

Abstract
Metabolic reprogramming has been described in rapidly growing tumors, which are thought to mostly contain fast‐cycling cells (FCCs) that have impaired mitochondrial function and rely on aerobic glycolysis. Here, we characterize the metabolic landscape of glioblastoma (GBM) and explore metabolic specificities as targetable vulnerabilities. Our studies highlight the metabolic heterogeneity in GBM, in which FCCs harness aerobic glycolysis, and slow‐cycling cells (SCCs) preferentially utilize mitochondrial oxidative phosphorylation for their functions. SCCs display enhanced invasion and chemoresistance, suggesting their important role in tumor recurrence. SCCs also demonstrate increased lipid contents that are specifically metabolized under glucose‐deprived conditions. Fatty acid transport in SCCs is targetable by pharmacological inhibition or genomic deletion of FABP7, both of which sensitize SCCs to metabolic stress. Furthermore, FABP7 inhibition, whether alone or in combination with glycolysis inhibition, leads to overall increased survival. Our studies reveal the existence of GBM cell subpopulations with distinct metabolic requirements and suggest that FABP7 is central to lipid metabolism in SCCs and that targeting FABP7‐related metabolic pathways is a viable therapeutic strategy.
Keywords: brain cancer, cancer stem cells, glioblastoma, metabolism, slow‐cycling cells
Subject Categories: Cancer, Metabolism, Stem Cells
Introduction
Intratumoral heterogeneity, which manifests in genetic, transcriptional, and functional levels, is increasingly recognized as a determinant of therapy resistance and disease recurrence. Indeed, tumor recurrence results from the ability of specific tumor subpopulations to resist treatment and expand. As has been shown for several malignancies, including glioblastoma (GBM), conventional cancer therapies most effectively eliminate rapidly dividing cells while sparing slower proliferating populations (Graham et al, 2002; Dembinski & Krauss, 2009; Gao et al, 2010; Pece et al, 2010; Roesch et al, 2010; Moore et al, 2012; Campos et al, 2014; Zeuner et al, 2014; Oshimori et al, 2015). GBM represents a prototypical example of heterogeneous cancer and is one of the most lethal malignancies, with a median survival of approximately 15–18 months despite multimodal therapy (Stupp et al, 2005, 2015). This dismal prognosis is attributable to therapy‐resistant GBM cells that drive recurrence, and the identification and characterization of these cellular subpopulations and their dynamic properties are essential for the development of more effective treatments.
A recent study demonstrated a proliferative hierarchy in human GBM, with slow‐cycling, cancer stem‐like cells via asymmetric division giving rise to rapidly proliferating progenies, which in turn generate limited‐lived and non‐proliferative offspring (Lan et al, 2017). A similar hierarchy has been proposed in a mouse model of glioma, with TMZ‐resistant, slow‐dividing cancer stem cells driving long‐term tumor growth via the generation of a rapidly growing transient population of cells (Chen et al, 2012). These results were also confirmed in human GBM (Campos et al, 2014). Similarly, Vanner et al (2014) showed that quiescent, SOX2‐positive cells drive long‐term tumor propagation and relapse in a sonic hedgehog subgroup of medulloblastoma. Using single‐cell RNA sequencing, Tirosh et al (2016b) reported a similar cellular hierarchy that is driven by developmental programs in oligodendroglioma.
We have previously reported the existence, isolation, and functional characterization of fast‐cycling cells (FCCs) and slow‐cycling cells (SCCs) in GBM (Deleyrolle et al, 2011, 2012). We found that human GBM SCCs are consistently enriched in cancer stem cell markers in vitro. This SCC population is enriched in tumor‐initiating cells, leading to enhanced tumorigenicity compared to the overall tumor population. SCCs were also identified and isolated in vivo and demonstrated all the key functional and phenotypic characteristics defining cancer stem cells, thus making them a clinically relevant target for new GBM treatment approaches (Deleyrolle et al, 2011).
According to the Warburg hypothesis (Warburg, 1926), tumorigenesis is partly driven by an impairment of mitochondrial function and oxidative phosphorylation (OxPhos). These alterations result in the Warburg effect, which is characterized by cancer cells generating most of their energy from glucose fermentation, i.e., aerobic glycolysis, with a limited ability to perform nutrient oxidation (Koppenol et al, 2011). This metabolic reprogramming is thought to be an adaptation mechanism of rapidly growing tumor cells to cover their increasing energy demands. The intrinsic cellular heterogeneity of GBM raises the question as to whether the different cellular subpopulations (e.g., FCCs and SCCs) are restricted to glucose fermentation or other metabolic pathways for their survival and proliferation. Interestingly, recent studies have demonstrated residual activity of mitochondrial function in GBM cells (Marin‐Valencia et al, 2012; Mashimo et al, 2014; Lin et al, 2017), suggesting that some of these cells might utilize mitochondrial OxPhos. However, the precise nature of the GBM cellular compartments harboring various metabolic specificities still needs to be established.
Here, we demonstrate that functionally different GBM cell subpopulations depend on distinct metabolic pathways for their growth and survival. Importantly, GBM SCCs display unique phenotypic traits, chemoresistance, and metabolic profiles that are divergent from those of FCCs. SCCs engage metabolic pathways that overlap with those found in recurrent GBM. Our work uncovers a previously unidentified metabolic dichotomy in GBM, with FCCs depending on glucose metabolism and SCCs relying on oxidative phosphorylation and lipid metabolism for their growth and survival. We show that blocking the specific energy pathways utilized by GBM FCCs and SCCs inhibits overall tumor growth. Our studies also highlight the SCC subpopulation as a determinant for GBM's resistance to metabolic treatments targeting the Warburg effect and identify in this population new candidate therapeutic targets including FABP7.
Results
SCCs display migration, invasion, and chemoresistance characteristics that promote GBM recurrence
GBM SCCs are associated with greater tumorigenicity and therefore enriched in stem‐like cells (Deleyrolle et al, 2011). Using gene set enrichment analysis, we confirmed that SCCs overexpress a gene module defined as a stem cell signature (Fig EV1A; Wong et al, 2008). We have previously shown that GBM stem‐like cells are more migratory and invasive (Siebzehnrubl et al, 2013). Therefore, we tested whether GBM cell proliferation rates might be inversely correlated with migration/invasion potentials (“go or grow hypothesis”) in vitro and in vivo. For this purpose, we isolated SCCs and FCCs from patient‐derived primary GBM Line 0 (L0), Line 1 (L1), and Line 2 (L2) using a flow cytometry‐based label retention paradigm (Deleyrolle et al, 2011; Fig EV1B). All downstream experiments were conducted immediately after purifying the SCC and FCC populations. Quantifying the migration abilities of these cellular subpopulations using in vitro scratch assays (Siebzehnrubl et al, 2013), we found that SCC migration distances were significantly higher than FCCs’ for all three cell lines (Fig 1A), despite the higher proliferation rates of FCCs (Deleyrolle et al, 2011).
Figure EV1. Invasion and chemoresistance properties in SCCs.
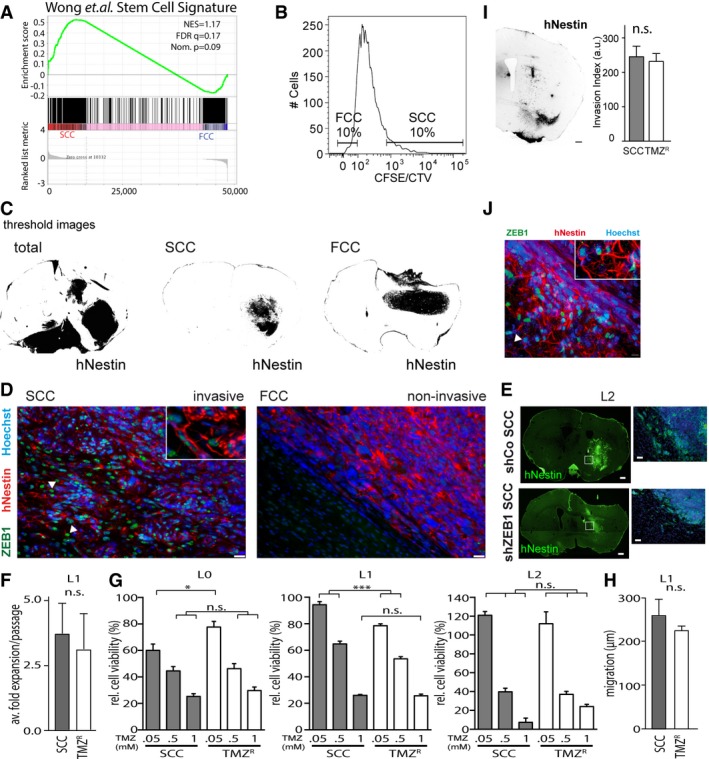
-
AGSEA of SCC (n = 3 biological replicates, L0‐1‐2) and FCC (n = 3 biological replicates, L0‐1‐2) RNA seq data sets for enrichment of the stem cell gene signature (Wong et al, 2008). FDR, false discovery rate; NES, normalized enrichment score; Nom., nominal.
