

Abstract
A set of glutamylases and deglutamylases controls levels of tubulin polyglutamylation, a prominent post‐translational modification of neuronal microtubules. Defective tubulin polyglutamylation was first linked to neurodegeneration in the Purkinje cell degeneration (pcd) mouse, which lacks deglutamylase CCP1, displays massive cerebellar atrophy, and accumulates abnormally glutamylated tubulin in degenerating neurons. We found biallelic rare and damaging variants in the gene encoding CCP1 in 13 individuals with infantile‐onset neurodegeneration and confirmed the absence of functional CCP1 along with dysregulated tubulin polyglutamylation. The human disease mainly affected the cerebellum, spinal motor neurons, and peripheral nerves. We also demonstrate previously unrecognized peripheral nerve and spinal motor neuron degeneration in pcd mice, which thus recapitulated key features of the human disease. Our findings link human neurodegeneration to tubulin polyglutamylation, entailing this post‐translational modification as a potential target for drug development for neurodegenerative disorders.
Keywords: cerebellum, cytosolic carboxypeptidase 1 (CCP1/AGTPBP1/NNA1), motor neuron, neurodegeneration, tubulin polyglutamylation
Subject Categories: Molecular Biology of Disease, Neuroscience
Introduction
The microtubule cytoskeleton is a key player in neuronal development, connectivity, plasticity, and function through regulating neuronal morphology (Liu & Dwyer, 2014), establishing and maintaining neuronal polarity (Craig & Banker, 1994), transporting cargo (Franker & Hoogenraad, 2013), and controlling signaling events (Dent & Baas, 2014). Dysfunctions of microtubules can lead to neurodevelopmental disorders and neurodegeneration, caused by aberrations of either tubulins (Jaglin & Chelly, 2009), the building blocks of microtubules, or microtubule‐associated proteins (Kielar et al, 2014; Wang et al, 2014). Tubulins undergo extensive posttranslational modifications (PTMs) including acetylation, phosphorylation, polyglycylation, detyrosination, deglutamylation, and polyglutamylation, which have been proposed to generate a “tubulin code”, a signaling mechanism that controls microtubule functions in cells (Gadadhar et al, 2017). Since some tubulin PTMs are particularly enriched in neurons (Janke & Kneussel, 2010), these modifications represent promising candidate mechanisms for neurodegenerative disorders. This hypothesis is supported by studies in mice linking tubulin acetylation (Dompierre et al, 2007; Outeiro et al, 2007) and polyglutamylation (Rogowski et al, 2010) to neurodegeneration, as well as observations of altered tubulin PTMs in diseased human brains (Zhang et al, 2015; Vu et al, 2017).
Tubulin PTMs are brought about by different specific enzymes (Gadadhar et al, 2017). One example is cytosolic carboxypeptidase 1 (CCP1; Kalinina et al, 2007; Rodriguez de la Vega et al, 2007) that removes glutamate residues from the carboxy‐termini of peptide chains (Rogowski et al, 2010; Berezniuk et al, 2012). With this specificity, CCP1 catalyzes two major types of tubulin PTMs: the removal of gene‐encoded, C‐terminal glutamate residues, which converts detyrosinated α‐tubulin into Δ2‐ and Δ3‐α‐tubulin (Rogowski et al, 2010; Aillaud et al, 2016), and the shortening of polyglutamate side chains (Rogowski et al, 2010; Berezniuk et al, 2012), which are posttranslationally added by polyglutamylases (Edde et al, 1990). Direct evidence for a critical role of CCP1 in the mammalian nervous system has been obtained in mice: The Purkinje cell degeneration (pcd) mouse lacks functional CCP1 (Fernandez‐Gonzalez et al, 2002) and displays massive cerebellar atrophy and ataxic behavior (Mullen et al, 1976). However, no human disease has so far been linked to CCP1 and the PTMs it mediates.
Here, we identify 13 patients from ten unrelated families with damaging biallelic variants in the gene encoding CCP1 and affected by infantile‐onset, progressive and frequently fatal neurodegeneration of the central and peripheral nervous systems.
Results
Identification of disease‐associated CCP1 variants
Whole‐exome sequencing (WES, Appendix Table S1) and array comparative genomic hybridization (array CGH) on ten families with genetically unresolved childhood‐onset neurodegeneration (Fig 1A) identified biallelic variants in the gene encoding CCP1 as a potential underlying cause [RefSeq NM_001330701 (mRNA) and NP_001317630 (protein), also known as ATP/GTP‐binding protein 1 (AGTPBP1) or nervous system nuclear protein induced by axotomy 1 (NNA1; Harris et al, 2000)]. We report six different loss‐of‐function variants, including one genomic deletion removing part of the gene but no other RefSeq sequence (Fig 1B, Appendix Fig S1) and one canonical splice site change resulting in the utilization of a cryptic splice site and disruption of the open reading frame (Fig 1C), as well as six distinct missense variants predicted to alter a single amino acid (Fig 1D). Variants segregated in accordance with autosomal recessive inheritance of the disease (Appendix Fig S2) and were absent or extremely rare (allele frequency below 0.00005) in databases on human genetic variation (Appendix Table S2). The Genome Aggregation Database (gnomAD) spanning exomes of 123,136 unrelated individuals (Lek et al, 2016) contained 98 CCP1 variants annotated as loss‐of‐function or missense with a predicted functional impact (according to PolyPhen‐2; Adzhubei et al, 2010) at least as deleterious as the mildest missense variant found in affected patients (Appendix Table S3). The probability of observing a homozygous or compound heterozygous genotype if an individual's alleles were randomly sampled from gnomAD is extremely low (P = 3.08e‐6). Our WES repositories contained eight biallelic CCP1 genotypes among 36,189 individuals with neurological disease which is exceptional (P = 5.39e‐13) even after a Bonferroni correction to account for the fact that CCP1 is only one of about 20,000 genes screened (P = 1.08e‐8).
Figure 1. Identification of individuals with CCP1 deficiency.
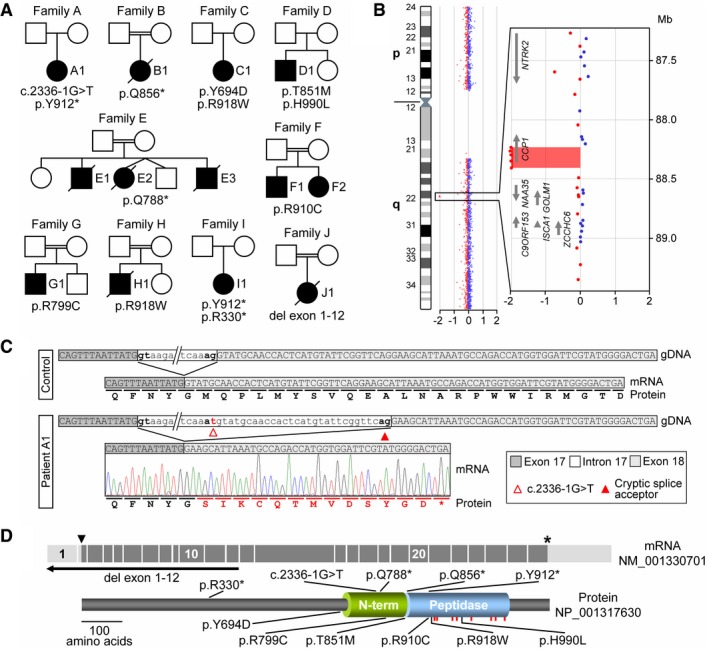
- Pedigrees of ten families with affected individuals carrying biallelic variants identified by WES or array CGH.
