |
| Congestive heart failure | 87 (5.7) | 52 (6.7) | 0.85 (0.61–1.19) | 0.35 |
| Ventricular tachycardia or ventricular fibrillation | 24 (1.6) | 20 (2.6) | 0.61 (0.34–1.10) | 0.10 |
| Atrial fibrillation | 8 (0.5) | 5 (0.6) | 0.82 (0.27–2.49) | 0.72 |
| Other clinically significant arrhythmia | 8 (0.5) | 3 (0.4) | 1.36 (0.33–8.00) | 0.92 |
For common outcomes (including for the primary outcome of arrhythmic death and for other outcomes with at least five events in each group), a simple Pearson chi-square test was used to obtain P values, and log-binomial regression was used to estimate a relative risk with confidence intervals. For rare outcomes (<5 events in one or both groups), P values and relative risks were estimated with the use of exact logistic regression under the assumption that odds ratios produced by logistic regression are good estimates of relative risk when the outcome is rare. P values were not corrected for multiple comparisons. Details regarding the deaths or events leading to death that occurred while the participant was wearing the device are provided in Table S12 in the Supplementary Appendix. NA denotes not applicable.
The primary outcome of arrhythmic death included sudden death and nonsudden death due to ventricular tachyarrhythmia.
Nonarrhythmic death included all the deaths that did not meet the criteria for either arrhythmic death or indeterminate death (see below).
§ When documentation was inadequate for characterization of deaths as arrhythmic or nonarrhythmic, they were categorized as being indeterminate. Details are provided in the Supplementary Appendix.
Site investigators were instructed to report all the hospitalization events occurring during follow-up if they were for conditions related to the heart or major arteries or to a trial procedure. These events were adjudicated for the presence of myocardial infarction, stroke, congestive heart failure, ventricular tachycardia or ventricular fibrillation, and other clinically significant arrhythmia with the use of standard criteria (see the Supplementary Appendix). Investigators were also asked to report the number of hospitalization events for any cause; these events were not adjudicated.
Figure 1. Time-to-Event Curves for the Primary Outcome and Two Secondary Outcomes.
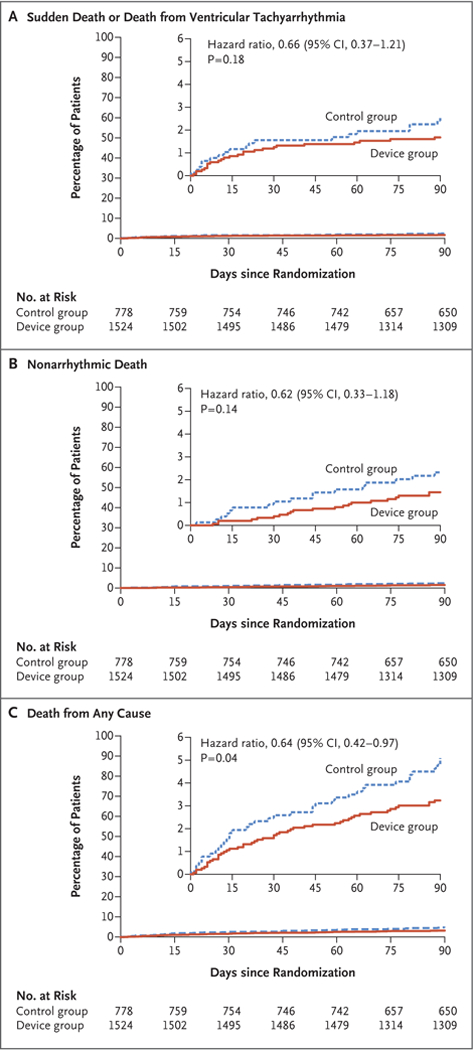
The primary outcome was a composite of sudden death or death due to ventricular tachyarrhythmia (Panel A). Secondary outcomes included nonarrhythmic death (Panel B) and death from any cause (Panel C).P values were not corrected for multiple comparisons. Insets show the same data on an enlarged y axis.
