

Abstract
Post‐translational modifications of tubulin can regulate the dynamics and mechanical properties of microtubules and their interactions with different proteins, such as molecular motors. Two studies now demonstrate that excessive accumulation of a specific modification, polyglutamylation, leads to neurodegeneration in mice and humans, likely due to defects in axonal microtubule‐based transport.
Subject Categories: Neuroscience; Post-translational Modifications, Proteolysis & Proteomics
Microtubules are cytoskeletal filaments controlling many aspects of cell architecture, such as cell shape and polarity, as well as the distribution and transport of various organelles. To support very diverse cellular functions, microtubule structure, organization, and dynamics can be adapted between cell types, developmental stages, and even different microtubule subsets within the same cell. Such microtubule specialization relies on the differential use of tubulin isotypes, microtubule‐associated proteins (MAPs), and post‐translational modifications of tubulin.
An important tubulin modification is (poly)glutamylation, the addition of polyglutamate tails to the C‐termini of α‐tubulin or β‐tubulin (Magiera et al, 2018b). Polyglutamylation is initiated by adding a glutamic acid residue to the γ‐carboxyl group of a gene‐encoded glutamate. The new branch of the polypeptide chain can then be elongated by further glutamate addition. Branch formation and extension are catalyzed by different members of the tyrosine tubulin ligase‐like (TTLL) family of enzymes, which have different specificities (Magiera et al, 2018b). Glutamylation is reversible—glutamate residues can be removed by the members of cytosolic carboxypeptidase (CCP) family (Rogowski et al, 2010). Tubulin is the major target of this post‐translational modification, but multiple other substrates are known (van Dijk et al, 2008).
Polyglutamylation affects the structure and increases the negative charge of the intrinsically disordered tubulin tails, which are important for microtubule interactions with a broad variety of MAPs. Tubulin polyglutamylation is detected on spindle and midbody microtubules during cell division and is strongly enriched on centrioles, cilia, and neuronal microtubules (Magiera et al, 2018b). The function of this modification can vary depending on the cell type and cellular compartment; for example, it is required for the motility and function of cilia (Magiera et al, 2018b).
The first indication that the levels of polyglutamylation are critical for neuronal function and survival came from the studies of the Purkinje cell degeneration (pcd) mouse model (Rogowski et al, 2010). This mouse lacks functional CCP1, and this defect leads to degeneration of cerebellar Purkinje cells. Since CCP1 is a negative regulator of polyglutamylation, this work showed that levels of this modification appear to be critical for neuronal survival. However, many important questions remained unanswered. CCP1 removes not only polyglutamate chains, but also gene‐encoded C‐terminal acidic residues from different proteins, including tubulin (Tanco et al, 2015; Magiera et al, 2018b), and the phenotype of the pcd mouse might be due to the lack of the latter activity. It also remained unclear whether the sensitivity to CCP levels is a general property of neuronal cells, and if so, what the underlying mechanism would be (Fig 1).
Figure 1. Effects of polyglutamylation on neurodegeneration.
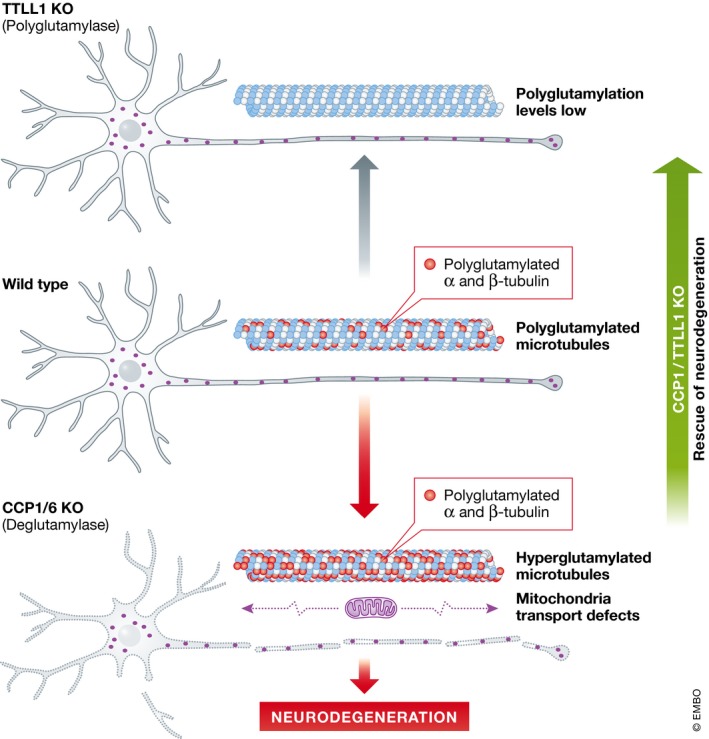
Neurons with reduced deglutamylase activity exhibit defects in mitochondrial motility and degenerate. This effect can be rescued by decreasing polyglutamylase levels, indicating that excessive polyglutamylation, for which microtubules represent a primary target in neurons, is the cause of cell death.
