

Abstract
Commonalities and, in some cases, pathological overlap between neurodegenerative diseases have led to speculation that targeting of underlying mechanisms might be of potentially shared therapeutic benefit. Alzheimer’s disease is characterized by the formation of plaques, composed primarily of the amyloid-β 1-42 (Aβ) peptide in the brain, resulting in neurodegeneration. Previously, we have shown that overexpression of the lysosomal-trafficking protein, human Vps41 (hVps41), is neuroprotective in a transgenic worm model of Parkinson’s disease, wherein progressive dopaminergic neurodegeneration is induced by α-synuclein overexpression. Here, we report the results of a systematic comparison of hVps41-mediated neuroprotection between α-synuclein and Aβ in transgenic nematode models of Caenorhabditis elegans. Our results indicate that an ARF-like GTPase gene product, ARL-8, mitigates endocytic Aβ neurodegeneration in a VPS-41-dependent manner, rather than through RAB-7 and AP3 as with α-synuclein. Furthermore, the neuroprotective effect of ARL-8 or hVps41 appears to be dependent on their colocalization and the activity of ARL-8. Additionally, we demonstrate that the LC3 orthologue, LGG-2, plays a critical role in Aβ toxicity with ARL-8. Further analysis of functional effectors of Aβ protein processing via the lysosomal pathway will assist in the elucidation of the underlying mechanism involving VPS-41-mediated neuroprotection. These results reveal functional distinctions in the intracellular management of neurotoxic proteins that serve to better inform the path for development of therapeutic interventions to halt neurodegeneration.
Introduction
Alzheimer’s disease (AD) is characterized by the formation of extracellular plaques, comprising aggregates of the amyloid precursor protein (APP) N-terminal cleavage product, β-amyloid (Aβ) and intracellular neurofibrillary tangles of tau protein (1,2). APP is directed to the plasma membrane through the secretory pathway where cleavage may occur but may also be internalized and trafficked to the early endosome (EE). In the EE, APP can undergo further processing or be directed to the Golgi through the retromer complex (3). Extracellular Aβ peptide stimulates internalization through interaction with various cell-surface proteins; it is thereby internalized by endocytosis and accumulates in endolysosomal compartments (4–6). The EE serves as a hub of vesicular trafficking for the trans-Golgi or plasma membrane (Fig. 1) and matures into a late-endosomal component termed the multivesicular body (MVB), which acidifies and accepts cargoes of digestive enzymes prior to lysosome fusion (7). Lysosomal acidity stabilizes Aβ oligomer formation, which destabilizes the MVB membrane, causing leakage of its contents into the cytosol (8,9). Thus, efficacious clearance of the MVB minimizes disruption by Aβ oligomers.
Figure 1.
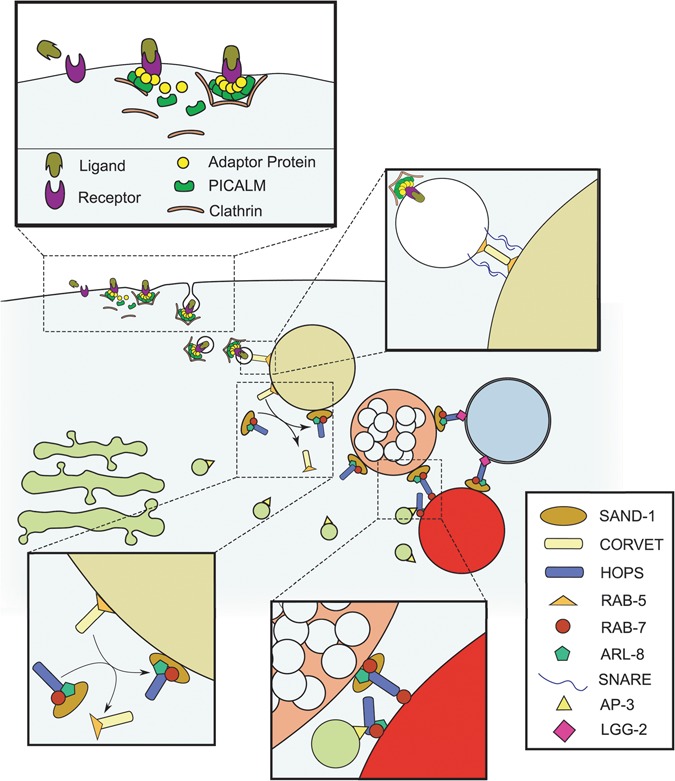
The endolysosomal trafficking system. Schematic diagram representing the proteins discussed and/or examined in this investigation is shown. Adaptor proteins stabilize the recruitment of clathrin to a ligand-receptor complex to stimulate its internalization by endocytosis. This process is facilitated by the PICALM/UNC-11 protein. Subsequently, internalized vesicles are trafficked to the EE, where SNARE-mediated tethering and fusion is facilitated by the CORVET complex, along with RAB-5. As the EE accumulates delivered cargoes, the increasing abundance of phosphatidylinositol-3-phosphate in the EE membrane stabilizes the recruitment of the SAND-1/Mon1-Ccz1 complex, which brings with it the HOPS complex (which includes VPS-41), RAB-7 and ARL-8. This process coincides with the progressive acidification of the endosome, in addition to the accumulation of intraluminal vesicles that define the LE. The HOPS complex tethers the LE with the lysosome through RAB-7 or ARL-8 and stabilizes the recruitment of SNARE proteins for homotypic fusion. On the lysosomal surface, the HOPS complex also mediates the tethering and fusion of autophagosomes with the lysosome through its interactions with LC3/LGG-2 and RAB-7 or ARL-8. Additionally, the AP3-interacting domain of VPS-41 of the HOPS complex on the lysosomal surface can be made accessible to AP3 coating the vesicles arriving from the trans-Golgi.
Fusion of the MVB to the lysosome requires cooperation of Rab GTPases and the homotypic fusion and vacuolar protein sorting (HOPS) complex (Fig. 1). Previously, we showed that deficiency in the HOPS complex protein VPS-41 increased α-synuclein-induced degeneration of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease (PD). Furthermore, overexpression of human Vps41 (hVps41) attenuated neurodegeneration and was dependent on the AP3 complex, which coats trans-Golgi vesicles targeted to the lysosome (10,11). The activity of endosomal system components in the context of disease-associated proteins (i.e. Aβ, α-synuclein) represents a molecular intersection clearly perturbed in neurodegenerative disorders (12,13). Precisely how these components function to defend the cell from Aβ toxicity is poorly understood.
Aβ has been observed directly in endosomal compartments and redistributed upon manipulation of specific endosomal components (6). Genome-wide association studies have identified a correlation between AD and the phosphatidylinositol-binding clathrin assembly protein (PICALM) in endocytosis (14–18). We previously reported that overexpression of PICALM and other endocytic factors in yeast, primary neuron cultures and C. elegans reduced Aβ toxicity (13). In cell cultures expressing defective Rab5, Aβ is dispersed within the cell, but with constitutively active Rab5, Aβ is trapped and accumulates within the EE (6). Accumulation of Aβ within the MVB is exacerbated by lysosomal dysfunction, but changes in Rab7 activity do not alter the total concentration of cell-associated Aβ, suggesting alternative late-endosomal pathways contribute to the lysosomal trafficking of Aβ.
Considering the role of endosomal trafficking in Aβ toxicity, we hypothesized that overexpression of hVps41 would reduce neurodegeneration. Because neuroprotection by hVps41 in the α-synuclein model was dependent on rab-7 and apd-3 (11), we sought to examine whether the neuroprotection conferred by hVps41 is similarly recapitulated in the Aβ model. Here, we employed transgenic C. elegans to dissect the functional role of VPS-41 in attenuating proteotoxicity of two different neurodegenerative disorders: PD and AD. C. elegans models have been proven informative in advancing mechanistic aspects of both AD (19) and PD (20) that have been recapitulated in mammalian and cell culture models. We demonstrate that overexpression of hVps41 rescues Aβ-mediated neurodegeneration in vivo, but neurodegeneration occurs independently from either rab-7 orapd-3. Unlike with α-synuclein, the Arf-like GTPase, ARL-8, appears to modulate Aβ toxicity. Here, we provide evidence to suggest that hVPS-41-mediated protection occurs at the intersection between endosomal, lysosomal, and autophagic machinery. Differences underlying the molecular pathology of neurodegenerative diseases are partly a manifestation of subtle distinctions in functional interactions between pathogenic proteins and their selective effects on intracellular trafficking.
Results
Vps41 modulates Aβ toxicity in vivo
In a systematic screen for modifiers of α-synuclein toxicity in C. elegans, we had identified vps-41 and found that overexpression of hVps41 reduced neurodegeneration in both C. elegans and mammalian models of PD (21). We further determined that this neuroprotective effect was dependent on rab-7 and interactions with AP3 (10,11). Because endocytic Aβ predominantly occupies and destabilizes the endosomes, we hypothesized that Vps41 also serves a critical function in mitigating Aβ toxicity. Since both amyloidogenic proteins, Aβ and α-synuclein, are prone to assemble into pore-forming oligomers in membranes (22–26), we considered that attenuation of Aβ by hVps41 required the same cellular mechanisms as the α-synuclein model.
As a preliminary examination of vps-41 in Aβ toxicity, a C. elegans model was used in which Aβ expression is restricted to the body-wall muscles to induce paralysis. Two variations of this model have been generated. In the first, Aβ expression is permitted by shifting the incubation temperature up from 16°C to 23°C (27). This background is useful for expedient assaying of targets and distinguishing rescuing effects of conditions. Using RNA interference (RNAi), we examined how depletion of vps-41 affected paralysis. Animals grown on empty vector (EV) control RNAi exhibited a steady increase in paralysis from 26 h post-upshift to 32 h post-upshift, with 50% of animals being paralyzed by 30 h, while with depletion of vps-41, more than 50% were paralyzed at 28 h post-upshift and followed by a significant decline until all animals were paralyzed by 32 h. (Fig. 2A). Conversely, we observed that overexpression of hVps41 significantly attenuated the paralysis phenotype, whereby there was little paralysis observable until approximately 32 h post-upshift, at which point about 30% remained not paralyzed.
Figure 2.