Results from the prespecified weighted sensitivity analyses to account for participants with unknown vital status or an indeterminate cause of death were similar to those of the primary analyses (Table S10 in the Supplementary Appendix). Analyses that were adjusted for the differences in length of follow-up owing to protocol changes were also similar to the main outcome analyses (Table S11 in the Supplementary Appendix). We found no significant be- tween-group differences in the rates of other secondary events (Table 3).
Among the 48 participants in the device group who died, 12 were wearing the device at the time of death, including 9 of the 25 participants who had arrhythmic death (Table S12 in the Supplementary Appendix). Of these 9 participants, 4 had had a ventricular tachyarrhythmia detected and had received appropriate shocks with conversion to sinus rhythm but with subsequent recurrent ventricular tachyarrhythmias or agonal rhythms. In the remaining participants, no tachyarrhythmias were recorded. One other participant received an appropriate shock and underwent ICD implantation but died 2 weeks later with ventricular tachyarrhythmia storm. A total of 6 participants who died while wearing the device had asystole events (>3-sec- ond pause) during death (in 2 participants, these were preceded by multiple ventricular tachyarrhythmia episodes and shocks), which may represent terminal rhythms.
An as-treated analysis showed a rate of arrhythmic death of 0.37 per 100 person-months of wearing the device, as compared with a rate of 0.86 per 100 person-months of not wearing the device (rate ratio, 0.43; 95% CI, 0.21 to 0.91; uncorrected P=0.03) (see the Supplementary Appendix). An as-treated analysis of total mortality showed a rate of 0.50 per 100 person-months of wearing the device, as compared with a rate of 1.91 per 100 person-months of not wearing the device (rate ratio, 0.26; 95% CI, 0.14 to 0.48; Bonferroni corrected P<0.001). Adjustment for age, education, ejection fraction, and revascularization had minimal effects. Potential biases in the as-treated analyses are discussed in the Supplementary Appendix.
SAFETY AND ADVERSE EVENTS
During a total of 1,765,772 hours of wearable cardioverter-defibrillator wear time, participants received 57,451 alarms for possible arrhythmias (as determined by the device algorithms); the average rate (number of alarms ÷ total wear time in hours) was 0.033 alarms per hour. With adjustment for clustering of alarms according to day and within participant, the chance that a participant would have at least 1 arrhythmia alarm during 24 hours of wear time was 10.8% (95% CI, 9.8 to 11.9). Overall, accounting for crossovers and variable time worn, arrhythmia alarms (both false and true detections) occurred in 72% of the participants in the device group and in 2% of those in the control group, with 9.6% of participants in the device group being exposed to more than 100 alarms over the 90-day period (Table 4). The median duration of the arrhythmia alarm was 7 seconds (interquartile range, 3 to 12).
Table 4.
Wearable Cardioverter-Defibrillator Therapies and Alarms.*
| Variable | Device Group (N = 1524) | Control Group (N = 778) | P Value |
|---|---|---|---|
| no. of participants with event (%) | |||
| No. of total shocks | <0.001 | ||
| 0 | 1495 (98.1) | 777 (99.9) | |
| 1 | 20 (1.3) | 0 | |
| ≥2 | 9 (0.6) | 1 (0.1) | |
| No. of appropriate shocks | 0.008 | ||
| 0 | 1504 (98.7) | 777 (99.9) | |
| 1 | 13 (0.9) | 0 | |
| ≥2 | 7 (0.5) | 1 (0.1) | |
| No. of inappropriate shocks | 0.12 | ||
| 0 | 1515 (99.4) | 778 (100) | |
| 1 | 7 (0.5) | 0 | |
| ≥2 | 2 (0.1) | 0 | |
| No. of shocks aborted by pressing response button† | <0.001 | ||
| 0 | 1455 (95.5) | 777 (99.9) | |
| 1 | 43 (2.8) | 1 (0.1) | |
| 2–5 | 11 (0.7) | 0 | |
| >5 | 15 (1.0) | 0 | |
| No. of alarms indicating arrhythmia | <0.001 | ||
| 0 | 432 (28.3) | 762 (97.9) | |
| 1 | 115 (7.5) | 1 (0.1) | |
| 2–5 | 252 (16.5) | 2 (0.3) | |
| 6–100 | 579 (38.0) | 12 (1.5) | |
| >100 | 146 (9.6) | 1 (0.1) | |
| No. of alarms indicating asystole | <0.001 | ||
| 0 | 1483 (97.3) | 777 (99.9) | |
| 1 | 22 (1.4)‡ | 0 | |
| ≥2 | 19 (1.2)‡ | 1 (0.1) | |
Shown are the numbers of participants who had an event over the entire 90-day period. The wearable cardioverter– defibrillator was used by 20 participants in the control group, who received the device outside the protocol. Percentages may not total 100 because of rounding. P values were not corrected for multiple comparisons.