- Array CGH ratio profile of chromosome 9 from patient J1. Left: chromosome 9 ideogram with the log2ratios of probes plotted as a function of chromosomal position. Right: zoomed‐in region of interest containing the presumed homozygous deletion encompassing the N‐terminal exons of the CCP1 gene. Arrows point in the direction of transcription.
- Characterization of the splice acceptor site variant in family A. At the mRNA level, the intron 17 variant c.2336‐1G >T resulted in activation of a cryptic splice site, removing 29 nt from the mRNA causing a frameshift (p.M780fs) of the CCP1 open reading frame.
- Schematic representation of the mRNA (exons 1–26), the encoded CCP1 protein, and positions of disease‐associated variants. Arrowhead: ATG start codon, asterisk: stop codon. Peptidase: carboxypeptidase domain, N‐term: conserved N‐terminal domain, red bars: localizations of residues required for CCP1 substrate binding and catalytic activity.
Biallelic CCP1 variants cause early‐onset neurodegenerative phenotypes
The clinical presentations and characteristics of 13 affected individuals are summarized in Appendix Table S4. Patients generally presented with muscular hypotonia, global developmental delays, and cerebellar atrophy on brain imaging (Fig 2A) from early infancy, followed by progressive worsening of neurological function and a fatal course in six individuals. Five individuals who underwent nerve conduction studies, electromyography, muscle ultrasound, or muscle histology had signs of peripheral nerve and spinal motor neuron degeneration (Fig 2B and C), which were also suggested clinically in five further individuals due to muscle atrophy or absent deep tendon reflexes. Additional constantly observed features were microcephaly, eye movement abnormalities, feeding difficulties, ataxia, spasticity, and dystonia. We recognized two phenotypic extremes, critically ill infants (A1, B1, E1, E2, E3, H1, J1) with profound impairment of motor and cognitive development as opposed to one 14‐year‐old individual (D1) with a spastic–ataxic movement disorder and only mild intellectual disability. Other individuals (C1, F1, F2, G1, I1) represented an intermediate phenotype with longer survival, but still severe developmental delays and progressive neurological deficits resulting in significant disability. Patients with fatal and severe forms of the disease usually had biallelic protein‐truncating (nonsense, frameshift, splice, exon‐deletion) variants (except for H1) while those with milder phenotypes tended to have biallelic missense variants (with the exception of I1). No facial dysmorphisms and no involvement of organs other than the nervous system were documented in our case series. A synopsis of the relative frequencies of symptoms and signs of the CCP1‐related human disease is provided in Fig 2D.
Figure 2. Clinical presentation of patients with CCP1‐associated neurodegeneration.
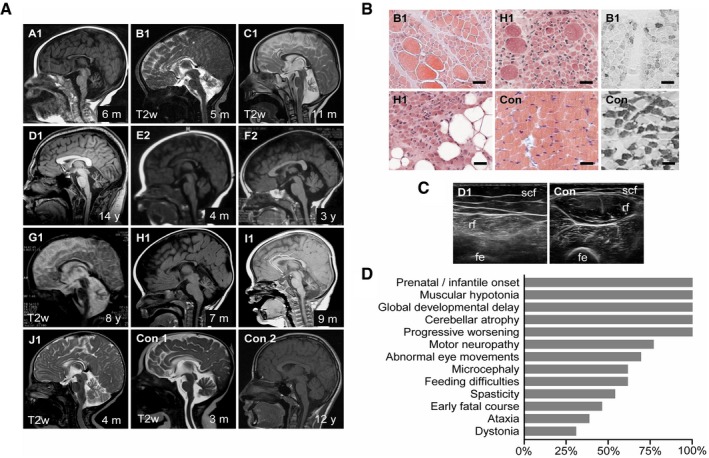
- Cerebellar atrophy (severe: A1, B1, C1, D1, G1, H1, I1; moderate: E2, F2, J1) and corpus callosum dysplasia (A1, B1, E2, J1). Magnetic resonance scans, T1w except where indicated. Con1‐2: non‐disease controls. Age at examination is given in months (m) or years (y).
- Chronic denervation with group fiber atrophy, interspersed hypertrophic fibers and fatty replacement (hematoxylin–eosin staining, left), and type 1 fiber predominance (fast myosin heavy‐chain staining, right). M. quadriceps biopsies at age 5 (B1) and 7 months (H1). Con: non‐disease control. Scale bar, 50 μm.
- Chronic denervation indicated by atrophy and increased echogenicity with granular and streaky pattern. M. rectus femoris (rf) ultrasound at age 14 years (D1). Con: healthy individual. Scf: subcutaneous fat, fe: femur.
- Relative frequencies of signs and symptoms of CCP1‐associated neurodegeneration based on 13 affected individuals. For details, see Appendix Table S4.
Disease‐associated variants lead to the absence of functional CCP1
As expected, nonsense, frameshift, and splice variants behaved like null alleles by completely abolishing expression of functional CCP1 (individuals A1 and B1 in Fig 3A), presumably either through nonsense‐mediated mRNA decay or early protein truncation. The genomic deletion (individual J1) removes upstream regulatory regions and the initiation codon (Fig 1B and D) and is predicted to result in no mRNA production or an N‐terminally truncated protein if an alternative promoter and transcription and translation initiation sites were used. Consistent with the absence of CCP1, polyglutamylated tubulin, generated by the continuous activity of polyglutamylases, appeared to accumulate in a diagnostic skeletal muscle biopsy specimen obtained from one of the patients (individual B1) with biallelic loss‐of‐function variants (Fig 3B). Missense variants clustered in or close to known functionally relevant domains of CCP1 (Kalinina et al, 2007; Fig 1D), affected residues that are highly conserved through evolution (Fig 3C), and probably had deleterious effects on the protein as predicted by computational structural analyses (Fig 3D) and 13/13 in silico algorithms (Appendix Table S3). Whenever tested, missense variants were associated with strongly reduced but still measurable amounts of CCP1 protein (individual D1 in Fig 3A and mutant‐transfected cells in Fig 3E). Levels of CCP1 missense mutants increased substantially after proteasome inhibition (Appendix Fig S3), again indicating that the variants may destabilize the protein structure, resulting in misfolded proteins susceptible to proteolysis. In further agreement with a loss‐of‐function disease mechanism, patient‐derived missense mutants appeared to have lost their catalytic activity as they could not generate detectable levels of Δ2‐tubulin when overexpressed in cells (Fig 3F).
Figure 3. Molecular consequences of disease‐related CCP1 variants.
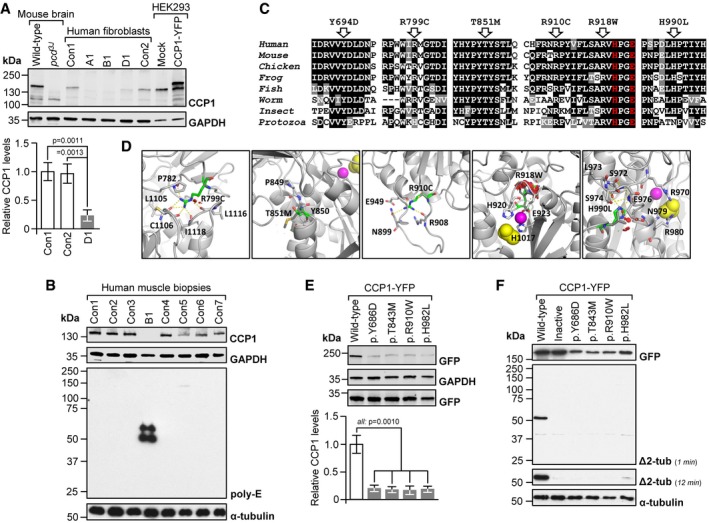
- Absent (A1, B1) or low‐level CCP1 (D1) in patient‐derived skin fibroblasts. Con1‐2: healthy donors. No signal in pcd brain and a higher‐molecular‐weight band in CCP1‐YFP‐expressing cells demonstrated specificity of the CCP1 antibody. Graphs: mean ± SD, n = 3. P‐values: one‐way ANOVA, Tukey post‐test.