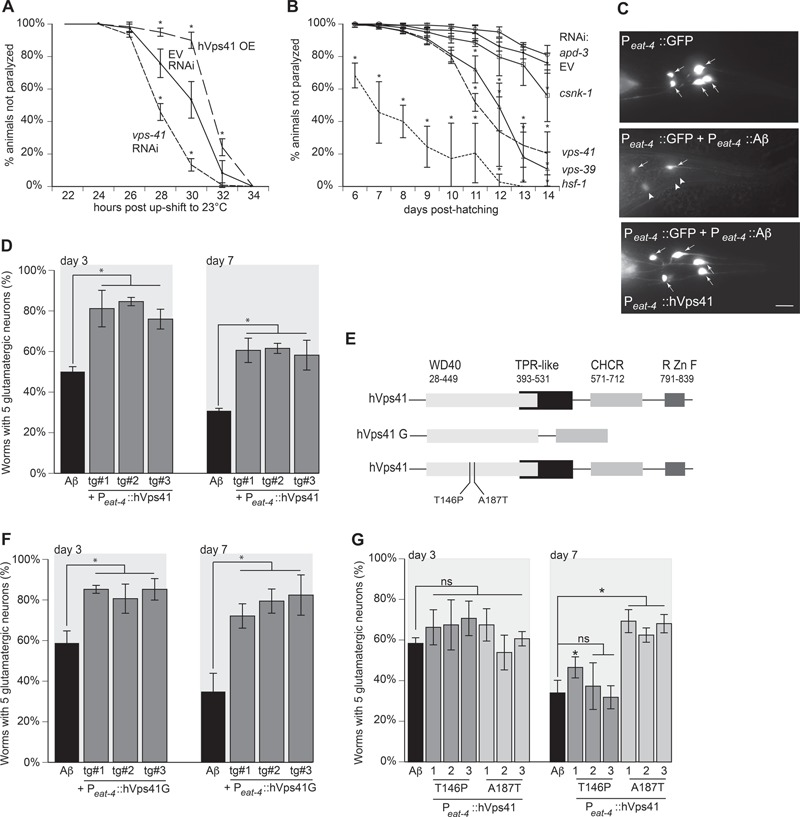
VPS-41 modulates Aβ toxicity. (A) Inducible overexpression of Aβ peptide in C. elegans body wall muscles occurs in worm strain CL4176 ((smg-1(cc546ts) I; dvIs27 [Pmyo-3::Aβ + rol-6(su1006)) whereby a temperature upshift from 20°C to 23°C induces rapid paralysis. Co-overexpression of hVps41 reduces paralysis (F(12,36) = 24.99, P < 0.0001 at 30 h post-upshift compared to the EV control; two-way ANOVA with a Tukey's post-hoc analysis), whereas vps-41 RNAi increases paralysis, P < 0.0001 at 30 h post-upshift compared to EV control. (B) In a separate transgenic strain, CL2006 [dvIs2[pCL12(Punc-54::Aβ) + pRF4], where constitutive overexpression of Aβ in C. elegans body wall muscle cells results in progressive paralysis; RNAi depletion of vps-41 or vps-39 results in increased paralysis over time (F(40,108) = 7.861, P < 0.0001 for both, at day 12, one-way ANOVA with a Tukey’s post-hoc analysis) compared to EV RNAi control. Additionally, depletion of hsf-1, which encodes a gene product that regulates Aβ-interacting chaperones, also displayed increased paralysis as a result of enhanced Aβ toxicity (P < 0.0001), while RNAi knockdown of apd-3 and csnk-1 did not exhibit increased paralysis (ns, not significantly different compared to EV control; P = 0.777, P = 0.933). (C) Worms overexpressing Aβ and GFP in glutamatergic neurons (UA198 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP])) have significant degeneration that was rescued with the overexpression of hVps41 in the glutamatergic neurons. The five tail glutamatergic neurons (one PVR, two LUA and two PLM neurons) were scored for neurodegeneration, as shown in these representative images of GFP alone, GFP + Aβ, or GFP + Aβ + full-length hVps41 (isoform 1). Arrows indicate the normal neurons, and arrowheads indicate degenerating neurons. Scale bar represents 10 μm. (D) Quantitation of three separate transgenic lines overexpressing Aβ and GFP in glutamatergic neurons (UA198 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP])). All three lines were significantly different from Aβ control on day 3 [tg#1 P = 0.0002; tg#2 P < 0.0001; tg#3 P = 0.0003 (n = 90 for each line)]. Likewise, on day 7, the transgenic lines were also significantly different from Aβ control [tg#1 P = 0.0006; tg#2 P = 0.0005; tg#3 P = 0.001 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. (E) Schematic diagram illustrating the domains of hVps41 full-length isoform 1, which consists of the AP3 interaction (WD40), tetratricopeptide repeat-like motif, CHCR, and RING zinc finger and hVps41 truncate G (11). Two SNPs found within the WD40 domain are also denoted. (F) Worms overexpressing Aβ and GFP in glutamatergic neurons (UA198 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP])) have significant degeneration that was rescued with the overexpression of hVps41 truncate G into the glutamatergic neurons. The five tail glutamatergic neurons were scored for neurodegeneration. All three transgenic lines were significantly different from Aβ control on day 3 [tg#1 P = 0.0003; tg#2 P = 0.0012; tg#3 P = 0.0003 (n = 90 for each line)]. Likewise, on day 7, the transgenic lines were also significantly different from Aβ control [tg#1 P = 0.0005; tg#2 P = 0.0001; tg#3 P < 0.0001 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. (G) Worms overexpressing Aβ and GFP in glutamatergic neurons (UA198 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP])) have significant degeneration. These strains were crossed with animals expressing hVps41 alleles T146P or A187T to create the following two strains: UA345 (baEx191[Peat-4::hVPs41 T146P, Punc-54::tdTomato]; baIn34([Peat-4:: Aβ,Pmyo-2::mCherry];adIs1240[Peat-4::GFP]) and UA346 (baEx192[Peat-4::hVPs41 A187T, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ,Pmyo-2::mCherry];adIs1240[Peat-4::GFP]). The five tail glutamatergic neurons were scored for neurodegeneration. All three transgenic T146P hVps41 lines were non-significantly different from Aβ control on day 3 [tg#1 P = 0.2609; tg#2 P = 0.1994; tg#3 P = 0.0898]. Similarly, all three A187T hVps41 transgenic lines were non-significant from Aβ control at day 3 [tg#1 P = 0.747; tg#2 P = 0.199; tg#3 P = 0.518 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. On day 7, one of the three T146P hVps41 transgenic lines displayed rescue, while the other two were still non-significantly different from Aβ control [tg#1 P = 0.046; tg#2 P = 0.569; tg#3 P = 0.714 (n = 90 for each line)]; however, the three A187T hVps41 transgenic lines were significantly different from Aβ control on day 7 [tg#1 P = 0.003; tg#2 P < 0.0001; tg#3 P = 0.0002 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. These data are reported as mean + S.D., and * denotes statistical significance as detailed within each section of this figure legend.
In the second paralysis model, we evaluated Aβ-induced paralysis in conjunction with the variable of aging, since this assay occurs across developmental stages rather than over a few hours/one larval stage (28). As a preliminary probe to distinguish the role of vps-41 in this Aβ model, RNAi was used to deplete vps-41, vps-39, and the AP3 δ subunit, apd-3 (Fig. 2B). By 11 days post-hatching, depletion of vps-39 or vps-41 significantly increased paralysis by at least 20% compared to EV and further increased by as much as 50% by 13 days post-hatching. Though depletion of apd-3 previously increased α-synuclein-mediated neurodegeneration (11), we did not observe an increase in Aβ-induced paralysis upon depletion of apd-3, suggesting the role of HOPS in the Aβ model diverges from its function in the α-synuclein model. Similarly, depletion of the casein kinase 1, Yck3/csnk-1, which phosphorylates Vps41 to allow it to interact with AP3 (29), did not increase Aβ-induced paralysis (Fig. 2B).
Vps41 overexpression attenuates Aβ-dependent glutamatergic neurodegeneration
Considering the limitations of modeling a toxic neural protein in muscle cells, we turned our attention to a neuronal model of Aβ in order to further distinguish how vps-41 functions in Aβ toxicity compared to α-synuclein toxicity. Because glutamatergic neurons are highly susceptible to Aβ toxicity in AD, we utilized a model of Aβ neurotoxicity in which secreted Aβ expression is driven from the glutamatergic neuron-specific eat-4 promoter in transgenic C. elegans. The five neurons of the posterior glutamatergic circuitry provide an expedient output for scoring neurodegeneration, as they are anatomically distinct, readily observable and quantifiable, and begin to exhibit progressive degeneration early in life (13). By day 3, only approximately 50% of animals expressing Aβ exhibit all five glutamatergic posterior neurons, and by day 7, as few as 30% of the population express the normal neuronal complement. In comparison, at either day 3 or day 7, glutamatergic Aβ animals also overexpressing hVps41 exhibited at least 25% less neurodegeneration than the Aβ-only populations (Fig. 2C and D). Previously, we examined which domains of hVps41 were minimally necessary for producing the rescuing phenotype in the α-synuclein model. We found that a truncate (hVPS41G), which possessed only the WD40 and clathrin heavy chain repeat (CHCR) domains, still robustly rescued α-synuclein-induced neurodegeneration (Fig. 2E) (11). Similarly, overexpression of the hVps41G truncate also rescued Aβ-induced neurodegeneration by at least 25% at both days 3 and 7 (Fig. 2F). Likewise, we previously examined two hVps41 variants, T146P and A187T (Fig. 2E), and discerned that both single nucleotide polymorphisms (SNPs) negated the neuroprotective property afforded by hVps41 in a C. elegans model of α-synuclein-induced neurodegeneration (11). When probed for their effect on Aβ toxicity, the T146P variant also failed to rescue at day 3 and produced a marginal difference at day 7; however, A187T was significantly protective at day 7 (Fig. 2G). Together, these results show that vps-41 impairment significantly increases toxicity of Aβ and that overexpression of hVps41 attenuates Aβ toxicity across multiple C. elegans tissue models.
Evaluation of Aβ-induced neurodegeneration using neuron-specific RNAi
To further compare the functional role of hVps41 in the Aβ model to the α-synuclein model, we generated an RNAi-sensitive Aβ model in which genetic depletion is restricted to neurons (11). A null mutant of the gene encoding the dsRNA transporter, sid-1, is resistant to RNAi. By driving expression of sid-1 from the pan-neuronal unc-119 promoter in sid-1 mutant animals, the neurons are made conditionally sensitive to RNAi (30). This selectively RNAi-sensitive strain was crossed with the neuronal Aβ model, producing a genetic background that allowed us to examine neurodegeneration as a response to RNAi restricted to the nervous system. This strategy facilitates analysis of post-developmental functions of gene products, including those that are otherwise systemically essential (11).
To distinguish increases in neurodegeneration in response to depletion of targets in the newly developed RNAi strain, we analyzed differences in neurodegeneration between treatment groups 48 h post-egg-lay. As a control for effective RNAi, we examined neurodegeneration as a product of depletion of heat shock factor 1 (hsf-1) or Aβ. Not only has depletion of hsf-1 been shown to increase Aβ-mediated paralysis (Fig. 2B), it has also been shown to regulate the expression of chaperone proteins that disaggregate Aβ oligomers (31,32). In the neuronal RNAi model of Aβ-mediated glutamatergic neurodegeneration, all five posterior glutamatergic neurons were scored as normal in 45% of control animals. Depletion of Aβ decreases neurodegeneration in the population by about 20%, and depletion of hsf-1 increased neurodegeneration by about 20% (Fig. 3A). Thus, this represents an effective background for screening genetic modulators of Aβ toxicity.
Figure 3.
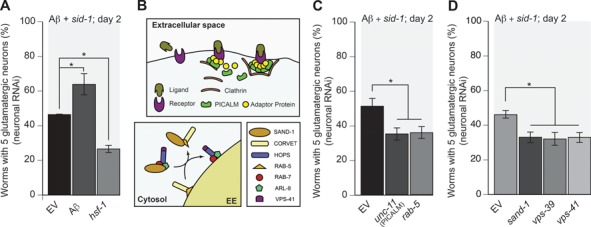
Aβ toxicity is mitigated by the endosomal pathway. (A, C, D) Selective RNAi knockdown of endosomal components in C. elegans that also express Aβ in glutamatergic neurons using the pan-neuronal RNAi-sensitive strain UA338. (A) Depletion of Aβ or hsf-1 confirms the efficacy of RNAi in neurons of a C. elegans model of Aβ-induced neurodegeneration (UA338 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321); uIS69 [Pmyo-2::mCherry; Punc-119::sid-1])) (P = 0.0341; P = 0.0158, respectively; n = 90 for each line; one-way ANOVA with a Fisher’s LSD test). (B) An illustration of the roles and positions in the endocytic pathway of the respective genes targeted by RNAi. (C). Depletion of rab-5 and PICALM/unc-11, which has been shown to rescue Aβ-induced neurodegeneration when overexpressed, increases neurodegeneration compared to the EV RNAi control (P = 0.0303; P = 0.0307, respectively; n = 90 for each line; one-way ANOVA with a Fisher’s LSD test). (D) Suppressing the recruitment of RAB-7 and maturation of the EE into the late endosome by depletion of sand-1 increased neurodegeneration (P = 0.0455; n = 90; one-way ANOVA with a Fisher’s LSD test). Depletion of vps-39 and vps-41, key components of the HOPS complex, also increased neurodegeneration (P = 0.0195; P = 0.025, respectively; n = 90 for each line; one-way ANOVA with a Fisher’s LSD test). These data are reported as mean + S.D., and * denotes statistical significance as detailed within each section of this figure legend.
The early endosomal pathway significantly impacts Aβ toxicity
Despite the association of amyloid plaques with AD, insoluble amyloid plaques do not exhibit significant toxicity. Rather, oligomers formed by intracellular Aβ at the endosomes or lysosomes are extremely toxic (8,9,33,34). Notwithstanding, internalization of Aβ by endocytosis has proven highly relevant to disease mechanisms, as evidenced by Aβ binding to surface proteins to stimulate endocytosis and alterations in gene expression (4–6,13,35–43). In cell culture, apoptosis induced by exogenous supplementation of Aβ is dependent on endocytosis (35–37). Using superresolution fluorescence imaging, extracellular Aβ was found to be rapidly internalized and localized to lysosomes and endosomal systems (43). Notably, overexpression of PICALM/UNC-11, a protein associated with AD (15,17,40), significantly reduced neurodegeneration in yeast, animal and cell culture models (13,15,17,41). PICALM/UNC-11 stabilizes clathrin assembly and adaptor protein recruitment at the cell surface to encourage endocytic bud formation (Fig. 3B). Shortly after internalization, an endocytic vesicle is trafficked to the EE for sorting. Tethering and fusion of the endocytic vesicle is coordinated by the class C core vacuole/endosome tethering (CORVET) complex with RAB-5, a GTPase that decorates the surface of the EE. In cell culture, colocalization of Aβ to the EE is dependent on Rab5 activity. With expression of a Rab5 dominant negative mutant, Aβ is internalized, but is dispersed (6). Similarly, we observed increased neurodegeneration in our Aβ transgenic nematodes with depletion of either PICALM/unc-11 or rab-5 (Fig. 3C).
As phosphatidylinositol-3-phosphate abundance in the EE membrane increases, recruitment of the RAB7 GEF component SAND-1/Mon1p, which is complexed with Ccz1, to the endosomal membrane is stabilized, bringing with it the Rab7 GTPase and the HOPS complex (Fig. 3B). The Rab5 guanosine exchange factor, Rabex-5, is displaced by SAND-1/Mon1-Ccz1p and the recruitment of HOPS promotes Rab7 activity, thus destabilizing Rab5 and the CORVET presence at the endosome (7). In vivo studies of these complexes and analysis of their potential impact on neurodegeneration have previously been limited. Here, through the selective post-developmental knockdown made possible in our Aβ model, we demonstrated that depletion of sand-1, vps-39 or vps-41 increased neurodegeneration (Fig. 3D). Because SAND-1/Mon1 is necessary for proper late endosome (LE) development and RAB-7 function, perturbation of SAND-1/Mon1p putatively precludes HOPS and RAB-7 function (44). However, depletion of any of these targets in an RNAi-sensitive background without Aβ did not exhibit neurodegeneration (data not shown).
