

ABSTRACT
Mitotic arrest can result in cell death through the process of apoptosis. We have shown by live-cell imaging that the ubiquitin-proteasome dependent proteolysis of the apoptotic regulator Mcl-1 under the control of the anaphase-promoting complex or cyclosome (APC/C) provides a timing mechanism that distinguishes prolonged mitotic arrest from normal mitosis.
Keywords: apoptosis, mitosis, Mcl-1, mitotic cell death, proteolysis
Induction of the intrinsic pathway of apoptosis in cells arrested in mitosis results in mitotic cell death. This process is thought to remove cells that have failed mitosis and thereby protects an organism against the propagation of chromosomal defects. Mitotic cell death can be induced pharmacologically (at least in vitro) by microtubule poisons such as paclitaxel that arrest cells in mitosis through prolonged activation of the mitotic or spindle assembly checkpoint. Cells arrested in mitosis appear to be particularly sensitive to BH3 (Bcl-2 homology domain 3) mimetic drugs that target anti-apoptotic proteins of the Bcl-2 family, namely Bcl-2 (BCL2), Bcl-xL (BCL2L1) and Mcl-1 (MCL1).1–3 Phosphorylation of Bcl-2, Bcl-xL and the apoptosis inhibitor XIAP (X-linked inhibitor of apoptosis) is thought to reduce their protective activity during mitosis, but these modifications do not appear to change during mitotic arrest,4–6 suggesting that another mechanism is responsible for the initiation of mitotic cell death.
Mcl-1 is a particularly interesting player in the control of responses to the disruption of mitosis. Mcl-1 is an unstable, cell cycle-regulated protein that accumulates during interphase and reaches a peak around late G2 phase (ref 4; see Figure 1). In mitosis, the level of Mcl-1 protein declines, and this loss has been proposed to promote apoptosis in cells held in mitotic arrest.1–5,7 Indeed, ablation of Mcl-1 promotes mitotic cell death while enhanced expression of the protein inhibits this process.2,3 Phosphorylation of Mcl-1 at Thr92 by the mitotic protein kinase CDK1-cyclin B promotes degradation of the protein,4,7 indicating a link between entry into mitosis and destabilisation of the protein, but the mechanism that controls Mcl-1 destruction during mitotic arrest has been controversial.
Figure 1.
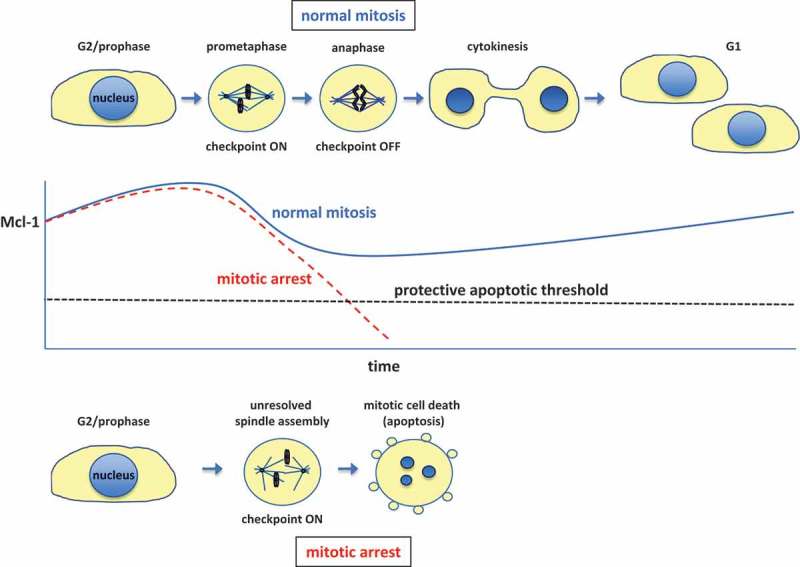
Cell cycle–dependent regulation of Mcl-1 levels and mitotic cell death.
The levels of the anti-apoptotic protein Mcl-1 (MCL1) vary during the cell cycle, reaching a peak around G2 and declining during mitosis due to ubiquitin proteasome-mediated degradation initiated by the APC/C (anaphase-promoting complex or cyclosome). The activity of the APC/C towards Mcl-1 is not affected by the spindle assembly checkpoint, which holds cells in a prometaphase-like state until proper chromosome attachment to spindle microtubules has been completed. Mcl-1 levels drop below a critical level during a prolonged mitotic arrest caused by unresolved spindle assembly, for instance due to disruption of microtubules by a drug, resulting in mitotic cell death by the intrinsic apoptotic pathway.
