

Abstract
The genetic basis of human neural tube defects (NTDs), such as anencephaly and spina bifida (SB), is complex and heterogeneous. Grainyhead-like genes represent candidates for involvement in NTDs based on the presence of SB and exencephaly in mice carrying loss-of-function alleles of Grhl2 or Grhl3. We found that reinstatement of Grhl3 expression, by bacterial artificial chromosome (BAC)-mediated transgenesis, prevents SB in Grhl3-null embryos, as in the Grhl3 hypomorphic curly tail strain. Notably, however, further increase in expression of Grhl3 causes highly penetrant SB. Grhl3 overexpression recapitulates the spinal NTD phenotype of loss-of-function embryos, although the underlying mechanism differs. However, it does not phenocopy other defects of Grhl3-null embryos such as abnormal axial curvature, cranial NTDs (exencephaly) or skin barrier defects, the latter being rescued by the Grhl3-transgene. Grhl2 and Grhl3 can form homodimers and heterodimers, suggesting a possible model in which defects arising from overexpression of Grhl3 result from sequestration of Grhl2 in heterodimers, mimicking Grhl2 loss of function. This hypothesis predicts that increased abundance of Grhl2 would have an ameliorating effect in Grhl3 overexpressing embryo. Instead, we observed a striking additive genetic interaction between Grhl2 and Grhl3 gain-of-function alleles. Severe SB arose in embryos in which both genes were expressed at moderately elevated levels that individually do not cause NTDs. Furthermore, moderate Grhl3 overexpression also interacted with the Vangl2Lp allele to cause SB, demonstrating genetic interaction with the planar cell polarity signalling pathway that is implicated in mouse and human NTDs.
Introduction
Although neural tube defects (NTDs) are among the most common birth defects worldwide and have a strong genetic component, the specific contributors to genetic risk are not well understood (1,4). Identifying the molecular determinants of human NTDs is hindered by their complex, multifactorial nature and likely heterogeneity between cases. Moreover, most cases are sporadic rather than familial and de novo mutations may also play a significant role in spina bifida (SB) causation (5,6). Patterns of recurrence risk support oligogenic or polygenic models in which most NTDs result from a combination of one or more genetic factors with contribution from environmental risk factors, both positive and negative (7). It is anticipated that large-scale whole-exome and whole-genome sequencing efforts will provide a greater understanding of the genetic basis of NTDs but analysis of these large data sets and assignment of causation to coding variants and/or potential regulatory mutations will not be trivial.
Candidate genes for human NTDs may be indicated by knowledge of environmental risk factors, such as folate status and maternal diabetes, and causative genes in genetic models of which several hundred have been identified in mice (2,8). Mouse models have demonstrated a crucial role for members of the grainyhead-like family of transcription factors in neural tube closure. Grhl3-null embryos develop fully penetrant SB (9,10) and a hypomorphic allele of Grhl3 is the main genetic cause of SB in the curly tail strain (11). Each of these strains also exhibit a low frequency of exencephaly, which results from incomplete closure of the cranial neural tube and leads to anencephaly in late gestation.
Analysis of Grhl3-null and tissue-specific knockout embryos indicates that the initial defect leading to failure of spinal neurulation is localized to the surface ectoderm component of the closing neural folds, corresponding with its prominent early expression in this cell layer (12). Grhl3 is also expressed in the node-streak border/caudo-lateral epiblast and, later and transiently, in the neuroepithelium as well as in the gut endoderm (9,11,12). Among these tissues, knockout of Grhl3 in the gut endoderm causes spinal NTDs, but with later onset than in null embryos. These defects result from excess ventral curvature of the body axis, as in curly tail (Grhl3ct) mutant embryos (12,13). Hence, Grhl3 deficit leads to tissue-specific abnormalities that inhibit closure at two successive stages of spinal neurulation.
The extent to which GRHL3 mutations may contribute to human NTDs is not yet clear. Both de novo and rare inherited missense mutations of GRHL3 have been reported in SB cases at a frequency that suggests a role in determining NTD predisposition (6,14). GRHL3 mutations have also been reported in individuals with cleft lip and palate and individuals with syndromic (Van der Woude syndrome) and non-syndromic isolated cleft palate (15,17), consistent with GRHL3 expression in the oral ectoderm. Sharing of a missense mutation in independent SB and cleft palate cases supports the concept that GRHL3 may contribute to both defects (6,15).
The potential for regulatory mutations in GRHL3 to contribute to causation of human NTDs has been largely unexplored to date. In mice, a regulatory mutation likely underlies the diminished expression of Grhl3, which causes SB in the curly tail mouse (11). Tissues from human fetuses with NTDs have revealed hypomethylation of CpG islands within the 5′UTR and introns of GRHL3 (18), perhaps suggesting that GRHL3 misexpression may lead to NTDs. In the current study we examined this possibility in mouse models. Notably, we found that overexpression of Grhl3 causes a high frequency of severe SB. Moreover, even moderately elevated abundance of Grhl3 was found to cause SB when in combination with mutant alleles of other NTD genes: Grhl2 or Vangl2. Hence, insufficient or excess levels of Grhl3 can both cause SB.
Results
Overexpression of Grhl3 causes NTDs
Reinstatement of Grhl3 expression, mediated by bacterial artificial chromosome (BAC) transgenesis, prevents spinal NTDs in curly tail mice that have partial loss of function of Grhl3 (11). We investigated the potential effect of increasing levels of Grhl3 expression by intercross of hemizygous Grhl3 BAC-transgenic mice (Grhl3ct/ct;TgGrhl3/0) to generate litters that include embryos carrying the BAC in homozygosity (Grhl3ct/ct;TgGrhl3/TgGrhl3). Litters were genotyped by BAC-specific polymerase chain reaction (PCR) and quantitative genomic PCR (GqPCR). As in our previous study (11), Grhl3ct/ct;TgGrhl3/0 embryos (i.e. ct/ct embryos also hemizygous for the Grhl3-BAC) did not display spinal NTDs, whereas a proportion of Grhl3ct/ct embryos developed SB and/or tail flexion defects (TFDs) (Fig.1). Remarkably, we observed SB in 67% of Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos, suggesting that overexpression of Grhl3 prevents neural tube closure (Fig. 1).
Figure 1.
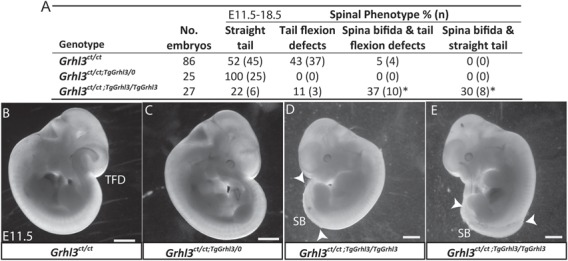
Spinal NTDs in Grhl3-transgenic mice. (A) Occurrence of SB among offspring of matings between Grhl3ct/ct;TgGrhl3/0 (BAC-hemizygous) and Grhl3ct/ct mice analysed at E11.5–18.5 (*, significant difference from other genotypes; P < 0.001, Chi-square). (B–E) Embryos of the three genotypes at E11.5. All Grhl3ct/ct;TgGrhl3/0 embryos appeared normal with a straight caudal region (C), whereas a proportion of Grhl3ct/ct (B) and Grhl3ct/ct;TgGrhl3/TgGrhl3 (D, E) exhibited TFDs and/or SB (extent of open region indicated by arrowheads). Scale bar represents 1 mm.
The NTD phenotype of Grhl3 overexpressing embryos could theoretically result from homozygous insertion of the transgene into an essential endogenous locus. To investigate this, the genomic location of the BAC was determined using inverse PCR (Fig. 2A and B). Sequence analysis indicated that the transgene insertion site is at position 3,005,382 on chromosome 18, in a low-complexity region. This was confirmed by PCR amplification of genomic DNA, using a series of primer pairs located in the transgene and putative chromosome 18 location (Fig. 2C and Supplementary Material, Fig. S1A). This site is more than 100 kb from the nearest recognized gene (vmn1r238) and 266 kb from the nearest gene (Crem) that has detectable embryonic expression at E10.5 (Supplementary Material, Fig. S1B). Fluorescence in situ hybridization (FISH) analysis on whole blood cultures confirmed that the BAC is present in only one location (Supplementary Material, Fig. S1D and E). We conclude that insertional mutagenesis is very unlikely to explain the NTDs observed in Grhl3 overexpressing embryos.
Figure 2.