Depletion of rab-7 or AP3 alone is insufficient to reduce Aβ toxicity
Previous work in yeast identified Vps41 as a protein involved in the tethering and fusion of the LE and AP3-coated vesicles of the trans-Golgi to the vacuole (45,46). Vps41 is stabilized in the HOPS complex by positive feedback from the nucleotide transfer and GTPase functions from the interactions of Vps39 and Rab7, respectively, whereupon it stimulates membrane fusion of the vesicle and vacuole (46–48). At the lysosome, Vps41 also mediates the tethering and fusion of AP3-coated vesicles (Fig. 4A). Vesicle–lysosome fusion through HOPS has been predominantly observed as dependent on Rab7. Despite this, we did not observe an increase in neurodegeneration with RNAi depletion of rab-7 in glutamatergic neurons expressing Aβ (Fig. 4B). To confirm this, we analyzed Aβ animals that were crossed with a rab-7 null mutant background. Similarly, we observed no significant increase in neurodegeneration of rab-7 null mutants compared to the control (Fig. 4C). This bears stark contrast to the C. elegansα-synuclein model. As noted in our previous study, depletion of rab-7 increased α-synuclein-induced dopaminergic neurodegeneration (11). Furthermore, endocytic α-synuclein has been found to colocalize with Rab5 and Rab7, while Rab7 overexpression expedited α-synuclein clearance (49,50). Similarly, we observed that α-synuclein-mediated neurodegeneration was exacerbated by the rab-7 mutation (Fig. 4D and E). Our observations suggest that although hVps41 is able to rescue neurodegeneration in two separate models of proteotoxicity, the precise mechanism by which it may function in its protective capacity is different between them.
Figure 4.
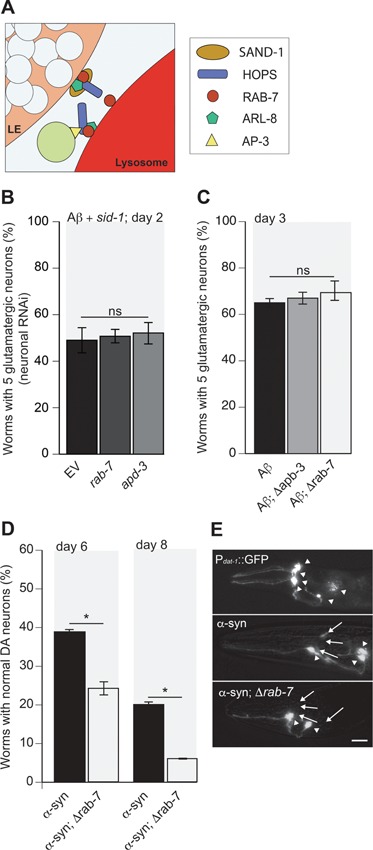
AP3 and RAB-7 do not alleviate Aβ toxicity. (A) An illustration of the roles and positions of AP3 and RAB-7 in the endocytic pathway. (B) Depletion of either rab-7 or apd-3 via pan-neuronal RNAi knockdown in worm strain UA338 did not significantly increase Aβ-mediated glutamatergic neurodegeneration (P = 0.6312, P = 0.2623, respectively; n = 90 for each line; one-way ANOVA with a Fisher’s LSD test). (C) Worms expressing Aβ and GFP in glutamatergic neurons (UA198 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry];adIs1240[Peat-4::GFP])) were crossed with rab-7 or apd-3 mutant animals to produce UA340 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; apb-3[ok429)]) and UA341 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]); (ok511)/mIn1 [mIs14 dpy-10(e128)]), respectively. Resulting homozygous animals were synchronized and analyzed at day 3 post-hatching. Loss of either AP3 by a null mutation of the AP3 β-subunit, apb-3 (Δapb-3), or rab-7 (Δrab-7) failed to impact neurodegeneration in the Aβ background (T(5) = 0.888, P = 0.4332; T(5) = 1.04, P = 0.3484, respectively; n = 90 for each line; Student’s t-test). (D) The rab-7 mutant was also crossed with animals expressing α-synuclein (α-syn) specifically in the dopaminergic neurons using worm strain UA44 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]) to create UA342 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]; (ok511)/mIn1 [mIs14 dpy-10(e128)]). Degeneration of dopaminergic neurons was enhanced by loss of rab-7 compared to α-syn-expressing neurons alone. On days 6 and 8, T(5) = 3.76, P = 0.013 and T(5) = 5.422, P = 0.003, respectively; n = 90 in both cases; Student’s t-test. (E) Representative images of GFP-labeled dopamine neurons (top) and α-syn-induced neurodegeneration (middle), which is enhanced in the rab-7 mutant background (bottom). Arrowheads indicate normal neurons while arrows indicate either degenerating neurons or regions where neurons are completely missing; scale bar represents 20 μm. These data are reported as mean + S.D., and * denotes statistical significance as detailed within each section of this figure legend.
Importantly, in our model of α-synuclein-mediated neurodegeneration, loss of the AP3 complex activity, either through depletion of apd-3 or loss of the δ subunit apb-3, increased neurodegeneration and negated the neuroprotective effect of hVps41 overexpression (11). In contrast to the α-synuclein model, neither depletion of apd-3 nor loss of apb-3 had an effect on Aβ-induced neurodegeneration (Fig. 4B and C). We therefore considered that, in rescue of Aβ-mediated neurodegeneration, hVps41 is further divergent from its function in the α-synuclein model. Depletion of vps-41 increased neurodegeneration, however. Because both AP3 and rab-7, key effectors of vps-41, appeared to not affect Aβ-mediated neurodegeneration, we turned our attention to other interactors of vps-41.
Coordination of vps-41 with autophagic protein function to attenuate Aβ neurotoxicity
In cell, mouse and clinical models of Aβ neurodegeneration, autophagy participates in attenuating Aβ toxicity. Autophagy is the process whereby large portions of the cytoplasm are engulfed by membrane for recycling at the lysosome. Envelopment of particles and delivery of the autophagosome to the lysosome is mediated by the Atg8/LC3/LGG-2 protein. Upon delivery to the lysosome, tethering of autophagosomes to lysosomes is coordinated by the HOPS complex. In C. elegans embryos, proper delivery and vesicular fusion were found to be contingent upon the colocalization of VPS-41 and the worm LC3 orthologue LGG-2 (51). We therefore hypothesized that vps-41 in our model might be coordinating with lgg-2 to attenuate Aβ toxicity. Accordingly, RNAi depletion of lgg-2 or another key autophagic component, atg-7, increased neurodegeneration by approximately 20% (Fig. 5A).
Figure 5.
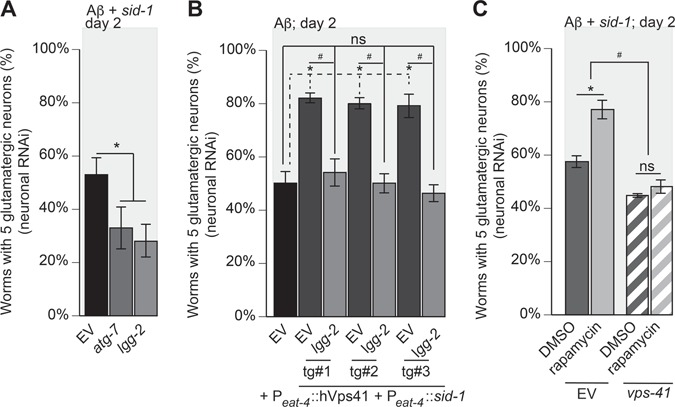
Vps41-mediated neuroprotection from Aβ-induced neurodegeneration is dependent on autophagy. (A) RNAi depletion of atg-7 or lgg-2 was performed specifically within neurons of C. elegans that also express Aβ in strain UA338 (baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321); uIS69 [Pmyo-2::mCherry; Punc-119::sid-1]). Here, there was a significantly increased neurodegeneration compared to the EV RNAi control (P = 0.008; P = 0.003; one-way ANOVA with a Fisher’s LSD test; n = 90 for both). (B) Worms overexpressing Aβ and GFP in glutamatergic neurons, designated as strain UA337 (baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321)), exhibit significant degeneration that was rescued with the overexpression of hVps41 and sid-1 following the creation of the RNAi-sensitive strain (UA329 (baEx195[Peat-4::hVps41, Peat-4::sid-1, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321)). In this background, overexpression of hVps41 significantly (*) rescues Aβ neurodegeneration compared to Aβ control when treated with EV RNAi in all three transgenic lines [tg#1 P = 0.0008; tg#2 P = 0.0008; tg#3 P = 0.0058]. RNAi depletion of lgg-2 in these transgenic lines negates (ns) the rescuing phenotype of hVps41 overexpression in all three transgenic lines when compared to Aβ alone [tg#1 P = 0.972; tg#2 P > 0.999; tg#3 P = 0.992]. The knockdown of lgg-2 in the hVps41 background also reduced glutamatergic neuronal rescue significantly (#) in all three transgenic lines [tg#1 P = 0.004; tg#2 P = 0.0005; tg#3 P = 0.002 (F(6,14) = 17.13; n = 90 for each line; one-way ANOVA with a Tukey’s post hoc analysis)]. (C) RNAi knockdown of EV or vps-41 was performed specifically within glutamatergic neurons of C. elegans that also express Aβ in strain UA338 ((baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321); uIS69 [Pmyo-2::mCherry; Punc-119::sid-1]). Worms were then treated with 0.5 μM rapamycin in 0.01% DMSO or 0.01% DMSO only. In the EV RNAi control, treatment with rapamycin significantly (*) reduced neurodegeneration (F(3,8) = 39.13; P = 0.002), whereas in animals treated with vps-41 RNAi, rapamycin exposure does not rescue neurodegeneration (P = 0.745). Consistent with previous observations, vps-41 RNAi increased (#) neurodegeneration compared to EV RNAi control (P = 0.0197; one-way ANOVA with a Tukey’s post-hoc analysis; n = 90 for each treatment). These data are reported as mean + S.D. while * and # denote statistical significance as detailed within each section of this legend.
To test the dependency of hVps41-mediated rescue on lgg-2, we generated a transgenic strain in which both hVps41 and sid-1 expression were restricted to the glutamatergic neurons. When lgg-2 was depleted with overexpression of hVps41, hVps41 failed to rescue (Fig. 5B). We thus hypothesized the converse, that the neuroprotective effects of increased autophagy would be negated by depletion of vps-41. To stimulate autophagy, we used rapamycin, an effective pharmacological agent that has been used to induce autophagy in multiple model systems. Through increased autophagy, rapamycin attenuates cognitive deficits and decreases plaque formation in mouse and cell culture models of AD (52,53). Likewise, we observed that treatment with rapamycin decreased Aβ-induced neurodegeneration; however, when vps-41 was depleted, rapamycin presented no rescuing effect (Fig. 5C). Despite this, previous work has shown that tethering and fusion of the autophagosome to the lysosome through the HOPS complex is dependent on RAB-7. Because of our previous results with RAB-7, we sought other possible effectors of the HOPS complex.
ARL-8 participates with VPS-41 in attenuating Aβ toxicity
In mammalian studies, Aβ has been shown to still associate with Rab7-labeled endosomes even with the loss of Rab7 function. Therefore, Aβ trafficking may occur through some alternative post-LE pathway (6). To discern how VPS-41 may be functioning independently from RAB-7 to coordinate clearance of Aβ, we turned our attention to other interactors of the HOPS complex. Evidence exists that the HOPS complex functions with Arl8 rather than Rab7, including for the tethering and fusion of autophagosomes to the lysosome (54). The Arf-like GTPase ARL-8 promotes the delivery of presynaptic cargo and large endocytic macromolecules to the lysosome (59,60). When presenting phagocytic material to the lysosome for degradation, ARL-8 directly interacts with VPS-41 (Fig. 6A) (57). We observed that Aβ-mediated neurodegeneration is exacerbated when arl-8 is depleted (Fig. 6B).
Figure 6.