To address these questions, Magiera et al now combined conditional Purkinje cell‐specific knockouts in genes encoding CCP1 and TTLL1, a major brain polyglutamylase (Magiera et al, 2018a). Whereas the loss of CCP1 causes Purkinje cell death soon after birth, the concomitant deletion of TTLL1 fully reversed this defect. TTLL1‐deficient mice had strongly reduced levels of polyglutamylation of tubulin. This result leads to several important conclusions. First, the loss of CCP1 activity is toxic due to the increased polyglutamylation and not because of the increased removal of gene‐encoded glutamates of tubulin or other proteins. Second, increased but not decreased levels of polyglutamylation compromise neuronal survival. This finding fits with the observation that blocking of polyglutamylation of the predominant α‐tubulin isoform in flies does not compromise their viability (Jenkins et al, 2017). Third, hyperglutamylation causes neuronal death in a cell‐autonomous manner.
If excessive polyglutamylation causes neuronal death, why does not this defect manifest itself in the cerebral cortex and hippocampus? The answer might lie in the fact that the neurons in these brain regions co‐express another abundant CCP enzyme, CCP6. Indeed, Magiera et al have found that the knockout mouse lacking both CCP1 and CCP6 displayed normal brain development but showed strong signs of progressive neuronal degeneration in cerebral cortex. Sensitivity to increased polyglutamylation seems to be a general neuronal property.
What is the mechanism underlying neuronal cell death in CCP knockout neurons? Histological analysis revealed clear signs of axonal degeneration, whereas the myelin sheets were preserved. A potential candidate to induce this phenotype is spastin—a microtubule‐severing enzyme, which is strongly stimulated by microtubule glutamylation (Valenstein & Roll‐Mecak, 2016). Magiera et al tested spastin involvement but found that spastin knockout did not prevent Purkinje cell degeneration in pcd animals, indicating that it is not caused by excessive spastin‐mediated microtubule fragmentation. It should be noted, however, that the response of spastin to microtubule glutamylation levels is complex: in vitro, tubulin glutamylation can promote the severing activity of the enzyme, but becomes inhibitory after a certain threshold (Valenstein & Roll‐Mecak, 2016). Therefore, spastin activity might not be significantly altered or in fact could be decreased in pcd mice, a possibility that might be worth exploring since the loss of spastin also causes a neurodegenerative disorder, hereditary spastic paraplegia (Millecamps & Julien, 2013).
Polyglutamylation is also known to affect molecular motors, such as kinesin‐1 and flagellar dyneins (Sirajuddin et al, 2014; Kubo & Oda, 2017). Therefore, Magiera et al next tested whether microtubule‐based transport is affected in CCP‐deficient cells. Interestingly, they found that a reduction in deglutamylase activity strongly diminished the overall motility of mitochondria in axons, although not the speed or processivity of mitochondria movement. Defects in microtubule‐based transport were implicated in many neurodegenerative diseases (Millecamps & Julien, 2013), and it is thus likely that transport perturbation is indeed the cause of neuronal death in CCP knockout cells. In agreement with this view, a very recent study demonstrated that neurons lacking CCP1 have fragmented mitochondria, which exhibit axonal motility defects (Gilmore‐Hall et al, 2018).