-
BSCCs and FCCs were separated 6–8 days after (CFSE or CTV) CellTrace loading. Gates were set as 10% CellTracehi versus CellTracelo).
-
CThreshold images of human‐specific nestin staining were used for quantification of tumor invasion (n = 5 animals per group, see Materials and Methods).
-
DImmunofluorescence imaging revealed notable differences between invasive/SCC‐derived, and non‐invasive/FCC‐derived, tumors. Invasive SCC‐derived tumors were positive for ZEB1 (green), while this marker was absent in the tumor masses derived from FCCs. Tumor cells were labeled with hNestin (red) and nuclei with Hoechst (blue). Scale bars, 10 μm. Arrowheads indicate infiltrative Zeb1+ GBM cells.
-
EFluorescence imaging showed that the invasion of ZEB1 knockdown (shZEB1) SCC‐derived tumors from orthotopic xenografts was greatly reduced compared to that of control SCC‐derived tumors (shCo). Scale bars, 10 μm.
-
F–JTemozolomide‐resistant cells (TMZR) and SCCs derived from the L1 patient‐derived GBM line displayed similar growth rates (n = 3) (F), TMZ sensitivities (n = 6–8, *P < 0.05, ***P < 0.001, one‐way ANOVA with Tukey post‐test) (G), as well as migration (H) and invasion capabilities (scale bars, 500 μm) (I) (n = 3–12). These results were accompanied by the detection of high expression levels of ZEB1 in TMZR‐derived tumors. The arrowhead indicates infiltrative Zeb1+ GBM cell (J). Values are mean ± SEM.
Figure 1. Invasiveness and chemoresistance as hallmarks of SCCs in GBM .
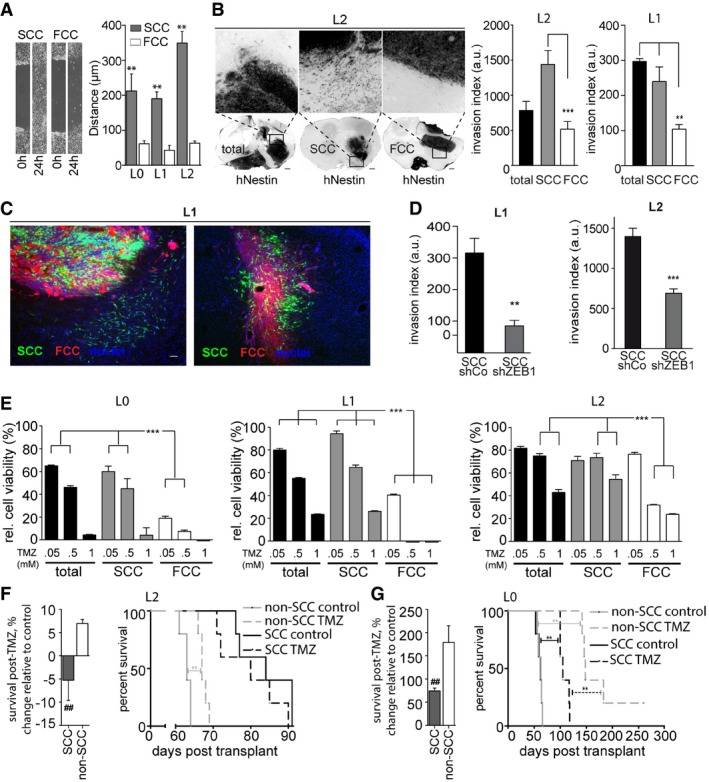
- Scratch assays demonstrated a greater migratory potential for SCCs than for FCCs within 24 h (mean ± SEM, n = 6, **P < 0.01, t‐test).
- Following murine xenografts of L1 or L2 patient‐derived cell lines, SCCs produced invasive tumors, while FCCs produced confined masses. Ten weeks after implantation, SCC‐derived tumors, and total population‐derived tumors for L1, exhibited greater invasion indices than FCC‐derived tumors (mean ± SEM, n = 3–12, **P < 0.001, ***P < 0.001, one‐way ANOVA with Tukey post‐test). Scale bars, 500 μm.
- Representative fluorescence microscopy images of tumor sections derived from intracranial xenografts of lentivirally transduced green fluorescent protein (GFP)‐labeled SCCs and red fluorescent protein (RFP)‐expressing FCCs in a 1:1 ratio, 6 weeks after implantation (n = 5). SCCs (in green) generated a network of cells infiltrating the brain parenchyma while FCCs (red) remained contained, forming tight masses. Images are from two mice out of five. Scale bar, 10 μm.
- The invasion of tumors derived from orthotopic xenografts of ZEB1 knockdown SCCs was significantly lower than that of control SCC‐derived tumors (mean ± SEM, n = 6, **P < 0.01, ***P < 0.001, t‐test).
- SCCs were found to be significantly more resistant to TMZ than the total or FCC populations in vitro using MTT assays (mean ± SEM, n = 6–8, ***P < 0.001, one‐way ANOVA with Tukey post‐test).
- In vivo TMZ treatment yielded no survival benefit following SCC xenograft of the most TMZ‐resistant GBM line, whereas TMZ treatment of animals xenografted with the non‐SCC population resulted in significantly prolonged survival (mean ± SEM, n = 5, ## P < 0.01, t‐test).
- TMZ treatment of animals xenografted with SCCs from a more TMZ‐sensitive line increased overall survival, but to a lesser degree than for non‐SCC‐implanted animals (mean ± SEM, n = 5, ## P < 0.01, t‐test).
Importantly, 10 weeks after intracranial xenotransplantation in mice (which represents the approximate survival endpoint for FCC‐ and non‐SCC‐implanted animals), SCCs had generated more invasive tumors than FCCs, for two patient‐derived GBM cell lines (Figs 1B and EV1C). In order to directly compare the invasion potentials of SCCs and FCCs in vivo, we lentivirally transduced SCCs with GFP and FCCs with RFP and intracranially implanted these together at a ratio of 1:1 into recipient mice. This labeling enabled us to assess whether the observed greater invasion of SCCs was due to slower tumor growth and thus smaller tumor size, whether FCC‐derived tumors would appear more invasive when observed at earlier stages of tumor formation, and/or whether invasion of FCCs would be influenced by SCCs in their environment. We analyzed these tumors 6 weeks after implantation to differentiate tumor mass expansion from tumor invasion. In all animals, SCCs generated a network of invasive cells infiltrating the brain parenchyma and extending long processes that were consistent with tumor microtubes (Osswald et al, 2015), while FCCs generated more contained tumor masses (Fig 1C). These results show that invasion is intrinsic to SCCs, while FCCs generate non‐infiltrating tumor masses.
Epithelial‐to‐mesenchymal transition (EMT), and in particular the EMT transcription factor zinc‐finger E‐box‐binding homeobox 1 (ZEB1), has been frequently associated with a loss of cell‐to‐cell contact and the distant spreading of tumors (Singh & Settleman, 2010; Siebzehnrubl et al, 2013). Moreover, ZEB1 promotes cancer cell stemness (Aigner et al, 2007; Shimono et al, 2009; Wellner et al, 2009; Chaffer et al, 2013), and the co‐expression of SOX2, OLIG2, and ZEB1 transforms tumor‐suppressor‐deficient murine astrocytes into glioma‐initiating cells in the absence of an upstream oncogene (Singh et al, 2017). Therefore, we hypothesized that ZEB1 may regulate SCC invasion and tested whether this transcription factor was differentially expressed in SCCs and FCCs in vivo. We found that FCC‐derived, non‐invasive tumors were devoid of ZEB1, while ZEB1‐immunoreactive cells were consistently found throughout SCC‐derived invasive tumors (Fig EV1D). To determine whether the higher ZEB1 levels in SCCs are linked to these cells’ greater capability for migration and invasion, we isolated SCC and FCC populations from control and ZEB1‐knockdown cells. The invasion of tumor derived from ZEB1 knockdown SCC in orthotopic xenografts was greatly reduced compared to control SCC‐derived tumors (Figs 1D and EV1E). These results demonstrate that ZEB1 is necessary for migration and invasion of SCCs.