- Excess tubulin polyglutamylation in CCP1‐deficient human skeletal muscle (M. quadriceps). B1: individual B1, Con1‐7: non‐disease controls. Poly‐E: polyglutamylated tubulin generated through the activity of polyglutamylases. Presence of CCP1 in human muscle was confirmed using the anti‐CCP1 antibody.
- Alignments of partial sequences of CCP1 from multiple species. Arrows: positions of CCP1 missense mutations observed in patients. Residues printed in red are required for enzymatic activity. Human (H. sapiens): NP_001317630, mouse (M. musculus): NP_075817, chicken (G. gallus): NP_001292036, frog (X. laevis): XP_018099357, fish (D. rerio): NP_001019616, worm (C. elegans): NP_491674, insect (A. mellifera): XP_006571511, and protozoa (T. brucei): XP_011778470.
- Possible effects of missense mutants on a predicted 3D structure of human CCP1 (based on 4b6z). Green stick models: mutated residues, sphere models: zinc ion (magenta) and acetate ion (substrate mimic, yellow), and dashed yellow lines: hydrogen (H) bonds. R799 points toward the protein core where it is engaged in H‐bonds. The p.R799C mutation is predicted to abolish H‐bond formation, which may destabilize the overall protein structure. The p.T851M mutation alters a conserved P‐F/Y‐S/T motif that is required for the folding of the N‐terminal domain in 4b6z. The larger and more hydrophobic methionine could impair stability of the immediate environment and thus interfere with protein folding. R910 resides in an outwardly projecting loop. Loss of R910 H‐bonds due to the p.R910C mutation may destabilize the loop region. The p.R918W substitution affects a loop containing residues (H920, E923) coordinating the zinc ion in the active site. The bulkier tryptophan may cause steric clashes (red), disrupting the shape of the active site. The p.H990L substitution affects a residue that contributes to a network of H‐bonds, possibly important for maintaining the structure of the close‐by active site (acetate ion).
- Low levels of missense‐mutant CCP1 in HEK293 cells transfected with expression vectors for mouse wild‐type and mutant CCP1‐YFP. Residues Y686, T843, R910, and H982 align to human Y694, T851, R918, and H990. Graphs: mean ± SD, n = 3. P‐values: one‐way ANOVA, Tukey post‐test.
- Impaired deglutamylase activity of overexpressed CCP1 missense mutants in HEK293 cells. Inactive: catalytically dead p.H912S + E915Q13. Δ2‐tub: Δ2‐α‐tubulin generated through CCP1‐mediated removal of the gene‐encoded C‐terminal glutamate residue from α‐tubulin.
A CCP1‐deficient mouse model recapitulates key features of the human disease
Mice with inactivating variants in the gene for CCP1 (pcd) (Fernandez‐Gonzalez et al, 2002) display cerebellar degeneration (Mullen et al, 1976) and dysregulated tubulin polyglutamylation (Rogowski et al, 2010), paralleling cerebellar atrophy in affected humans (Fig 2A) and accumulation of polyglutamylated tubulin in patient‐derived biomaterial (Fig 3B). However, peripheral nerve and spinal motor neuron involvement had not been reported in pcd mice despite the fact that CCP1 seems to be required for motor neuron function (Harris et al, 2000; Zhao et al, 2012) and is robustly expressed in peripheral nerve (Fig 4A and B) and spinal cord (Fig 4A and C). We therefore reinvestigated the pcd mouse model, confirmed brain pathology and abnormal tubulin PTMs (Fig 5A and B), but also noticed additional overt peripheral nerve degeneration in mutant mice. Analysis of wild‐type and pcd motoric femoral quadriceps nerves revealed reduced total nerve calibers (Fig 5C), an about 20–25% loss of myelinated axons (Fig 5D), perturbed axon morphology (Fig 5E), and concomitant macrophage activation (Fig 5F), indicating an ongoing degenerative process. The sensory saphenous nerve appeared normal in terms of total nerve calibers and numbers of axons (Appendix Fig S4), suggesting that the neuropathy in mice was pure motor or motor predominant, closely resembling the findings in human patients. Motor axon degeneration in pcd nerves indicated that the corresponding motor neuron cell bodies in the spinal cord could also be affected. Indeed, quantification of motor neurons in the ventral horns on consecutive sections of lumbar spinal cords revealed an approximately 50% reduction in the number of motor neurons in pcd mice (Fig 5G), accompanied by dysregulated tubulin polyglutamylation (Fig 5H).
Figure 4. Detection of CCP1 in normal mouse peripheral nerve and spinal cord.
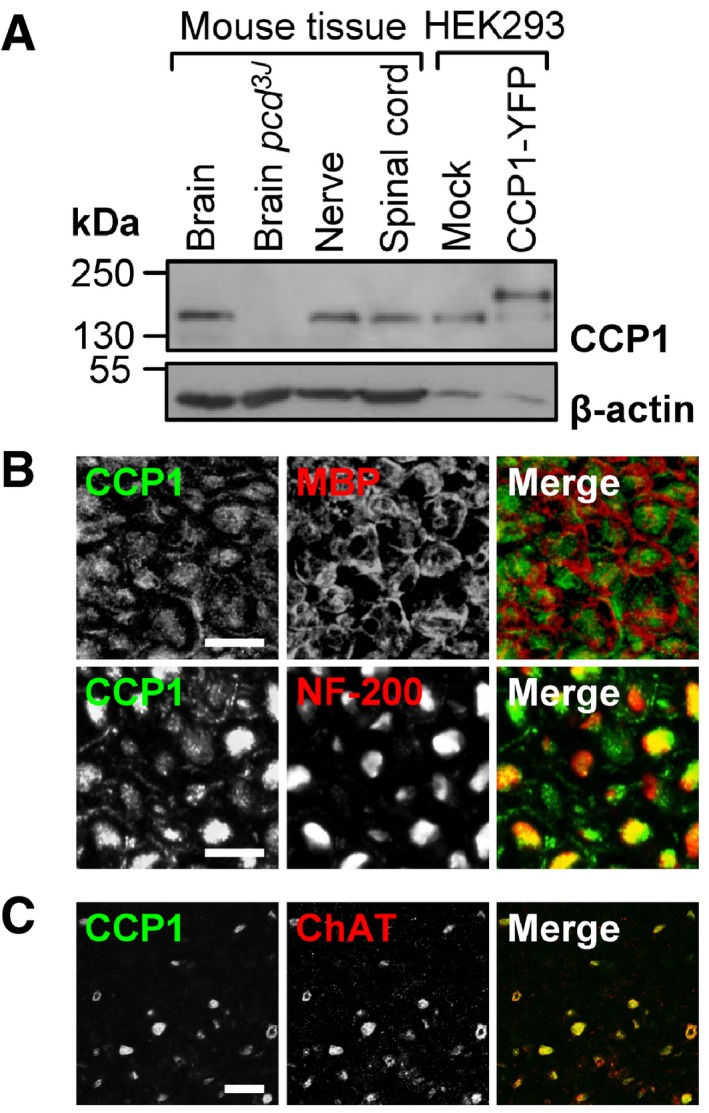
- Detection of CCP1 in protein extracts from brain, spinal cord, and peripheral nerve. Specificity of the anti‐CCP1 antibody was verified by no signal in pcd mouse brain and an extra band in HEK293 cells transfected with a CCP1‐YFP fusion protein.