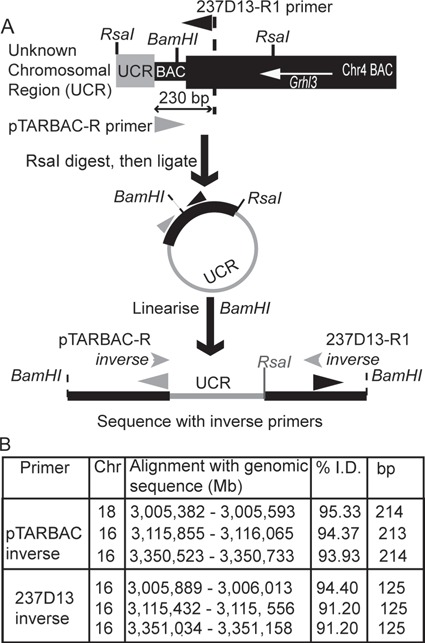
Localization of BAC-transgene in Grhl3ct/ct;TgGrhl3/0 embryos. (A) Inverse PCR was used to isolate genomic fragments adjacent to the insertion site of the Grhl3-containing BAC. (B) Sequence tags most closely aligned to the reference genomic sequence in a region on chromosome 18 with insertion of the BAC at 18: 3,005,382 in a low complexity repeat. Inverse PCR fragments also show homology to regions on chromosome 16 but with lower identity.
Further evidence that Grhl3 overexpression causes NTDs was provided by backcross of the Grhl3 transgene onto a wild-type BALB/c genetic background. NTDs did not occur among wild-type embryos (n = 20), but among hemizygous (+/+TgGrhl3/0) embryos we observed a low frequency of SB or TFDs (3/20; 15%). These defects are predicted to arise because the level of endogenous Grhl3 expression from the wild-type allele is higher than in the hypomorphic ct strain such that overexpression mediated by a single copy of the BAC is sufficient to exceed the level that is compatible with neural tube closure. Consistent with this, all homozygous (+/+TgGrhl3/TgGrhl3) embryos developed SB on this genetic background (18/18; 100%).
SB in Grhl3 overexpressing embryos results from early failure of posterior neuropore closure
In addition to a higher frequency of SB than in Grhl3ct/ct embryos, the size of the open lesion was also greater in Grhl3 overexpressing embryos at E11.5–13.5 (Fig. 1 and Supplementary Material, Fig. S2A–D). Previous studies showed that the posterior neuropore (PNP) length of Grhl3ct/ct embryos becomes larger than genetically matched wild-type embryos from the 25–27somite stage (E10.5) (11). In order to assess the stage at which spinal neurulation fails in Grhl3 overexpressing embryos, we collected litters generated by intercross of Grhl3ct/ct;TgGrhl3/0 mice. Among embryos analysed at E9–10.5, the PNP length was already larger among Grhl3ct/ct; TgGrhl3/TgGrhl3 embryos than Grhl3ct/ct littermates from the 12–15 somite stage (E9) onwards (Fig. 3A and Supplementary Material, Fig. S2E). This earlier failure of closure explains why the extent of the open SB lesion is greater among Grhl3 overexpressing fetuses than in their hypomorphic littermates.
Figure 3.
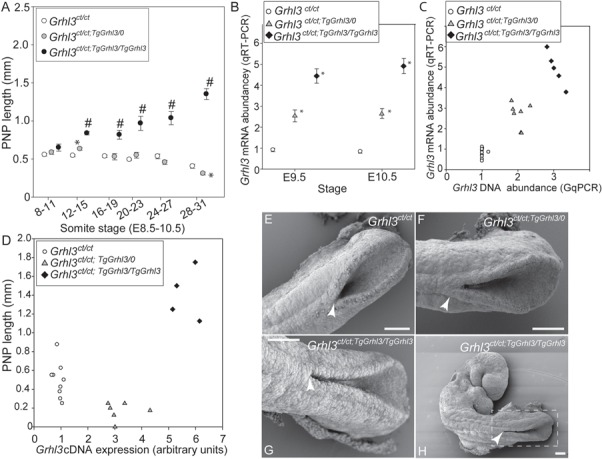
Excess expression of Grhl3 results in spinal NTDs owing to failure of PNP closure. (A) Among litters from Grhl3ct/ct;TgGrhl3/0 matings analysed at E8.5–10.5, the PNP was significantly enlarged among Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos (n = 64) from the 12–15 somite stage onwards (#, significant difference from both other genotypes, P < 0.001). PNP closure was normalized in Grhl3ct/ct;TgGrhl3/0 embryos (n = 123) compared with Grhl3ct/ct littermates (n = 176) at 28–31 somites (*, significantly different from Grhl3ct/ct, P < 0.001). Mean ± SEM values; n = 4–77 embryos/genotype/stage (see Supplementary, Fig. S2 for plot of individual data). (B) Abundance of Grhl3 mRNA varies significantly with genotype at E9.5 and E10.5 (*P < 0.0001 ANOVA; Holm-Sidak pairwise analysis). For context, we previously found that Grhl3 expression in ct/ct embryos at E10.5 was ∼50% of that in partially congenic wild-type (+ct) embryos (11). (C) Analysis of individual embryos at E10.5 (28–29 somites) shows that increased abundance of Grhl3 genomic DNA (in transgenic embryos) results in gene dosage-dependent increase in Grhl3 mRNA expression. qG-PCR signal corresponds to 2, 3 and 4 copies of Grhl3 in ct/ct, hemizygous and homozygous transgenics, respectively. (D) Moderate overexpression of Grhl3 normalizes PNP closure in individual Grhl3ct/ct;TgGrhl3/0 embryos, compared with Grhl3ct/ct, whereas excess expression prevents PNP closure, in Grhl3ct/ct;TgGrhl3/TgGrhl3. (E–H) Scanning electron micrographs at E9 (13 somite stage) show typical appearance of the closing PNP. Neural folds are elevated and apposed in all three genotypes (E–G) but failure of closure progression is already apparent in Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos (G,H), leaving a narrow unclosed region (compare region adjacent to white arrow E–H). Scale bars represent 0.1 mm.
Among embryos at E9.5 and E10.0, Grhl3 mRNA was upregulated by 2–3-folds in the caudal region of Grhl3ct/ct; TgGrhl3/0 embryos and 4–6-folds in Grhl3ct/ct; TgGrhl3/TgGrhl3, compared with Grhl3ct/ct (Fig. 3B). This equates to ∼1.3–1.5- and 2.5–3-fold higher expression than in wild-type embryos with a similar genetic background (based on our finding that Grhl3 abundance in ct/ct embryos is ∼50% of that in partially congenic (+ct/+ct) wild types (11). Hence, Grhl3 mRNA abundance (by qRT-PCR) correlated with the number of copies of the Grhl3 gene, determined by GqPCR (Fig. 3C). We conclude that during spinal neurulation the PNP becomes enlarged in Grhl3ct/ct hypomorphic embryos compared with Grhl3ct/ct; TgGrhl3/0 embryos that have mildly elevated Grhl3 expression, but a further increase in Grhl3 expression results in an even larger PNP in Grhl3ct/ct; TgGrhl3/TgGrhl3 embryos (Fig. 3D), resulting in SB. Scanning electron microscopy showed that the enlarged PNP of Grhl3 overexpressing embryos was characteristically very narrow (compare Fig. 3E–F with Fig. 3G–H), suggesting that NTDs do not result from a defect in elevation or bending of the neural folds.
Balance between excess and insufficient expression of Grhl3 in spinal neurulation
Homozygous embryos for Grhl3-null (Grhl3-/-) or gain-of-function (Grhl3ct/ct; TgGrhl3/TgGrhl3) alleles develop severe spinal NTDs. In order to further investigate the correlation between Grhl3 expression level and neural tube closure we asked whether transgenic expression was sufficient to rescue Grhl3-null NTDs and/or vice versa. Grhl3+/- and Grhl3ct/ct;TgGrhl3/0 mice were crossed and offspring with genotype Grhl3ct/-; TgGrhl3/0 were intercrossed to generate experimental litters carrying combinations of the null allele and the Grhl3-BAC (Table 1). Grhl3-/- and Grhl3ct/ct; TgGrhl3/TgGrhl3 fetuses developed SB as expected. The majority of embryos with Grhl3ct/- genotype exhibited TFDs, with SB also present in 50% (5 of 10) of embryos, intermediate between that normally observed in Grhl3ct/ct hypomorphs and Grhl3-/- embryos. Hemizygosity for the transgene normalized neural tube closure irrespective of the endogenous Grhl3 genotype, with no NTDs arising in Grhl3ct/ct;TgGrhl3/0 or Grhl3ct/-;TgGrhl3/0 embryos, or most notably in Grhl3-/-;TgGrhl3/0 that lacks endogenous Grhl3 (Table 1). SB occurred in only three of eight homozygous null embryos that were also homozygous for the Grhl3-BAC (Grhl3-/-;TgGrhl3/TgGrhl3; Table 1). Overall, these findings suggest that overexpression of Grhl3 is sufficient to rescue the null phenotype and that nullizygosity for Grhl3 can partially rescue NTDs resulting from Grhl3 overexpression, highlighting the importance of Grhl3 gene dosage.