Because ZEB1 and other EMT regulators have been shown to induce chemoresistance in GBM (Qi et al, 2012; Siebzehnrubl et al, 2013; Depner et al, 2016), we next tested whether GBM SCCs, which are enriched in ZEB1, are more resistant to therapy than FCCs, as has been demonstrated for other quiescent subsets of GBM cells (Chen et al, 2012; Campos et al, 2014). We evaluated the in vitro effects of the standard‐of‐care chemotherapeutic drug temozolomide (TMZ) on the cell viabilities of the total tumor cell populations as well as FCCs and SCCs using MTT assays. While all three L0, L1, and L2 total cell populations displayed some sensitivity to TMZ, L0 was the most sensitive and L2 the most resistant line. Importantly, the SCCs from all three patient‐derived GBM cell lines showed higher resistance to TMZ than the corresponding cell line's FCCs (Fig 1E). Moreover, by repeatedly exposing these primary GBM lines to TMZ, we selected for TMZ‐resistant cell populations (TMZR) with expansion rates and TMZ resistance profiles similar to SCCs’ (Fig EV1F and G). TMZR and SCCs also showed comparable migration and invasion potentials (Fig EV1H–J). These results further underscore the link between GBM cell proliferation rate, invasiveness, and chemoresistance.
We next tested whether SCCs were more chemoresistant than the rest of the GBM cell population in vivo. We treated tumor‐bearing animals, orthotopically grafted with either SCCs or FCCs, with clinically relevant TMZ concentrations (20 mg/kg; Zhou et al, 2007). TMZ treatment prolonged the median survival of animals implanted with L2 FCCs but did not improve the survival of the L2 SCC‐implanted group (Fig 1D). Similarly, TMZ treatment lengthened the survival of L0 FCC‐grafted animals but had significantly smaller effects in L0 SCC‐grafted animals (Fig 1E). These results further demonstrate that GBM SCCs are more resistant to TMZ than the rest of the tumor cell population and thus more likely to escape standard‐of‐care therapy (Fig EV1I). Together, our findings support critical roles of SCCs in both GBM invasion and chemoresistance and thus in tumor recurrence.
Treatment‐resistant/recurrent tumors share metabolic gene signatures with SCCs
Based on the hypothesis that SCCs might contribute to tumor recurrence, and to identify the molecular mechanisms involved in their survival and growth, we investigated the molecular pathways that are characteristic of recurrent GBM tumors. We compared the RNA sequencing data of 153 primary and 14 recurrent GBM patient tumors from the TCGA database (Cancer Genome Atlas Research Network, 2008) and identified several genes that were significantly up‐regulated in recurrent tumors compared to primary tumors (Fig 2A and Table EV1). Using the String database (Szklarczyk et al, 2015), we found that lipid metabolism was one of the top 5 most significantly enriched gene pathway groups in GBM recurrent tumors (GO:0033993, Fig EV2A and Table EV2). Interestingly, lipid metabolism constitutes the main source for mitochondrial energy production, and we found significantly higher mRNA expression levels of multiple genes involved in mitochondrial OxPhos, the tricarboxylic acid (TCA) cycle, and pyruvate and antioxidant metabolism in recurrent GBM (fold change > 2, Mann–Whitney test, P < 0.05; Fig EV2B and C, and Table EV3). Notably, GBM SCCs displayed these specific metabolic signatures, further supporting SCCs’ influential presence and role in tumor recurrence (Fig 2B). We also found that the mRNA expression levels of genes involved in the glycolytic/gluconeogenesis pathways were down‐regulated in recurrent tumors (fold change > 2, Mann–Whitney test, P < 0.05; Table EV4). Together, these data support the presence of metabolic heterogeneity and plasticity in GBM.
Figure 2. Shared metabolic gene signature between recurrent GBM and SCCs.
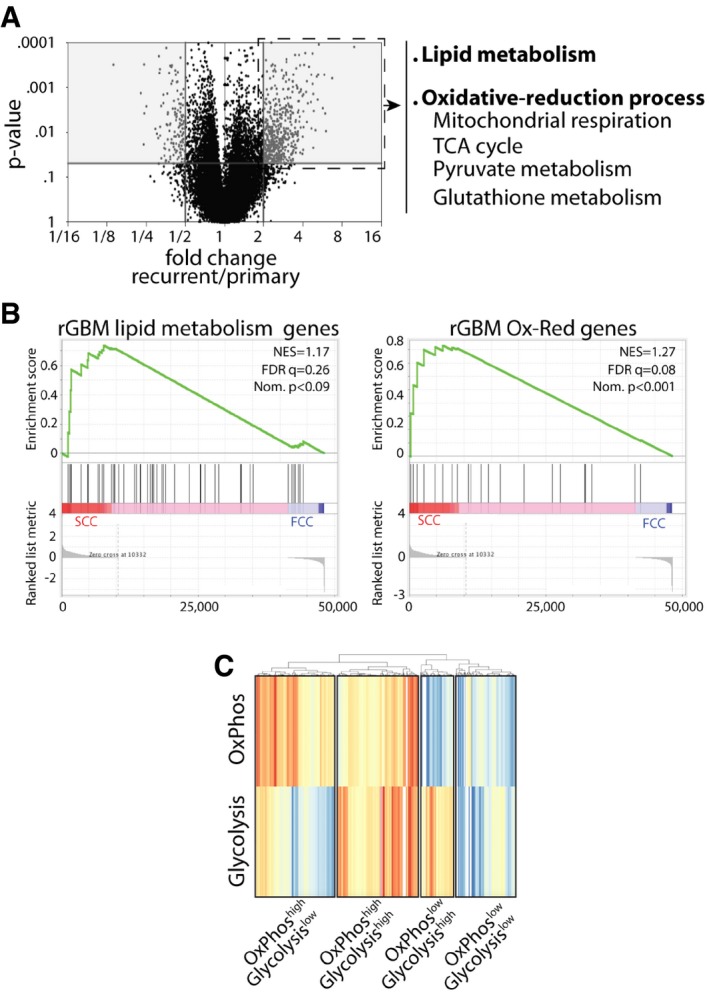
- Volcano plot representation of 20,530 genes that were identified in primary and recurrent human GBMs using the TCGA database and showing differentially expressed genes between the two groups. Gray areas denote significant increases or decreases in gene expression. Recurrent tumors show a significant increase in the expression of genes involved in lipid metabolism, mitochondrial respiration, TCA cycle, as well as pyruvate and antioxidant metabolism (fold change > 2 and P < 0.05, Mann–Whitney U‐test, Subio platform). These genes were then clustered into two signatures representing lipid metabolism and oxidative‐reduction (Ox‐Red) genes.
- GSEA of SCC (n = 3 biological replicates, L0‐1‐2) and FCC (n = 3 biological replicates, L0‐1‐2) RNA seq data sets for enrichment of these recurrent glioblastoma (rGBM) gene signatures. FDR, false discovery rate; NES, normalized enrichment score; Nom., nominal.
- Different metabolic signatures were identified from GBM single‐cell RNA sequencing data (Venteicher et al, 2017). These results, generated by GenePattern ssGSEA and visualized with pheatmap (Euclidean algorithm), highlight the metabolic heterogeneity in GBM, with various clusters of cells demonstrating gene signatures for OxPhos and glycolysis.
Figure EV2. Differentially regulated pathways between SCCs and FCCs.