- Association of CCP1 with myelinated axons (neurofilament (NF‐200)‐positive) but not compact myelin (myelin basic protein (MBP)‐positive) in peripheral nerve. Cross sections, scale bar, 10 μm.
- Presence of CCP1 in motor neurons (choline acetyltransferase (ChAT)‐positive) in the ventral horn of the lumbar spinal cord. Cross sections, scale bar, 50 μm.
Figure 5. Extended phenotype of CCP1 ablation in mice.
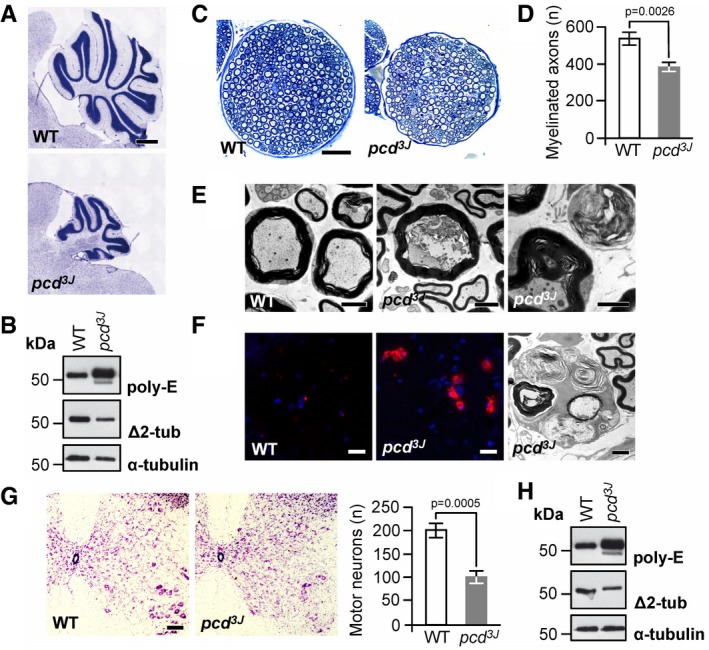
- Cerebellar atrophy in pcd mice. Nissl staining of mid‐sagittal sections of cerebella at P150, displaying massively reduced size of the cerebellar vermis in pcd mice relative to wild type (WT). Scale bar, 500 μm.
- Excess tubulin polyglutamylation (poly‐E) and decreased Δ2‐tubulin (Δ2‐tub) generation in pcd cerebellum. WT: wild type.
- Reduction in nerve diameter and numbers of myelinated fibers in pcd mice. Cross sections of femoral quadriceps nerves, toluidine blue staining, scale bar, 50 μm.
- Quantification of myelinated axons in the femoral quadriceps nerves of WT and pcd mice. Graphs: mean ± SD, n = 3. P‐value: Student's unpaired two‐tailed t‐test.
- Disorganized axoplasm (middle), reduced axon diameter with preserved myelin profile and contorted myelin devoid of an axon (right). Left: normal myelinated axons (WT mouse). Cross sections of femoral quadriceps nerves, electron microscopy, scale bar, 2 μm.
- Activated endoneurial macrophages in pcd mice. CD68 immunostaining (macrophage marker, left and middle image) and electron microscopy (rounded macrophages containing axonal fragments and myelin debris, right image). Scale bars, 20 μm (left and middle image), 2 μm (right image).
- Reduction in the number of motor neurons (large cells in the lower right corners) in ventral horns of pcd spinal cord. Transverse sections, Nissl staining, scale bar, 100 μm. Graphs: mean ± SD, n = 3. P‐values: Student's unpaired two‐tailed t‐test.
- Excess tubulin polyglutamylation (poly‐E) and decreased Δ2‐tubulin (Δ2‐tub) generation in pcd spinal cord. WT: wild type.
Discussion
In this study, we have shown that rare biallelic variants in the gene encoding the protein deglutamylase CCP1 cause degeneration of the central and peripheral nervous systems in humans. The salient features in affected individuals were infantile onset of developmental delays, progressive, often fatal neurological decline, cerebellar atrophy, and motor neuropathy. We noted that patients with protein‐truncating variants usually had more severe disease while those with missense variants tended to have milder phenotypes. While this observation is intriguing, it is probably too early to claim any definitive genotype–phenotype correlations as the number of reported cases is still small. Moreover, we did not find a molecular correlate as both, protein‐truncating and missense changes, were uniformly associated with the absence or low expression of catalytically dead CCP1.
The pcd mouse recapitulates key features of the human disease, including the absence of functional CCP1, abnormal tubulin polyglutamylation, cerebellar atrophy, and a previously unrecognized involvement of spinal motor neurons and peripheral nerves. However, the onset seems later in mice, and we could not yet obtain direct evidence for Purkinje cell degeneration in human patients as no postmortem examinations were conducted. Moreover, the characteristic retinal degeneration of the pcd mice (LaVail et al, 1982) has not been documented in affected humans; the eye movement abnormalities in the humans are likely to be the result of muscle weakness and cerebellar dysfunction, with no known evidence of retinal degeneration. This may indicate species differences and other deglutamylating enzymes may counterbalance the loss of CCP1 function in photoreceptors. It has indeed been shown that another deglutamylase, CCP5, is essential for photoreceptors in the human retina as CCP5 deficiency has been linked to retinitis pigmentosa (Kastner et al, 2015).
While the CCP1‐associated human disease reported here represents a rare monogenic condition, dysregulation of tubulin polyglutamylation could also be involved in other neurodegenerative disorders. In a manuscript appearing in the same issue of this journal, Magiera and colleagues recognized neurodegeneration as a general response to impaired tubulin deglutamylation in mice with global ablation of neuronal deglutamylases activity (Magiera et al, 2018). In their manuscript, Magiera et al pinpointed a potential disease mechanism in tubulin deglutamylase‐deficient neurons: They observed disturbed axonal transport, which is a common theme in age‐related neurodegenerative conditions (Millecamps & Julien, 2013). It is tempting to speculate that dysregulation of tubulin glutamylation could be a so‐far unrecognized risk factor that accelerates neuronal death in conjunction with other influences over the long life span of humans, ultimately contributing to common, usually sporadic late‐onset neurodegeneration. Indeed, first indications for such links have recently emerged when altered tubulin PTMs were detected in brain tissue of probands with Alzheimer's disease (Zhang et al, 2015; Vu et al, 2017).
Finally, the discovery of CCP1 deficiency as a cause of human neurodegeneration also entails a promising perspective that these disorders could become therapeutically accessible. Notably, Purkinje cell degeneration in pcd mice can be halted by counterbalancing tubulin glutamylation by genetic approaches (Rogowski et al, 2010; Berezniuk et al, 2012; Magiera et al, 2018). Since PTMs and their modifying enzymes are accessible targets for drug development (Huq & Wei, 2007), agents specifically modulating levels of tubulin polyglutamylation will probably soon become available. They could then be directly tested in already existing, relevant mouse models to explore their potential to treat often disabling and fatal neurodegenerative disorders.