Table 1.
Phenotypes of embryos carrying combinations of Grhl3 loss of function and overexpressing alleles
| Spinal phenotype % (n) | |||||
|---|---|---|---|---|---|
| Genotype | No. embryos | Straight tail | TFDs | SB & TFDs | P |
| Grhl3ct/ct | 13 | 77 (10) | 15 (2) | 8 (1) | |
| Grhl3 ct/ct;TgGrhl3/0 | 8 | 100 (8) | 0 (0) | 0 (0) | |
| Grhl3 ct/ct;TgGrhl3/TgGrhl3 | 9 | 11 (1) | 11 (1) | 78 (7) | <0.001 |
| Grhl3ct/- | 10 | 20 (2) | 30 (3) | 50 (5) | |
| Grhl3 ct/-;TgGrhl3/0 | 10 | 100 (12) | 0 (0) | 0 (0) | |
| Grhl3 ct/-;TgGrhl3/TgGrhl3 | 10 | 30 (3) | 0 (0) | 70 (7) | <0.001 |
| Grhl3-/- | 11 | 0 (0) | 0 (0) | 100 (11) | |
| Grhl3 -/-;TgGrhl3/0 | 12 | 100 (12) | 0 (0) | 0 (0) | |
| Grhl3 -/-;TgGrhl3/TgGrhl3 | 8 | 62 (5) | 0 (0) | 38 (3) | <0.001 |
Offspring of Grhl3ct/-;TgGrhl3/0 intercrosses were assessed at E11.0–18.5 for the presence of NTDs and/or TFDs. NTDs did not arise among embryos heterozygous for the Grhl3 transgene irrespective of the endogenous genotype. Differences in the number (0, 1 or 2) of Grhl3 transgene copies was associated with significant variation in the distribution of spinal phenotypes among Grhl3ct/ct, Grhl3ct/- and Grhl3-/- embryos (P < 0.001; Chi-square).
Abnormal function of the surface ectoderm is implicated in causation of spinal NTDs in Grhl3 overexpressing embryos
Partial rescue of NTDs resulting from excess Grhl3 by deletion of the endogenous allele suggested that the deleterious effect of Grhl3 overexpression on neural tube closure was localized to one or more of the endogenous sites of expression, as opposed to ectopic activity. At E8.5, Grhl3 is expressed in the surface ectoderm and in the posterior part of the embryo corresponding to the node-streak border and caudo-lateral epiblast (11). At E9.5, Grhl3 mRNA is also detected in the neural plate of the PNP and then, at E10–10.5, also in the hindgut endoderm. Whole mount in situ hybridization (WMISH)of Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos confirmed Grhl3 expression to be more intense but localized in the same tissues as in wild-type and hemizygous Grhl3-transgenic embryos (Fig. 4A and B) (11,12). Analysis of Grhl3 expression in the context of lack of the endogenous allele further confirmed expression of the transgene in the normal expression domain (Fig. 4C).
Figure 4.
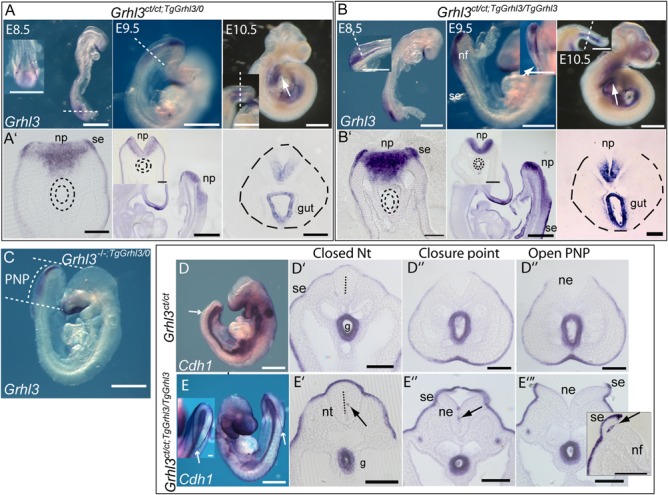
Overexpression of Grhl3 and Cdh1 in Grhl3-transgenic embryos. (A and B) Grhl3 expression in hemizygous (A) and homozygous (B) Grhl3-BAC transgenics detected by WMISH. Expression is detected in the expected domains in the surface ectoderm (se), neural plate (np) and hindgut (arrows in whole mounts; dotted lines in sections). Transverse sections are at the level of white dotted line on corresponding whole mount images; sagittal sections are shown at E9.5. (C) On a Grhl3-null background, the expression pattern of Grhl3 (entirely from the transgene), resembles the previously reported endogenous expression pattern. (D and E) Cdh1 shows the expected expression in surface ectoderm (se) and gut endoderm (g) in Grhl3ct/ct embryos (D). In Grhl3-transgenic embryos (E), Cdh1 expression appears more intense in the surface ectoderm (se) and occasional Cdh1-postive cells (arrows in E’-E”’) are present in the recently closed neural tube (nt; L) and the neuroepithelial component (ne) of the open neural folds (E”’, inset). Figure shows representative embryos at E9.5 (19–20 somite stage; D,E) with site of neural fold closure shown by white arrow in whole mounts. Scale bar = 0.5 mm in whole mounts; 0.1 mm in sections.
In Grhl3-/- embryos, the timing of closure failure and the phenotype of conditional knockouts suggest that the earliest abnormality of spinal neural tube closure results from a defect in the surface ectoderm (12). The surface ectoderm is the precursor of the epidermis, in which Grhl3 regulates terminal differentiation, barrier formation and repair (10,19–21). We asked whether expression of epithelial/epidermal genes was altered at neurulation stages. Using qRT-PCR, we found that known Grhl3 targets in late-fetal epidermis, Lor and Tgm1, were already upregulated in the caudal region of Grhl3 overexpressing embryos at E10.5 (Supplementary Material, Table S1). At E9, the stage at which neurulation begins to fail in Grhl3 mutant embryos, Trp63 (encoding TAp63) (22), a key transcriptional regulator of epidermal specification, was also upregulated in Grhl3ct/ct;TgGrhl3/TgGrhl3 (Supplementary Material, Table S2), suggesting abnormal specification of the surface ectoderm. This correlates with the finding that Trp63 is downregulated in Grhl3 null embryos at the same stage (12).
Cdh1 (encoding E-cadherin) is a key marker of surface ectoderm, a direct target of Grhl3 in mouse mammary gland cells (23) and upregulated in skin of Grhl3-transgenics at E18.5 (Supplementary Material, Fig. S6C). In the caudal region of Grhl3-transgenic embryos at E9.5 and 10.5, we observed a non-significant trend towards upregulation of Cdh1 (Supplementary Material, Tables S1 and S2). As expression in the surface ectoderm comprises only a subset of the Cdh1 expression domain we further examined expression by in situ hybridization at E8.5–10.5. Expression was observed at expected sites in the surface ectoderm and hindgut, with noticeably more intense staining in the surface ectoderm in Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos compared with Grhl3ct/ct (Fig. 4D–E). Cells expressing Cdh1 were also present at ectopic locations in the neuroepithelium of the PNP and tailbud of Grhl3ct/ct;TgGrhl3/0 and most notably Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos (Fig. 4D–E and Supplementary Material, Fig. S3). Ectopic E-cadherin protein expression was also confirmed by immunohistochemistry (Supplementary Material, Fig. S3). We hypothesize that earlier overexpression of Grhl3 in the node-streak border and caudo-lateral epiblast, which contains precursors of the neuroepithelium and mesoderm (neuromesodermal progenitors) (24), and in the neural plate may lead to persistent expression of Cdh1, such that expression is not downregulated in a subset of neuroepithelial cells. This idea is consistent with the observation of Cdh1-positive cells in ectopic locations within the tailbud. Given the sparse nature of Cdh1-expressing cells in the neural plate it seems unlikely that they have a significant deleterious effect on closure, particularly as elevation of the neural folds does not appear compromised in Grhl3 overexpressing embryos.
The premature upregulation of markers such as Trp63, Lor and Tgm1, and the abnormal expression of Cdh1, suggest that NTDs caused by Grhl3 gain of function could result primarily from dysregulation of gene expression in the surface ectoderm. A contribution from other Grhl3 expressing tissues appears unlikely as follows: overexpression of Grhl3 in the neural plate occurs prior to closure failure but there is no obvious defect in neural fold elevation (Fig. 3). Grhl3 is expressed in the node-streak border and caudo-lateral epiblast (Fig. 4A) but we found no evidence for altered patterning of the caudal region in Grhl3 overexpressing embryos (Supplementary Material, Fig. S4). Loss of function of Grhl3 in the hindgut alone is sufficient to prevent closure at late stages of spinal neurulation (12). However, in Grhl3 overexpressing embryos, closure fails prior to onset of hindgut expression (Figs 3 and 4) and there is no increase in ventral curvature (Supplementary Material, Fig. S5), the mechanism by which insufficient Grhl3 expression in the hindgut inhibits closure (13).
