

Summary
Early onset epileptic encephalopathy (EOEE) has been used to encompass Ohtahara syndrome (early infantile epileptic encephalopathy [EIEE]), early myoclonic epilepsy, and many others. Multiple genes have been established to cause epileptic encephalopathy in the immature brain, and next‐generation sequencing has accelerated the process of novel gene discovery. Many of the previously published candidate genes are still pending confirmatory reports or functional studies. Although most of the genes involved are ion channels (channelopathies), multiple other pathways have been implicated as well. NECAP1 is a key element in clathrin‐mediated endocytosis and has been reported previously to cause EOEE in a Saudi family. We report another family with the same variant confirming the pathogenicity of this variant as a Saudi founder mutation, further delineate its phenotype, and propose that it causes EOEE instead of EIEE.
Keywords: Encephalopathy, Early onset encephalopathy, Early onset epileptic encephalopathy, Epilepsy, NECAP1, Early infantile epileptic encephalopathy, Clathrin‐mediated endocytosis
Short Communication
Epileptic encephalopathy has been defined by the International League Against Epilepsy (ILAE) as when the epileptic activity itself may lead to severe cognitive and behavioral impairments above and beyond what might be expected from the underlying pathology alone.1 Early onset epileptic encephalopathies (EOEEs), on the other hand, are severe disorders that affect the development of the young brain with onset in early infancy. Myriad of novel genes were discovered in recent years after the introduction of next‐generation sequencing.2 Yet, many of these proposed candidate genes are awaiting confirmation.3, 4 Identification of the pathways involved in epileptic encephalopathy is important to guide novel drug discovery and epilepsy management in the future. Alazami et al.5 has previously reported that the NECAP1 gene variant in a multiplex Saudi family resulted in early infantile epileptic encephalopathy (EIEE). This gene has been proposed because it is highly expressed in the brain and plays a crucial role in clathrin‐mediated endocytosis (CME). CME is a major cellular mechanism in eukaryotic cells for the internalization of nutrients, receptors, and other substances that are important for homeostasis.6 Multiple proteins work in synchrony to deform the bilayer plasma membrane and lead to vesicle formation. It was previously shown that NECAP1 through its WxxF motif at its C‐terminus interacts with adaptor protein 2 (AP‐2) and works as a negative regulator of AP‐2 to control the vesicle number, size, and content.7, 8 As far as we know, neither confirmatory functional studies nor a second family has been reported. We identified a second Saudi simplex family that is not related to the previous family, albeit with the same variant. The index is a 41‐month‐old girl who was born at full term via normal spontaneous vaginal delivery to a healthy 29‐year‐old G3P2 mother following an uneventful pregnancy. The parents are a first‐degree consanguineous couple with 2 other healthy daughters, and there is no family history of a similar condition. However, there is a history of an abortion at 5 weeks of gestation and a stillbirth at 37 weeks in 2 succeeding pregnancies. The perinatal course was unremarkable apart from mild desaturation that resolved spontaneously, and she was discharged on the second day after delivery. Nevertheless, the mother noticed that since birth the child had a weak cry, and was not following, fixating, or able to support her neck. She was also noted to have increased respiratory secretions, asthma, congenital exotropia, and a premature closure of the anterior fontanelle. At the age of 3 months, she started to have uprolling of the eyes without any other abnormal movements, which was brought to medical attention only a few months later when her generalized tonic seizures commenced. She was started on phenytoin, and her seizures decreased in frequency. She was then referred to our institution at the age of 8 months when she had her first electroencephalography (EEG) study. The first reading of her EEG was reported as normal. She was then weaned from phenytoin and started on levetiracetam, and she became seizure free for 3 months. At the age of 12 months, she started to have flexor hemispasms in her right side with a frequency of 4–5 per day, so EEG was repeated and showed generalized slow waves at a frequency of 2–3 delta activity without evidence of hypsarrhythmia or epileptiform discharges. She was then started on vigabatrin while levetiracetam was decreased gradually, but her seizures worsened, and she had generalized tonic–clonic seizures. Vigabatrin was subsequently stopped, and she was started on clobazam. Her generalized seizures ceased, but she continued to have hemispasms. Her parents refused adrenocorticotropic hormone (ACTH) treatment, and she was started on topiramate and the frequency of her spasms decreased. She has been on physiotherapy since the age of 8 months that helped decrease her contractures. At the age of 3 years, she was found to have gastroesophageal reflux, and she underwent laparoscopic fundoplication. She continued to have breakthrough generalized tonic–clonic seizures and occasional hemispasms. In terms of her development, she is nonverbal and has a profound global developmental delay. At the age of 41 months, she could not follow, fixate, support her neck, rollover, say baba and mama, or recognize her parents. Her growth parameters at 41 months of age were weight 9.9 kg (−3.22 standard deviation [SD]), height 90 cm (−1.72 SD), and head circumference 43 cm (−3.78 SD). Physical examination revealed axial hypotonia, appendicular hypertonia, hyperreflexia, scaphocephaly, and exotropia. She had unpurposeful movements of all limbs, and she was not following or fixating. She did not have any stigmata of a neurocutaneous syndrome. The examination of other systems was within normal limits. Brain magnetic resonance imaging (MRI) at 15 months of age showed delayed myelination for the patient's age, benign enlargement of the subarachnoid spaces, moderate supratentorial ventriculomegaly, and thinning of the corpus callosum (Fig. 1). A renal ultrasound at 3 years of age showed bilateral echogenic kidney with bilateral grade 1 hydronephrosis that is more prominent on the right side. An ascending urethrogram showed left grade III vesicoureteral reflex. Routine laboratory workup as well as plasma amino acids, urine organic acids, serum ammonia, newborn screening, blood creatine kinase, serum homocysteine, serum carbohydrate deficient transferrin, urine creatinine, blood lactic acid, serum very long chain fatty acids, urine mucopolysaccharides, urine oligosaccharides, serum biotinidase, karyotyping, microarray comparative genomic hybridization, visual evoked potential, electroretinogram, and brainstem auditory potentials were all within normal limits.
