

ABSTRACT
Estrogen receptor alpha gene (ESR1) fusion transcripts have been identified in breast cancer but their role in breast cancer is not completely understood. Here, we report a causal role for ESR1 fusions in driving both endocrine therapy resistance and metastasis, and describe a therapeutic strategy to target ESR1 fusion-induced growth.
Keywords: ESR1 fusions, breast cancer, endocrine therapy resistance, EMT, metastasis
The majority of breast cancers express the nuclear hormone receptor, estrogen receptor alpha (ER) and therefore are fueled by estrogen and downstream ER signaling. Despite the tremendous success endocrine therapies, resistance and the development of lethal metastatic disease is common and a major clinical problem.
Dysregulation of the estrogen receptor alpha gene (ESR1) is an established mechanism of inducing endocrine therapy resistance in ER positive (ER+) breast cancer. Recurrent point mutations clustering around the ligand binding domain (LBD) of ESR1 that cause single amino acid residue changes have been found in up to 40% of treatment-refractory, metastatic ER+ breast cancer patients (reviewed in 1). These activating ESR1 point mutations confer constitutive, hormone-independent activity of ER and are often enriched in breast tumors after aromatase inhibitor (AI) treatment.
Emerging evidence now suggests genomic rearrangement events involving ESR1 producing ESR1 fusion genes are another class of somatic mutation that is associated with endocrine therapy resistance. One class of recurrent ESR1 fusion transcripts have been found in a subset of Luminal B primary breast tumors that retain the first two non-coding exons of ESR1 (ESR1-e2) fused to various sequences from a nearby gene, coiled-coil domain containing 170, CCDC170 (ESR1-e2>CCDC170), potentially generated by tandem-duplication, based on the observed orientation of the fusion sequences.2 This fusion gene forms a promoter trap driving aberrant expression of CCDC170 since this gene is now in the context of the ESR1 promoter, producing stable truncated forms of CCDC170 protein (ΔCCDC170). Specific forms of ΔCCDC170 leads to reduced tamoxifen sensitivity in experimental models.2 Recently, an additional ESR1-e2 fusion with the acidic residue methyltransferase 1 gene, C6orf211 (ESR1-e2>C6orf211) fusion as well as ESR1-e2>CCDC170 fusions have been identified in AI resistant breast tumors, but more studies are required to test whether these ESR1-e2 fusions produce pathogenic proteins.3 Taken together, these ESR1 fusion events have the potential to generate pathogenic, truncated forms of fusion partner proteins, but does not produce ESR1 fusion proteins (Figure 1B).
Figure 1.
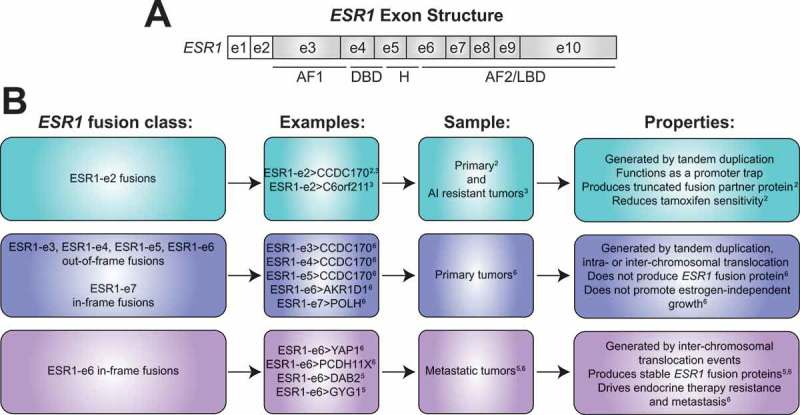
Landscape of ESR1 fusions in estrogen receptor positive breast cancer. (A) Structure of the estrogen receptor alpha gene (ESR1) containing 10 exons (e). The first two non shaded boxes represent non-coding exons (e1 and e2) followed by shaded boxes representing exons encoding for activation function 1 domain (AF1), DNA-binding domain (DBD), hinge region (H), and the activation function 2 (AF2)/ligand binding domain (LBD). (B) Three major classes of ESR1 fusion transcripts identified in estrogen receptor positive breast tumors. The first class arises from tandem duplication of ESR1promoter sequences including the first two exons of ESR1 (ESR1-e2) and a nearby gene found in primary and aromatase inhibitor (AI) resistant tumors, producing truncated forms of fusion partner protein, and therefore potentially driving disease pathogenesis by functioning as a promoter trap. The second class consists of the first 3–6 exons of ESR1 (ESR1-e3, ESR1-e4, ESR1-e5, ESR1-e6) fused out-of-frame or the first 7 exons of ESR1 (ESR1-e7) fused in-frame to C-terminal sequences from the partner gene. These fusion genes are found in treatment naïve primary breast tumors and do not produce stable ESR1 fusion proteins nor induce growth in estrogen-deprived conditions. The third class involves the first 6 exons of ESR1 (ESR1-e6) fused in-frame to C-terminal partner gene sequences provided by inter-chromosomal translocation found in metastatic breast tumors that have been extensively pretreated with endocrine agents. ESR1-e6>YAP1 and ESR1-e6>PCDH11X have been functionally characterized in detail and have been shown to drive endocrine therapy resistance and metastasis in experimental models. Coiled-coil domain containing protein 170, CCDC170; acidic residue methyltransferase 1, C6orf211; aldo-keto reductase family 1 member D1, AKR1D1; DNA polymerase eta, POLH; yes associated protein 1, YAP1; protocadherin 11 X-linked, PCDH11X; disabled homolog 2, DAB2; glycogenin 1, GYG1.