Overexpression of Grhl3 does not exacerbate cranial NTDs or cause skin barrier defects
In addition to SB, loss of function of Grhl3 has been shown to cause cranial NTDs (exencephaly) and epidermal defects (9,10). We therefore asked whether overexpression of Grhl3 recapitulates these phenotypes.
In Grhl3-/- models, the frequency of exencephaly was reported as 2% and 14% (9,10). Unlike SB this is not markedly higher than in the curly tail strain, in which exencephaly typically affects 6–8% of embryos (25–27). Among Grhl3ct/ct;TgGrhl3/0 intercrosses, exencephaly occurred in a similar proportion of transgene-negative Grhl3ct/ct (13/181; 7.2%) and transgene-positive (Grhl3ct/ct;TgGrhl3/0 and Grhl3ct/ct;TgGrhl3/Grhl3) embryos (39/487; 8.0%). Among 147 of these transgene-positive embryos genotyped by gPCR, the proportion of exencephalic embryos did not differ between BAC-hemizygotes and homozygotes. Hence, in contrast to the striking effect on SB, overexpression of Grhl3 in the cranial region of Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos (confirmed by qRT-PCR; Supplementary Material, Fig. S1C) did not cause a significant increase in frequency of cranial NTDs. Moreover, as we found previously, cranial NTDs in the ct strain were not rescued by upregulation of Grhl3 in the cranial region of Grhl3ct/ct;TgGrhl3/0 embryos (27). These findings support the hypothesis that diminished Grhl3 expression is not the main cause of cranial NTDs in the ct strain and that the underlying mechanism therefore differs from Grhl3-null embryos. Instead, the principal contribution may be from other deleterious variants in the ct genetic background, Lmnb1 and Mthfd1L having been identified as potential modifiers of NTDs in the ct strain (25,27).
Grhl3 is required for differentiation and repair of the epidermal barrier at late fetal stages (10) and for post-natal repair of the epidermal barrier after injury (21). Skin histology of Grhl3-null fetuses becomes abnormal by E16.5, while the epidermal permeability barrier, which normally begins to form at E16–17, fails to develop by E18.5 (10). In the Grhl3 null/transgenic crosses in the current study, we confirmed that the epidermal barrier fails to form in Grhl3-/- fetuses, using a dye penetration assay (Fig. 5A and B). The skin barrier was complete by E18.5 in 6/6 wild-type and 16/16 Grhl3+/- fetuses but not in Grhl3-/- littermates (0/5). Transgenic expression of Grhl3 was sufficient to rescue the epidermal barrier defect phenotype in Grhl3-/- fetuses, both in hemizygous (Grhl3-/-;TgGrhl3/0) and homozygous (Grhl3+/-;TgGrhl3/TgGrhl3) transgenics (3/3 of each genotype tested) (Fig. 5). Known Grhl3 targets in the epidermis were upregulated in skin at E8.5 (Fig. 5D). However, in contrast to the NTDs produced by overexpression of Grhl3, we did not observe epidermal abnormalities in Grhl3+/+;TgGrhl3;TgGrhl3 fetuses (barrier complete in 3/3; Fig. 5). The histological appearance of skin in Grhl3-overexpressing fetuses was also comparable to controls at E18.5, with normal staining for the basal marker p63 (Supplementary Material, Fig. S6).
Figure 5.
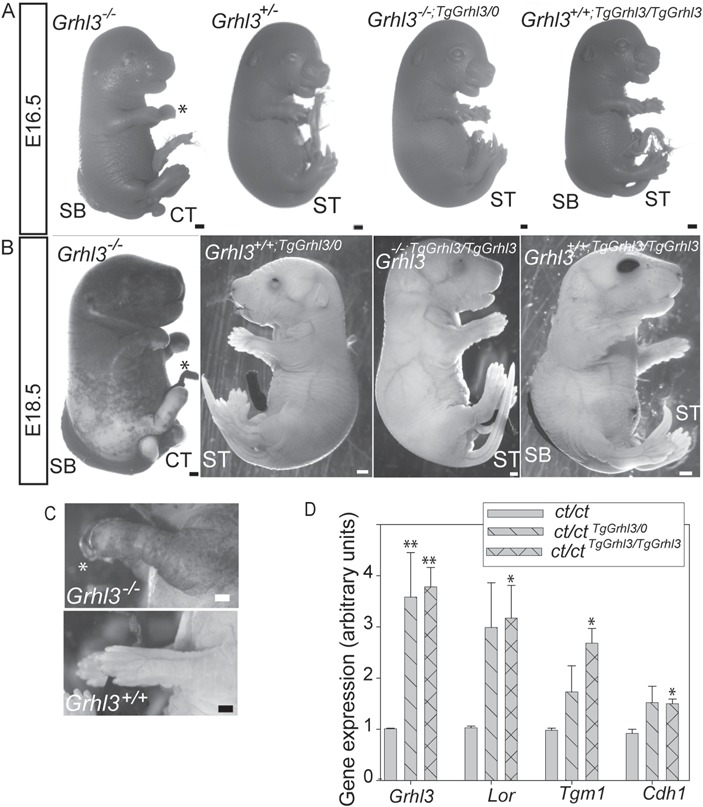
Establishment of the epidermal barrier is not compromised by Grhl3 overexpression. (A–C) Integrity of the permeability barrier was determined by dye penetration assay. At E16.5 (A), all genotypes show dye penetration, whereas at E18.5 (B) Grhl3-/- is the only genotype with incomplete epidermal barrier. Note that barrier formation is rescued by transgenic expression of Grhl3 in Grhl3-/-;TgGrhl3/TgGrhl3 fetuses (in parallel with rescue of NTDs). Although Grhl3 overexpression causes SB, the epidermal barrier is established by E18.5 in hemizygous and homozygous transgenics. Note that limbs appear normal in transgenic fetuses, unlike in Grhl3-/- (*, abnormal forelimbs in B,C). Moreover, partially or completely open eyelids are visible in the transgenic fetuses (B). Scale bar represents 1 mm; CT: curly tail; ST: straight tail. (D) Analysis of gene expression in E18.5 skin samples in litters from compound mutant/transgenic or Grhl3ct/ct;TgGrhl3/0 intercrosses shows that Grhl3 overexpression is associated with increased expression of Lor, Tgm1 and Cdh1 (significant differences from Grhl3ct/ct: *P < 0.05, **P < 0.001; ANOVA).
Additional phenotypes reported in Grhl3-null fetuses include the presence of open eyelids at E18.5 and an abnormal limb phenotype (28). During normal development, the digits become separated by E15 and then undergo a temporary epithelial fusion, with displacement of intervening periderm cells (29). Digit fusion appears to occur normally in Grhl3-null fetuses but the distal limb appears swollen (28) (Fig. 5C). Consistent with normal barrier formation in Grhl3 overexpressing embryos, limb development was also apparently normal (Fig. 5B). In contrast, the open-eye phenotype that accompanies SB in Grhl3-null fetuses was also present in Grhl3 transgenics with SB at E18.5, whether wild type or mutant at the endogenous Grhl3 locus.
Genetic interaction of Grhl3 and Grhl2 overexpression alleles
Grhl2 and Grhl3 both form homodimers but also exhibit protein–protein interactions to form heterodimers (30,31), although the functional role of these interactions is unknown. We speculated that excessive abundance of Grhl3 could disturb the relative abundance of Grhl2 and Grhl3 proteins and favour formation of heterodimers, thereby inhibiting function of Grhl2 homodimers and causing spinal NTDs (as observed in Grhl2-null embryos) (32). Similarly, it could be predicted that spinal NTDs caused by overexpression of Grhl2 in Axd mutants could result from suppression of Grhl3 function. These models predict that overexpressing Grhl2 in Grhl3-transgenic embryos would normalize spinal neural tube closure by compensating for excess of Grhl3. To test this hypothesis we generated embryos that overexpress both Grhl2 and Grhl3, by intercross of Axd/+ and +/+TgGrhl3/0 mice. Spinal NTDs occurred only at very low frequency in single heterozygotes as expected (Fig. 6A). Remarkably, however, a large SB was observed in 100% (n = 19) of doubly heterozygous Axd/+; +/+TgGrhl3 embryos (Fig. 6A–C). Analysis of a further series of embryos collected at E9.0–10.5 showed that doubly heterozygous embryos exhibit failure of closure, with a significantly enlarged PNP from E9.5 (15–19 somites stage) onwards (Fig. 6D). We confirmed that expression of Grhl2 and Grhl3 was elevated in the embryos carrying the Grhl2Axd allele and Grhl3 transgene, respectively (Fig. 6E), without evidence of reciprocal regulation. Hence, occurrence of SB results from an additive genetic interaction of the Grhl2 and Grhl3 alleles, and we can rule out a mechanism of NTDs based on mutual repression of function by gene overexpression.