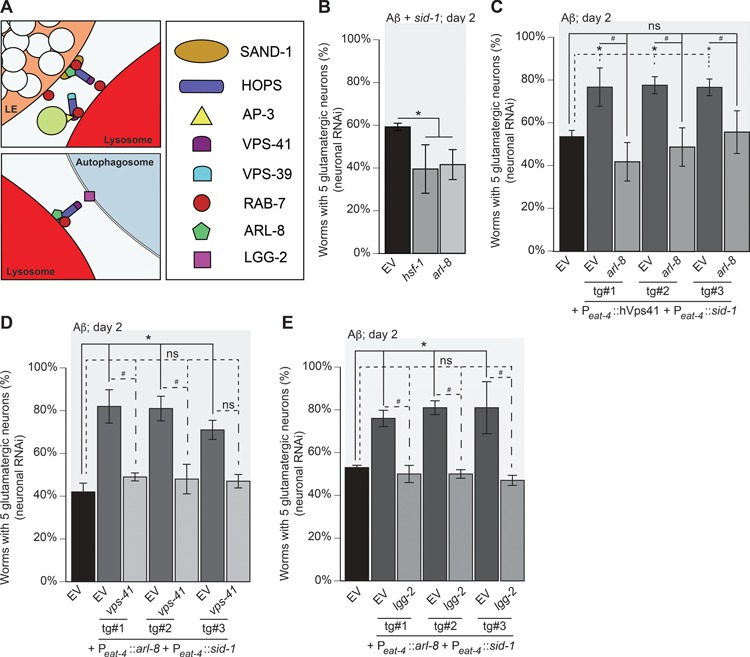
hVps41 neuroprotection from in Aβ neurotoxicity requires arl-8 in conjunction with autophagy. (A) An illustration of the respective targets in the process of tethering homotypic and vesicular fusion. (B) Pan-neuronal RNAi knockdown of components within C. elegans that also express Aβ in glutamatergic neurons in strain UA338 (baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321); uIS69 [Pmyo-2::mCherry; Punc-119::sid-1]). Depletion of hsf-1 (control) and arl-8 increase neurodegeneration compared to the EV RNAi control (P = 0.017, P = 0.028, respectively; n = 90 for each line; one-way ANOVA with a Fisher’s LSD test). (C) Worms overexpressing Aβ and GFP in glutamatergic neurons, designated as strain UA337 (baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321)), have significant degeneration that was rescued with the overexpression of hVps41 and sid-1 following the creation of an RNAi strain (UA329 (baEx195[Peat-4::hVps41, Peat-4::sid-1, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321)). In this background, overexpression of hVps41 significantly (*) rescues Aβ neurodegeneration compared to Aβ control when treated with EV RNAi in all three transgenic lines [tg#1 P = 0.035; tg#2 P = 0.0026; tg#3 P = 0.039]. RNAi depletion of arl-8 in these transgenic lines negates (ns) the rescuing phenotype of hVps41 overexpression in all three transgenic lines when compared to Aβ alone [tg#1 P = 0.414; tg#2 P = 0.971; tg#3 P > 0.999]. The knockdown of arl-8 in the hVps41 background also reduced glutamatergic neuron rescue significantly in all three transgenic lines (#): [tg#1 P = 0.0009; tg#2 P = 0.005; tg#3 P = 0.047 (F(6,14) = 11.88; n = 90 for each line; one-way ANOVA with a Tukey’s post hoc analysis)].(D) Worms overexpressing Aβ and GFP in glutamatergic neurons, designated as strain UA337 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321)), have significant degeneration that was rescued with the overexpression of arl-8 and sid-1 following the creation of an RNAi strain UA330 (baEx196[Peat-4::arl-8, Peat-4::sid-1, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321)). In this background, overexpression of arl-8 significantly (*) rescues Aβ neurodegeneration compared to Aβ control when treated with EV RNAi in all three transgenic lines examined [tg#1 P = 0.001; tg#2 P = 0.001; tg#3 P = 0.014]. RNAi depletion of vps-41 in these transgenic lines negates (ns) the rescuing phenotype of arl-8 overexpression in all three lines when compared to Aβ alone [tg#1 P = 0.955; tg#2 P = 0.962; tg#3 P = 0.983]. The knockdown of vps-41 in the arl-8 background also reduced glutamatergic neuronal rescue significantly in two of the three transgenic lines (#):[tg#1 P = 0.005; tg#2 P = 0.007; tg#3 P = 0.059 (F(6,14) = 11.65; n = 90 for each line; one-way ANOVA with a Tukey’s post hoc analysis)]. (E) Worms overexpressing Aβ and GFP in glutamatergic neurons (strain UA337, as above) exhibit significant neurodegeneration that was rescued with the overexpression of arl-8 and sid-1 using RNAi strain UA330. In this background, overexpression of arl-8 significantly (*) rescued Aβ neurodegeneration compared to Aβ control when treated with EV RNAi in all three transgenic lines [tg#1 P = 0.026; tg#2 P = 0.006; tg#3 P = 0.016]. RNAi depletion of lgg-2 in these transgenic lines negates (ns) the rescuing phenotype of arl-8 overexpression in all three transgenic lines when compared to Aβ alone [tg#1 P = 0.998; tg#2 P = 0.966; tg#3 P = 0.91]. The knockdown of lgg-2 in the arl-8 background also reduced glutamatergic neuronal rescue significantly in all three transgenic lines (#) [tg#1 P = 0.01; tg#2 P = 0.001; tg#3 P = 0.002 (F(6,14) = 13.43; n = 90 for each line; one-way ANOVA with a Tukey’s post hoc analysis)]. These data are reported as mean + S.D. while * and # denote statistical significance as detailed within each section of this figure legend.
To evaluate whether hVps41-mediated rescue of neurodegeneration is dependent on arl-8, we utilized the conditionally RNAi-sensitive transgenic Aβ strain in which hVps41 and sid-1 expression is restricted to the glutamatergic neurons. While hVps41 once again rescued neurodegeneration by 20–30% compared to the control, this rescue was negated by depletion of arl-8 (Fig. 6C). Conversely, we generated genetic lines overexpressing arl-8 in the glutamatergic neurons of conditionally RNAi-sensitive Aβ animals. In this background, arl-8 co-overexpression decreased neurodegeneration by 20–40%, but this was negated by depletion of vps-41 (Fig. 6D). We observed a similar effect when lgg-2 was depleted with arl-8 overexpression (Fig. 6E). Together, these data suggest alternative functions for VPS-41 in autophagy and the endosomal system in mitigation of Aβ toxicity. Furthermore, they demonstrate a distinct divergence in the machinery utilized to attenuate Aβ in contrast to α-synuclein.
Overexpression of arl-8 rescues Aβ, but not α-synuclein, neurodegeneration
In a cell model of α-synuclein aggregation and toxicity, overexpression of ARL-8 resulted in increased aggregation corresponding with decreased autophagy, while knockdown of ARL-8 had the opposite effect (58). Because mammalian ARL-8 also stabilizes VPS-41 recruitment, we sought to further distinguish its role in the Aβ model from the α-synuclein model. To do this, we generated transgenic lines expressing arl-8 pan-neuronally and crossed them into both the Aβ and α-synuclein backgrounds. Because the same transgenic lines were used to cross into both models, this enabled a direct comparison between different neurodegenerative backgrounds. Pan-neuronal overexpression of arl-8 reduced Aβ-mediated neurodegeneration by approximately 10–15% at day 3 and 40% at day 7 (Fig. 7A). In contrast, overexpression of the same arl-8 transgenes in the α-synuclein background yielded no rescue effect (Fig. 7B). These data indicate that arl-8 operates distinctly in the context of Aβ toxicity but lacks impact on α-synuclein-induced neurodegeneration in vivo.
Figure 7.
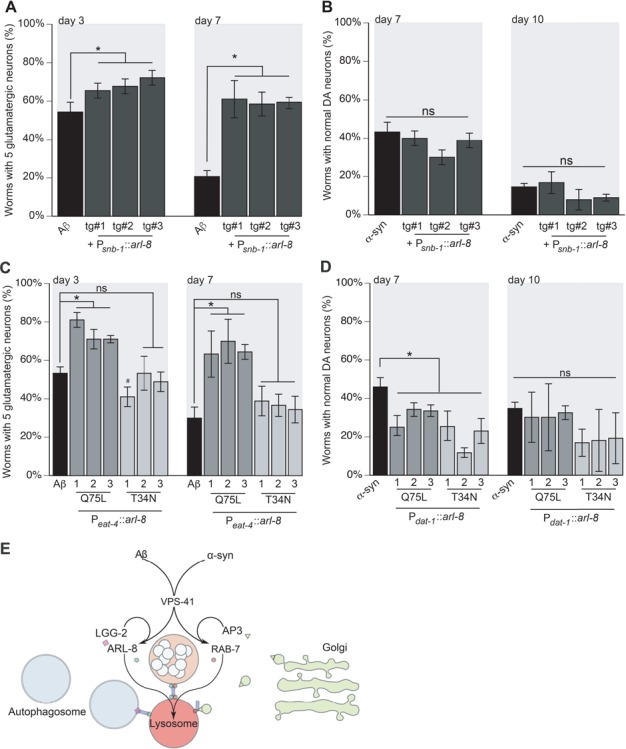
arl-8 overexpression can rescue Aβ but not α-syn-dependent neurotoxicity. (A, B) To examine the same arl-8 overexpression constructs in both glutamatergic and dopaminergic neurons, the arl-8 cDNA was cloned under a pan-neuronal promoter (snb-1) and injected into wild-type N2 animals. The same lines were then crossed with both strains expressing either Aβ in glutamatergic neurons or α-synuclein in dopaminergic neurons. (A) Worms expressing Aβ and arl-8 in glutamatergic neurons were analyzed using transgenic strain UA331 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry];adIs1240[Peat-4::GFP]; baEx190[Psnb-1::arl-8, Punc-54::tdTomato]). All three lines were significantly different from Aβ control on day 3, tg#1 P = 0.012; tg#2 P = 0.002; tg#3 P = 0.0003 (n = 90 for each line). Likewise, on day 7, the transgenic lines were also significantly different from Aβ control, tg#1 P = 0.0001; tg#2 P = 0.0004; tg#3 P = 0.0002 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test). (B) Worms expressing α-synuclein and arl-8 in dopaminergic neurons were analyzed (UA334 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]; baEx190[Psnb-1::arl-8, Punc-54::tdTomato])). All three lines were non-significantly different from α-synuclein control on day 3, [tg#1 P = 0.619; tg#2 P = 0.072; tg#3 P = 0.412 (n = 90 for each line)]. Likewise, on day 7, the transgenic lines were also non-significantly different from α-synuclein control, [tg#1 P = 0.524; tg#2 P = 0.081; tg#3 P = 0.13 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. (C, D) The neuroprotective effects of hVps41 and arl-8 are dependent on nucleotide-bound state. Dominant mutants of arl-8 mimic GTP-bound (Q75L) and GDP-bound (T34N) states in Aβ and α-synuclein expressing neurons. (C) Worms overexpressing Aβ and Q75L arl-8 were analyzed, as strain UA332 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; baEx193[Psnb-1::arl-8 Q75L, Punc-54::tdTomato]). All three lines were significantly different from Aβ control on day 3: [tg#1 P = 0.0001; tg#2 P = 0.0008; tg#3 P = 0.0008 (n = 90 for each line)]. Likewise, on day 7, the transgenic lines were also significantly different from Aβ control, [tg#1 P = 0.0002; tg#2 P < 0.0001; tg#3 P = 0.0001 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. Worms overexpressing Aβ and T34N arl-8 were also analyzed (strain UA333 (baIn34[Peat4::Aβ,Pmyo2::mCherry]; adIs1240[Peat-4::GFP]; baEx194[Psnb-1::arl-8 T34N, Punc-54::tdTomato])). On day 3, one transgenic line showed an increase in neurodegeneration while the other two were non-significantly different from Aβ control animals [tg#1 #P = 0.011; tg#2 P > 0.999; tg#3 P = 0.308 (n = 90 for each line)]. Similarly, on day 7, the transgenic lines were not significantly different from the Aβ control [tg#1 P = 0.204; tg#2 P = 0.334; tg#3 P = 0.516 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. (D) The same arl-8 mutants were examined in worms expressing α-synuclein in the dopaminergic neurons. Specifically, the arl-8 Q75L GTP-bound mutant expressing worms were examined (UA335 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]; baEx193[Psnb-1::arl-8 Q75L, Punc-54::tdTomato])) as well as the arl-8 T34N GDP-bound mutant (UA336 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]; baEx194[Psnb-1::arl-8 T34N, Punc-54::tdTomato]). On day 7, all transgenic lines displayed enhanced neurodegeneration compared to controls. For the Q75L: [tg#1 P = 0.018; tg#2 P = 0.044; tg#3 P = 0.024 (n = 90 for each line)] and for the T34N: [tg#1 P = 0.004; tg#2 P = 0.0003; tg#3 P = 0.004 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. On day 10, all transgenic lines were non-significantly different compared to α-synuclein only controls. For the Q75L: [tg#1 P = 0.642; tg#2 P = 0.642; tg#3 P = 0.822 (n = 90 for each line)] and for the T34N: [tg#1 P = 0.087; tg#2 P = 0.107; tg#3 P = 0.131 (n = 90 for each line; one-way ANOVA with a Fisher’s LSD test)]. These data are reported as mean + S.D. while * and # denote statistical significance as detailed within each section of this figure legend. (E) An experimental model of the distinct roles for VPS-41 in attenuating proteotoxicity between Aβ and α-synuclein. Previously, we have shown that VPS-41 participates in the intracellular management of α-synuclein toxicity, and here we have shown that it also modulates Aβ toxicity. However, VPS-41 appears to be the primary point of divergence between the two endosomal mechanisms of mitigation. Participation with AP3 and RAB-7 is the central means by which VPS-41 executes its activity in the α-synuclein model, by tethering AP3-coated trans-Golgi vesicles to the lysosome and promoting SNARE recruitment for membrane fusion. In this model, the RAB-7 GTPase modulates the tethering activity of VPS-41 and promotes retrograde trafficking. In contrast, the Arf-like GTPase ARL-8 promotes anterograde trafficking while stimulating VPS-41 activity. We have shown that AP3 and RAB-7 are dispensable in modulating Aβ toxicity and that ARL-8 and LGG-2, the C. elegans LC-3 orthologue, instead cooperate with VPS-41 to attenuate Aβ toxicity. Because ARL-8 activity affects autophagosome positioning and lysosomal acidity, alterations of arl-8 expression may reflect a change in autophagosomal activity responsible for Aβ clearance. Alternatively, internalized Aβ might be targeted to the lysosome by ARL-8. Further, ARL-8 is responsible for the distribution of vesicles at the synaptic terminus, implicating its anterograde activity in the delivery of Aβ-containing compartments to the membrane for secretion. Autophagy also participates in secretion of Aβ aggregates into extracellular deposits, suggesting that ARL-8 may coordinate with LGG-2 for the secretion of neurotoxic aggregates.