This analysis included arrhythmia alarms lasting more than 30 seconds that were aborted when the participant pressed the shock-suppression button.
Among 41 participants with an alarm indicating asystole, 6 events (all in the device group) were adjudicated as having had a true asystole event.
A total of 29 participants in the device group received at least one shock from the wearable cardioverter-defibrillator (Table 4); 20 participants (1.3%) received at least one appropriate shock, and 9 (0.6%) received at least one inappropriate shock. Of the 21 participants who received an appropriate shock (20 in the device group and 1 in the control group), 6 died (all in the device group). A total of 69 participants in the device group aborted shocks by pressing the patient-response buttons during an alarm; 3 of these participants subsequently received appropriate shocks within a few minutes but died, and 1 other participant died 12 hours later, after an appropriate shock (Table S12 in the Supplementary Appendix).
Four adverse events were potentially related to the wearable cardioverter-defibrillator (Table S13 in the Supplementary Appendix). Three were hospitalizations (two for aborted shocks and one for an inappropriate shock), and one was a death while the participant was wearing the device, which was deemed likely to not be an arrhythmic death (no tachyarrhythmia was recorded by the device and emergency medical technicians noted pulseless electrical activity on arrival).
A higher proportion of participants in the device group than in the control group reported itch and rash (P<0.001). A lower proportion of participants in the device group than in the control group reported shortness of breath (P = 0.004). Details are provided in Table S14 in the Supplementary Appendix.
DISCUSSION
VEST compared the use of a Wearble cardio-verter-defibrillator plus guidelibne-directed medical therapy with guideline-directed medical therapy alone in patients who presented with an acute myocardial infarction with an ejection fraction of 35% or less. During follow-up, we observed cardiac event rates that were similar to those in previous studies.3,4,6,11,12 The wearable cardioverter-defibrillator did not lead to a rate of arrhythmic death during the first 90 days — the primary outcome of the trial — that was significantly lower than the rate with guideline- directed medical therapy alone.
The trial may have been underpowered to detect a beneficial effect of the wearable cardioverter-defibrillator on the primary outcome. Our power calculation anticipated a 58% lower rate of arrhythmic death with the device than without it. The power was, in part, reduced because 5% of the deaths were adjudicated as being of indeterminate cause and were thus removed from the primary analysis. Misclassification of the adjudicated cause of death may have further reduced the power for the primary outcome. It is difficult to determine an arrhythmic cause of death accurately for unwitnessed deaths or deaths with limited documentation. In the Valsartan in Acute Myocardial Infarction (VALIANT) trial, only half the patients with sudden death who underwent autopsy were found to have died from arrhythmic death.34 In a recent study that used a definition of sudden death that was similar to the definition in our trial but that also used autopsy as a standard for determining cause of death, only 56% of the presumed sudden cardiac deaths were found to be of arrhythmic origin.35 In our trial, five of nine participants with adjudicated arrhythmic death who were wearing the device during the event had no ventricular tachyarrhythmias (adjudicators were unaware of the arrhythmia data from the device).
The original primary outcome of the trial was death from any cause; for this outcome, the uncorrected P value for comparison was 0.04 in favor of the wearable cardioverter-defibrillator. However, this result was not corrected for multiple testing, and given the use of most such corrections, the difference between the device and control groups would not be significant. Thus, the conservative interpretation is that this result was a chance finding. As with the primary outcome, the trial may have been underpowered to detect a beneficial effect of the device with regard to all-cause mortality. Although there is no clear mechanism to explain a benefit of the wearable cardioverter-defibrillator on non- arrhythmic death, it is often difficult to determine an arrhythmic cause of death, as noted above.