Figure 6.
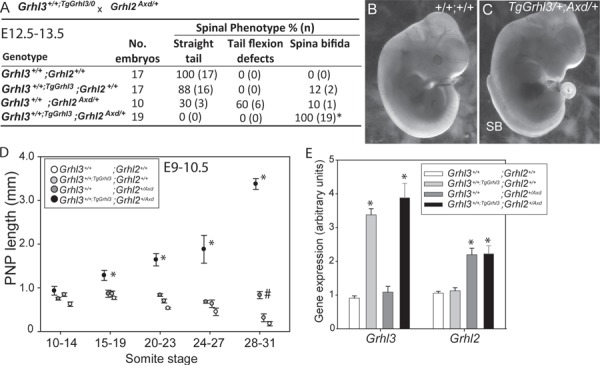
Genetic interaction between Grhl2 and Grhl3 overexpressing alleles results in severe SB. (A–C) Among experimental litters generated by intercross of Grhl2Axd/+ and Grhl3+/+:TgGrhl3/0 mice, all compound heterozygous fetuses exhibited severe SB. Note the very large size of the SB lesion that extends as far rostrally as the forelimb bud at E13.5 (C) (*P < 0.001; significant difference among genotypes, Chi-square). (D) Analysis of PNP length among embryos at E9–10.5 shows that compound heterozygous, Grhl2Axd/+;Grhl3+/+:TgGrhl3/0 embryos have significantly enlarged PNPs from E9.5 (15–19 somite stage) onwards (*, significant difference from all other genotypes, P < 0.001). At E10.5 (28–31 somites) the PNP length of Grhl2Axd/+;Grhl3+/+;TgGrhl3/0 embryos is also greater than in wild type (#, P < 0.01). (E) In the caudal region of E9.5 embryos, mRNA abundance of Grhl2 and Grhl3 corresponds with the presence of the Grhl2Axd and Grhl3TgGrhl3 alleles, with no indication of reciprocal regulation (*, differences from wild-type expression level, P < 0.01).
The Vangl2Lp allele genetically interacts with Grhl3 overexpression to cause SB
We next asked whether moderate overexpression of Grhl3 interacts with another genetic risk factor for NTDs, the loop-tail (Lp) mutation in Vangl2, encoding a core component of the PCP signalling pathway. Homozygosity for Vangl2Lp causes the severe NTD craniorachischisis, (33,34), while heterozygosity for Vangl2 can also cause craniorachischisis or SB when in combination with mutant alleles of other genes including Fzd2, Ptk7, Scrib and Sdc4 (35–37). Previous studies have also demonstrated genetic interaction of Vangl2Lp with the loss of function Grhl3ct- and Grhl3-null alleles (38,39). We intercrossed Vangl2Lp/+ mice with Grhl3ct/ct;TgGrhl3/0 transgenics and analysed litters of embryos at E11.5 (Table 2). TFDs occurred in almost all Vangl2Lp/+;Grhl3ct/+ double heterozygotes, correlating with the almost complete penetrance of this defect in Vangl2 heterozygotes in the loop-tail strain (37). Strikingly, SB developed in 77% of Vangl2Lp/+;Grhl3ct/+;TgGrhl3/0 embryos, but not among embryos of any other genotype, demonstrating additive interaction of Vangl2Lp and the Grhl3-transgene (Table 2a).
Table 2.
Genetic interaction between Grhl3 and Vangl2Lp alleles results in severe SB
| Spinal Phenotype % (n) | ||||
|---|---|---|---|---|
| Genotype | No. embryos | Straight tail | TFDs | SB & TFDs |
|
||||
| Vangl2+/+;Grhl3ct/+ | 4 | 100 (4) | 0 (0) | 0 (0) |
| Vangl2+/+;Grhl3ct/+;TgGrhl3/0 | 11 | 55 (6) | 45 (5) | 0 (0) |
| Vangl2Lp/+;Grhl3ct/+ | 12 | 8 (1) | 92 (11) | 0 (0) |
| Vangl2Lp/+;Grhl3ct/+ TgGrhl3 | 13 | 0 (0) | 23 (3) | 77 (10)* |
| ||||
| Vangl2+/+;Grhl3ct/+ | 14 | 86 (12) | 14 (2) | 0 (0) |
| Vangl2+/+;Grhl3ct/ct | 11 | 55 (6) | 45 (5) | 0 (0) |
| Vangl2+/+;Grhl3ct/+;TgGrhl3/0 | 9 | 78 (7) | 22 (2) | 0 (0) |
| Vangl2+/+;Grhl3ct/ct;TgGrhl3/0 | 8 | 88 (7) | 12 (1) | 0 (0) |
| Vangl2Lp/+;Grhl3ct/+ | 6 | 0 (0) | 100 (6) | 0 (0) |
| Vangl2Lp/+;Grhl3ct/ct | 9 | 0 (0) | 22 (2) | 78 (7) |
| Vangl2Lp/+;Grhl3ct/+;TgGrhl3 | 11 | 0 (0) | 82 (9) | 18 (2) |
| Vangl2Lp/+;Grhl3ct/ct;TgGrhl3 | 11 | 0 (0) | 55 (6) | 45 (5) |
(a) Among experimental litters generated by intercross of Grhl3ct/ct;TgGrhl3/0 with Vangl2Lp/+, SB occurred at high frequency in embryos carrying both the Grhl3 transgene and the Vangl2Lp allele, but was not observed among embryos carrying either allele alone (*P < 0.05; Chi square). (b) Embryos that are heterozygous or homozygous for the Grhl3ct allele but are wild type for Vangl2 display partially penetrant TFDs. Doubly heterozygous (Vangl2Lp/+;Grhl3ct/+) embryos display TFDs, correlating with 100% penetrance of this defect among Lp/+ mice. SB occurs with high frequency among Lp/+;Grhl3ct/ct embryos as reported previously (38). Unlike our finding in Grhl3ct/ct (Gustavsson et al., 2007) and Grhl3-/- embryos (Table 1), this defect is not rescued by the Grhl3 transgene supporting the hypothesis that SB in embryos carrying the Vangl2Lp and Grhl3 transgene, results from an additive genetic interaction. Litters analysed at E11.5 (n = 6 and 9 litters in a and b, respectively).
In a second experimental cross, Vangl2Lp/+;Grhl3ct/+ double heterozygotes were intercrossed with Grhl3ct/ct;TgGrhl3/0 transgenics (Table 2b). Among litters analysed at E11.5 a high frequency of SB occurred in Vangl2Lp/+;Grhl3ct/ct embryos, confirming previous findings (38). However, in contrast to the preventive effect of hemizygosity for the Grhl3-BAC on NTDs in Grhl3ct/ct and Grhl3-/- embryos (Table 1), the presence of the Grhl3-transgene did not prevent the NTDs in Vangl2Lp/+;Grhl3ct/ct embryos (Table 2b). These findings provide further evidence for an exacerbating combinatorial effect of Vangl2 mutation and Grhl3 overexpression on spinal neural tube closure. This does not appear to involve direct regulation of Vangl2 expression by Grhl3 as altered Vangl2 expression was not associated with Grhl3 loss of function in our previous transcriptomic analyses (10,21,27) or in qRT-PCR analysis of Grhl3+/+ and Grhl3-/- embryos at E9.5 (P > 0.05; three or more per genotype). Similarly, Vangl2 expression did not differ between Grhl3ct/ct, Grhl3ct/ct;TgGrhl3/0 or Grhl3ct/ct;TgGrhl3/TgGrhl3 embryos at E9.5 (P > 0.05; three or more per genotype). Moreover, Grhl3 binding sites were not identified in the 10 kb region upstream of Vangl2 (39).
Discussion
A requirement for sufficient Grhl3 expression to enable spinal neural tube closure is shown by the presence of SB in Grhl3-null embryos (10), hypomorphic Grhl3ct/ct embryos (11) and in tissue-specific knockouts (12). In the current study we found that overexpression of Grhl3 also causes SB, emphasizing the exquisite sensitivity of the closure process to the abundance of Grhl3 (Table 3). The induction of SB by excess Grhl3 appears to involve a defect in the surface ectoderm: PNP closure fails soon after initiation of closure when Grhl3 is strongly expressed in the surface ectoderm bordering the PNP and is not associated with a defect of neural fold elevation or altered molecular patterning of the caudal region. Moreover, Grhl3 overexpressing embryos do not exhibit the excess body curvature that results from Grhl3 loss of function in the hindgut and prevents the later stages of PNP closure (12).