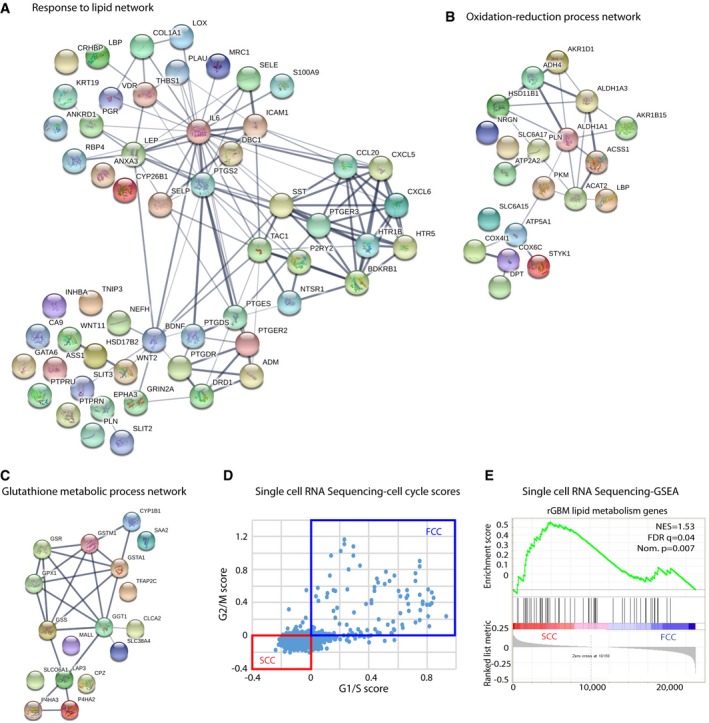
-
AThe use of Search Tool in the Retrieval of Interacting Genes/Proteins database (STRING) indicated an overrepresentation of genes associated with lipid metabolism in recurrent GBMs. Red nodes designate the response to lipid genes (n = 56) that are significantly overexpressed in recurrent GBMs within the entire network of genes.
-
B, CGene networks involved in oxidation–reduction (B) and antioxidant processes (C) are up‐regulated in recurrent tumors.
-
DCells from single‐cell RNA sequencing data were classified into slow‐ (SCC) and fast‐cycling (FCC) clusters based on the relative expression of cell cycle G1/S (x‐axis) and G2/M (y‐axis)‐associated gene sets.
-
EGSEA of SCC (n = 3 biological replicates, L0‐1‐2) and FCC (n = 3 biological replicates, L0‐1‐2) RNA seq data sets for enrichment of the lipid gene signature identified in TCGA rGBM and our three GBM‐derived SCCs. FDR, false discovery rate; NES, normalized enrichment score; Nom., nominal.
In addition, we performed in silico analysis of single‐cell RNA sequencing data from existing glioma databases (Venteicher et al, 2017), to further confirm GBM's intratumoral heterogeneity, and identified various clusters of cells demonstrating gene signatures for OxPhos and glycolysis (Fig 2C and Table EV5). We propose that the cluster “OxPhos high/Glycolysis high” represents cells undergoing glycolysis via glucose oxidation, and the cluster “OxPhos high/Glycolysis low” indicates cells oxidizing nutrients other than glucose, such as lipids or amino acids. The “OxPhos low/Glycolysis high” cluster reveals cells that follow the Warburg effect, while cells with the “OxPhos low/Glycolysis low” signature use alternative metabolic pathways (Fig 2C). We classified cells from this single‐cell RNA sequencing data into slow‐ and fast‐cycling clusters based on the relative expression of cell cycle G1/S (x‐axis) and G2/M (y‐axis)‐associated gene sets (Fig EV2D), as previously described (Patel et al, 2014; Tirosh et al, 2016a,b). We then evaluated the lipid signature identified in recurrent GBM tumors and our GBM cell line SCCs and found overrepresentation of the lipid gene set in the slow‐cycling cluster defined from the single‐cell RNA sequencing data (Fig EV2E).
Together, our analyses demonstrate the existence of metabolic heterogeneity in GBM, in which common and specific metabolic pathways, particularly related to OxPhos and lipid metabolism, are overrepresented in SCCs and recurrent tumors.
SCCs display increased mitochondrial activity and OxPhos
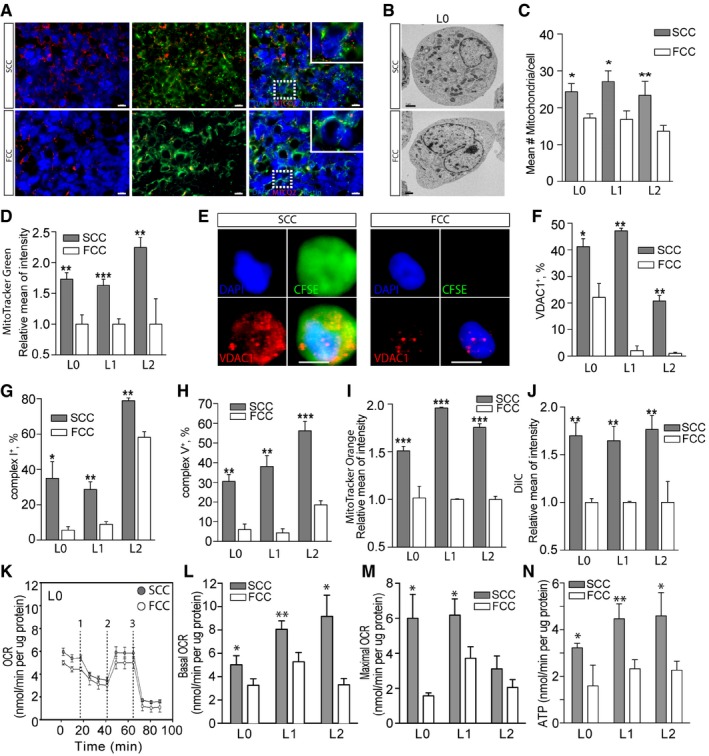
As our results suggest that GBM SCCs contribute to the metabolic gene signature of recurrent tumors (i.e., increased mitochondrial and lipid pathway genes), we next compared the amount, components, and activity of SCC and FCC mitochondria. First, in vivo tumors derived from SCC or FCC xenografts were immunostained with the mitochondrial marker MTCO2 and showed a higher number of mitochondria in SCC‐derived tumors (Fig 3A). This finding was confirmed by electron microscopy, which demonstrated more mitochondria per cell in SCCs than in FCCs (Figs 3B and C, and EV3A). We also found that MitoTracker Green accumulated significantly more in GBM SCCs than FCCs (Figs 3D and EV3B), indicating that SCCs possess a higher mitochondrial mass (De Paepe, 2012).
Figure 3. Enhanced mitochondrial activity in SCCs.
-
AFluorescence microscopy images of tumor sections, derived from intracranial xenografts of L1 SCCs or FCCs and immunostained with the mitochondrial marker MTCO2, showed a higher number of mitochondria in SCC‐derived tumors. Scale bars, 10 μm.
-
B, CElectron microscopy analysis (B) and quantification (n = 6–11) (C) revealed a higher number of mitochondria per cell in SCCs than in FCCs for L0, L1, and L2. Scale bar, 1 μm.
-
DQuantification of MitoTracker Green staining in SCCs and FCCs derived from hGBM L0 (n = 10), L1 (n = 13), and L2 (n = 6) consistently showed higher mitochondrial content in SCCs.
-
EFluorescence microscopy images of single cells labeled for VDAC1 (red), DAPI (blue), and CFSE (green). Scale bar, 5 μm.
-
FFlow cytometry quantification of VDAC1 in SCCs and FCCs showed increased VDAC1 levels in SCCs (n = 2 for all lines).
-
G, HFlow cytometry quantification of mitochondrial complex I (n = 3 for all lines) (G) and complex V (L0, n = 3; L1, n = 3; L2, n = 6) (H) revealed an increase in these mitochondrial electron transport chain components in SCCs when compared with FCCs.
-
IQuantification of MitoTracker Orange staining showed significantly higher mitochondrial reactive oxygen species production in SCCs than in FCCs (L0, n = 9; L1, n = 2; L2, n = 6).
-
JSCC and FCC mitochondrial membrane potentials were analyzed with the MitoProbe DilC1 assay (L0, n = 4; L1, n = 2; L2, n = 5). Carbonyl cyanide m‐chlorophenyl hydrazone (CCCP) was added as control mitochondrial membrane uncoupler.