As described previously,15–17 the wearable cardio- verter-defibrillator was effective at converting ventricular tachyarrhythmias, with successful conversion in all 20 participants in the device group who received an appropriate shock, 14 of whom survived to 90 days (Table S12 in the Supplementary Appendix). Nonadherence to wearing the device may have reduced the power of the trial to show the effectiveness of this treatment strategy for the prevention of arrhythmic death. The power calculation assumed a deviceadherence rate of 70%, a goal that was met or exceeded in the first 2 weeks after randomization but that waned over time (Fig. S4 in the Supplementary Appendix). It is also evident that some patients who are successfully treated with an appropriate shock subsequently die; not all successful defibrillations prolong survival. However, in an as-treated analysis, a significantly lower percentage of patients died when they were wearing the wearable cardioverter-defibrillator than when they were not, a finding that remained significant even after the most conservative correction for multiple comparisons. Although this result is subject to bias, it suggests a benefit to wearing the device (see the Supplementary Appendix) and implies that low adherence to wearing the device may be a limiting factor in the potential benefit of the wearable cardioverter-defibrillator.
Guidelines for primary prevention of sudden death with ICD implantation recommend waiting 40 days after an acute myocardial infarction and 90 days after revascularization. Randomized trials have shown no benefit to ICD implantation early after an acute myocardial infarction.11,12 However, mortality was high during this vulnerable period, even with guideline-directed medical therapy and revascularization. We observed that mortality at 90 days was 4.9% in the control group, despite 84% of the participants having undergone PCI for acute myocardial infarction and more than 85% being treated with guideline-directed medical therapy. It remains unclear how to reduce the risk of arrhythmic death definitively, beyond what is possible with appropriate medical therapy, in the early period after myocardial infarction before ICDs are indicated.
In conclusion, in this trial, we compared the use of a wearable cardioverter-defibrillator plus guideline-directed medical therapy with guideline-directed medical therapy alone in patients who presented with an acute myocardial infarction with an ejection fraction of 35% or less. The wearable cardioverter-defibrillator did not result in a significantly lower rate of arrhythmic death than medical therapy during the first 90 days.
Supplementary Material
Acknowledgments
Supported by grants (U01HL089458 and U01HL089145) from the National Institutes of Health and by Zoll Medical.
Dr. Olgin reports receiving consulting fees from Novartis; Dr. Morin, receiving lecture fees from Zoll Medical, Biotronik, and Medtronic and grant support from Boston Scientific; Dr. Zwei- bel, receiving lecture fees and consulting fees from Medtronic; Dr. Buxton, receiving grant support from Medtronic and Biosense Webster and honoraria from Boston Scientific; Dr. Chung, receiving consulting fees from AliveCor; Dr. Rashba, receiving grant support from Medtronic, Boston Scientific, St. Jude Medical, and Biotronik and lecture fees from Pfizer and Bristol-Myers Squibb; and Ms. Maguire, receiving travel support from Zoll Medical. No other potential conflict of interest relevant to this article was reported.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
Contributor Information
Jeffrey E. Olgin, The Division of Cardiology, Department of Medicine, the UCSF Center for the Prevention of Sudden Death
Mark J. Pletcher, The Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco
Eric Vittinghoff, The Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco
Jerzy Wranicz, Department of Electrocardiology, Medical University of Lodz, Lodz, Poland
Rajesh Malik, McLeod Regional Medical Center, Florence, SC
Daniel P. Morin, Ochsner Medical Center and Ochsner Clinical School, University of Queensland School of Medicine, New Orleans
Steven Zweibel, Hartford Healthcare Heart and Vascular Institute and University of Connecticut School of Medicine, Hartford
Alfred E. Buxton, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston
Claude S. Elayi, Gill Heart Institute, University of Kentucky, and Veterans Affairs Medical Center, Lexington