Table 3.
Summary of the relationship between spina bifida (SB) frequency and Grhl3 abundance among embryos carrying different combinations of endogenous Grhl3 allele and Grhl3 transgene
| Genotype & NTDs | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Grhl3 locus | -/- | ct/- | ct/ct | +/- | +/+ | ct/- | ct/ct | +/+ | -/- | ct/- | ct/ct |
| Grhl3 transgene | 0/0 | 0/0 | 0/0 | 0/0 | 0/0 | Tg/0 | Tg/0 | Tg/0 | Tg/Tg | Tg/Tg | Tg/Tg |
| SB (%) | 100 | 50 | 10 | 0 | 0 | 0 | 0 | 15 | 38 | 70 | 70 |
| Increasing Grhl3 expression → | |||||||||||
| Grhl3 expression | 0 | ∼0.5 | ∼0.5 | 1 | ∼1.5 | ∼2 | 2.5-3 | ||||
Indicative expression levels (approximate) are based on collation of qRT-PCR data within differing experimental crosses. The frequency of spina bifida (SB) refers to approximate percentage of embryos with SB among each specific genotype.
The Grhl3 transgene is expressed in the endogenous domain during neural tube closure and rescue of SB by the presence of the transgene in Grhl3ct/ct and Grhl3-/- embryos shows that the target genes necessary for neurulation are appropriately regulated. Similarly, rescue of skin barrier defects shows that key epidermal genes are activated in the correct spatiotemporal manner. Grhl3 is known to regulate numerous genes in the epidermis (derived from the surface ectoderm) at late-fetal and post-natal stages, including cell adhesion molecules, lipid metabolizing enzymes and structural proteins of the stratum corneum (10,19–21).
The genome-wide binding pattern of Grhl3 appears dynamic with regulation of distinct sets of genes depending on the functional state of the epidermis. For example, comparison of epidermis during differentiation and during re-establishment of the epidermal barrier following post-natal injury shows overlap of Grhl3 targets, but more than half of the binding events differed (21). The potential for context-dependent and diverse transcriptional regulatory activity of Grhl3 suggests that the requirement during neural tube closure could involve overlapping and/or distinct functions compared with later epidermal differentiation and repair. Notwithstanding the possible difference in Grhl3 targets at different stages, we found a subset of epidermal and epithelial markers to be upregulated in Grhl3 overexpressing embryos at neurulation stages, suggesting a potential dysregulation of surface ectoderm properties. Such an effect could be incompatible with closure, consistent with the site of initial contact of the neural folds being mediated by surface ectoderm cells at the border with the neural plate (40).
The finding that both insufficient and excess abundance of Grhl3 cause SB is similar to observations with Grhl2, null alleles of which cause cranial and spinal NTDs (32,41,42), while upregulation causes SB (32). Interestingly, while Grhl2- and Grhl3-null embryos exhibit multiple defects alongside NTDs (e.g. skin barrier, urothelial differentiation, kidney and placental defects) (10,41,43,44), we found that their overexpression counterparts display isolated SB that more closely resembles the corresponding human condition.
Overall, findings in mouse models suggest that GRHL2 and GRHL3 represent strong candidates for potential involvement in human NTDs, with the consideration that not only loss-of-function variants but also regulatory and gain-of-function mutations could plausibly play a role. In addition to the development of NTDs in homozygous transgenic embryos it is notable that even moderate overexpression of Grhl3 was sufficient to cause SB when in combination with heterozygous mutations in Grhl2 or Vangl2. Such gene–gene interactions appear likely to more closely resemble the multigenic etiology of human NTDs than single gene mutants.
Materials and Methods
Mice
Curly tail mice were maintained as a homozygous, closed random-bred colony. The transgenic curly tail line Grhl3ct/Grhl3ct;Tg(Grhl3)1Ndeg (MGI:3794067), here denoted as Grhl3ct/ct;TgGrhl3/0, carries a BAC that encompasses the Grhl3 gene (11). Homozygous transgenics were generated by intercross of Grhl3ct/ct;TgGrhl3/0 mice. The mutant line carrying a conditional (floxed) allele of Grhl3 (designated Grhl3f/+) has been described (10). These mice were crossed to β-actin-Cre mice to generate heterozygous-null, Grhl3+/-, mice used in subsequent experimental matings. To generate embryos carrying combinations of Grhl3 alleles with the Grhl3-BAC we crossed Grhl3+/- and Grhl3ct/ct;TgGrhl3/0 mice. The offspring with genotype Grhl3ct/-;TgGrhl3/0 were intercrossed to generate experimental litters.
To transfer the Grhl3-BAC onto a partial BALB/c genetic background, Grhl3ct/ct;TgGrhl3/0 mice were backcrossed with wild-type BALB/c mice for three generations. Grhl3+/+;TgGrhl3/0 were either intercrossed to generate experimental litters or crossed with Axd/+ mice on a BALB/c genetic background (32), to generate litters containing double heterozygotes. Mice carrying the loop-tail allele of Vangl2 were on a CBA/Ca background.
Animal studies were carried out under regulations of the Animals (Scientific Procedures) Act 1986 of the UK Government, and in accordance with guidance issued by the Medical Research Council, UK in Responsibility in the Use of Animals for Medical Research (July 1993). Mice were used for experimental matings from 6 weeks of age. Litters were generated by timed matings in which mice were paired overnight and the day of finding a copulation plug was designated embryonic day 0.5 (E0.5). Pregnant females were killed by cervical dislocation. The uterus was removed and transferred to Dulbecco’s Modified Eagles Media (DMEM; Invitrogen) containing 10% fetal bovine serum (heat inactivated at 58°C; Invitrogen).
The PNP length of embryos at E8.5–E10.5 was measured using an eyepiece graticule. The PNP was defined as the distance from the tip of the tail bud to the rostral limit of the open neural folds. For measurements of ventral curvature, the caudal region of E9.5–10.5 embryos was isolated from the body, photographed, and curvature of the caudal region measured as described (13,32). Briefly, as shown in Supplementary Material, Figure S6, the angle was determined in side views of the caudal region between a line tangential to the ventral edge of the penultimate somite and a line drawn along the midline of the tail bud, parallel to and equidistant from the ventral and dorsal surfaces.
Genotyping
Mice were genotyped by PCR of genomic DNA, as described (10, 11,45). Mice carrying the Grhl3 BAC-transgene were genotyped by PCR using a BAC-specific primer (11) and gene dosage determined by quantitative real-time genomic PCR (qG-PCR) on DNA isolated from individual yolk sacs amplified using RealTime PCR Mesa Blue qPCR Master Mix Plus for SYBR assay (EUROGENTEC), or iTAQ Universal SYBR Green Supermix assay (Bio-Rad). A Grhl3 intronic sequence was amplified with normalization to a reference gene (Grhl2), whose copy number is unchanged between strains. Each assay experiment included at least one Grhl3ct/ct sample as calibrator and one hemizygous transgenic sample, for which the genotype was known (BAC-positive embryos from crosses between Grhl3ct/ct and Grhl3TgGrhl3/0 mice). These samples provided an index level for the copy number of Grhl3 in hemizygous transgenic, for comparison with the ‘unknown’ hemizygous or homozygous transgenic BAC-positive samples. PCR amplification of the transgene appeared more efficient than the endogenous Grhl3 locus such that relative signal was 1 (2 endogenous copies), 2 (2 endogenous + 1 transgene) and 3 (2 endogenous + 2 transgene) (Fig. 3C).
BAC localization by inverse PCR
DNA was extracted from transgenic embryos using the QIAamp DNA Mini kit (Qiagen), digested with RsaI (Invitrogen) or HaeIII (Fermentas), circularized and re-linearized with BamHI (Promega). The linearized product was then amplified by PCR with BAC inverse primers (327D13-R1 inverse_5′-CCCTAATGATGACCACGTGA-3′ and pTARBAC-R inverse_5′-TAGTGTCACCTAAATGTCGAC-3′). PCR products were separated on a 1% agarose gel and all obvious bands were excised from the gel and purified using QIAquick Gel Extraction kit (Qiagen). DNA was eluted in water for subsequent sequencing with each of the inverse primers (BigDye Terminator Cycle Sequencing kit, Applied Biosystems).
Sequence tags did not show 100% identity with a specific chromosomal location owing to variation between the unique curly tail genetic background and the C57BL/6 reference sequence. However, sequences generated from inverse PCR were aligned to the reference genomic sequence, with closest homology to a region on chromosome 18 that indicates insertion of the BAC at 18: 3,005,382 in a low complexity repeat. A series of primers (R1–R5; Supplementary Material, Table S5) complementary to chromosome 18 were used to amplify genomic DNA with the BAC-specific primer (pTARBAC-Rinv) (Supplementary Material, Fig. S1). To confirm localization genomic PCR was performed using a BAC specific primer (pTARBAC-R inverse) with a series of primers located in chromosome 18 (Supplementary Material, Table S4).