Materials and Methods
Clinical studies and biomaterials
Study procedures were consistent with the policies of local institutional review boards at the Duke University, LMU Munich, Radboud University, National Institute of Neurological Disorders and Stroke, the University of California San Diego, King Saud bin Abdulaziz University for Health Sciences, Baylor College of Medicine, and Istituto Giannina Gaslini. Written informed consent was obtained from all study participants or their legal guardians. All experiments involving human samples and data conformed to the principles set out in the World Medical Association's Declaration of Helsinki and the Department of Health and Human Services Belmont Report. Probands were recruited after being seen in clinic by authors. If appropriate, further family members were invited to participate in the study. All examinations and diagnostic procedures were performed by experienced neurologists, pediatricians, child neurologists, and clinical geneticists. Primary human skin fibroblasts from individuals A1, B1, and D1 and tissue of a muscle biopsy from individual B1 were leftovers from routine diagnostic procedures performed in the past. Control samples were derived from healthy individuals or individuals with unrelated diseases and were obtained through the biobanks of the Friedrich‐Baur‐Institute (Munich Tissue Culture collection) and the National Institute of Neurological Disorders and Stroke.
Reagents
If not stated otherwise, reagents were purchased from Sigma‐Aldrich.
DNA sequencing
Details of WES procedures and data analysis employed by the seven independent teams at the Duke University, LMU Munich, Radboud University, National Institute of Neurological Disorders and Stroke, the University of California San Diego, King Saud bin Abdulaziz University for Health Sciences, and Baylor College of Medicine are given in Appendix Table S1. For validation of WES results and segregation studies, DNA sequences containing variants of interest were PCR‐amplified using oligonucleotide primer pairs which were designed based on the Human Genome Browser genomic sequence (GRCh37/hg19). Oligonucleotide primer sequences and PCR conditions are available upon request. Sequences of purified PCR products were determined by automated fluorescent cycle sequencing and capillary electrophoresis using ABI BigDye chemistry (Applied Biosystems) and ABI instruments (Applied Biosystems). Chromatograms were aligned to the reference genome sequence, and variants were detected by visual inspection.
Comparative genomic hybridization (CGH) and fluorescence in situ hybridization (FISH)
The team at the Istituto Giannina Gaslini identified a homozygous genomic deletion in a further family, family J, by using array CGH. DNA from patient J1 and her parents was analyzed with the Agilent SurePrint G3 Human CGH 8 × 60 K Microarray Kit (Agilent Technologies) according to the manufacturer's instructions. Data were evaluated using Cytogenomics software (Agilent Technologies) with genome coordinates according to human genome build GRCh37/hg19 and the following analysis settings: aberration algorithm ADM‐2; threshold 6.0; window size 0.2 Mb; filter 4 probes, and DLRS < 0.25. FISH with BAC clone RP11‐30E21, selected from the human library RPCI‐11 according to human genome build GRCh37/hg19, and a 9p subtelomere probe (Cytocell) was used for confirmation of a putative genomic deletion encompassing exons encoding the N‐terminus of CCP1. To further characterize this variant, we performed PCR for fragments corresponding to exonic sequences located near the presumed deletion breakpoint (exons 10, 11, 12, 13, 14, and 15; transcript NM_001330701) with DNA samples from the proband, her parents, and healthy control individuals. Because the deletion variant was homozygous in the patient according to array CGH and FISH experiments, we expected consistent failure of PCR amplification of fragments contained within the deletion region when using patient's DNA as template. A β‐actin primer set was used to ensure appropriate PCR set‐up and sufficient template DNA quality and quantity. The primer sequences and PCR conditions are available upon request. Correct amplification of target sequences was verified by Sanger sequencing of PCR products as described above (see “DNA sequencing”).
Genomic matchmaking
After identification of CCP1 variants, research teams posted the gene name in GeneMatcher (Sobreira et al, 2015), a service that enables contact between researchers who are interested in the same gene, and got connected through the platform.
Allele frequencies in case and control subjects
For allele frequencies of disease‐related CCP1 variants in several control data sets, see Appendix Table S2. We found 98 CCP1 alleles in gnomAD (Lek et al, 2016) with loss‐of‐function annotation or representing missense variants with a predicted functional impact at least as deleterious as the least deleterious variant observed in our families (missense p.H990L; PolyPhen‐2 score 0.95; Appendix Table S3). Assuming linkage equilibrium, which seems adequate as these variants are extremely rare, the probability of finding an individual with homozygous or compound heterozygous genotypes comprised of these variants is given by
where f k is the frequency of the k th variant in the set of 98 gnomAD variants. This probability was then used to compute the probability of observing at least eight individuals with such genotypes in 36,189 cases with neurological disease in WES repositories across seven investigative groups, using the tail of a binomial distribution.
RT–PCR of whole‐blood mRNA
Total RNA of individual A1 was isolated from a whole‐blood sample using the PAXgene Blood RNA System according to the manufacturer's instructions (PreAnalytix). Total RNA (1 μg) was then subjected to DNase I treatment and subsequently reverse transcribed with the Omniscript Kit (Qiagen) containing a 1‐to‐1 mixture of random hexamers and oligo‐dT primers. The PCR was performed using the Maxima Hot Start Green mix (Thermo Fisher Scientific) with 4 μl of the first‐strand cDNA and primers to amplify the region of the CCP1 transcript encompassing exon 16 to exon 20. The primer sequences and PCR conditions are available upon request. PCR products were electrophoresed on agarose gels; individual fragments were purified using QIA quick gel extraction kit (Qiagen) and analyzed by Sanger sequencing as described above (see “DNA sequencing”).
In silico protein analysis
We used PROVEAN (Choi et al, 2012), SIFT (Kumar et al, 2009), PolyPhen‐2 (Adzhubei et al, 2010), CADD (Kircher et al, 2014), LRTnew (Chun & Fay, 2009), SNAP2 (Hecht et al, 2015), MutationAssessor (Reva et al, 2011), VEST3 (Carter et al, 2013), MutationTaster2 (Schwarz et al, 2010), PMut (López‐Ferrando et al, 2017), GERP++ (Davydov et al, 2010), PhastCons, and PhyloP (Pollard et al, 2010) to analyze the effects of amino acid substitutions on CCP1. We gathered sequences for homologs to human CCP1 (NP_001317630) by running PSI‐BLAST with default settings against the NCBI non‐redundant database and created a multiple sequence alignment using ClustalW with default settings. For computational structural analysis of missense mutants, 3D models of human wild‐type and mutant CCP1 were predicted using the RaptorX server (Kallberg et al, 2014) and Burkholderia cenocepacia metallocarboxypeptidase (PDB entry 4b6z) (Rimsa et al, 2014) as template. Models were visualized, and effects of mutations were inspected using PyMOL (https://pymol.org).
Cloning of expression constructs
A mammalian expression construct containing wild‐type mouse CCP1 cDNA (NM_023328) subcloned into pcDNA 3.1‐eYFP (with N‐terminal located YFP) and the same construct but coding for a double mutation p.H912S + E915Q producing enzymatically inactive CCP1 have been published previously (Rogowski et al, 2010). Disease‐associated missense mutants were generated by overlap extension PCR (Ho et al, 1989) using the wild‐type construct as template and outer primers yielding PCR products containing unique restriction endonuclease sites of the CCP1 open reading frame (one 5′ and one 3′ of the site of the mutation). The primer sequences and PCR conditions are available upon request. PCR products were digested with EcoRV and SacII for generation of p.Y686D or BamHI and SacII (all from New England Biolabs) for generation of p.T843M, p.R910W, and p.H982L. After gel purification, fragments were subcloned into the corresponding restriction sites of the wild‐type CCP1 construct. For each construct, sequences were verified by Sanger sequencing as described above (see “DNA sequencing”). Amino acids Y686, T843, R910, and H982 of mouse CCP1 correspond to Y694, T851, R918, and H990 of human CCP1.