Figure 1.
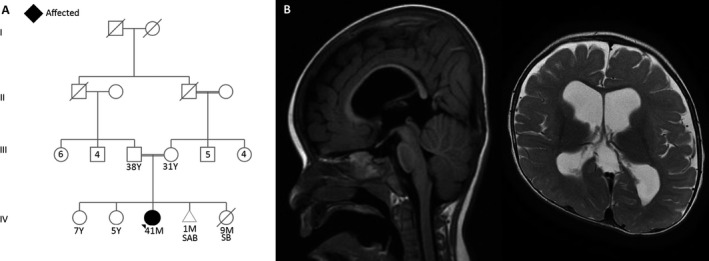
(A) Family pedigree of the current cohort. (B) Sagittal and axial brain MRI showing delayed myelination for the patient's age, benign enlargement of the subarachnoid spaces, moderate supratentorial ventriculomegaly, and thinning of the corpus callosum.
After obtaining signed consent from the parents, trio whole‐exome sequencing (WES) was carried out. We did not find any suspicious variant in known epilepsy genes or intellectual disability (ID); however, it revealed a homozygous nonsense truncating pathogenic variant in the NECAP1 gene [NM_015509.3: c.142C>T (p.Arg48Ter)] in the index, with both parents being heterozygous carriers. This variant was absent in our 1200 ethnically matched exome database (Majeen), publicly available genomic catalogs, Centogene database, and Saudi Human Genome Program database (2,000 exomes). Further segregation analysis of other siblings supported the pathogenicity of this variant. Although the seizure semiology reported by Alazami et al. was different from that of the current report, there is a significant phenotypic overlap.
Of interest, DNM1, which is another gene involved in the CME pathway, has been reported to cause epileptic encephalopathy by multiple cohort studies.4,9–18 DNM1 gene encodes for dynamin‐1 protein, which is essential for the scission of newly formed CME vesicles.19 Patients with DNM1 mutation have severe ID and early onset intractable seizures (Table 1).
Table 1.
Comparison between NECAP1 patients and DNM1 patients
| Current report and Alazami et al. | PMID: 29427836; 29588952; 23934111; 26648591; 25262651; 25533962; 27806796; 27476654; 26611353; 28667181 | |
|---|---|---|
| Gene | NECAP1 | DNM1 |
| Inheritance | AR | AD |
| Number of patients | 7 | 29 |
| Seizure type at onset | Generalized tonic and clonic, generalized tonic, hemispasms | Infantile spasms, absence seizures, myoclonic, generalized tonic and clonic, head dropping |
| Age of onset | 3 months, NA | 0–13 months, 4.5 years |
| Antiepileptic drug response | + | + |
| EGG | Generalized slowing and frequent generalized epileptiform discharges | Slow background, multifocal discharges, hypsarrhythmia, modified hypsarrhythmia |
| GDD | 100% | 100% |
| Hypotonia | + | + |
| MRI |
|
|
AR, autosomal recessive; AD, autosomal dominant; EEG, electroencephalography; GDD, global developmental delay; MRI, magnetic resonance imaging; NA, not available.
Early infantile epileptic encephalopathy (or EIEE) is an age‐dependent syndrome with the age at onset confined to the neonatal period or very early in infancy; more than 75% manifests before the first month of life. It also has the characteristic EEG pattern of suppression‐burst.20 Although the previously reported NECAP1 phenotype was similar to the current index (Table 2), we here show that our patient's course along with previously published information is not enough to label this syndrome as one of the EIEE causes. Thus, we suggest classifying it as one of the early onset epileptic encephalopathy syndromes until more reports delineate the phenotype.