Preparation of blood cultures and FISH
Interphase nuclei were prepared on slides using peripheral blood from curly tail and hemizygous Grhl3 transgenic male mice. FISH analysis was performed on DAPI-stained interphase nuclei spreads on slides according to standard procedures using the BAC probe RP24-327D13 (BACPAC Resources Center at Childrens Hospital Oakland Research Institute).
Whole mount in situ hybridization was performed as previously reported (25). For sectioning, embryos were embedded in albumin-gelatine and 40 μm sections obtained on a vibratome (Leica VT 1000S, Leica Microsystems). Photography of whole embryos was performed on a stereo microscope (Leica MZFLIII microscope) using a Leica DC500 camera. Bright-field image acquisition of sections was performed with an Axiophot 2 microscope (Zeiss) with Leica DC500 camera software (AxioVision). Images were processed using Photoshop (Version 6.0) for cropping and figures were prepared using Adobe Illustrator software.
Cdh1-positive cells were counted on serial 40 μm sections after WMISH for Cdh1. For each embryo the total number of ectopic cells was divided by the total number of sections. The mean and the standard error of the mean (mean ± SEM) were plotted for different genotypes. To analyse the axial distribution of Cdh1 positive cells in the neuroepithelium, four regions were defined and for each region the total number of positive cells per section was calculated as a percentage of the total number of sections (all embryos combined at each stage).
Whole-mount antibody staining
4% PFA-fixed, methanol-dehydrated whole E8.5–E9.0 embryos were post-fixed (methanol/DMSO) overnight at 4°C. After bleaching (methanol/DMSO/30% H2O2) at room temperature for 4 h, embryos were blocked (phosphate buffered saline (PBS) containing 10% heat-inactivated Sheep serum/2% Bovine Serum Albumin/0.5% Triton X-100) for 4 h. Primary purified mouse anti-Ecadherin (BD Transduction Lab, 1:150 dilution) and relevant secondary (Alexa Fluor 488 goat anti-mouse, Life Technologies, 1/500) antibodies were applied in the same blocking solution overnight at 4°C. All the washes were performed with PBS 0.5% Triton X-100. Embryos were counterstained with DAPI for 2 h at room temperature. Prior to imaging, embryos were incubated in Scale A2 for clearing the tissue. Embryos were positioned in ‘wells’ cut into 4% agarose gels such that the PNP faced upwards. All images were captured on a Zeiss Examiner LSM880 confocal microscope using a 20x/NA1.0 Plan Apochromat dipping objective immersed in PBS. Low-magnification images were captured at 0.7× zoom with a pixel size of 0.6 μm and a Z step of 2 μm. High-magnification images were taken at 2× zoom with a pixel size of 0.1 μm and a Z step of 2 μm. Salt and pepper noise was subtracted, brightness and contrast were adjusted evenly across each image and maximum intensity projections were obtained in Fiji (ImageJ, NIH, PMID 22743772). Digital reslicing of confocal Z-stacks was also performed in Fiji, as previously (46).
Quantitative real-time PCR
RNA was isolated from the caudal region of E8.5 (10–14ss, cut at somite 10), E9.5 (15–16ss, cut at somite 12) and E10.5 (26–31ss, cut at somite 14) embryos, and the cranial region, rostrally from the level of the otocyst (excluding branchial arches). Total RNA was isolated using TRIzol Reagent (Gibco) followed by DNase treatment (DNA-free, Ambion). cDNA was generated using the SuperScript VILO kit (Invitrogen) or SuperScript II Reverse Transcriptase (RT) kit (Invitrogen). Normalization was performed using glyceraldehyde-3-phosphate dehydrogenase (Gapdh) as (11). Quantitative RT-PCR was performed using iTAQ Universal SYBR Green Supermix assay (Bio-Rad) on a CFX96 system (Bio-Rad) with analysis using Bio-Rad CFX Manager software (see Supplementary Material, Table S5 for primer sequences). For each experiment a calibrator sample was chosen to normalize levels of cDNA expression. Individual experiments were combined and analysed using SigmaStat v 3.5 software (ANOVA or t-test).
Supplementary Material
Acknowledgements
The mouse Cdh1 plasmid was provided by Marc Stemmler (Institute of Experimental Medicine I, Nikoloaus-Fiebiger Center for Molecular Medicine).
Conflict of Interest statement. None declared.
Funding
Medical Research Council (G0802163, J003794 to N.G. and A.C.); Child Health Research CIO (to N.G.); Wellcome Trust (087525 to A.C. and N.G., 107474 to G.L.G.); National Institutes of Health Grant (R01AR44882 to B.A.); N.G., P.S. and A.C. are supported by Great Ormond Street Hospital Children's Charity and the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London.
References
- 1. Greene N.D.E., Stanier P. and Copp A.J. (2009) Genetics of human neural tube defects. Hum. Mol. Genet., 18, R113–R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greene N.D. and Copp A.J. (2014) Neural tube defects. Annu. Rev. Neurosci., 37, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ishida M., Cullup T., Boustred C., James C., Docker J., English C., Lench N., Copp A.J., Moore G.E., Greene N.D.E. and Stanier P. (2017) A targeted sequencing panel identifies rare damaging variants in multiple genes in the cranial neural tube defect anencephaly. Clin. Genet., 93, 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ross M.E., Mason C.E. and Finnell R.H. (2017) Genomic approaches to the assessment of human spina bifida risk. Birth Defects Res., 109, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boyles A.L., Hammock P. and Speer M.C. (2005) Candidate gene analysis in human neural tube defects. Am. J. Med. Genet. C. SeminMed. Genet., 135, 9–23. [DOI] [PubMed] [Google Scholar]
- 6. Lemay P., Guyot M.C., Tremblay E., Dionne-Laporte A., Spiegelman D., Henrion E., Diallo O., De M.P., Merello E., Massicotte C. et al. (2015) Loss-of-function de novo mutations play an important role in severe human neural tube defects. J. Med. Genet., 52, 493–497. [DOI] [PubMed] [Google Scholar]
- 7. Harris M.J. and Juriloff D.M. (2007) Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res. A Clin Mol. Teratol., 79, 187–210. [DOI] [PubMed] [Google Scholar]
- 8. Nikolopoulou E., Galea G.L., Rolo A., Greene N.D. and Copp A.J. (2017) Neural tube closure: cellular, molecular and biomechanical mechanisms. Development, 144, 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ting S.B., Wilanowski T., Auden A., Hall M., Voss A.K., Thomas T., Parekh V., Cunningham J.M. and Jane S.M. (2003) Inositol- and folate-resistant neural tube defects in mice lacking the epithelial-specific factor Grhl-3. Nature Med., 9, 1513–1519. [DOI] [PubMed] [Google Scholar]
- 10. Yu Z., Lin K.K., Bhandari A., Spencer J.A., Xu X., Wang N., Lu Z., Gill G.N., Roop D.R., Wertz P. and Andersen B. (2006) The Grainyhead-like epithelial transactivator Get-1/Grhl3 regulates epidermal terminal differentiation and interacts functionally with LMO4. Dev. Biol., 299, 122–136. [DOI] [PubMed] [Google Scholar]
- 11. Gustavsson P., Greene N.D., Lad D., Pauws E., Castro S.C., Stanier P. and Copp A.J. (2007) Increased expression of Grainyhead-like-3 rescues spina bifida in a folate-resistant mouse model. Hum. Mol. Genet., 16, 2640–2646. [DOI] [PubMed] [Google Scholar]
- 12. De Castro S.C.P., Hirst C.S., Savery D., Rolo A., Lickert H., Andersen B., Copp A.J. and Greene N.D.E. (2018) Neural tube closure depends on expression of Grainyhead-like 3 in multiple tissues. Dev. Biol., 435, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brook F.A., Shum A.S.W., Van Straaten H.W.M. and Copp A.J. (1991) Curvature of the caudal region is responsible for failure of neural tube closure in the curly tail (ct) mouse embryo. Development, 113, 671–678. [DOI] [PubMed] [Google Scholar]
- 14. Lemay P., De M.P., Emond A., Spiegelman D., Dionne-Laporte A., Laurent S., Merello E., Accogli A., Rouleau G.A., Capra V. and Kibar Z. (2017) Rare deleterious variants in GRHL3 are associated with human spina bifida. Hum. Mutat., 38, 716–724. [DOI] [PubMed] [Google Scholar]