Neuroprotective effect of ARL-8 depends on its nucleotide-bound state
Dominant mutations in ARL-8 that mimic nucleotide-bound states alter its activity and localization with hVps41 (59,60). In a cellular model of the α-synuclein variant A53T, overexpression of ARL-8 decreased turnover of α-synuclein, whereas depletion of ARL-8 increased turnover of α-synuclein (58). We therefore expected that the nucleotide-bound states would have inverse effects between the two models, such that the dominant-positive Q75L mutant that mimics the GTP-bound state would be neuroprotective in the Aβ model but would increase neurodegeneration in the α-synuclein model. Conversely, the dominant-negative T34N mutant that mimics the GDP-bound state would increase neurodegeneration in the Aβ model but be neuroprotective in the α-synuclein model. To test this, the arl-8 genetic variants were cloned into a pan-neuronal expression construct driving expression from the snb-1 promoter. The expression constructs were injected into wild-type N2 animals, and isolated stable lines were crossed with either Aβ or α-synuclein backgrounds. As a result, we were able to compare respective stable lines between the two neurodegenerative models. Expression of the dominant-positive Q75L variant reduced neurodegeneration in the Aβ model (Fig. 7C), but the dominant-negative variant T34N produced no effect on neurodegeneration distinguishable from control (Fig. 7C). The same stable lines of both Q75L and T34N increased neurodegeneration in the α-synuclein model on day 7 but had no effect on day 10 (Fig. 7D), indicating that the nucleotide-bound state of ARL-8 plays a significant role in mitigating Aβ toxicity, but not in α-synuclein toxicity.
Discussion
The structural properties inherent to Aβ stabilize the formation of oligomers, which have been observed to perforate and rupture digestive compartments in the cell. Similarly, α-synuclein has been observed to form oligomers that form pores in the membranes of digestive compartments. Additionally, both α-synuclein and Aβ invoke autophagy, ER stress, oxidative stress, and unfolded protein responses, overtly suggesting that Aβ and α-synuclein share overlapping mechanisms in the molecular etiology of AD and PD, respectively. As a means to delineate the underlying intracellular functional manifestations of these pathogenic peptides, we have employed a suite of transgenic C. elegans models that recapitulate the progressive time-dependent neurodegeneration associated with these diseases (19,20).
Using a C. elegans model of α-synuclein-induced neurodegeneration, we previously reported that overexpression of the hVps41 attenuated neurodegeneration, and this effect was lost upon depletion or loss of endosomal effectors AP3, csnk-1, or rab-7 (11). Considering α-synuclein and Aβ are amyloidogenic proteins associated with functional impacts on vesicular and endolysosomal trafficking, we hypothesized that hVps41 overexpression would rescue Aβ-induced neurodegeneration through the same elements of endosomal trafficking we identified in the α-synuclein model. Here, we show that overexpression of hVps41 also mitigates Aβ-induced neurodegeneration of glutamatergic neurons in a C. elegans model. The recombinant Aβ peptide in the C. elegans model predictably forms toxic aggregates within the cytoplasm of cells (32,61,62), consistent with observations in mammalian and cell culture models that demonstrate that Aβ toxicity typically arises from cytoplasmic species (4–6,8,9,13,15,17,33,35–43). Previously, we and others have demonstrated that overexpression of PICALM/unc-11 and other endocytosis-associated proteins decreased Aβ-induced neurodegeneration in mice, cell culture, and C. elegans models, indicative of the conserved functional utility of the nematode Aβ model (13). We also show that depletion of PICALM/unc-11 increased Aβ-induced neurodegeneration and that components of the endosomal system modulate Aβ neurotoxicity. Further, the overexpression of a truncated hVps41 (hVps41G), a minimal functional unit engineered for viral delivery in ongoing gene therapy studies that contain only WD40 and CHCR domains, was able to rescue neurodegeneration in both the Aβ and α-synuclein models. Contrary to our initial hypothesis, however, Aβ toxicity was not contingent upon the clathrin-adaptor protein complex AP3, thus distinguishing the two neurodegenerative models (Fig. 7E). Because the loss of rab-7 increases α-synuclein-induced neurodegeneration, this distinction was further supported by the previous observation that localization of endocytic Aβ is altered upon depletion of rab-5 and the HOPS complex, but not by depletion of rab-7 (6). Similarly, we observed an increase in Aβ-induced toxicity upon depletion of either rab-5 or two other major components of the HOPS complex, vps-39 and vps-41, in both paralysis and neuronal C. elegans models of Aβ toxicity, but not rab-7. We further distinguish these worm AD and PD models by showing that overexpression of arl-8 rescues neurodegeneration in the Aβ but not the α-synuclein, background.
One of the defining features of the LE is RAB-7, which decorates its surface and regulates motor protein attachment and homotypic tethering and fusion through its nucleotide binding states. However, though depletion of rab-7 was expected to arrest the transport of Aβ in the EE, Aβ was nonetheless observed in the LEs (6). For this reason, we expected that loss of rab-7 would delimit transport of endocytic Aβ to EEs and increase neurodegeneration. As SAND-1 is responsible for recruitment of RAB-7 to the endosomal surface, loss or depletion of sand-1 would be expected to recapitulate phenotypes of rab-7 loss or depletion. However, we observed no effect from loss of rab-7 despite increased neurodegeneration with sand-1 RNAi, thus implicating a distinct mechanism for Aβ toxicity compared to α-synuclein (Fig. 7E).
Large macromolecules engulfed by the cell for degradation are coordinated with the lysosome through ARL-8, which is recruited to the late endosomal and lysosomal structures through SAND-1 activity (63,64). Because ARL-8 is recruited with RAB-7, they work together towards differential targeting of cargoes. In particular, ARL-8 promotes recruitment of hVps41 to the lysosome and the T146P SNP in hVps41 obstructs this behavior. In the α-synuclein model, overexpression of hVps41 T146P failed to rescue neurodegeneration (11). Another SNP, A187T, maintains colocalization in cell culture (60) yet is unable to rescue neurodegeneration in the C. elegans model of α-synuclein-mediated neurodegeneration (11). However, we found that overexpression of arl-8 did not decrease α-synuclein-induced neurodegeneration in dopaminergic neurons, suggesting little functional relationship between hVps41 and ARL-8 in modulating α-synuclein toxicity. In contrast, overexpression of arl-8 decreased Aβ-induced neurodegeneration. In the Aβ model, it appears that arl-8 and vps-41 have a functional relationship independent of rab-7 and AP3 (Fig. 7E).
Though it has been reported that autophagosomes failed to fuse with lysosomes upon depletion of RAB-7, other sources have found that, in some conditions, ARL-8 participates with HOPS in the tethering and fusion of autophagosomes to the lysosomes (64,65). Similarly, allocation of SNARE proteins involved in the formation of autophagosomes is largely affected by changes in PICALM/unc-11 expression (55), which likewise altered neurodegenerative phenotypes in our model. We found that the neuroprotective effect of either hVps41 or arl-8 overexpression failed when lgg-2 was depleted, suggesting that vps-41 may coordinate with arl-8 in autophagy to mitigate Aβ toxicity (Fig. 7E). Similar circumstances have been observed, in which accumulation of huntingtin protein was reduced by a complex interaction between ARL-8 isoforms and the autophagosome for lysosomal degradation (58). In contrast, protein levels of α-synuclein changed in direct proportion to Arl8 expression, further supporting our hypothesis that Aβ and α-synuclein are attenuated distinctly (58). Additionally, loss of Rab5 hampers autophagosomal flux by impeding upon lysosome acidification (66). Thus, we propose that the toxicity of Aβ presents a circumstance in which ARL-8 and VPS-41 coordinate to attenuate neurodegeneration with autophagy (Fig. 7E).
The manner by which ARL-8 coordinates these efforts with HOPS, however, remains elusive. When recruited to the LE by SAND-1/Mon1-Ccz1, Rab7 is paired with the Rab-interacting lysosomal protein (RILP) to mediate interactions with dynein for retrograde transport (67). Recruitment of human Arl8 is concomitant with Rab7, but paired with the SifA and kinesin-interacting protein (SKIP) for anterograde transport of lysosomes or presynaptic vesicles (68–70). Together, these protein pairs modulate endosomal and lysosomal migration towards proper localization of cargo. As a result, ARL-8 overexpression enhances mTORC activity and lysosomes are mislocalized towards the cellular periphery, hampering the initiation of autophagy and delivery to the lysosomes (58,71). In these conditions, autophagy and the degradation of α-synuclein are diminished.
Rapamycin treatment increased clearance of α-synuclein through autophagy, in a manner dependent on LE formation (56). Congruently, we observed that overexpression of arl-8 did not improve α-synuclein-mediated neurodegeneration. In contrast, neuroprotection afforded by arl-8 overexpression in the Aβ model was negated by depletion of lgg-2, indicating a positive relationship between arl-8 and autophagy in our model. This diverges from the model that the effect of arl-8 overexpression is manifested through alterations in autophagy. If the initiation and progression of autophagy are significantly hampered by the mislocalization of mTORC to the cellular periphery, it would be expected that the phenotype from overexpression would imitate depletion of lgg-2. Furthermore, since Rab7–RILP yields retrograde translocation that antagonizes the anterograde movement produced by Arl8–SKIP, loss of Rab7 would functionally mimic Arl8 overexpression. Though we observed a marginal decrease in Aβ-mediated neurodegeneration with loss of rab-7, it was not statistically significant, suggesting that if ARL-8 and VPS-41 are coordinating with autophagy and anterograde transport, it does so independently from starvation responses associated with ARL8-mediated mislocalization of autophagosomes. Indeed, there may be an independent role of autophagy with anterograde transport, as loss of autophagy exacerbates intracellular accumulation of toxic Aβ aggregates (72,73) and is associated with AD in mice and humans (74).
As a GTPase, Arl8 activity in humans was shown to be modulated by its GTP-bound state. Constitutively bound to GTP, Arl8 supports colocalization of hVps41, but hVps41 fails to colocalize with Arl8 when mutated to lock Arl8 in a GDP-bound state (60). Furthermore, the interaction of hVps41 with Arl8 is abrogated by a T146P mutation, but not an A187T mutation (60). Though neither T146P nor A187T mutations had protective effects in the nematode α-synuclein model (11), the A187T variant was protective against Aβ-mediated neurodegeneration (Fig. 2G). Overexpression of the GTP-locked arl-8 variant attenuated neurodegeneration by Aβ (Fig. 7C) but not α-synuclein (Fig. 7D), indicating that the activity of ARL-8 and its recruitment of HOPS are pivotal distinctions in how HOPS mollifies different toxic aggregates. Similarly, aggregation of the huntingtin protein in SQSTM1/p62-positive autophagosomes is exacerbated with the loss of the Arl8 effector, BORC. Since Aβ oligomers also accumulate in SQSTM1/p62-positive autophagosomes (75), it is possible that the Aβ and huntingtin proteins are subject to similar clearance mechanisms (76).
One such possible clearance mechanism is autophagic secretion. Not only is BORC necessary for anterograde transport of lysosomes to the terminus for degradation of autophagosomes (77), it is also necessary for the transport of non-degradative lysosomes and late endosomal components to the periphery for the secretion of cargoes (78). In transgenic APP mice, disrupting autophagy resulted in decreased plaque burden but increased intracellular loads of toxic oligomers (72). Conversely, stimulating autophagy increased plaque formation but decreased neurodegeneration (72). Similarly, HeLa and mouse neuroblastoma cells have been observed to release Aβ in exosomes (79), and glutamatergic signaling stimulated release of endosomal compartments from oligodendrocytes to be received by neuronal axons (80). Indeed, the release of protein aggregates, including Aβ and huntingtin, is quantifiably detectable in C. elegans neurons (81). Similarly, endosomal compartments containing huntingtin protein are released from mouse neuroblastoma, striatal cells and rat primary cortical neurons (76). Whether hVps41 plays a role in autophagic or exosomal secretion is unclear, but considering the propagation of Aβ by exosomes (82) and the detection of lysosomal proteins in plasma as potential preclinical markers for AD (83,84), it is worth exploring the role of hVps41 in this phenomenon in prospect of a potential therapeutic target.
Whether this activity is related to Aβ trafficking or autophagy is unclear, but further analysis of these properties can elucidate the role of arl-8 with vps-41 in Aβ-mediated neurodegeneration. The cellular machinery between humans and C. elegans is remarkably conserved, such that C. elegans is a foremost platform for the investigation of aging, development, and apoptosis (85–87). It should come as no surprise, then, that C. elegans shares significant homology with a multitude of human genes associated with AD (88). These, together with the tools developed for C. elegans, have made it a predictive neurodegenerative model for human diseases, such as Parkinson’s, Huntington’s, amyotrophic lateral sclerosis, dystonia and ataxia (20,89–91). An important feature to distinguish about the C. elegans model is that it is obviously limited with respect to modeling human cognition and behavior as a consequence of Aβ toxicity; rather, its cellular conservation of function, in conjunction with its complete neuronal connectivity map (92–94), renders the nematode system as an unparalleled tool for probing how Aβ affects mechanistic aspects of neurodegeneration in vivo. Given the pivotal axis represented by Vps41 function, distinguishing the divergent protective effects of hVps41 and associated molecules in different neurodegenerative models will provide a better understanding towards more effectively identifying therapeutic strategies designed to combat neurodegenerative disease.