Eugene H. Chung, The Department of Internal Medicine, University of Michigan, Michigan Medicine, Ann Arbor
Eric Rashba, Stony Brook Medicine, Stony Brook, NY
Martin Borggrefe, First Department of Medicine-Cardiology, University Medical Center Mannheim, Mannheim, and DZHK (German Center for Cardiovascular Research), Heidelberg — both in Germany
Trisha F. Hue, The Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco
Carol Maguire, Division of Cardiology, Department of Medicine, the UCSF Center for the Prevention of Sudden Death
Feng Lin, The Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco
Joel A. Simon, The Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco
Stephen Hulley, The Department of Epidemiology and Biostatistics, University of California, San Francisco, San Francisco
Byron K. Lee, The Division of Cardiology, Department of Medicine, the UCSF Center for the Prevention of Sudden Death
REFERENCES
- 1.Berger CJ, Murabito JM, Evans JC, Anderson KM, Levy D. Prognosis after first myocardial infarction: comparison of Q-wave and non-Q-wave myocardial infarction in the Framingham Heart Study. JAMA 1992;268:1545–51. [DOI] [PubMed] [Google Scholar]
- 2.Kannel WB, Sorlie P, McNamara PM. Prognosis after initial myocardial infarction: the Framingham study. Am J Cardiol 1979;44:53–9. [DOI] [PubMed] [Google Scholar]
- 3.Solomon SD, Zelenkofske S, McMurray JJV, et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med 2005;352:2581–8. [DOI] [PubMed] [Google Scholar]
- 4.Adabag AS, Therneau TM, Gersh BJ, Weston SA, Roger VL. Sudden death after myocardial infarction. JAMA 2008;300: 2022–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Julian DG, Camm AJ, Frangin G, et al. Randomised trial of effect of amiodarone on mortality in patients with left-ventricular dysfunction after recent myocardial infarction: EMIAT. Lancet 1997;349:667–74. [DOI] [PubMed] [Google Scholar]
- 6.Pitt B, White H, Nicolau J, et al. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005;46:425–31. [DOI] [PubMed] [Google Scholar]
- 7.Zaman S, Kovoor P. Sudden cardiac death early after myocardial infarction: pathogenesis, risk stratification, and primary prevention. Circulation 2014;129: 2426–35. [DOI] [PubMed] [Google Scholar]
- 8.Buxton AE, Lee KL, Fisher JD, Joseph- son ME, Prystowsky EN, Hafley G. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med 1999;341:1882–90. [DOI] [PubMed] [Google Scholar]
- 9.Moss AJ, Hall WJ, Cannom DS, et al. Improved survival with an implanted defibrillator in patients with coronary disease at high risk for ventricular arrhythmia. N Engl J Med 1996;335:1933–40. [DOI] [PubMed] [Google Scholar]
- 10.Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 2002;346:877–83. [DOI] [PubMed] [Google Scholar]
- 11.Hohnloser SH, Kuck KH, Dorian P, et al. Prophylactic use of an implantable cardioverter-defibrillator after acute myocardial infarction. N Engl J Med 2004;351: 2481–8. [DOI] [PubMed] [Google Scholar]
- 12.Steinbeck G, Andresen D, Seidl K, et al. Defibrillator implantation early after myocardial infarction. N Engl J Med 2009;361: 1427–36. [DOI] [PubMed] [Google Scholar]
- 13.Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017. AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol 2017. October 25 (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 14.Epstein AE, DiMarco JP, Ellenbogen KA, et al. 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/ HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2013;127(3):e283–e352. [DOI] [PubMed] [Google Scholar]
- 15.Chung MK, Szymkiewicz SJ, Shao M, et al. Aggregate national experience with the wearable cardioverter-defibrillator: event rates, compliance, and survival. J Am Coll Cardiol 2010;56:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldman AM, Klein H, Tchou P, et al. Use of a wearable defibrillator in terminating tachyarrhythmias in patients at high risk for sudden death: results of the WEARIT/BIROAD. Pacing Clin Electrophysiol 2004;27:4–9. [DOI] [PubMed] [Google Scholar]
- 17.Kutyifa V, Moss AJ, Klein H, et al. Use of the wearable cardioverter defibrillator in high-risk cardiac patients: data from the Prospective Registry of Patients Using the Wearable Cardioverter Defibrillator (WEARIT-II Registry). Circulation 2015; 132:1613–9. [DOI] [PubMed] [Google Scholar]
- 18.Lee BK, Olgin JE. The wearable car- dioverter-defibrillator: is it now the standard of care? Circulation 2016;134:644–6. [DOI] [PubMed] [Google Scholar]