Can these findings be extended to neurodegeneration in humans? Shashi et al found biallelic mutations that disrupted CCP1 function and tubulin deglutamylation in 13 patients with infantile‐onset neurodegeneration (Shashi et al, 2018). A careful comparison of the pcd mouse and the pathology observed in human patients showed striking similarities, including abnormalities in the cerebellum, spinal motor neurons, and peripheral nerves. Although one cannot fully discount deglutamylase substrates other than tubulin, these data strongly suggest that an impaired balance in microtubule polyglutamylation has a major effect on neuronal survival in humans. It would be interesting to know whether such imbalance occurs in other neurodegenerative conditions. Since the enzymes controlling this modification are potentially druggable, modulation of their activity might be an interesting avenue for inhibiting neurodegeneration in human patients.
The EMBO Journal (2018) 37: e101023
See also: https://doi.org/10.15252/embj.2018100440 (December 2018) and
https://doi.org/10.15252/embj.2018100540 (December 2018)
References
- van Dijk J, Miro J, Strub JM, Lacroix B, van Dorsselaer A, Edde B, Janke C (2008) Polyglutamylation is a post‐translational modification with a broad range of substrates. J Biol Chem 283: 3915–3922 [DOI] [PubMed] [Google Scholar]
- Gilmore‐Hall S, Kuo J, Ward JM, Zahra R, Morrison RS, Perkins G, La Spada AR (2018) CCP1 promotes mitochondrial fusion and motility to prevent Purkinje cell neuron loss in pcd mice. J Cell Biol 10.1083/jcb.201709028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins BV, Saunders HAJ, Record HL, Johnson‐Schlitz DM, Wildonger J (2017) Effects of mutating alpha‐tubulin lysine 40 on sensory dendrite development. J Cell Sci 130: 4120–4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Oda T (2017) Electrostatic interaction between polyglutamylated tubulin and the nexin‐dynein regulatory complex regulates flagellar motility. Mol Biol Cell 28: 2260– 2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Bodakuntla S, Žiak J, Lacomme S, Sousa PM, Leboucher S, Hausrat TJ, Bosc C, Andrieux A, Kneussel M, Landry M, Calas A, Balastik M, Janke C (2018a) Excessive tubulin polyglutamylation causes neurodegeneration and perturbs neuronal transport. EMBO J 37: e100440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magiera MM, Singh P, Gadadhar S, Janke C (2018b) Tubulin posttranslational modifications and emerging links to human disease. Cell 173: 1323–1327 [DOI] [PubMed] [Google Scholar]
- Millecamps S, Julien JP (2013) Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci 14: 161–176 [DOI] [PubMed] [Google Scholar]
- Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme JC, Bosson A, Peris L, Gold ND, Lacroix B, Bosch Grau M, Bec N, Larroque C, Desagher S, Holzer M, Andrieux A, Moutin MJ, Janke C (2010) A family of protein‐deglutamylating enzymes associated with neurodegeneration. Cell 143: 564–578 [DOI] [PubMed] [Google Scholar]
- Shashi V, Magiera MM, Klein D, Zaki M, Schoch K, Rudnik?Schöneborn S, Norman A, Lopes Abath Neto O, Dusl M, Yuan X, Bartesaghi L, De Marco P, Alfares AA, Marom R, Arold ST, Guzmán‐Vega FJ, Pena LDM, Smith EC, Steinlin M, Babiker MOE et al (2018) Loss of tubulin deglutamylase CCP1 causes infantile‐onset neurodegeneration. EMBO J 37: e100540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirajuddin M, Rice LM, Vale RD (2014) Regulation of microtubule motors by tubulin isotypes and post‐translational modifications. Nat Cell Biol 16: 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanco S, Tort O, Demol H, Aviles FX, Gevaert K, Van Damme P, Lorenzo J (2015) C‐terminomics screen for natural substrates of cytosolic carboxypeptidase 1 reveals processing of acidic protein C termini. Mol Cell Proteomics 14: 177–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenstein ML, Roll‐Mecak A (2016) Graded control of microtubule severing by tubulin glutamylation. Cell 164: 911–921 [DOI] [PMC free article] [PubMed] [Google Scholar]