Materials and Methods
Plasmid construction
hVps41 cDNA was obtained from Open Biosystems and mutations in hVps41 (G truncate, T146P and A187T) were generated as previously described (11). All of these hVps41 variants were subcloned into the pDEST-Peat-4 and the pDEST-Punc-54 expression vectors. The arl-8 and sid-1 expression clones were generated using Gateway Technology (Invitrogen, Carlsbad, CA). Briefly, primers 5ʹ-ggggacaagtttgtacaaaaaagcaggctccatgttggctatggtgaataaggtt-3ʹ and 5ʹ-ggggaccactttgtacaagaaagctgggtcttagcgttgagctttcgagtga-3ʹ were used to amplify arl-8 and primers 5ʹ-ggggacaagtttgtacaaaaaagcaggctcccctcatttttccaggttcacaatg-3ʹ and 5ʹ-ggggaccactttgtacaagaaagctgggtccagaaaggtgtcatggtctagtgg-3ʹ were used to amplify sid-1 using Phusion high-fidelity Taq polymerase [New England Biolabs (NEB), Ipswich, MA] from genomic DNA extracted from N2 Bristol nematodes. Amplified clones were recombined with pDONR221 by BP reactions to create entry clones of each, which were subsequently recombined with expression constructs for tissue- or cell-type-specific expression in animals. The arl-8 Q75L allele was generated in pDONR221 by Q5 Site-Directed Mutagenesis (NEB) using the primers 5ʹ-ggggcctgccacggttcc-3ʹ and 5ʹ-cgatatcccataatttgatcgtca-3ʹ, using the arl-8 pDONR entry clone as template. The arl-8 T34N allele was generated by polymerase chain reaction (PCR) fusion using primers 5ʹ-cattgacaaatgtattttttccagag-3ʹ and 5ʹ-ctctggaaaaaatacatttgtcaatg-3ʹ with primers attB_arl-8_R and attB_arl-8_F, respectively. All the resulting amplica were recombined into the Gateway pDONR221 vector according to the manufacturer’s guidelines and then confirmed by DNA sequencing. These entry clones were then cloned into respective pDEST expression vectors. Expression constructs were injected into Bristol N2 animals, unless otherwise described, at 50 ng/μL with the co-injection marker transgene (Punc-54::tdTomato) at 50 ng/μL. At least three stable independent lines were generated and analyzed for each C. elegans transgenic construct.
C. elegans strains
The strains VC308 (rab-7(ok511)/mIn1 [mIs14 dpy-10(e128)]), RB662 (apb-3[ok429]), DA1240 (adIs1240[Peat-4::GFP + lin-15(+)]), CL2006 (dvIs2[pCL12(Punc-54::Aβ1–42) + pRF4), and CL4176 ((smg-1(cc546ts) I; dvIs27 [Pmyo-3::Aβ + rol-6(su1006)) were provided by the Caenorhabditis Genetics Center. Strain BY250 (vtIs7[Pdat-1::GFP)]) was a generous gift from Randy Blakely (Florida Atlantic University), while TU3401 (sid-1(pk3321); uIS69 [Pmyo-2::mCherry; Punc-119::sid-1]) was a gift from Martin Chalfie (Columbia University) (Table 1).
Table 1.
A summary of all the C. elegans strains utilized in this publication
| Strain | Genotype |
|---|---|
| Neuronal Models | |
| DA1240 | adIs1240[Peat-4::GFP + lin-15(+)] |
| UA198 | baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP] |
| BY250 | vtIs7[Pdat-1::GFP] |
| UA44 | baIn11[Pdat-1::α-syn, Pdat-1::GFP] |
| Paralysis models (body wall muscle expression of Aβ) | |
| CL4176 | smg-1(cc546ts) I; dvIs27 [Pmyo-3::Aβ + rol-6(su1006)] |
| UA343 | baIn49[Punc-54::hVps41; Pmyo-2::mCherry] |
| UA344 | baIn49[Punc-54::hVps41, Pmyo-2::mCherry]; smg-1(cc546) I; dvIs27[pAF29(Pmyo-3::Aβ::let UTR) + pRF4(rol-6(su1006))] |
| CL2006 | dvIs2[pCL12(Punc-54::Aβ) + pRF4] |
| Mutant strains and crosses | |
| RB662 | apb-3[ok429] |
| VC308 | rab-7(ok511)/mIn1 [mIs14 dpy-10(e128)] |
| UA340 | baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; apb-3[ok429] |
| UA341 | baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]); (ok511)/mIn1 [mIs14 dpy-10(e128)] |
| UA342 | baIn11[Pdat-1::α-syn, Pdat-1::GFP]; (ok511)/mIn1 [mIs14 dpy-10(e128)] |
| Glutamatergic expression of hVps41 variants | |
| UA322 | baEx189[Peat-4::hVPs41, Punc-54::tdTomato] |
| UA347 | baEx189[Peat-4::hVPs41, Punc-54::tdTomato]; baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP] |
| UA348 | baEx197[Peat-4::hVPs41 G, Punc-54::tdTomato] |
| UA349 | baEx197[Peat-4::hVPs41 G, Punc-54::tdTomato]; baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP] |
| UA324 | baEx191[Peat-4::hVPs41 T146P, Punc-54::tdTomato] |
| UA325 | baEx192[Peat-4::hVPs41 A187T, Punc-54::tdTomato] |
| UA345 | baEx191[Peat-4::hVPs41 T146P, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ, Pmyo-2::mCherry];adIs1240[Peat-4::GFP]) |
| UA346 | baEx192[Peat-4::hVPs41 A187T, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ, Pmyo-2::mCherry];adIs1240[Peat-4::GFP] |
| Pan-neuronal RNAi-sensitivity in Aβ model | |
| UA328 | baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321) |
| UA338 | baIn34[Peat-4-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321); uIS69 [Pmyo-2::mCherry; unc-119::sid-1] |
| hVps41 and arl-8 over-expression and RNAi sensitivity in glutamatergic neurons | |
| UA329 | baEx195[Peat-4::hVps41, Peat-4::sid-1, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ, Pmyo-2::mCherry] |
| UA330 | baEx196[Peat-4::arl-8, Peat-4::sid-1, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ, Pmyo-2::mCherry] |
| Pan-neuronal expression of arl-8 variants in Aβ and α-syn models | |
| UA323 | baEx190[Psnb-1::arl-8, Punc-544::tdTomato] |
| UA326 | baEx193[Psnb-1::arl-8 Q75L, Punc-54::tdTomato] |
| UA327 | baEx194[Psnb-1::arl-8 T34N, Punc-54::tdTomato] |
| UA331 | baIn34[Peat-4::Aβ, Pmyo-2::mCherry];adIs1240[Peat-4::GFP]; baEx190[Psnb-1:: arl-8, Punc-54::tdTomato] |
| UA332 | baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; baEx193[Psnb-1::arl-8 Q75L, Punc-54::tdTomato] |
| UA333 | baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; baEx194[Psnb-1::arl-8 T34N, Punc-54::tdTomato] |
| UA334 | baIn11[Pdat-1::α-syn, Pdat-1::GFP]; baEx190[Psnb-1::arl-8, Punc-54::tdTomato] |
| UA335 | baIn11[Pdat-1::α-syn, Pdat-1::GFP]; baEx193[Psnb-1::arl-8 Q75L, Punc-54::tdTomato] |
| UA336 | baIn11[Pdat-1::α-syn, Pdat-1::GFP]; baEx194[Psnb-1::arl-8 T34N, Punc-54::tdTomato]) |
The isogenic strain UA44 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]) expresses α-synuclein and green fluroescent protein (GFP) in the dopaminergic neurons. UA198 (baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]) was generated as previously described (13) and expresses GFP and Aβ in the glutamatergic neurons. The following strains were produced by injecting N2 animals with constructs generated by Gateway Cloning (Invitrogen; described in the section below) to overexpress genes of interest from eat-4 or snb-1 promoters: UA322 (baEx189[Peat-4::hVPs41, Punc-54::tdTomato]), UA348 (baEx197[Peat-4::hVPs41 G, Punc-54::tdTomato]), UA323 (baEx190[Psnb-1::arl-8, Punc-54::tdTomato]), UA324 (baEx191[Peat-4::hVPs41 T146P, Punc-54::tdTomato]), UA325 (baEx192[Peat-4::hVPs41 A187T, Punc-54::tdTomato]),UA326 (baEx193[Psnb-1::arl-8Q75L, Punc-54::tdTomato]), and UA327 (baEx194[Psnb-1::arl-8 T34N, Punc-54::tdTomato]). The Peat-4::sid-1 plasmid was co-injected together with either the hVps41 or the arl-8 plasmids into a sid-1 mutant-containing UA198 strain (UA328; see description in following section) to create the following strains: UA329 (baEx195[Peat-4::hVps41, Peat-4::sid-1, Punc-54::tdTomato]; baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP] ; sid-1(pk3321)), and UA330 (baEx196[Peat-4::arl-8, Peat-4::sid-1, Punc-54::tdTomato]; baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321)) (Table 1).
Genetic crosses
N2 males were crossed with stable Peat-4::arl-8 strains, UA323 (arl-8 WT), UA326 (arl-8 Q75L) and UA327 (arl-8 T34N), to generate males that were then crossed with UA198 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]) or UA44 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]). Theoretically, all F1s are heterozygous for either UA198 or UA44, thus hermaphroditic F1s were collected and allowed to self-propagate. Individual F2s possessing each respective co-injection marker were then isolated to individual plates and allowed to self-propagate. Populations homozygous for UA198 or UA44 backgrounds were identified by complete expression of the transgenic indicator (mCherry or GFP, respectively). This generated strains UA331 to UA333 for arl-8/UA198 combinations and strains UA334 to UA336 for arl-8/UA44 combinations (Table 1).
Strains overexpressing Aβ and GFP in glutamatergic neurons (UA198 (baIn34[Peat4::Aβ,Pmyo2::mCherry]; adIs1240[Peat-4::GFP])) were crossed with animals expressing hVps41 alleles WT (UA322), T146P (UA324), A187T (UA325), or the G truncate (UA348) to create the following strains: UA347 (baEx189[Peat-4::hVPs41, Punc-54::tdTomato]; baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]), UA345 (baEx191[Peat-4::hVPs41 T146P, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]), UA346 (baEx192[Peat-4::hVPs41 A187T, Punc-54::tdTomato]; baIn34[Peat-4:: Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]), and UA349 (baEx197[Peat-4::hVPs41 G, Punc-54::tdTomato]; baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]) (Table 1).
To generate pan-neuronal RNAi sensitive strains with and without Aβ in the glutamatergic neurons, the following procedure was employed. Both UA198 (baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]) and DA1240 (adIs1240[Peat-4::GFP + lin-15(+)]) hermaphrodites crossed with NL3321 (sid-1(pk3321)) males to generate UA328 and UA337, respectively. TU3401 males were generated by heat shock and then crossed with UA328. The presence of the Punc-119::sid-1 and Aβ transgenes were confirmed by PCR, since they both used the same transgenic marker. PCR for these transgenes used the following primers: 5ʹ-agttcccaggaaatttatgatgaaaac-3ʹ and 5 -cgcggcgactttggttaaat-3
-cgcggcgactttggttaaat-3 ; and 5
; and 5 -atgcataaggttttgctggcac-3ʹ and 5
-atgcataaggttttgctggcac-3ʹ and 5 -cgctatgacaacaccgccc-3
-cgctatgacaacaccgccc-3 , respectively. Two continuous generations were probed to identify homozygotes. During the duration of the cross, worms were kept on plc-3 RNAi to select for the sid-1(pk3321) mutation. plc-3 RNAi has a fully penetrant and consistent sterile phenotype. Only worms with homozygous sid-1(pk3321) mutation can give normal brood size in at least two continuous generations. The final strain created was UA338 (baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321); uIS69 [Pmyo-2::mCherry; Punc-119::sid-1]) (Table 1).
, respectively. Two continuous generations were probed to identify homozygotes. During the duration of the cross, worms were kept on plc-3 RNAi to select for the sid-1(pk3321) mutation. plc-3 RNAi has a fully penetrant and consistent sterile phenotype. Only worms with homozygous sid-1(pk3321) mutation can give normal brood size in at least two continuous generations. The final strain created was UA338 (baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; sid-1(pk3321); uIS69 [Pmyo-2::mCherry; Punc-119::sid-1]) (Table 1).
To generate AP3 mutant animals, UA198 was crossed with strain RB662 [apb-3 (ok429)]. Heterozygous F1 progeny were isolated and allowed to self-propagate. Individual hermaphroditic F2 progeny were isolated to individual 35 mm plates and allowed to self-propagate. Homozygous populations were identified by complete inheritance of the co-injection marker and PCR of the apb-3 locus using the following primers: 5 -gttgctcaattgaagtgcactgtg-3
-gttgctcaattgaagtgcactgtg-3 and 5
and 5 -ccgagaaatcaacgtcaatcagc-3
-ccgagaaatcaacgtcaatcagc-3 . This generated the strain UA340 (baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; apb-3[ok429]) (Table 1).
. This generated the strain UA340 (baIn34[Peat-4::Aβ, Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; apb-3[ok429]) (Table 1).
Because animals homozygous for the rab-7 mutant allele, ok511, lay dead eggs, UA198 or UA44 were crossed with the balanced strain, VC308 ((ok511)/mIn1 [mIs14dpy-10(e128)]), to create strains UA341 (baIn34[Peat-4::Aβ,Pmyo-2::mCherry]; adIs1240[Peat-4::GFP]; (ok511)/mIn1 [mIs14 dpy-10(e128)]) and UA342 (baIn11[Pdat-1::α-syn, Pdat-1::GFP]; (ok511)/mIn1 [mIs14 dpy-10(e128)]), respectively. Heterozygous F1s were allowed to self-propagate, and individual F2s were isolated to individual plates to self-propagate. Homozygous inheritance of the UA198 or UA44 background was determined by complete inheritance of the co-injection marker. To identify inheritance of the VC308 background, populations were selected that had equal distribution of Pmyo-2::GFP; dumpy (Dpy), WT, Pmyo-2::GFP and WT phenotypes. Inheritance of the ok511 allele was confirmed by collecting WT animals without the Pmyo-2::GFP marker onto plates and allowing them to lay eggs. If the ok511 allele had been properly inherited within the population, the eggs would fail to hatch. This generated two strains, UA341 and UA342, carrying the Aβ and α-syn transgenes, respectively (Table 1). The rab-7 mutation was maintained by picking heterozygous animals, and homozygous ok511 progeny were analyzed for neurodegeneration.