Expression levels of wild‐type and disease‐mutant CCP1
Primary human skin fibroblasts (from individuals A1, B1, and D1 and non‐disease controls) and HEK293 cells were cultured in DMEM containing 10% FCS, 2 mM L‐glutamine, 40 U/ml penicillin, and 0.04 mg/ml streptomycin (all from Life Technologies). HEK293 cells were co‐transfected with YFP‐tagged mouse CCP1 expression constructs and a GFP expression plasmid (to control for transfection efficiency) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. To explore a potential effect of proteasomal degradation on CCP1 turnover, parallel HEK293 cultures were treated with 5 μM MG132 or vehicle only (DMSO). Subconfluent cells were collected and lysed in lysis buffer (1% SDS, 10 mM Tris (pH 7.4)). Deep‐frozen human skeletal muscle biopsy specimens (M. quadriceps) from individual B1 and controls were homogenized with a rotor–stator homogenizer in 10 mM Tris, pH 7.4 containing 1% SDS. Proteins from cell extracts and tissue samples were resolved by SDS–PAGE and blotted onto nitrocellulose membranes. Immunoblots were developed by incubation with rabbit anti‐CCP1 (Proteintech #14067‐1‐AP, 1:1,500; for fibroblast and muscle tissue samples), rabbit anti‐GFP (Abcam #ab6556, 1:2,500 or Torrey Pines Biolabs #TP401, 1:5,000; for HEK293 cells), and mouse anti‐GAPDH antibodies (Millipore #AB2302, 1:500), followed by IRDye 800CW‐conjugated donkey anti‐rabbit IgG and IRDye 680RD‐conjugated donkey anti‐mouse IgG antibodies (both from LI‐COR, 1:10,000). Signals were obtained using an Odyssey Fc Imaging System (LI‐COR). Densitometry for quantification of CCP1 expression levels was performed using ImageJ software (developed by the National Institutes of Health). Signals obtained for CCP1 were normalized to signals from GAPDH bands (and GFP bands when transfected cells were used). Statistical significance of differences was determined using one‐way ANOVA and Tukey post‐test.
Detection of CCP1‐related tubulin PTMs
HEK293 cells were cultured in DMEM containing 10% FCS, 2 mM L‐glutamine, 40 U/ml penicillin, and 0.04 mg/ml streptomycin. Cells were transfected with YFP‐tagged mouse CCP1 expression constructs using JetPei (Polyplus) according to the manufacturer's instructions. Cells were lysed in MEM buffer (50 mM MES/NaOH, pH 6.8, 1 mM EGTA, 1 mM MgCl2 without protease inhibitors). Cerebellum and spinal cord were dissected from 3‐week‐old pcd 3J (on a C57BL/6N background) and C57BL/6N wild‐type mice and homogenized in 2.5× Laemmli buffer (225 mM DTT, 5% SDS, 200 mM Tris–HCl pH 6.8, 25% glycerol, bromophenol blue) using an Eppendorf tube potter. Deep‐frozen human muscle biopsy specimens (M. quadriceps) from individual B1 and controls were homogenized with a rotor–stator homogenizer in 10 mM Tris, pH 7.4 containing 1% SDS. Samples were run on 10% SDS–PAGE gels under conditions allowing separation of α‐ and β‐tubulins (TUB gels) (Magiera & Janke, 2013) and blotted onto nitrocellulose membranes. Immunoblots were probed with rabbit anti‐∆2‐tubulin [∆2‐tub, detecting removal of the penultimate C‐terminal glutamate residue of α‐tubulin (Parturle‐Lafanechere et al, 1994), 1:2,000], rabbit anti‐polyglutamate chain (poly‐E, detecting polyglutamate side chains consisting of at least three residues, Adipogen #IN105, 1:5,000), and mouse anti‐α‐tubulin antibodies (clone 12G10, Developmental Studies Hybridoma Bank, 1:500). HRP‐conjugated goat anti‐mouse and goat anti‐rabbit IgG antibodies (both from Santa Cruz, 1:1,000) were used as secondary antibodies. Signals were revealed using Clarity Western ECL (Bio‐Rad). Films were developed using a Curix 60 developer (Agfa) and scanned using a Perfection V750 PRO scanner (Epson).
Detection of CCP1 in mouse peripheral nerve and spinal cord
For Western blotting, sciatic nerve and spinal cord samples were taken from young adult (P56) C57BL/6N wild‐type mice and lysed in lysis buffer (95 mM NaCl, 25 mM Tris (pH 7.5), 10 mM EDTA, 2% SDS, 1 mM NaF, 1 mM NaVO4, and one tablet protease inhibitor cocktail (Roche) per 10 ml solution). Samples were centrifuged at 16,000 g for 5 min., and 50 μg of whole‐protein lysate from the supernatant was resolved by SDS–PAGE and blotted onto PVDF membranes. Immunoblots were developed by incubation with rabbit anti‐CCP1 (Proteintech #14067‐1‐AP, 1:1,500) and mouse anti‐β‐actin antibodies (Sigma‐Aldrich #A2228, 1:1,000) followed by HRP‐conjugated goat anti‐rabbit IgG and HRP‐conjugated goat anti‐mouse IgG antibodies (both from Dako, 1:2,000). Signals were obtained using ECL (GE Healthcare). Lysates from HEK293 cells overexpressing wild‐type YFP‐CCP1 (or not) and protein extracts from whole brains of wild‐type and homozygous pcd 3J mice served as controls. For immunofluorescence microscopy, sciatic nerve and spinal cord samples were dissected from P56 C57BL/6N wild‐type mice, embedded in Tissue Tek OCT Compound (Sakura), and deep‐frozen in liquid nitrogen before cryosectioning. Ten‐μm‐thick nerve sections and 20‐μm‐thick spinal cord sections were fixed with 4% paraformaldehyde in PBS, permeabilized with cold methanol (only for spinal cord samples), and then blocked in 5% BSA, 1% donkey serum, and 0.3% Triton X‐100 in PBS. Sections were incubated with primary antibodies rabbit anti‐CCP1 (Proteintech #14067‐1‐AP, 1:100) and either rat anti‐MBP (Millipore #MAB386, 1:200) or mouse anti‐NF‐200 (Sigma‐Aldrich #N2912, 1:400) for sciatic nerve sections or goat anti‐ChAT (Millipore #AB144P, 1:100) for spinal cord sections. Donkey anti‐rabbit IgG conjugated to Alexa Fluor 488, goat anti‐rat IgG conjugated to Alexa Fluor 647, goat anti‐mouse IgG conjugated to Alexa Fluor 594, and donkey anti‐goat IgG conjugated to Alexa Fluor 568 (all from Thermo Fisher Scientific, 1:500) were used as secondary antibodies. Images were captured with an LSM 800 confocal microscope (Zeiss) with ZEN 2.1 imaging software (Zeiss) and processed and pseudocolored in Photoshop (Adobe). Specificity of the signals was ensured by control experiments where none or only one of the primary antibodies was used.