-
KSeahorse experiments were conducted to compare the mitochondrial function between SCC and FCC populations. The oxygen consumption rate (OCR) responses were quantified using the Seahorse XF Cell Mito Stress Test. The dashed lines indicate the timing of oligomycin (1), FCCP (2), and rotenone with antimycin A (3) injections.
-
L–NBasal (n = 9–21) (L) and maximal (n = 8–17) (M) oxygen consumption rates (OCR) as well as ATP production (n = 4–14) (N) were significantly higher in SCCs than in FCCs for L0, L1, and L2.
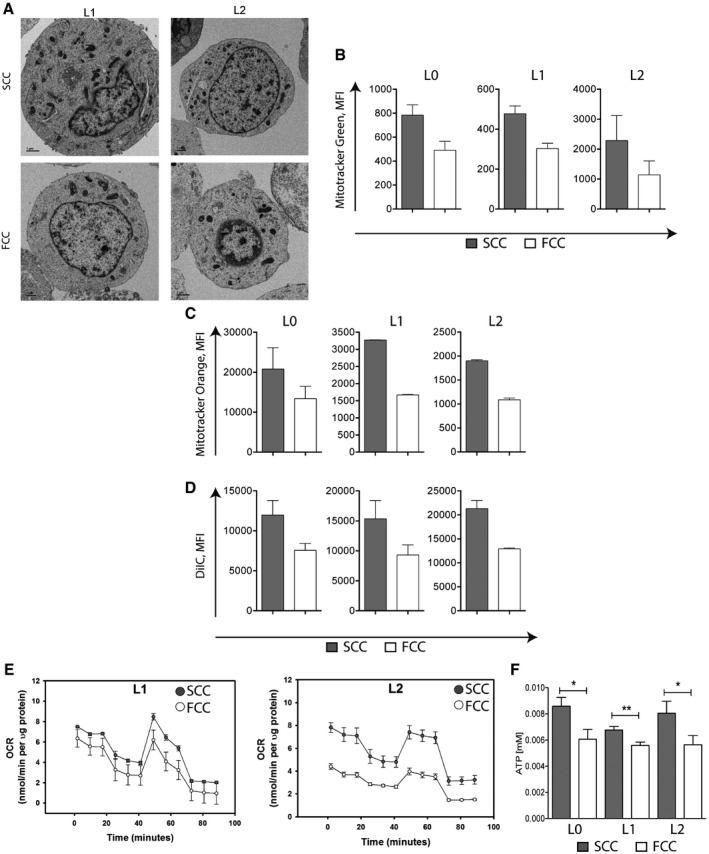
Figure EV3. SCCs and FCCs show different mitochondrial properties.
-
AElectron microscopy images of SCCs and FCCs from L1 and L2 patient‐derived GBM cell lines showing greater number of mitochondria in SCCs than FCCs. Scale bar, 1 μm.
-
BRaw values of mean fluorescence intensity (MFI) following MitoTracker Green staining in SCCs and FCCs derived from hGBM L0 (n = 10), L1 (n = 13), and L2 (n = 6). Values are mean ± SEM.
-
CRaw values of mean fluorescence intensity (MFI) following MitoTracker Orange staining in SCCs and FCCs (mean ± SEM, L0, n = 9; L1, n = 2; L2, n = 6).
-
DRaw values of mean fluorescence intensity (MFI) in SCCs and FCCs following the MitoProbe DilC1 assay (mean ± SEM, L0, n = 4; L1, n = 2; L2, n = 5).
-
E, FOxygen consumption rate (OCR) measured using the XF Cell Mito Stress Assay (E) and ATP production measured using the luciferase‐based ATP‐lite assay (F) in SCCs and FCCs (mean ± SEM, L0, n = 18; L1, n = 15; L2, n = 15, *P < 0.05, **P < 0.01, t‐test).
Second, we compared the mitochondria components between GBM SCCs and FCCs and examined the voltage‐dependent anion‐selective channels (VDACs) in these two populations. VDACs are a class of porin ion channels that are located on the outer mitochondrial membrane and play a key role in regulating metabolic and energetic flux across that membrane (Hoogenboom et al, 2007). VDACs are involved in the transport of ATP, ADP, pyruvate, malate, and other metabolites, thus interacting extensively with enzymes from various metabolic pathways (Blachly‐Dyson & Forte, 2001). Notably, VDACs are important regulators of Ca2+ transport in and out of the mitochondria, and because Ca2+ is a co‐factor for metabolic enzymes such as pyruvate dehydrogenase and isocitrate dehydrogenase, energetic production through OxPhos and homeostasis are both affected by VDACs’ permeability to Ca2+ (Shoshan‐Barmatz & Gincel, 2003). Of the three VDAC isoforms, VDAC1 is the main Ca2+ ion transport channel and the most abundantly transcribed (Chu et al, 2014). Interestingly, we found consistent VDAC1 staining in GBM SCCs by immunofluorescence (Fig 3E) and flow cytometry (Fig 3F), while FCCs displayed significantly weaker VDAC1 expression. Of note, we also found an increased expression of electron transport chain (ETC) enzymatic complexes, particularly NADH dehydrogenase (complex I) and ATP synthase (complex V), in SCCs compared to FCCs (Fig 3G and H), suggesting higher OxPhos activity in GBM SCCs than in FCCs.
We next compared mitochondrial respiratory activities between GBM SCCs and FCCs. Mitochondrial respiration functional assays with MitoTracker Orange, a dye that accumulates in active mitochondria where it gets oxidized, revealed significantly higher levels of reactive oxygen species (ROS) in SCCs than in FCCs (Figs 3I and EV3C). Of note, even though the majority of cellular ROS are normally produced by mitochondria, approximately a quarter can be produced from protein and lipid metabolism in the endoplasmic reticulum (ER) of glioma cells (Salazar‐Ramiro et al, 2016). Therefore, the differences in ROS levels observed from our MitoTracker Orange assays could have been due to either higher mitochondrial OxPhos activity or greater ER protein/lipid metabolism in SCCs than in FCCs. However, greater mitochondrial OxPhos activity is supported by our experimental results, which show elevated mitochondrial membrane potential and higher levels of mitochondrial respiratory energetics in SCCs compared to FCCs with the MitoProbe DilC1 assay (Figs 3J and EV3D). In addition, we conducted Seahorse experiments to directly compare the metabolic activities between SCC and FCC populations. Basal and maximal oxygen consumption rates, as well as ATP production, were significantly higher in SCCs compared to FCCs for L0, L1, and L2 (Figs 3K–N and EV3E and F). Together, our data show that SCCs display heightened mitochondrial and OxPhos activities compared to FCCs.
A metabolic dichotomy exists between GBM SCCs and FCCs
Maintaining mitochondrial OxPhos activities may confer SCCs broader metabolic capacities beyond those of aerobic glycolysis. We had previously shown that GBM cells were sensitive to glucose deprivation (Martuscello et al, 2016) and next examined whether GBM SCCs and FCCs displayed the same glucose dependencies. We tested the response of SCCs and FCCs to high (500 mg/dl) or lowered/physiological (90–110 mg/dl) glucose levels in normoxic conditions. Flow cytometry revealed increased cell death in FCCs, but not SCCs, following glucose restriction (Fig 4A and Appendix Fig S1A), indicating that SCCs do not rely on glycolysis for energy production and survival. In addition, we found that SCCs are less sensitive than FCCs to the pharmacological inhibitor of glycolysis, 2‐deoxyglucose (2‐DG), complementing the results obtained using glucose restriction (Fig 4B and Appendix Fig S1B). In support of these observations, we found, using quantitative RT‐PCR, that the expression of the lactate dehydrogenase (LDH) A, B, and C enzymes, which catalyze the conversion of the final product of glycolysis, pyruvate, to lactate, was down‐regulated in SCCs compared to FCCs (Appendix Fig S1C). Conversely, unlike FCCs, SCCs were sensitive to OxPhos inhibition, as demonstrated by a significant increase in apoptotic cell death after treatment with rotenone or metformin, which are pharmacological inhibitors of the mitochondrial ETC complex I (Fig 4C and D, and Appendix Fig S1D and E).