Another mechanism that can produce ESR1 fusions is inter- or intra-chromosomal translocation events where the ESR1 gene is fused to more distant locations in the genome. Our group described the first stable and functional ESR1 fusion protein produced by a fusion gene involving the first six exons of ESR1 (ESR1-e6) fused in-frame to C-terminal sequences of the yes associated protein 1 gene, YAP1 (ESR1-e6>YAP1) provided by an inter-chromosomal translocation event.4 This was identified in a patient with endocrine therapy-resistant, metastatic ER+ disease and in a matched patient-derived xenograft (PDX). A recent study described additional ESR1-e6 in-frame, inter-chromosomal translocation events involving the disabled homolog 2 gene, DAB2, and the glycogenin 1 gene, GYG1 (ESR1-e6>DAB2 and ESR1-e6>GYG1) producing stable in-frame ESR1 fusion proteins at metastatic sites in endocrine therapy refractory ER+ breast cancer patients.5 All of these ESR1-e6 fusions retain the N-terminus of ESR1 encoding the DNA binding and nuclear localization domains, suggesting some functionality. However, detailed functional characterization and studies demonstrating a causal role for ESR1 fusions in endocrine therapy resistance and metastasis has been lacking (Figure 1B).
Our current study investigated the role of ESR1 fusion genes in driving therapeutic resistance and metastasis in ER+ breast cancer.6 We identified a variety of in-frame and out-of-frame translocations involving ESR1 from RNA-seq analysis of primary and metastatic ER+ breast samples. In-frame fusions included inter-chromosomal ESR1 translocations with the YAP1 gene (ESR1-e6>YAP1) as previously described,4 the protocadherin 11 X-linked gene, PCDH11X (ESR1-e6>PCDH11X) and the nucleolar protein 2 homolog gene, NOP2 (ESR1-e6>NOP2), and two intra-chromosomal translocations with the A-kinase anchoring protein 12 gene, AKAP12 (ESR1-e6>AKAP12) and the DNA polymerase eta gene, POLH (ESR1-e7>POLH). However, only inter-chromosomal ESR1 fusions, ESR1-e6>YAP1, ESR1-e6>PCDH11X, and ESR1-e6>NOP2, produced stable in-frame ESR1 fusion proteins in vitro. Of these, the two ESR1 fusions identified from endocrine therapy-refractory, ER+ metastatic disease, ESR1-e6>YAP1 and ESR1-e6>PCDH11X, drove endocrine therapy resistant proliferation in experimental models, while ESR1-e6>NOP2 from an endocrine therapy naïve primary tumor did not. In addition, none of the out-of-frame ESR1-e3, ESR1-e4, ESR1-e5, and ESR1-e6 containing fusions nor even an in-frame ESR1-e7 fusion, all identified in primary tumors, were able to drive growth under estrogen-deprived conditions (Figure 1B).
To explore transcriptional properties of the ESR1 fusions that produced stable ESR1 fusion proteins, chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq) was performed along with RNA-seq in T47D cell lines to examine regulation of ESR1 fusion bound genes. The ESR1-e6>YAP1 and ESR1-e6>PCDH11X drove constitutive expression of these ER target genes in the absence of estrogen, demonstrating strong estrogen-independent transcriptional activation. The ESR1-e6>NOP2 bound relatively few sites compared to the other ESR1 fusions, potentially explaining the weak functional activity in proliferation assays. Interestingly, a cluster of genes was found to be strongly and uniquely up-regulated by the ESR1-e6>YAP1 and ESR1-e6>PCDH11X. Pathway analysis revealed enrichment of epithelial-to-mesenchymal transition (EMT) genes, including induction of an established EMT gene, snail family transcriptional repressor 1, SNAI1. Subsequent functional studies showed the transcriptionally active ESR1-e6>YAP1 and ESR1-e6>PCDH11X fusions induced cell motility in vitro and drove metastasis to the lung in xenograft models.