Table 2.
Comparison between reported cases and the current cohort
| Case | Current study | Published PMID: 24399846 |
|---|---|---|
| Variant | c.142C>T | c.142C>T |
| Number of affected | 1 | 6 |
| Gender | F | F and M |
| Consanguinity | + | + |
| Growth parameters at birth | Normal | Normal |
| GDD | + | + |
| Seizure age of onset | 3 months | NA |
| Seizures’ semiology |
Generalized tonic Hemispasms |
Generalized tonic and clonic |
| Pharmacoresistant seizures | + | + |
| Height | −1.72 SD | NA |
| Weight | −3.22 SD | NA |
| Head Circumference | −3.78 SD | NA |
| Axial hypotonia | + | + |
| Appendicular Hypertonia | + | + |
| Brain MRI |
|
• Non‐specific generalized brain atrophy |
| EEG | • Generalized slowing |
|
EEG, electroencephalography; F, female; GDD, global developmental delay; M, male; MRI, magnetic resonance imaging; NA, not available.
Our institution's standardized consent was obtained from the parents of the index to participate in this study. The study received ethical approval from King Abdullah International Medical Research Center (KAIMRC), Riyadh, Saudi Arabia.
Disclosure
The authors declare no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Biography
Dr. Saud Alsahli, MD is a PGY‐1 Child Neurology resident and a junior researcher.

References
- 1. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010;51:676–685. [DOI] [PubMed] [Google Scholar]
- 2. McTague A, Howell KB, Cross JH, et al. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 2016;15:304–316. [DOI] [PubMed] [Google Scholar]
- 3. de Kovel CG, Brilstra EH, van Kempen MJ, et al. Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med 2016;4:568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. EuroEPINOMICS‐RES Consortium , Epilepsy Phenome/Genome Project , Epi4K Consortium . De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014;95:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alazami AM, Hijazi H, Kentab AY, et al. NECAP1 loss of function leads to a severe infantile epileptic encephalopathy. J Med Genet 2014;51:224–228. [DOI] [PubMed] [Google Scholar]
- 6. Murshid A, Srivastava A, Kumar R, et al. Characterization of the localization and function of NECAP 1 in neurons. J Neurochem 2006;98:1746–1762. [DOI] [PubMed] [Google Scholar]
- 7. Ritter B, Murphy S, Dokainish H, et al. NECAP 1 regulates AP‐2 interactions to control vesicle size, number, and cargo during clathrin‐mediated endocytosis. PLoS Biol 2013;11:e1001670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beacham GM, Partlow EA, Lange JJ, et al. NECAPs are negative regulators of the AP2 clathrin adaptor complex. Elife 2018;7:e32242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Asinof SK, Sukoff Rizzo SJ, Buckley AR, et al. Independent neuronal origin of seizures and behavioral comorbidities in an animal model of a severe childhood genetic epileptic encephalopathy. PLoS Genet 2015;11:e1005347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. von Spiczak S, Helbig KL, Shinde DN, et al. DNM1 encephalopathy: a new disease of vesicle fission. Neurology 2017;89:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakashima M, Kouga T, Lourenco CM, et al. De novo DNM1 mutations in two cases of epileptic encephalopathy. Epilepsia 2016;57:e18–e23. [DOI] [PubMed] [Google Scholar]
- 12. Fung CW, Kwong AK, Wong VC. Gene panel analysis for nonsyndromic cryptogenic neonatal/infantile epileptic encephalopathy. Epilepsia Open 2017;2:236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kolnikova M, Skopkova M, Ilencikova D, et al. DNM1 encephalopathy – atypical phenotype with hypomyelination due to a novel de novo variant in the DNM1 gene. Seizure 2018;56:31–33. [DOI] [PubMed] [Google Scholar]
- 14. Epi4K Consortium , Epilepsy Phenome/Genome Project , Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allen NM, Conroy J, Shahwan A, et al. Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia 2016;57:e12–e17. [DOI] [PubMed] [Google Scholar]
- 16. Deciphering Developmental Disorders Study . Large‐scale discovery of novel genetic causes of developmental disorders. Nature 2015;519:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Deng XL, Yin F, Zhang CL, et al. [Dynamin‐1‐related infantile spasms: a case report and review of literature]. Zhonghua Er Ke Za Zhi 2016;54:856–859. [DOI] [PubMed] [Google Scholar]
- 18. Epi KC. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet 2016;99:287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marks B, Stowell MH, Vallis Y, et al. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 2001;410:231–235. [DOI] [PubMed] [Google Scholar]
- 20. Ohtahara S, Yamatogi Y. Ohtahara syndrome: with special reference to its developmental aspects for differentiating from early myoclonic encephalopathy. Epilepsy Res 2006;70(Suppl 1):S58–S67. [DOI] [PubMed] [Google Scholar]