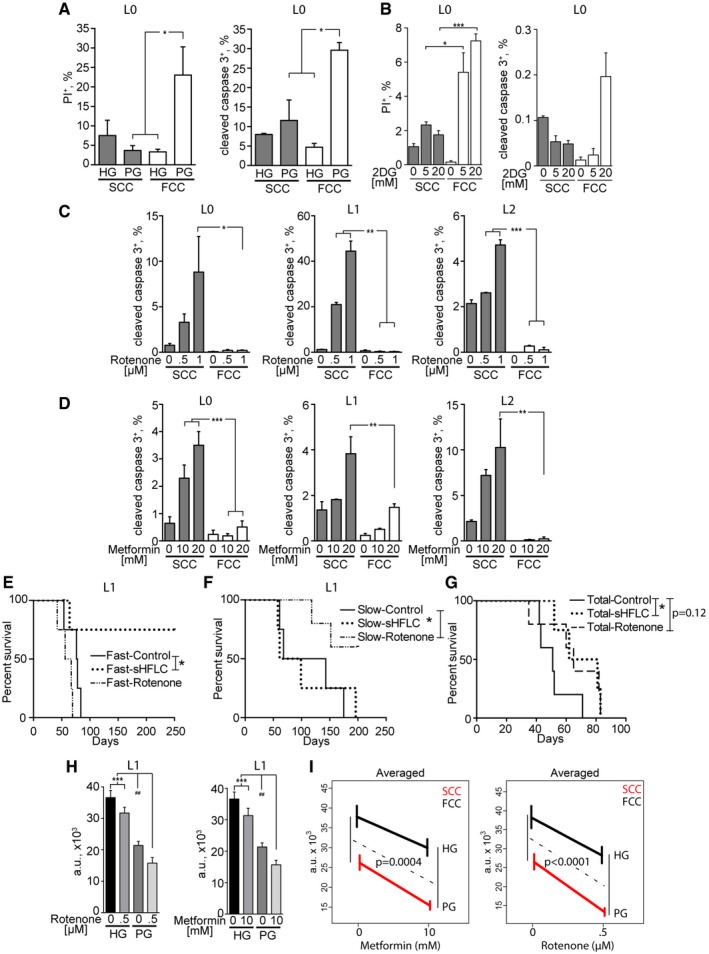
Figure 4. Metabolic dichotomy in GBM .
-
ASCCs and FCCs were cultured in high glucose (HG, > 500 mg/dl) or physiological glucose (PG, 90–110 mg/dl) conditions for 24 h. Cell death was quantified by flow cytometry through propidium iodide (PI) incorporation (L0, n = 3–5) and cleaved caspase‐3 expression (L0, n = 2–3). *P < 0.05, 1‐way ANOVA.
-
BSCCs and FCCs were cultured in 0, 5, or 20 mM 2‐deoxyglucose (2DG) for 24 h. Cell death was quantified by flow cytometry through propidium iodide (PI) incorporation (L0, n = 3) and cleaved caspase‐3 expression (L0, n = 3). *P < 0.05, **P < 0.01, 1‐way ANOVA.
-
C, DQuantification of cleaved caspase‐3 and CellTrace signals in cultured hGBM cells after 24 h of treatment with rotenone (L0, n = 4–5; L1–L2, n = 3; L2) (C) or metformin (L0, n = 4; L1, n = 3–4; L2, n = 2) (D). *P < 0.05, **P < 0.001, ***P < 0.0005, one‐way ANOVA with Tukey post‐test.
-
E–GKaplan–Meier curves showing the survival of animals intracranially implanted with SCCs or FCCs (n = 4–5) and treated with rotenone (0.5 mg/kg) or fed with a glucose‐restricted/supplemented high‐fat, low‐carbohydrate diet (sHFLC; *P ≤ 0.05, log‐rank test).
-
HThe combinatorial effects of administering glucose restriction along with mitochondrial targeting with rotenone (n = 16, left) or metformin (n = 16, right) were measured using CyQUANT assays after 24 h of treatment (***P < 0.001, one‐way ANOVA with Tukey post‐test, ## P < 0.01, t‐test).
-
IMean responses predicted by the generalized linear model (GLM). The effect of lowering glucose on cell viability (HG‐PG vertical differences) depended on the presence/absence of metformin or rotenone. F tests were used to measure the significance of the interactions between glucose and metformin or rotenone effects. Interaction P‐values are indicated.
We next set to validate the metabolic differences between SCCs and FCCs in vivo. For in vivo glucose restriction, we implemented a custom high‐fat/low‐carbohydrate dietary regimen supplemented with a specialized fat source composed of medium‐chain triglycerides (sHFLC), as previously reported (Martuscello et al, 2016). Subjecting animals bearing orthotopic xenotransplants of GBM patient‐derived L1 SCCs or FCCs to this sHFLC diet significantly improved the survival of FCC‐implanted, but not SCC‐implanted, animals (Fig 4E and F), indicating that SCC‐derived tumors are insensitive to glucose restriction (Fig 4F).
Based on our in vitro data showing SCCs’ heightened sensitivity to mitochondrial inhibition, we then treated SCC‐ and FCC‐implanted animals with rotenone. Compared with the vehicle‐treated group, SCC‐implanted animals that were treated with rotenone showed a significant increase in survival (Fig 4F), while animals implanted with FCCs did not gain any survival benefit from the same treatment (Fig 4E).
Together, our in vitro and in vivo data indicate that FCCs mostly utilize aerobic glycolysis and SCCs mitochondrial OxPhos for their survival and proliferation. To demonstrate the overall effect of the metabolic interventions on tumor growth, animals were also implanted with total cell populations from each cell line and treated with sHFLC or rotenone (Fig 4G). Inhibiting glycolysis significantly improved the survival of the animals. We observed a trending increased in survival with rotenone treatment; however, significance was not achieved. For both treatments, no long‐term survivors were observed. This result further supports the existence of tumor heterogeneity and suggests metabolic plasticity as an escape mechanism to the applied treatments. We hypothesized that the combinatorial inhibition of glycolysis and mitochondrial OxPhos would have a greater effect on GBM cell proliferation than either treatment alone through impeding metabolic adaptation and targeting both the SCC and FCC phenotypes. Thus, we cultured GBM cells in normal or glucose‐restricted conditions and in the presence or absence of rotenone or metformin. Glucose concentrations were maintained stable over time to prevent glucose supply exhaustion, especially in the rapidly growing FCC cultures. We confirmed a synergistic effect between glucose deprivation and mitochondria inhibition (Fig 4H and I, and Appendix Fig S1D and E). Using the generalized linear models (GLM, see Materials and Methods), we found that inhibiting mitochondria with metformin or rotenone increased the overall effect of glucose deprivation by 36 and 47%, respectively. These results demonstrate that a functional metabolic dichotomy exists in GBM, in which a subpopulation of cells is capable of switching from mitochondrial OxPhos to glycolysis (e.g., SCCs) and is sensitized to glycolytic inhibition through targeting of their mitochondria.
Lipid metabolite levels are increased in SCCs
In order to investigate the metabolic pathways fueling mitochondrial OxPhos in SCCs, we performed comprehensive metabolic profiling of SCCs and FCCs isolated from L0, L1, and L2 GBM cell lines cultured in nutrient‐replete conditions using ultra‐high‐performance liquid chromatography coupled with high‐resolution quantitation mass spectrometry (UHPLC/HRQMS). Multivariate principal component analysis (PCA) and partial least squares‐discriminant analysis (PLS‐DA) of metabolite profiles showed a segregation between FCCs and SCCs (Fig 5A). Pathway analysis of the metabolites that were up‐regulated by at least twofold in the SCCs consistently showed elevated metabolic intermediates specifically involved in lipid metabolism pathways. Interestingly, more than 60% of these lipid metabolites were unsaturated (Fig 5B; see Appendix Fig S2 for the full list of pathways; see Table EV6 for the full list of identified metabolites).
Figure 5. Elevated lipid metabolite levels and preferential storage of lipid droplets specifically metabolized in response to reduced glucose levels in SCCs.