Since the formation of these ESR1 fusion proteins lacks the LBD of ESR1, all known endocrine therapies that target the LBD are likely to be ineffective. Therefore, we targeted downstream ER signaling events by using a United States Food and Drug Administration approved cyclin-dependent kinase (CDK) 4/6 inhibitor for metastatic ER+ breast cancer, palbociclib, based on our previous observation that palbociclib antagonized tumor growth driven by ESR1 point activating mutations. Palbociclib suppressed growth driven by ESR1-e6>YAP1 and ESR1-e6>PCDH11X in vitro, and at primary and metastatic sites in a PDX model naturally harboring the ESR1-e6>YAP1 fusion.
Taken together, these results further our understanding of the mechanisms underlying endocrine therapy resistance and metastasis in ER+ breast cancer. Transcriptionally active in-frame ESR1-e6 fusions such as ESR1-e6>YAP1 and ESR1-e6>PCDH11X constitutively drives expression of not only ER target genes leading to endocrine therapy-resistant proliferation but also induces EMT genes leading to metastasis (Figure 1B). Therefore, formation of ESR1 fusion genes links these two processes together, potentially explaining the lethal outcomes in the patients these fusions were identified.
These findings also have important therapeutic and diagnostic implications. Since ESR1 fusion driven growth remained sensitive to CDK4/6 inhibition, the presence of an in-frame ESR1 fusion could be used as a biomarker to stratify patients for CDK4/6 inhibitor therapy. Also, a pattern of ESR1 fusions is emerging in metastatic ER+ breast cancer, in which the first six exons of ESR1 are fused in-frame to C-terminal partner sequences provided by the partner gene. This finding could potentially drive targeted sequencing approaches to efficiently identify additional active ESR1 fusions by using a 3ʹ exon sequence against ESR1 exon 6 as bait.
In conclusion, ESR1 fusions are a new class of recurrent somatic mutations that drive endocrine therapy resistance and metastasis in ER+ breast cancer. This study adds to the catalog of actionable ESR1 alterations and furthers our understanding of how ER+ breast cancer gives rise to lethal metastatic disease.
Funding Statement
This work was supported by a Susan G. Komen Promise Grant (PG12220321 to MJE); a Cancer Prevention Institute of Texas (CPRIT) Recruitment of Established Investigators Award (RR140033 to MJE); a Breast Cancer Research Foundation Grant (BCRF ELFF-16-003 to MJE); and by a National Institutes of Health Training Grant (T32-GM088129 to JTL).
Disclosure statement
MJE received consulting fees from Abbvie, Sermonix, Pfizer, AstraZeneca, Celgene, NanoString, Puma, and Novartis, and is an equity stockholder, consultant, and Board Director member of BioClassifier, and inventor on a patent for the Breast PAM50 assay. These interests have been fully disclosed to Taylor & Francis and there is an approved plan for managing any potential conflicts arising from this arrangement.
References
- 1.Pejerrey SM, Dustin D, Kim JA, Gu G, Rechoum Y, Fuqua SAW.. The impact of ESR1 mutations on the treatment of metastatic breast cancer. Hormones Can. 2018;43(1). doi: 10.1007/s12672-017-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veeraraghavan J, Tan Y, Cao -X-X, Kim JA, Wang X, Chamness GC, Maiti SN, Cooper LJN, Edwards DP, Contreras A, et al. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun. 2014;5:4577. doi: 10.1038/ncomms5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV, Nixon MJ, Du L, Sanchez V, Ericsson PG, et al. Genomic profiling of ER(+) breast cancers after short-term estrogen suppression reveals alterations associated with endocrine resistance. Sci Transl Med. 2017; 9(402):eaai7993.doi: 10.1126/scitranslmed.aai7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013; 4(6):1116–1130.doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartmaier RJ, Trabucco SE, Priedigkeit N, Chung JH, Parachoniak CA, Borre PV, Morley S, Rosenzweig M, Gay LM, Goldberg ME, et al. Recurrent hyperactive ESR1 fusion proteins in endocrine therapy resistant breast cancer. Ann Oncol. 2018; 29(4):872–880.doi: 10.1093/annonc/mdy025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lei JT, Shao J, Zhang J, Iglesia M, Chan DW, Cao J, Anurag M, Singh P, He X, Kosaka Y, et al. Functional annotation of ESR1 Gene fusions in estrogen receptor-positive breast cancer. Cell Rep. 2018; 24(6):1434–1444.e1437.doi: 10.1016/j.celrep.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]