The body wall muscle promoter-hVPS41construct (Punc-54::hVps41) was injected into N2 worms. This strain was then chromosomally integrated using standard methods (85) to create the strain UA343 (baIn49[Punc-54::hVPS41; Pmyo-2::mCherry]). This strain was then crossed to CL4176 to generate the strain UA344 (baIn49[Punc-54::hVps41, Pmyo-2::mCherry]; smg-1(cc546) I; dvIs27[pAF29(Pmyo-3::Aβ 1-42::let UTR) + pRF4(rol-6(su1006))]) to use in paralysis assays (Table 1).
Neurodegeneration analysis in C. elegans
Animals for analysis were synchronized by a 3 h egg-lay. To examine the neurons, animals were immobilized using 3 mm levamisole on glass cover slips and inverted onto 2% agarose pads on microscope slides. Each analysis was replicated at least three times with 30 animals per condition (30 animals × 3 trials = 90). For each transgenic construct, 30 animals from each of three stables, independent lines were examined for neurodegeneration at least three times (30 animals × 3 lines × 3 trials = 270). C. elegans glutamatergic neurons were analyzed for neurodegeneration as previously described (13,95,96). Briefly, animals were scored for glutamatergic neurodegeneration at days 3 and 7 post-hatching, as reported in the Results and Figure Legends. An animal was scored as normal if all five tail neurons were present and without malformities such as distention, apoptotic swelling, axon breaks, separation of the soma, or loss of fluorescence. C. elegans dopaminergic neurons were also analyzed for neurodegeneration as previously described (11). Briefly, animals were scored for neurodegeneration at days 7 to 10, as reported in Results and Figure Legends. An animal was scored as normal if all six anterior dopaminergic neurons were present and without malformities such as neurite blebbing, cell body rounding, cell loss, and dendrite or axon loss.
RNAi
RNAi feeding clones were cultivated initially on LB solid media containing tetracycline (5 μg/mL) and ampicillin (100 μg/mL), and then individual colonies were grown overnight in liquid LB media containing 50 μg/mL carbenicillin. IPTG was spread on plates for a final concentration of 1 mm, seeded with RNAi feeding clones, and allowed to dry. Induction of dsRNA occurred during a 14–18 h incubation at 20°C. Adult hermaphrodites were allowed to lay eggs for 3 h on RNAi feeding clones to produce a synchronized population. Glutamatergic neurons of synchronized progeny were analyzed at least 2 days after hatching, as described above.
Paralysis assays
Temperature-restricted Aβ-induced paralysis assays were performed on C. elegans strain CL4176 as previously described (97). Briefly, gravid animals were permitted to lay eggs on EV (plasmid L4440) or vps-41 RNAi bacterial lawns for 2 h at 16°C, at which point they were removed and then synchronized progeny were incubated at 16°C. 48 h post-hatching, the populations were shifted to 23°C. Beginning 24 h post-upshift, animals were scored for paralysis every 2 h until all animals in each condition exhibited paralysis. Animals were considered paralyzed when they no longer responded to posterior or anterior gentle prodding with a platiunum wire (worm pick).
Paralysis assays from constitutive expression of Aβ in animal muscles in C. elegans strain CL2006 were performed as previously described (28). Briefly, gravid animals were permitted to lay eggs on EV (plasmid L4440) or RNAi bacterial-feeding clones, as described in the Fig. 2 legend, for 3 h at 20°C, at which point they were removed, and then synchronized progeny were incubated at 20°C. After 3 days post-hatching, animals were transferred every day and scored for paralysis from days 6 to 14 post-hatching. Animals were considered paralyzed when they no longer responded to posterior or anterior gentle prodding with a platiunum wire (worm pick).
Chemical treatments
Rapamycin (Alfa Aesar, Haverhill, Massachusetts, USA) was dissolved in 100% DMSO at 5 mm and 1:100 dilutions produced working stock solutions of rapamycin at 50 μM in 1% DMSO. Synchronized embryos were collected in 100 μL of 50 μM rapamycin solution and applied to a bacterial lawn on 60 mm worm plate (with 10 mL NGM solid media) for a final concentration of 0.5 μM rapamycin, with a final concentration of 0.01% DMSO. Vehicle control animals were collected in 1% DMSO and applied to a bacterial lawn on NGM for a final concentration of 0.01% DMSO.
Experimental design and statistical analysis
Hermaphrodites were analyzed, which is standard in the C. elegans field. For all experiments where transgenic constructs were created, we used three independently generated extrachromosomal transgenic lines per construct to control for gene copy number. In all cases, sample sizes (30 animals per transgenic line, for a total of 90 animals) were standardized within each experiment and examined in a uniform fashion. All experiments used at least three independent replicates per experiment per variable to generate a mean experimental value and a standard deviation. The null hypothesis assumes that tested conditions do not significantly alter measured values observed in negative controls, which generally consisted of GFP-only controls in an N2 background when comparing animals or solvent-only controls when analyzing drug effects. In experiments using one independent variable across multiple tested effects (e.g. neuron cell death as a function of construct type), a one-way analysis of variance (ANOVA) series was used with a multiple-comparisons post hoc test (Tukey's). However, if values were being compared only with a control variable, a Fisher’s LSD test was used instead. In cases in which there were two independent variables (e.g. paralysis as a function of time and condition), a two-way ANOVA series was used with a multiple-comparisons post hoc test (Sidak’s). A P < 0.05 was the absolute minimum threshold for statistical significance. Statistics were performed using GraphPad Prism software.
Acknowledgements
We thank Laura Berkowitz, Bryan Martinez, and Sam Scopel for assistance and/or expert advice in the progress of this project. This research was initiated with support of a Target Validation grant awarded by the Michael J. Fox Foundation for Parkinson’s Research [to G.A.C. and K.A.C.] and funded in part by a grant from the National Institutes of Health [R15 NS075684-01 to G.A.C.]. Some strains were provided by Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs [P40 OD010440].
Conflict of Interest statement. None declared.
References
- 1. Thinakaran G. and Koo E.H. (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem., 283, 29615–29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haass C. and Selkoe D.J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol., 8, 101–112. [DOI] [PubMed] [Google Scholar]
- 3. Trousdale C. and Kim K. (2015) Retromer: structure, function, and roles in mammalian disease. Eur. J. Cell Biol., 94, 513–521. [DOI] [PubMed] [Google Scholar]
- 4. Snyder E.M., Nong Y., Almeida C.G., Paul S., Moran T., Choi E.Y., Nairn A.C., Salter M.W., Lombroso P.J., Gouras G.K. et al. (2005) Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci., 8, 1051–1058. [DOI] [PubMed] [Google Scholar]
- 5. Hu X., Crick S.L., Bu G., Frieden C., Pappu R.V. and Lee J.-M. (2009) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA, 106, 20324–20329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li J., Kanekiyo T., Shinohara M., Zhang Y., LaDu M.J., Xu H. and Bu G. (2012) Differential regulation of amyloid-β endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem., 287, 44593–44601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huotari J. and Helenius A. (2011) Endosome maturation. EMBO J., 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takahashi R.H., Milner T.A., Li F., Nam E.E., Edgar M.A., Yamaguchi H., Beal M.F., Xu H., Greengard P. and Gouras G.K. (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol., 161, 1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang A.J., Chandswangbhuvana D., Margol L. and Glabe C.G. (1998) Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Aβ1-42 pathogenesis. J. Neurosci. Res., 52, 691–698. [DOI] [PubMed] [Google Scholar]
- 10. Ruan Q., Harrington A.J., Caldwell K.A., Caldwell G.A. and Standaert D.G. (2010) VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson’s disease. Neurobiol. Dis., 37, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Harrington A.J., Yacoubian T.A., Slone S.R., Caldwell K.A. and Caldwell G.A. (2012) Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J. Neurosci., 32, 2142–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooper A.A., Gitler A.D., Cashikar A., Haynes C.M., Hill K.J., Bhullar B., Liu K., Xu K., Strathearn K.E., Liu F. et al. (2006) Alpha-synuclein blocks ER–Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science, 313, 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Treusch S., Hamamichi S., Goodman J.L., Matlack K.E.S., Chung C.Y., Baru V., Shulman J.M., Parrado A., Bevis B.J., Valastyan J.S. et al. (2011) Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science, 334, 1241–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harold D., Abraham R., Hollingworth P., Sims R.,Gerrish A., Hamshere M.L., Pahwa J.S., Moskvina V., Dowzell K., Williams A. et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet., 41, 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naj A.C., Jun G., Beecham G.W., Wang L.-S., Vardarajan B.N., Buros J., Gallins P.J., Buxbaum J.D., Jarvik G.P., Crane P.K. et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet., 43, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen X. and Burgoyne R.D. (2012) Identification of common genetic modifiers of neurodegenerative diseases from an integrative analysis of diverse genetic screens in model organisms. BMC Genomics, 13, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu G., Zhang S., Cai Z., Ma G., Zhang L., Jiang Y., Feng R., Liao M., Chen Z., Zhao B. et al. (2013) PICALM gene rs3851179 polymorphism contributes to Alzheimer’s disease in an Asian population. NeuroMolecular Med., 15, 384–388. [DOI] [PubMed] [Google Scholar]
- 18. Mukherjee S., Russell J.C., Carr D.T., Burgess J.D., Allen M., Serie D.J., Boehme K.L., Kauwe J.S.K., Naj A.C., Fardo D.W. et al. (2017) Systems biology approach to late-onset Alzheimer’s disease genome-wide association study identifies novel candidate genes validated using brain expression data and Caenorhabditis elegans experiments. Alzheimer’s Dement., 13, 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Griffin E.F., Caldwell K.A. and Caldwell G.A. (2017) Genetic and pharmacological discovery for Alzheimer’s disease using Caenorhabditis elegans. ACS Chem. Neurosci., 8, 2596–2606. [DOI] [PubMed] [Google Scholar]
- 20. Martinez B.A., Caldwell K.A. and Caldwell G.A. (2017) C. elegans as a model system to accelerate discovery for Parkinson disease. Curr. Opin. Genet. Dev., 44, 102–109. [DOI] [PubMed] [Google Scholar]
- 21. Hamamichi S., Rivas R.N., Knight A.L., Cao S., Caldwell K.A. and Caldwell G.A. (2008) Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA, 105, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rockenstein E., Nuber S., Overk C.R., Ubhi K., Mante M., Patrick C., Adame A., Trejo-Morales M., Gerez J., Picotti P. et al. (2014) Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain, 137, 1496–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsigelny I.F., Sharikov Y., Wrasidlo W., Gonzalez T., Desplats P.A., Crews L., Spencer B. and Masliah E. (2012) Role of α-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J., 279, 1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee H.-J., Khoshaghideh F., Patel S. and Lee S.-J. (2004) Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci., 24, 1888–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Di Scala C., Yahi N., Boutemeur S., Flores A., Rodriguez L., Chahinian H., Fantini J., Hardy J.A., Higgins G.A., Irvine G.B. et al. (2016) Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci. Rep., 6, 28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim H.-Y., Cho M.-K., Kumar A., Maier E., Siebenhaar C., Becker S., Fernandez C.O., Lashuel H.A., Benz R., Lange A. et al. (2009) Structural properties of pore-forming oligomers of α-synuclein. J. Am. Chem. Soc., 131, 17482–17489. [DOI] [PubMed] [Google Scholar]
- 27. Link C.D., Taft A., Kapulkin V., Duke K., Kim S., Fei Q., Wood D.E. and Sahagan B.G. (2003) Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol. Aging, 24, 397–413. [DOI] [PubMed] [Google Scholar]
- 28. Fonte V., Kapulkin W.J., Kapulkin V., Taft A., Fluet A., Friedman D. and Link C.D. (2002) Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc. Natl. Acad. Sci. USA, 99, 9439–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cabrera M., Langemeyer L., Mari M., Rethmeier R., Orban I., Perz A., Bröcker C., Griffith J., Klose D., Steinhoff H.-J. et al. (2010) Phosphorylation of a membrane curvature-sensing motif switches function of the HOPS subunit Vps41 in membrane tethering. J. Cell Biol., 191, 845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Calixto A., Chelur D., Topalidou I., Chen X. and Chalfie M. (2010) Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods, 7, 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinkraus K.A., Smith E.D., Davis C., Carr D., Pendergrass W.R., Sutphin G.L., Kennedy B.K. and Kaeberlein M. (2008) Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell, 7, 394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cohen E., Bieschke J., Perciavalle R.M., Kelly J.W. and Dillin A.. (2006). Opposing activities protect against age-onset proteotoxicity. Science (80), 313, 1604–1610. [DOI] [PubMed] [Google Scholar]
- 33. Burdick D., Soreghan B., Kwon M., Kosmoski J., Knauer M., Henschen A., Yates J., Cotman C. and Glabe C. (1992) Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem., 267, 546–554. [PubMed] [Google Scholar]
- 34. Yang A.J., Chandswangbhuvana D., Shu T., Henschen A. and Glabe C.G. (1999) Intracellular accumulation of insoluble, newly synthesized abetan-42 in amyloid precursor protein-transfected cells that have been treated with Abeta1-42. J. Biol. Chem., 274, 20650–20656. [DOI] [PubMed] [Google Scholar]
- 35. Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A. and Yuan J. (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature, 403, 98–103. [DOI] [PubMed] [Google Scholar]