Histology of pcd mice
Cerebella were dissected from paraformaldehyde‐perfused 5‐month‐old C57BL/6N wild‐type and homozygous pcd 3J mutant mice (on a C57BL/6N background), embedded in Tissue Tek OCT Compound and deep‐frozen in isopentane pre‐cooled with liquid nitrogen. Blocks were then cut into 40‐μm sagittal cryosections which were subsequently Nissl‐stained using cresyl violet. Samples were analyzed by light microscopy. Femoral nerves were dissected from glutaraldehyde‐perfused 6‐ to 7‐month‐old wild‐type (n = 3) and homozygous pcd 3J mice (n = 3) and embedded in epoxy resin according to published protocols (Groh et al, 2012). Blocks were cut into semithin sections, stained with toluidine blue, and analyzed by light microscopy using an Axiophot 2 microscope (Zeiss) equipped with a CCD camera (Visitron Systems). Spinal cords were removed from paraformaldehyde‐perfused 6‐ to 9‐month‐old wild‐type (n = 3) and homozygous pcd 3J mutant mice (n = 3), embedded in Tissue Tek OCT Compound and deep‐frozen in isopentane pre‐cooled with liquid nitrogen. Blocks were then cut into serial cross sections (20 sections à 30 μm per lumbar spinal cord; segments L3–L5) and Nissl‐stained using cresyl violet. Slides were studied by light microscopy, and motor neurons (defined as neurons with cell bodies with a diameter > 30 μm) were counted. Numbers were corrected for cells sectioned more than once according to Abercrombie (Abercrombie, 1946), and statistical significance was determined using Student's unpaired two‐tailed t‐test.
Electron microscopy of pcd mouse nerve
Femoral nerves taken from glutaraldehyde‐perfused mice were processed as described above, cut into ultrathin sections, and studied by electron microscopy according to published protocols (Groh et al, 2012). Electron micrographs were taken by a ProScan Slow Scan CCD camera (ProScan) mounted to a Leo 906 E electron microscope (Zeiss). For quantification of axonal loss, total numbers of myelinated axons were counted on composed electron micrographs of femoral nerve cross sections from three wild‐type and three pcd 3J mutant mice. Statistical significant differences between wild‐type and mutant animals were determined using Student's unpaired two‐tailed t‐test.
Detection of activated endoneurial macrophages
Femoral nerves were dissected from paraformaldehyde‐perfused six‐month‐old wild‐type and homozygous pcd 3J mice, embedded in Tissue Tek OCT Compound, and deep‐frozen in isopentane pre‐cooled with liquid nitrogen. Cryosections were processed for immunohistochemistry of macrophages using a rat anti‐CD68 antibody (Bio‐Rad # MCA1957, 1:500) as described earlier (Groh et al, 2010).
Author contributions
Conceptualization: VSh, MMM, RMart, CJ, and JS; formal analysis: MMM, STA, FJG‐V, NS, TMS, and CJ; investigation: VSh, MMM, DK, OLAN, MD, XY, LB, VSt, IMW, RS, RC, TMS, E‐JK, RMart, and JS; resources: VSh, MZ, KS, SR‐S, AN, OLAN, PDM, AAA, RMaro, LDMP, ECS, MSt, MOEB, PM, ARF, SD, RK, PSG, VSt, DM, CN, CM, BK, MSc, ET, PP, PD, CF, MYI, AAlas, AAlah, AG, PL, YY, BE‐W, PGK, ASA, DBG, BS, KMR, MA, VC, E‐JK, CGB, and JGG; writing—original draft: VSh, MMM, CJ, and JS; writing—review and editing: VSh, MMM, DK, LB, VSt, RC, JGG, RMart, CJ, and JS; supervision: VSh, MMM, DK, RMart, CJ, and JS; project administration: VSh, MMM, RMart, CJ, and JS; funding acquisition: VSh, MMM, DK, SR‐S, STA, DBG, RC, CGB, JGG, RMart, CJ, and JS.
Conflict of interest
The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered at Baylor Genetics. IMW is employed by and receives a salary from GeneDx.
Supporting information
Appendix
Review Process File
Acknowledgements
We thank the families for participating in this study. We are grateful to Jijumon A. S. (Institut Curie) for technical help and S. Lacomme, M. Landry, and A. Calas (Université Bordeaux) for data on pcd pathology. CGB thanks C. Mendoza and G. Averion for their help in the clinic. MMM received a fellowship from the European Molecular Biology Organization (ASTF 148‐2015), MD from the Bayerische Gleichstellungsförderung, and R. Marom from the Osteogenesis Imperfecta Foundation. This work was supported by the National Institutes of Health (NIH) Common Fund (1U01HG007672‐01; to VSh and DBG), the Fondation Vaincre Alzheimer (FR‐16055p; to MMM), the German Federal Ministry of Education and Research (BMBF) through the German Network for Charcot‐Marie‐Tooth neuropathies (CMT‐NET) (01GM1511B, 01GM1511D, 01GM1511F; to DK, SR‐S, RMart and JS), the King Abdullah University of Science and Technology (baseline fund and Award No FCC/1/1976‐21; to STA and FJGV), the Swedish StratNeuro program (to RC), the National Institute of Neurological Disorders and Stroke (to CGB), the NIH (R01NS098004, R01NS048453; to JGG), the French National Research Agency (ANR) (ANR‐12‐BSV2‐0007; to CJ), the program “Investissements d'Avenir” of the French government and ANR (ANR‐10‐LBX‐0038, ANR‐10‐IDEX‐0001‐02 PSL; to CJ), the Institut Curie (to CJ), the Institut National du Cancer (2013‐1‐PL BIO‐02‐ICR‐1, 2014‐PL BIO‐11‐ICR‐1; to CJ), the Fritz‐Thyssen‐Stiftung (Az.10.15.1.021MN; to JS), and the Friedrich‐Baur‐Stiftung (to JS). WES was funded through the Clinical Center Genomics Opportunity, sponsored by the National Human Genome Research Institute, the NIH Deputy Director for Intramural Research and the NIH Clinical Center, the Broad Joint Center for Mendelian Genomics (UM1 HG008900; to D. MacArthur and H. Rehm), and the Yale Center for Mendelian Disorders (U54HG006504; to R. Lifton, M. Günel, M. Gerstein and S. Mane). This study makes use of data shared through the Broad Institute matchbox repository. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The EMBO Journal (2018) 37: e100540
1–39 The list of affiliations appears at the end of this article
See also: https://doi.org/10.15252/embj.2018100440 (December 2018) and
https://doi.org/10.15252/embj.2018101023 (December 2018)
Contributor Information
Vandana Shashi, Email: vandana.shashi@duke.edu.
Carsten Janke, Email: carsten.janke@curie.fr.
Jan Senderek, Email: jan.senderek@med.uni-muenchen.de.