- 15. Peyrard-Janvid M., Leslie E.J., Kousa Y.A., Smith T.L., Dunnwald M., Magnusson M., Lentz B.A., Unneberg P., Fransson I., Koillinen H.K. et al. (2014) Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am. J. Hum. Genet., 94, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mangold E., Bohmer A.C., Ishorst N., Hoebel A.K., Gultepe P., Schuenke H., Klamt J., Hofmann A., Golz L., Raff R. et al. (2016) Sequencing the GRHL3 coding region reveals rare truncating mutations and a common susceptibility variant for nonsyndromic cleft palate. Am. J. Hum. Genet., 98, 755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Basha M., Demeer B., Revencu N., Helaers R., Theys S., Bou S.S., Boute O., Devauchelle B., Francois G., Bayet B. and Vikkula M. (2018) Whole exome sequencing identifies mutations in 10% of patients with familial non-syndromic cleft lip and/or palate in genes mutated in well-known syndromes. J. Med. Genet., 55, 449–458. [DOI] [PubMed] [Google Scholar]
- 18. Tian T., Wang L., Shen Y., Zhang B., Finnell R.H. and Ren A. (2018) Hypomethylation of GRHL3 gene is associated with the occurrence of neural tube defects. Epigenomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ting S.B., Caddy J., Hislop N., Wilanowski T., Auden A., Zhao L.L., Ellis S., Kaur P., Uchida Y., Holleran W.M. et al. (2005) A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science, 308, 411–413. [DOI] [PubMed] [Google Scholar]
- 20. Hopkin A.S., Gordon W., Klein R.H., Espitia F., Daily K., Zeller M., Baldi P. and Andersen B. (2012) GRHL3/GET1 and trithorax group members collaborate to activate the epidermal progenitor differentiation program. PLoS. Genet., 8, e1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon W.M., Zeller M.D., Klein R.H., Swindell W.R., Ho H., Espetia F., Gudjonsson J.E., Baldi P.F. and Andersen B. (2014) A GRHL3-regulated repair pathway suppresses immune-mediated epidermal hyperplasia. J. Clin. Invest, 124, 5205–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koster M.I., Dai D., Marinari B., Sano Y., Costanzo A., Karin M. and Roop D.R. (2007) p63 induces key target genes required for epidermal morphogenesis. Proc. Natl. Acad. Sci. U. S. A., 104, 3255–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alotaibi H., Basilicata M.F., Shehwana H., Kosowan T., Schreck I., Braeutigam C., Konu O., Brabletz T. and Stemmler M.P. (2015) Enhancer cooperativity as a novel mechanism underlying the transcriptional regulation of E-cadherin during mesenchymal to epithelial transition. Biochim. Biophys. Acta, 1849, 731–742. [DOI] [PubMed] [Google Scholar]
- 24. Henrique D., Abranches E., Verrier L. and Storey K.G. (2015) Neuromesodermal progenitors and the making of the spinal cord. Development, 142, 2864–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Castro S.C., Malhas A., Leung K.Y., Gustavsson P., Vaux D.J., Copp A.J. and Greene N.D. (2012) Lamin b1 polymorphism influences morphology of the nuclear envelope, cell cycle progression, and risk of neural tube defects in mice. PLoS Genet., 8, e1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leung K.Y., De Castro S.C., Savery D., Copp A.J. and Greene N.D. (2013) Nucleotide precursors prevent folic acid-resistant neural tube defects in the mouse. Brain, 136, 2836–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sudiwala S., De Castro S.C., Leung K.Y., Brosnan J.T., Brosnan M.E., Mills K., Copp A.J. and Greene N.D. (2016) Formate supplementation enhances folate-dependent nucleotide biosynthesis and prevents spina bifida in a mouse model of folic acid-resistant neural tube defects. Biochimie, 126, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu Z., Bhandari A., Mannik J., Pham T., Xu X. and Andersen B. (2008) Grainyhead-like factor Get1/Grhl3 regulates formation of the epidermal leading edge during eyelid closure. Dev. Biol., 319, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maconnachie E. (1979) A study of digit fusion in the mouse embryo. J. Embryol. Exp. Morphol., 49, 259–276. [PubMed] [Google Scholar]
- 30. Kudryavtseva E.I., Sugihara T.M., Wang N., Lasso R.J., Gudnason J.F., Lipkin S.M. and Andersen B. (2003) Identification and characterization of Grainyhead-like epithelial transactivator (GET-1), a novel mammalian Grainyhead-like factor. Dev. Dyn., 226, 604–617. [DOI] [PubMed] [Google Scholar]
- 31. Ting S.B., Wilanowski T., Cerruti L., Zhao L.L., Cunningham J.M. and Jane S.M. (2003) The identification and characterization of human Sister-of-Mammalian Grainyhead (SOM) expands the grainyhead-like family of developmental transcription factors. Biochem. J., 370, 953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brouns M.R., Castro S.C., Terwindt-Rouwenhorst E.A., Massa V., Hekking J.W., HIrst,C.S., Savery D., Munts C., Partridge D., Lamers W., et al. (2011) Over-expression of Grhl2 causes spina bifida in the Axial defects mutant mouse. Hum. Mol. Genet., 20, 1536–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murdoch J.N., Doudney K., Paternotte C., Copp A.J. and Stanier P. (2001) Severe neural tube defects in the loop-tail mouse result from mutation of Lpp1, a novel gene involved in floor plate specification. Hum. Mol. Genet., 10, 2593–2601. [DOI] [PubMed] [Google Scholar]
- 34. Kibar Z., Vogan K.J., Groulx N., Justice M.J., Underhill D.A. and Gros P. (2001) Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nature Genet., 28, 251–255. [DOI] [PubMed] [Google Scholar]
- 35. Juriloff D.M. and Harris M.J. (2012) A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res. A Clin. Mol. Teratol., 94, 824–840. [DOI] [PubMed] [Google Scholar]
- 36. Escobedo N., Contreras O., Munoz R., Farias M., Carrasco H., Hill C., Tran U., Pryor S.E., Wessely O., Copp A.J. and Larrain J. (2013) Syndecan 4 interacts genetically with Vangl2 to regulate neural tube closure and planar cell polarity. Development, 140, 3008–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Murdoch J.N., Damrau C., Paudyal A., Bogani D., Wells S., Greene N.D., Stanier P. and Copp A.J. (2014) Genetic interactions between planar cell polarity genes cause diverse neural tube defects in mice. Dis. Model. Mech., 7, 1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stiefel D., Copp A.J. and Meuli M. (2007) Fetal spina bifida: loss of neural function in utero. J. Neurosurg., 106, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caddy J., Wilanowski T., Darido C., Dworkin S., Ting S.B., Zhao Q., Rank G., Auden A., Srivastava S., Papenfuss T.A. et al. (2010) Epidermal wound repair is regulated by the planar cell polarity signaling pathway. Dev. Cell, 19, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rolo A., Savery D., Escuin S., De Castro S.C., Armer H.E., Munro P.M., Mole M.A., Greene N., Copp A.J. (2016) Regulation of cell protrusions by small GTPases during fusion of the neural folds. Elife, 5, e13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Werth M., Walentin K., Aue A., Schonheit J., Wuebken A., Pode-Shakked N., Vilianovitch L., Erdmann B., Dekel B., Bader M. et al. (2010) The transcription factor grainyhead-like 2 regulates the molecular composition of the epithelial apical junctional complex. Development, 137, 3835–3845. [DOI] [PubMed] [Google Scholar]
- 42. Rifat Y., Parekh V., Wilanowski T., Hislop N.R., Auden A., Ting S.B., Cunningham J.M. and Jane S.M. (2010) Regional neural tube closure defined by the Grainy head-like transcription factors. Dev. Biol, 345, 237–245. [DOI] [PubMed] [Google Scholar]
- 43. Yu Z., Mannik J., Soto A., Lin K.K. and Andersen B. (2009) The epidermal differentiation-associated Grainyhead gene Get1/Grhl3 also regulates urothelial differentiation. EMBO J., 28, 1890–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walentin K., Hinze C., Werth M., Haase N., Varma S., Morell R., Aue A., Potschke E., Warburton D., Qiu A. et al. (2015) A Grhl2-dependent gene network controls trophoblast branching morphogenesis. Development, 142, 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Copp A.J., Checiu I. and Henson J.N. (1994) Developmental basis of severe neural tube defects in the loop-tail (Lp) mutant mouse: Use of microsatellite DNA markers to identify embryonic genotype. Dev. Biol., 165, 20–29. [DOI] [PubMed] [Google Scholar]
- 46. Galea G.L., Cho Y.J., Galea G., Mole M.A., Rolo A., Savery D., Moulding D., Culshaw L.H., Nikolopoulou E., Greene N.D.E. and Copp A.J.. (2017). Biomechanical coupling facilitates spinal neural tube closure in mouse embryos. Proc. Natl. Acad. Sci. U. S. A., 114, E5177–E5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.