- 36. Okoshi T., Yamaguchi I., Ozawa D., Hasegawa K. and Naiki H. (2015) Endocytosed β2-microglobulin amyloid fibrils induce necrosis and apoptosis of rabbit synovial fibroblasts by disrupting endosomal/lysosomal membranes: a novel mechanism on the cytotoxicity of amyloid fibrils. PLoS One, 10, e0139330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cha M.-Y., Han S.-H., Son S.M., Hong H.-S., Choi Y.-J., Byun J. and Mook-Jung I. (2012) Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS One, 7, e34929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reinders N.R., Pao Y., Renner M.C., Silva-Matos C.M., Lodder T.R., Malinow R. and Kessels H.W. (2016) Amyloid-β effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl. Acad. Sci. USA, 113, E6526–E6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ulrich D. (2015) Amyloid-β impairs synaptic inhibition via GABAA receptor endocytosis. J. Neurosci., 35, 9205–9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang H.Y., Lee D.H., Davis C.B. and Shank R.P. (2000) Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J. Neurochem., 75, 1155–1161. [DOI] [PubMed] [Google Scholar]
- 41. Zhao Z., Sagare A.P., Ma Q., Halliday M.R., Kong P., Kisler K., Winkler E.A., Ramanathan A., Kanekiyo T., Bu G. et al. (2015) Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat. Neurosci., 18, 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kounnas M.Z., Moir R.D., Rebeck G.W., Bush A.I., Argraves W.S., Tanzi R.E., Hyman B.T. and Strickland D.K. (1995) LDL receptor-related protein, a multifunctional ApoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell, 82, 331–340. [DOI] [PubMed] [Google Scholar]
- 43. Esbjörner E.K., Chan F., Rees E., Erdelyi M., Luheshi L.M., Bertoncini C.W., Kaminski C.F., Dobson C.M. and Kaminski Schierle G.S. (2014) Direct observations of amyloid β self-assembly in live cells provide insights into differences in the kinetics of Aβ(1-40) and Aβ(1-42) aggregation. Chem. Biol., 21, 732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poteryaev D., Fares H., Bowerman B. and Spang A. (2007) Caenorhabditis elegans SAND-1 is essential for RAB-7 function in endosomal traffic. EMBO J., 26, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rehling P., Darsow T., Katzmann D.J. and Emr S.D. (1999) Formation of AP-3 transport intermediates requires Vps41 function. Nat. Cell Biol., 1, 346–353. [DOI] [PubMed] [Google Scholar]
- 46. Ostrowicz C.W., Bröcker C., Ahnert F., Nordmann M., Lachmann J., Peplowska K., Perz A., Auffarth K., Engelbrecht-Vandré S. and Ungermann C. (2010) Defined subunit arrangement and rab interactions are required for functionality of the HOPS tethering complex. Traffic, 11, 1334–1346. [DOI] [PubMed] [Google Scholar]
- 47. Peplowska K., Markgraf D.F., Ostrowicz C.W., Bange G. and Ungermann C. (2007) The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo-lysosomal biogenesis. Dev. Cell, 12, 739–750. [DOI] [PubMed] [Google Scholar]
- 48. Hickey C.M., Stroupe C. and Wickner W. (2009) The major role of the Rab Ypt7p in vacuole fusion is supporting HOPS membrane association. J. Biol. Chem., 284, 16118–16125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Desplats P., Lee H.-J., Bae E.-J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E. and Lee S.-J. (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci., 106, 13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dinter E., Saridaki T., Nippold M., Plum S., Diederichs L., Komnig D., Fensky L., May C., Marcus K., Voigt A. et al. (2016) Rab7 induces clearance of α-synuclein aggregates. J. Neurochem., 138, 758–774. [DOI] [PubMed] [Google Scholar]
- 51. Manil-Ségalen M., Lefebvre C., Jenzer C., Trichet M., Boulogne C., Satiat-Jeunemaitre B. and Legouis R. (2014) The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev. Cell, 28, 43–55. [DOI] [PubMed] [Google Scholar]
- 52. Majumder S., Richardson A., Strong R. and Oddo S. (2011) Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One, 6, e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spilman P., Podlutskaya N., Hart M.J., Debnath J., Gorostiza O., Bredesen D., Richardson A., Strong R. and Galvan V. (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One, 5, e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jia R., Guardia C.M., Pu J., Chen Y. and Bonifacino J.S. (2017) BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy, 13, 1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Moreau K., Fleming A., Imarisio S., Lopez Ramirez A., Mercer J.L., Jimenez-Sanchez M., Bento C.F., Puri C., Zavodszky E., Siddiqi F. et al. (2014) PICALM modulates autophagy activity and tau accumulation. Nat. Commun., 5, 4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Spencer B., Kim C., Gonzalez T., Bisquertt A., Patrick C., Rockenstein E., Adame A., Lee S.-J., Desplats P. and Masliah E. (2016) α-synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Hum. Mol. Genet., 25, 1100–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sasaki A., Nakae I., Nagasawa M., Hashimoto K., Abe F., Saito K., Fukuyama M., Gengyo-Ando K., Mitani S., Katada T. et al. (2013) Arl8/ARL-8 functions in apoptotic cell removal by mediating phagolysosome formation in Caenorhabditis elegans. Mol. Biol. Cell, 24, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Korolchuk V.I., Saiki S., Lichtenberg M., Siddiqi F.H., Roberts E.A., Imarisio S., Jahreiss L., Sarkar S., Futter M., Menzies F.M. et al. (2011) Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol., 13, 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Garg S., Sharma M., Ung C., Tuli A., Barral D.C., Hava D.L., Veerapen N., Besra G.S., Hacohen N. and Brenner M.B. (2011) Lysosomal trafficking, antigen presentation, and microbial killing are controlled by the Arf-like GTPase Arl8b. Immunity, 35, 182–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Khatter D., Raina V.B., Dwivedi D., Sindhwani A., Bahl S. and Sharma M. (2015) The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J. Cell Sci., 128, 1746–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fay D.S., Fluet A., Johnson C.J. and Link C.D. (1998) In vivo aggregation of beta-amyloid peptide variants. J. Neurochem., 71, 1616–1625. [DOI] [PubMed] [Google Scholar]
- 62. Florez-McClure M.L., Hohsfield L.A., Fonte G., Bealor M.T. and Link C.D. (2007) Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy, 3, 569–580. [DOI] [PubMed] [Google Scholar]
- 63. Nakae I., Fujino T., Kobayashi T., Sasaki A., Kikko Y., Fukuyama M., Gengyo-Ando K., Mitani S., Kontani K. and Katada T. (2010) The arf-like GTPase Arl8 mediates delivery of endocytosed macromolecules to lysosomes inCaenorhabditis elegans. Mol. Biol. Cell, 21, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Marwaha R., Arya S.B., Jagga D., Kaur H., Tuli A. and Sharma M. (2017) The Rab7 effector PLEKHM1 binds Arl8b to promote cargo traffic to lysosomes. J. Cell Biol., 216, 1051–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McEwan D., Popovic D., Gubas A., Terawaki S., Suzuki H., Stadel D., Coxon F., Miranda de Stegmann D., Bhogaraju S., Maddi K. et al. (2015) PLEKHM1 regulates autophagosome–lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell, 57, 39–54. [DOI] [PubMed] [Google Scholar]
- 66. Hegedűs K., Takáts S., Boda A., Jipa A., Nagy P., Varga K., Kovács A.L. and Juhász G. (2016) The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy.Mol. Biol. Cell, 27, 3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jordens I., Fernandez-Borja M., Marsman M., Dusseljee S., Janssen L., Calafat J., Janssen H., Wubbolts R. and Neefjes J. (2001) The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol., 11, 1680–1685. [DOI] [PubMed] [Google Scholar]
- 68. Rosa-Ferreira C. and Munro S. (2011) Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev. Cell, 21, 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Klassen M.P., Wu Y.E., Maeder C.I., Nakae I., Cueva J.G., Lehrman E.K., Tada M., Gengyo-Ando K., Wang G.J., Goodman M. et al. (2010) An Arf-like small G protein, ARL-8, promotes the axonal transport of presynaptic cargoes by suppressing vesicle aggregation. Neuron, 66, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Niwa S., Tao L., Lu S.Y., Liew G.M., Feng W., Nachury M.V. and Shen K. (2017) BORC regulates the axonal transport of synaptic vesicle precursors by activating ARL-8. Curr. Biol., 27, 2569–2578.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Poüs C. and Codogno P. (2011) Lysosome positioning coordinates mTORC1 activity and autophagy. Nat. Cell Biol., 13, 342–344. [DOI] [PubMed] [Google Scholar]
- 72. Nilsson P., Loganathan K., Sekiguchi M., Matsuba Y., Hui K., Tsubuki S., Tanaka M., Iwata N., Saito T. and Saido T.C. (2013) Aβ secretion and plaque formation depend on autophagy. Cell Rep., 5, 61–69. [DOI] [PubMed] [Google Scholar]
- 73. Barbero-Camps E., Roca-Agujetas V., Bartolessis I., Dios C., Fernández-Checa J.C., Marí M., Morales A., Hartmann T. and Colell A. (2018) Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy, 14, 1129–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Pickford F., Masliah E., Britschgi M., Lucin K., Narasimhan R., Jaeger P.A., Small S., Spencer B., Rockenstein E., Levine B. et al. (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest., 118, 2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rocchi A., Yamamoto S., Ting T., Fan Y., Sadleir K., Wang Y., Zhang W., Huang S., Levine B., Vassar R. et al. (2017) A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLOS Genet., 13, e1006962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Trajkovic K., Jeong H. and Krainc D. (2017) Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci., 37, 9000–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Farías G.G., Guardia C.M., De Pace R., Britt D.J. and Bonifacino J.S. (2017) BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci. USA, 114, E2955–E2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pu J., Schindler C., Jia R., Jarnik M., Backlund P. and Bonifacino J.S. (2015) BORC, a multisubunit complex that regulates lysosome positioning. Dev. Cell, 33, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rajendran L., Honsho M., Zahn T.R., Keller P., Geiger K.D., Verkade P. and Simons K. (2006) Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA, 103, 11172–11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Frühbeis C., Fröhlich D., Kuo W.P., Amphornrat J., Thilemann S., Saab A.S., Kirchhoff F., Möbius W., Goebbels S., Nave K.-A. et al. (2013) Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol., 11, e1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Melentijevic I., Toth M.L., Arnold M.L., Guasp R.J., Harinath G., Nguyen K.C., Taub D., Parker J.A., Neri C., Gabel C.V. et al. (2017) C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature, 542, 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sardar Sinha M., Ansell-Schultz A., Civitelli L., Hildesjö C., Larsson M., Lannfelt L., Ingelsson M. and Hallbeck M. (2018) Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol., 136, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Goetzl E.J., Boxer A., Schwartz J.B., Abner E.L., Petersen R.C., Miller B.L. and Kapogiannis D. (2015) Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology, 85, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hamlett E.D., Goetzl E.J., Ledreux A., Vasilevko V., Boger H.A., LaRosa A., Clark D., Carroll S.L., Carmona-Iragui M., Fortea J. et al. (2017) Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down syndrome. Alzheimer’s Dement., 13, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ellis H.M. and Horvitz H.R. (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell, 44, 817–829. [DOI] [PubMed] [Google Scholar]
- 86. Kimura K.D., Tissenbaum H.A., Liu Y. and Ruvkun G (1997) daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science (80), 277, 942–946. [DOI] [PubMed] [Google Scholar]
- 87. Sulston J.E., Schierenberg E., White J.G. and Thomson J.N. (1983) The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol., 100, 64–119. [DOI] [PubMed] [Google Scholar]
- 88. Vahdati Nia B., Kang C., Tran M.G., Lee D. and Murakami S. (2017) Meta analysis of human AlzGene database: benefits and limitations of using C. elegans for the study of Alzheimer’s disease and co-morbid conditions. Front. Genet., 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dexter P.M., Caldwell K.A. and Caldwell G.A. (2012) A predictable worm: application of Caenorhabditis elegans for mechanistic investigation of movement disorders. Neurotherapeutics, 9, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Thompson M.L., Chen P., Yan X., Kim H., Borom A.R., Roberts N.B., Caldwell K.A. and Caldwell G.A. (2014) TorsinA rescues ER-associated stress and locomotive defects in C. elegans models of ALS. Dis. Model. Mech., 7, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sorkaç A., Alcantara I.C., Hart A.C., Newman B., Thummel J. and Olesen S. (2016) In vivo modelling of ATP1A3 G316S-induced ataxia in C. elegans using CRISPR/Cas9-mediated homologous recombination reveals dominant loss of function defects. PLoS One, 11, e0167963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. White J.G., Southgate E., Thomson J.N. and Brenner S. (1986) The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B. Biol. Sci., 314, 1–340. [DOI] [PubMed] [Google Scholar]
- 93. Chalfie M., Sulston J., White J., Southgate E., Thomson J. and Brenner S. (1985) The neural circuit for touch sensitivity in Caenorhabditis elegans. J. Neurosci., 5, 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Brenner S. (1974) The genetics of Caenorhabditis elegans. Genetics, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Matlack K.E.S., Tardiff D.F., Narayan P., Hamamichi S., Caldwell K.A., Caldwell G.A. and Lindquist S. (2014) Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. USA., 111, 4013–4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tardiff D.F., Tucci M.L., Caldwell K.A., Caldwell G.A. and Lindquist S. (2012) Different 8-hydroxyquinolines protect models of TDP-43 protein, α-synuclein, and polyglutamine proteotoxicity through distinct mechanisms. J. Biol. Chem., 287, 4107–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dostal V. and Link C.D. (2010) Assaying β-amyloid toxicity using a transgenic C. elegans model. J. Vis. Exp ., 44, e2252. [DOI] [PMC free article] [PubMed] [Google Scholar]