References
- Abercrombie M (1946) Estimation of nuclear population from microtome sections. Anat Rec 94: 239–247 [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aillaud C, Bosc C, Saoudi Y, Denarier E, Peris L, Sago L, Taulet N, Cieren A, Tort O, Magiera MM, Janke C, Redeker V, Andrieux A, Moutin MJ (2016) Evidence for new C‐terminally truncated variants of alpha‐ and beta‐tubulins. Mol Biol Cell 27: 640–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezniuk I, Vu HT, Lyons PJ, Sironi JJ, Xiao H, Burd B, Setou M, Angeletti RH, Ikegami K, Fricker LD (2012) Cytosolic carboxypeptidase 1 is involved in processing alpha‐ and beta‐tubulin. J Biol Chem 287: 6503–6517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Douville C, Stenson PD, Cooper DN, Karchin R (2013) Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom 14(Suppl 3): S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7: e46688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC (2009) Identification of deleterious mutations within three human genomes. Genome Res 19: 1553–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Banker G (1994) Neuronal polarity. Annu Rev Neurosci 17: 267–310 [DOI] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++ . PLoS Comput Biol 6: e1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Baas PW (2014) Microtubules in neurons as information carriers. J Neurochem 129: 235–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci 27: 3571–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edde B, Rossier J, Le Caer JP, Desbruyeres E, Gros F, Denoulet P (1990) Posttranslational glutamylation of alpha‐tubulin. Science 247: 83–85 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Gonzalez A, La Spada AR, Treadaway J, Higdon JC, Harris BS, Sidman RL, Morgan JI, Zuo J (2002) Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy‐induced gene, Nna1. Science 295: 1904–1906 [DOI] [PubMed] [Google Scholar]
- Franker MA, Hoogenraad CC (2013) Microtubule‐based transport – basic mechanisms, traffic rules and role in neurological pathogenesis. J Cell Sci 126: 2319–2329 [DOI] [PubMed] [Google Scholar]
- Gadadhar S, Bodakuntla S, Natarajan K, Janke C (2017) The tubulin code at a glance. J Cell Sci 130: 1347–1353 [DOI] [PubMed] [Google Scholar]
- Groh J, Heinl K, Kohl B, Wessig C, Greeske J, Fischer S, Martini R (2010) Attenuation of MCP‐1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot‐Marie‐Tooth 1X. Hum Mol Genet 19: 3530–3543 [DOI] [PubMed] [Google Scholar]
- Groh J, Weis J, Zieger H, Stanley ER, Heuer H, Martini R (2012) Colony‐stimulating factor‐1 mediates macrophage‐related neural damage in a model for Charcot‐Marie‐Tooth disease type 1X. Brain 135: 88–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Morgan JI, Pecot M, Soumare A, Osborne A, Soares HD (2000) Regenerating motor neurons express Nna1, a novel ATP/GTP‐binding protein related to zinc carboxypeptidases. Mol Cell Neurosci 16: 578–596 [DOI] [PubMed] [Google Scholar]
- Hecht M, Bromberg Y, Rost B (2015) Better prediction of functional effects for sequence variants. BMC Genom 16(Suppl 8): S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site‐directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59 [DOI] [PubMed] [Google Scholar]
- Huq M, Wei LN (2007) Protein posttranslational modification: a potential target in pharmaceutical development In Handbook of Pharmaceutical Biotechnology, Gad SC. (ed), pp 417–441. Hoboken: John Wiley & Sons; [Google Scholar]
- Jaglin XH, Chelly J (2009) Tubulin‐related cortical dysgeneses: microtubule dysfunction underlying neuronal migration defects. Trends Genet 25: 555–566 [DOI] [PubMed] [Google Scholar]
- Janke C, Kneussel M (2010) Tubulin post‐translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci 33: 362–372 [DOI] [PubMed] [Google Scholar]
- Kalinina E, Biswas R, Berezniuk I, Hermoso A, Aviles FX, Fricker LD (2007) A novel subfamily of mouse cytosolic carboxypeptidases. FASEB J 21: 836–850 [DOI] [PubMed] [Google Scholar]
- Kallberg M, Margaryan G, Wang S, Ma J, Xu J (2014) RaptorX server: a resource for template‐based protein structure modeling. Methods Mol Biol 1137: 17–27 [DOI] [PubMed] [Google Scholar]
- Kastner S, Thiemann IJ, Dekomien G, Petrasch‐Parwez E, Schreiber S, Akkad DA, Gerding WM, Hoffjan S, Gunes S, Bagci H, Epplen JT (2015) Exome sequencing reveals AGBL5 as novel candidate gene and additional variants for retinitis pigmentosa in five Turkish families. Invest Ophthalmol Vis Sci 56: 8045–8053 [DOI] [PubMed] [Google Scholar]
- Kielar M, Tuy FP, Bizzotto S, Lebrand C, de Juan Romero C, Poirier K, Oegema R, Mancini GM, Bahi‐Buisson N, Olaso R, Le Moing AG, Boutourlinsky K, Boucher D, Carpentier W, Berquin P, Deleuze JF, Belvindrah R, Borrell V, Welker E, Chelly J et al (2014) Mutations in Eml1 lead to ectopic progenitors and neuronal heterotopia in mouse and human. Nat Neurosci 17: 923–933 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081 [DOI] [PubMed] [Google Scholar]
- LaVail MM, Blanks JC, Mullen RJ (1982) Retinal degeneration in the pcd cerebellar mutant mouse. I. Light microscopic and autoradiographic analysis. J Comp Neurol 212: 217–230 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce‐Hoffman E, Berghout J, Cooper DN et al (2016) Analysis of protein‐coding genetic variation in 60,706 humans. Nature 536: 285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Dwyer T (2014) Microtubule dynamics in axon guidance. Neurosci Bull 30: 569–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Ferrando V, Gazzo A, de la Cruz X, Orozco M, Gelpí JL (2017) PMut: a web‐based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res 45: W222–W228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Janke C (2013) Investigating tubulin posttranslational modifications with specific antibodies In Methods in Cell Biology. Correia JJ, Wilson L. (eds), pp 247–267. Burlington: Academic Press; [DOI] [PubMed] [Google Scholar]
- Magiera MM, Bodakuntla S, Ziak J, Lacomme S, Marques Sousa P, Leboucher S, Hausrat TJ, Bosc C, Andrieux A, Kneussel M, Landry M, Calas A, Balastik M, Janke C (2018) Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J 37: e100440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps S, Julien JP (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14: 161–176 [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Eicher EM, Sidman RL (1976) Purkinje cell degeneration, a new neurological mutation in the mouse. Proc Natl Acad Sci USA 73: 208–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, Young AB, Abagyan R, Feany MB, Hyman BT, Kazantsev AG (2007) Sirtuin 2 inhibitors rescue alpha‐synuclein‐mediated toxicity in models of Parkinson's disease. Science 317: 516–519 [DOI] [PubMed] [Google Scholar]
- Parturle‐Lafanechere L, Manier M, Trigault N, Pirollet F, Mazarguil H, Job D (1994) Accumulation of delta 2‐tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J Cell Sci 107: 1529–1543 [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C (2011) Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39: e118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimsa V, Eadsforth TC, Joosten RP, Hunter WN (2014) High‐resolution structure of the M14‐type cytosolic carboxypeptidase from Burkholderia cenocepacia refined exploiting PDB_REDO strategies. Acta Crystallogr D Biol Crystallogr 70: 279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez de la Vega M, Sevilla RG, Hermoso A, Lorenzo J, Tanco S, Diez A, Fricker LD, Bautista JM, Aviles FX (2007) Nna1‐like proteins are active metallocarboxypeptidases of a new and diverse M14 subfamily. FASEB J 21: 851–865 [DOI] [PubMed] [Google Scholar]
- Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme JC, Bosson A, Peris L, Gold ND, Lacroix B, Bosch Grau M, Bec N, Larroque C, Desagher S, Holzer M, Andrieux A, Moutin MJ, Janke C (2010) A family of protein‐deglutamylating enzymes associated with neurodegeneration. Cell 143: 564–578 [DOI] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 7: 575–576 [DOI] [PubMed] [Google Scholar]
- Sobreira N, Schiettecatte F, Valle D, Hamosh A (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36: 928–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu HT, Akatsu H, Hashizume Y, Setou M, Ikegami K (2017) Increase in alpha‐tubulin modifications in the neuronal processes of hippocampal neurons in both kainic acid‐induced epileptic seizure and Alzheimer's disease. Sci Rep 7: 40205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Gao X, Wang ZH (2014) The physiology and pathology of microtubule‐associated protein tau. Essays Biochem 56: 111–123 [DOI] [PubMed] [Google Scholar]
- Zhang F, Su B, Wang C, Siedlak SL, Mondragon‐Rodriguez S, Lee HG, Wang X, Perry G, Zhu X (2015) Posttranslational modifications of alpha‐tubulin in alzheimer disease. Transl Neurodegener 4: 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Onteru SK, Dittmer KE, Parton K, Blair HT, Rothschild MF, Garrick DJ (2012) A missense mutation in AGTPBP1 was identified in sheep with a lower motor neuron disease. Heredity 109: 156–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File