A key component in the mitotic destruction of Mcl-1 is the ubiquitin E3 ligase that catalyses the poly-ubiquitylation of Mcl-1 and primes it for recognition by the proteasome. Biochemical analyses of the pathways leading to mitotic cell death are restricted by the limited ability to follow the temporal regulation of the process and admixture of early G1 cells due to mitotic slippage. To counter these limitations, we recently employed a live-cell imaging approach pioneered in the analysis of mitotic protein destruction by Pines and others (e.g. Ref 8) in which the protein of interest is expressed as a fusion with a fluorescent protein derived from Aequorea and its degradation is monitored minute-by-minute during mitosis in individual cells. Caveats to this approach are that the fusion protein might be altered in its molecular interactions, regulation or localisation compared to the endogenous protein. We used an N-terminal Yellow Fluorescent Protein fusion with Mcl-1 (YFP-Mcl-1) which, although expressed at a higher level than the endogenous protein, was phosphorylated and degraded with similar kinetics to the endogenous protein during mitotic arrest, and was functional in inhibiting apoptosis.9 We demonstrated that YFP-Mcl-1 loss during mitotic arrest occurs relatively slowly compared to cyclin A, but is faster than the checkpoint-inhibited degradation of cyclin B. We confirmed our previous biochemical data (ref 4) that Mcl-1 degradation during mitotic arrest is dependent on the catalytic subunits of the ubiquitin E3 ligase APC/C (anaphase-promoting complex or cyclosome) and showed that it is not dependent on FBW7 (encoded by the FBXW7 gene, also known as hCDC4), a substrate recognition component of the SCF complex that regulates Mcl-1 stability in response to its phosphorylation by GSK3.7,10
By mutation of YFP-Mcl-1, we identified a D box motif similar to that found in other APC/C substrates that is required for efficient degradation during mitotic arrest. Revealingly, deletion of a C-terminal motif consisting of Isoleucine-Arginine (IR) changes the APC/C subunit requirements for Mcl-1 degradation and changes it from an atypical APC/C substrate that is not stimulated by the APC/C cofactor CDC20 (or the related cofactor CDH1) to one that is rapidly degraded in a conventional CDC20-dependent manner once the checkpoint is switched off, similar to cyclin B. We propose that the IR tail ensures that Mcl-1 is ubiquitylated slowly by basal APC/C activity, such that its rate of loss is not affected by the strength of the checkpoint and it is not catastrophically destroyed when the checkpoint is switched off. The level of Mcl-1 is reduced below the threshold to permit apoptosis only after a cell has become stuck in mitosis for a prolonged period and not during normal mitosis. This mechanism provides an opportunity to promote mitotic cell death by arresting cells in mitosis with a reagent like TAME (tosyl-L-arginine methyl ester) that inhibits CDC20-dependent APC/C activity towards cyclin B and while permitting Mcl-1 destruction. Our study raises a number of questions about the mechanism of Mcl-1 recognition and ubiquitination by the APC/C that will probably require reconstitution of the reaction in vitro for resolution.
Our work has shown that the steady rate of Mcl-1 destruction during mitosis provides an apoptotic timer that is independent of checkpoint strength. At the end of mitosis, Mcl-1 degradation is presumably stopped when CDK1-cyclin B activity is reduced, Mcl-1 becomes dephosphorylated at Thr92 and the APC/C is switched off. If spindle assembly cannot be completed successfully, however, then loss of Mcl-1 below the threshold required to restrain apoptosis may initiate cell death during mitosis (see Figure 1). Interestingly, reduction of Mcl-1 also appears to permit sub-apoptotic caspase activation in cells that do not undergo apoptosis, leading to an interphase stress response that determines subsequent cell fate.2 We speculate that this sustained response to the disruption of mitosis under the control of Mcl-1 may be as important as mitotic cell death for the clinical effects of microtubule poisons and BH3 mimetic drugs on tumours.
Abbreviations
APC/C anaphase-promoting complex or cyclosome.
BH3 Bcl-2 homology domain 3
CDK1 cyclin-dependent kinase 1
D box Destruction box
FBW7 F-Box and WD repeat domain containing protein 7
GSK3 glycogen synthase kinase 3
IR Isoleucine-Arginine.
Mcl-1 Myeloid Cell Leukemia 1
SCF SKP1-cullin-1-F-box complex
XIAP X-linked inhibitor of apoptosis
YFP Yellow Fluorescent Protein
Acknowledgments
Our work was funded by Cancer Research UK and Worldwide Cancer Research (formerly the Association for International Cancer Research).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was reported by the authors.
References
- 1.Shi J, Zhou Y, Huang HC, Mitchison TJ.. Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res. 2011;71(13):4518–4526. doi: 10.1158/0008-5472.CAN-10-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colin DJ, Hain KO, Allan LA, Clarke PR. Cellular responses to a prolonged delay in mitosis are determined by a DNA damage response controlled by Bcl-2 family proteins. Open Biol. 2015;5(3):140156. doi: 10.1098/rsob.140156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sloss O, Topham C, Diez M, Taylor S. Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget. 2016;7(5):5176–5192. doi: 10.18632/oncotarget.6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010;29(14):2407–2420. doi: 10.1038/emboj.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haschka MD, Soratroi C, Kirschnek S, Hacker G, Hilbe R, Geley S, Villunger A, Fava LL. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat Commun. 2015;6:6891. doi: 10.1038/ncomms7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hou Y, Allan LA, Clarke PR. Phosphorylation of XIAP by CDK1-cyclin-B1 controls mitotic cell death. J Cell Sci. 2017;130(2):502–511. doi: 10.1242/jcs.192310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471(7336):110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 8.Collin P, Nashchekina O, Walker R, Pines J. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol. 2013;15(11):1378–1385. doi: 10.1038/ncb2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allan LA, Skowyra A, Rogers KI, Zeller D, Clarke PR. Atypical APC/C-dependent degradation of Mcl-1 provides an apoptotic timer during mitotic arrest. EMBO J. 2018;pii:e96831. doi: 10.15252/embj.201796831 Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature. 2011;471(7336):104–109. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]