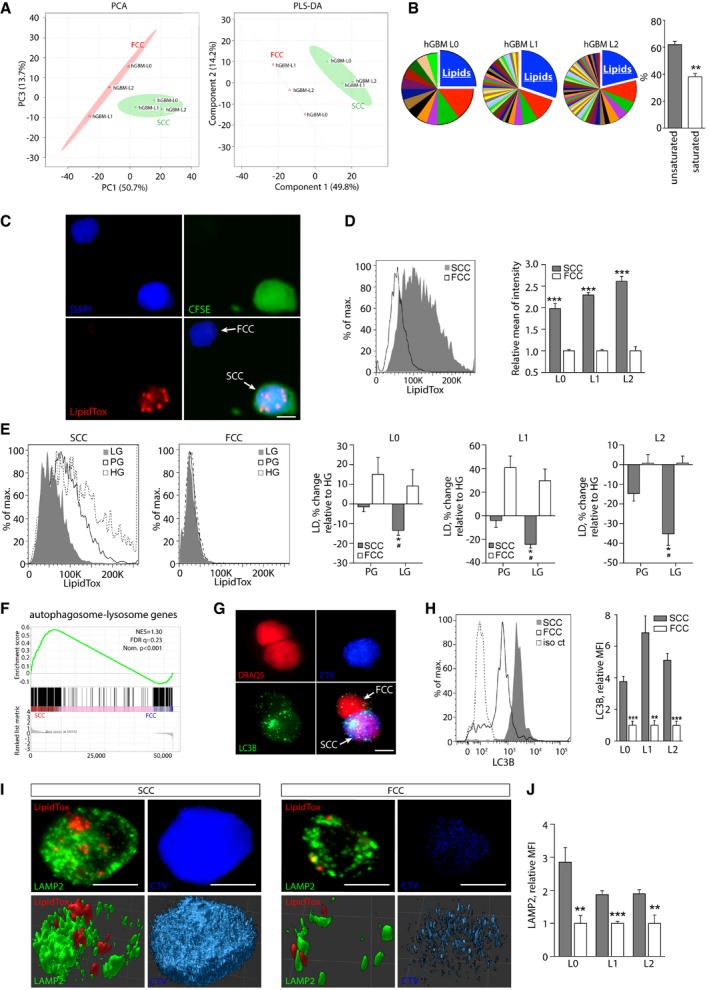
- PCA and PLS‐DA score plots derived from UHPLC/HRQMS metabolomics. Green cross: SCCs; red triangle: FCCs.
- Pathway analysis performed for each individual hGBM cell line revealed that lipids constitute the most represented metabolic intermediates that are up‐regulated in SCCs compared with FCCs. The majority of lipids up‐regulated in SCCs are unsaturated (mean ± SEM, n = 3, **P < 0.01, t‐test).
- Representative fluorescence microscopy images of single cells showing lipid droplets that were detected using LipidTox (red). SCCs were identified with CFSE‐CellTrace (green). Nuclei were stained with DAPI (blue). Scale bar, 5 μm.
- Flow cytometry quantification of lipid droplets in hGBM L0, L1, and L2 cell lines (mean ± SEM, n = 3–7, ***P < 0.001, t‐test).
- Using flow cytometry, lipid droplet contents were compared between cells cultured for 24 h in high (HG), physiological (PG), or low (LG, 65–80 mg/dl) glucose conditions. Results are represented as average percent change relative to HG (mean ± SEM, n = 3 for all lines; *P < 0.05, t‐test compared to HG; # P < 0.05, t‐test compared to PG).
- GSEA of SCC (n = 3 biological replicates, L0‐1‐2) and FCCs (n = 3 biological replicates, L0‐1‐2) RNA seq data sets for enrichment of the autophagosome–lysosome pathway. FDR, false discovery rate; NES, normalized enrichment score; Nom., nominal.
- Fluorescence microscopy images of LC3 (autophagosome marker) in CellTrace‐positive (blue) and negative cells. Nuclei were stained with DRAQ5 (red). Scale bar, 5 μm.
- Quantification of LC3 by flow cytometry demonstrated greater amounts of autophagosomes in SCCs than in FCCs (mean ± SEM, L0, n = 4; L1, n = 4; L2, n = 7, **P < 0.01, ***P < 0.001, t‐test).
- Confocal microscopy images of lipid droplets (LipidTox, red) and lysosomes (LAMP2, green) in a single SCC (CTVhigh, blue) and FCC (CTVlow). Top panels represent maximum z‐stack projections, and bottom panels show 3D reconstructions. Scale bar, 5 μm.
- LAMP2 expression was measured and compared between SCCs and FCCs by flow cytometry (mean ± SEM, L0, n = 3; L1, n = 4; L2, n = 4, **P < 0.01, ***P < 0.001, t‐test).
Lipid droplets constitute a form of energy storage in SCCs
Next, we examined whether/how these increased lipid intermediates might be stored in GBM SCCs. Fatty acids and their saturation status have been correlated with cancer stemness (Tirinato et al, 2015; Li et al, 2017; Noto et al, 2017), and the increased uptake of unsaturated fatty acids in cancer cells promotes the formation of triglyceride‐enriched lipid droplets, representing an efficient way of storing energy (Mei et al, 2011). To compare lipid droplet amounts between SCCs and FCCs, we used a lipid‐specific probe (LipidTox) that accumulates in intracellular lipid droplets. We observed consistent LipidTox staining in SCCs and a marked increase in LipidTox content in SCCs when compared to FCCs using flow cytometry analysis (Fig 5C and D, and Appendix Fig S3A).
We then quantified the amount of lipid droplets in response to glucose deprivation in order to assess whether these lipid droplets are utilized by SCCs or FCCs as a source of energy. We found a significant decrease in lipid droplet content in SCCs when these cells were cultured in low‐glucose conditions, whereas no change was observed in FCCs (Fig 5E). These data suggest that lipid droplets represent a form of energy storage that can be used by SCCs in response to metabolic stress.
We next investigated how those stored lipids might be catabolized by SCCs for energy production. Lipid droplets can be sequestered in autophagosomes that fuse with lysosomes following nutrient deprivation, leading to the breakdown of lipid droplet components by lysosomal enzymes in order to generate energy and meet the cells’ metabolic demands (Singh et al, 2009; Dong & Czaja, 2011; Velazquez & Graef, 2016). Interestingly, gene set enrichment analysis (GSEA) from RNA sequencing (RNA seq) data across the L0, L1, and L2 GBM cell lines revealed an elevated expression of autophagosome–lysosome genes in SCCs compared to FCCs (Fig 5F, Appendix Fig S3B and C, and Table EV7) for the full list of genes defining the autophagosome–lysosome pathway). We then set to confirm the presence of autophagosomes and lysosomes in SCCs using quantitative flow cytometry analysis and found a fivefold increase in the amount of autophagosomes, which were labeled with microtubule‐associated protein light chain 3 (LC3B), in SCCs compared to FCCs (Fig 5G and H, and Appendix Fig S3D). In addition, immunofluorescence staining for lysosomal membrane‐associated protein 2 (LAMP2) revealed a rich network of lysosomes surrounding lipid droplets in SCCs (Fig 5I), and flow cytometry analysis showed increased LAMP2 expression in SCCs when compared to FCCs (Fig 5J and Appendix Fig S3E). Together, our observations suggest that SCCs may engage in autophagy mechanisms to metabolize stored lipid, particularly in response to metabolic stress.
Fatty acid transport is facilitated in SCCs
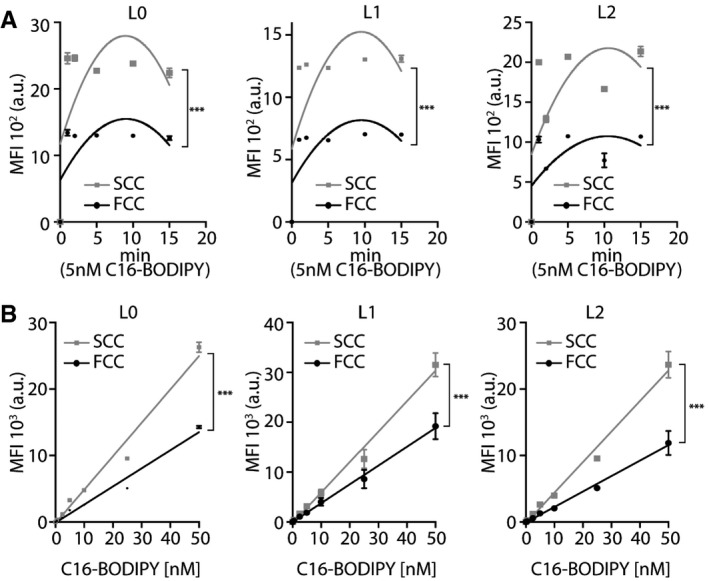
In order to investigate the mechanisms that might contribute to the increased lipid content observed in SCCs, we next compared the rate of lipid uptake/transport between SCCs and FCCs. To assess basal fatty acid uptake activities, we performed time‐course and dose–response studies for the three primary GBM L0, L1, and L2 cell lines using a fluorescently labeled fatty acid (C16‐BODIPY). C16‐BODIPY uptake was progressively stimulated with increasing exposure time, and significantly higher incorporation was observed in SCCs compared with FCCs (Fig 6A). The increase in lipid analog absorption was also dose‐dependent and significantly higher in SCCs than FCCs, by approximately 85% (Fig 6B). This increased fatty acid uptake in SCCs compared to FCCs is consistent with the SCCs’ elevated lipid metabolic intermediate content that we found using UHPLC/HRQMS and LipidTox analyses.