- 19.Zishiri ET, Williams S, Cronin EM, et al. Early risk of mortality after coronary artery revascularization in patients with left ventricular dysfunction and potential role of the wearable cardioverter defibrillator. Circ Arrhythm Electrophysiol 2013; 6:117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks GC, Lee BK, Rao R, et al. Predicting persistent left ventricular dysfunction following myocardial infarction: the PREDICTS study. J Am Coll Cardiol 2016; 67:1186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thygesen K, Alpert JS, White HD, et al. Universal definition of myocardial infarction. Circulation 2007;116:2634–53. [DOI] [PubMed] [Google Scholar]
- 22.Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non ST-Elevation Myocardial Infarction): developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons: endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. Circulation 2007;116(7): e148–e304. [DOI] [PubMed] [Google Scholar]
- 23.Anderson JL, Adams CD, Antman EM, et al. 2011 ACCF/AHA focused update in-corporated into the ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011;123(18): e426–e579. [DOI] [PubMed] [Google Scholar]
- 24.Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of Patients with Acute Myocardial Infarction). Circulation 2004;110(9):e82–e292. [PubMed] [Google Scholar]
- 25.Antman EM, Hand M, Armstrong PW, et al. 2007 focused update of the ACC/AHA 2004 guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the Canadian Cardiovascular Society endorsed by the American Academy of Family Physicians: 2007 Writing Group to Review New Evidence and Update the ACC/AHA 2004 Guidelines for the Management of Patients with ST-Elevation Myocardial Infarction, writing on behalf of the 2004 Writing Committee. Circulation 2008;117:296–329. [DOI] [PubMed] [Google Scholar]
- 26.Kushner FG, Hand M, Smith SC Jr, et al. 2009 Focused updates: ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction (updating the 2004 guideline and 2007 focused update) and ACC/AHA/SCAI guidelines on percutaneous coronary intervention (updating the 2005 guideline and 2007 focused update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009;120:2271–306. [DOI] [PubMed] [Google Scholar]
- 27.Levine GN, Bates ER, Blankenship JC, et al. 2015 ACC/AHA/SCAI focused update on primary percutaneous coronary intervention for patients with ST-elevation myocardial infarction: an update of the 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention and the 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Society for Cardiovascular Angiography and Interventions. Circulation 2016;133: 1135–47. [DOI] [PubMed] [Google Scholar]
- 28.Klein HU, Goldenberg I, Moss AJ. Risk stratification for implantable cardioverter defibrillator therapy: the role of the wearable cardioverter-defibrillator. Eur Heart J 2013;34:2230–42. [DOI] [PubMed] [Google Scholar]
- 29.Lee BK, Olgin JE. Role of wearable and automatic external defibrillators in improving survival in patients at risk for sudden cardiac death. Curr Treat Options Cardiovasc Med 2009;11:360–5. [DOI] [PubMed] [Google Scholar]
- 30.Epstein AE, DiMarco JP, Ellenbogen KA, et al. ACC/AHA/HRS 2008 guidelines for device-based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices): developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. Circulation 2008; 117(21):e350–e408. [DOI] [PubMed] [Google Scholar]
- 31.Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death): developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation 2006;114(10): e385–e484. [DOI] [PubMed] [Google Scholar]
- 32.Gould AL. Interim analyses for monitoring clinical trials that do not materially affect the type I error rate. Stat Med 1992;11:55–66. [DOI] [PubMed] [Google Scholar]
- 33.Gould AL. Sample size re-estimation: recent developments and practical considerations. Stat Med 2001;20:2625–43. [DOI] [PubMed] [Google Scholar]
- 34.Pouleur AC, Barkoudah E, Uno H, et al. Pathogenesis of sudden unexpected death in a clinical trial of patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. Circulation 2010;122:597–602. [DOI] [PubMed] [Google Scholar]
- 35.Tseng ZH, Olgin JE, Vittinghoff E, et al. Prospective countywide surveillance and autopsy characterization of sudden cardiac death: POST SCD study. Circulation 2018;137:2689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.