Figure 6. Enhanced exogenous fatty acid transport in SCCs.
-
A, BC16‐BODIPY uptake was measured using flow cytometry. (A) Time‐course, second‐order polynomial nonlinear regression fit, L0, n = 3; L1, n = 3; L2, n = 2, ***P < 0.0001, two‐way ANOVA, MFI: mean fluorescence intensity. (B) Dose–response, mean ± SEM, L0, n = 3; L1, n = 6; L2, n = 3, ***P < 0.0001, linear regression.
SCCs’ resistance to metabolic stress is driven by FABP7‐dependent exogenous fatty acid uptake
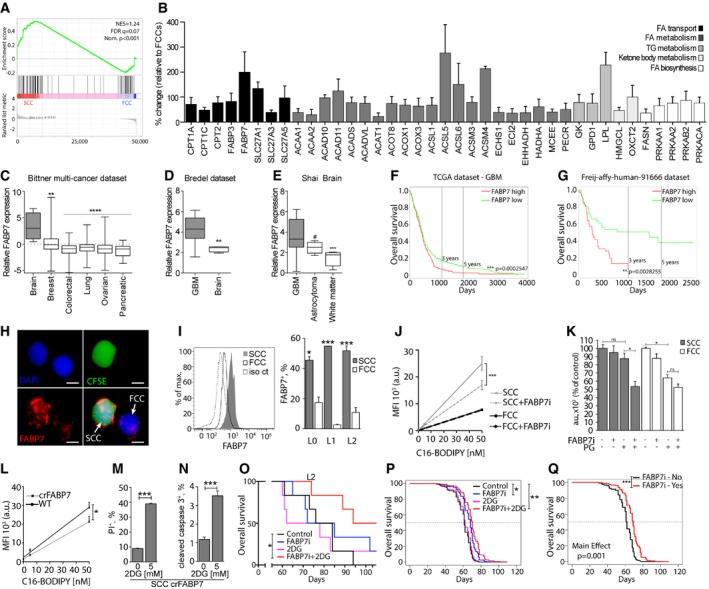
To identify potential candidates regulating fatty acid uptake in SCCs, we performed gene set enrichment analyses of a lipid metabolism gene signature (Table EV9 for gene set description) obtained from RNA sequencing comparing the gene expression of SCCs and FCCs isolated from L0, L1, and L2 GBM primary cell lines (Fig 7A). The results showed an enrichment of lipid metabolism pathways in SCCs, and the significant overexpression of several fatty acid transporters was validated by qRT‐PCR (Fig 7B). The results of this screen prompted us to focus on FABP7, which is expressed mostly by glial cells in normal brain tissue (Fig EV4A) and is up‐regulated in glioma (Fig EV4B; Uhlen et al, 2015). FABP7 is a radial glial marker enriched in glioma stem cells, associated with tumor invasiveness and poor prognosis in GBM, and a key protein involved in exogenous fatty acid uptake into cells and intracellular trafficking and storage (Liang et al, 2006; De Rosa et al, 2012; Morihiro et al, 2013; Bensaad et al, 2014). Moreover, siRNA knockdown of FABP7 expression significantly reduced cell proliferation and migration (De Rosa et al, 2012). Using the Bittner dataset (Rhodes et al, 2004), we compared the expression levels of the FABP7 gene in cell lines derived from multiple advanced cancers. We found that FABP7 expression is significantly elevated in brain tumor cell lines when compared to breast, colorectal, lung, ovarian, and pancreatic cancer cell lines (Fig 7C). In addition, using the Bredel dataset (Bredel et al, 2005), we confirmed that FABP7 is elevated in GBM versus non‐neoplastic brain tissues (Fig 7D). Oncomine analysis of the Shai dataset also revealed higher expression of FABP7 in GBM compared to lower grade astrocytomas (Fig 7E). Furthermore, we interrogated the TCGA and Freij‐affy‐human‐91666 datasets and found that higher FABP7 expression correlated with poorer patient prognosis (Fig 7F and G). These data suggest that FABP7 expression carries important prognostic information about patient survival. Interestingly, and in agreement with our qRT‐PCR data, we found increased FABP7 in SCCs compared to FCCs by immunofluorescence (Fig 7H) and flow cytometry (Fig 7I). To further link FABP7 expression to the SCC phenotype, we identified all genes that are positively correlated with FABP7 using the GlioVis data portal. Gene set enrichment analysis was then performed to compare the expression levels of FABP7‐correlated genes between the SCCs and FCCs isolated from L0, L1, and L2 (Fig EV4C, Table EV10). The results of this analysis support that FABP7 and positively correlated gene signatures are overexpressed in SCCs. Additionally, publicly available single‐cell RNA sequencing analyses (Venteicher et al, 2017) also confirmed the overexpression of FABP7 in the SCCs defined based on the expression of cell cycle genes as described in Tirosh et al (2016b; Table EV11).
Figure 7. Inhibition of lipid uptake and resistance to glucose restriction following FABP blockade in SCCs.
-
AGSEA comparing RNA sequencing results for the fatty acid metabolism gene signature between SCCs and FCCs (n = 3 biological replicates, L0‐1‐2) . FDR, false discovery rate; NES, normalized enrichment score; Nom., nominal.
-
BResults of qRT‐PCR of genes with statistical significant differential expression between SCCs and FCCs using the RT2 Profiler PCR Array for Human Fatty Acid Metabolism. Changes in transcript levels are given as percent change relative to FCCs (mean ± SEM, n = 3–8, P < 0.05, one‐sample t‐test).
-
CFABP7 expression in brain (n = 4), breast (n = 336), colorectal (n = 374), lung (n = 117), ovarian (n = 242), and pancreatic (n = 20) cancer cell lines (**P < 0.005, ****P < 0.0001, one‐way ANOVA). Boxes extend from 25th to 75th percentiles. The line in the boxes represents the median, and the whiskers go down to the smallest value and up to the largest.
-
DEvaluation of the Bredel dataset indicated higher expression of FABP7 in glioblastoma (GBM, n = 27) compared to non‐malignant (n = 4) brain tissue (**P < 0.005, t‐test). Boxes extend from 25th to 75th percentiles. The line in the boxes represents the median, and the whiskers go down to the smallest value and up to the largest.
-
EData from the Shai database showed greater FABP7 expression in GBM (n = 27) versus astrocytoma (n = 5; ***P < 0.001, one‐way ANOVA, # P < 0.05, t‐test). Boxes extend from 25th to 75th percentiles. The line in the boxes represents the median, and the whiskers go down to the smallest value and up to the largest.
-
F, GKaplan–Meier curves derived from TCGA (high expression, n = 217; low expression, n = 271) (F) and Freij‐affy‐human‐91666 (high expression, n = 42; low expression, n = 41) (G) datasets indicated that high expression of FABP7 (red) correlated with poorer patient survival compared to the low FABP7‐expressing group (green). **P < 0.01, ***P < 0.001, log‐rank test.
-
HImmunofluorescence microscopy images of a representative single SCC (CFSE‐CellTrace positive, green) and FCC (CellTrace negative) labeled for FABP7 (red). Nuclei were stained with DAPI (blue). Scale bar, 5 μm.
-
IFlow cytometry analysis demonstrated that SCCs overexpress FABP7 compared to FCCs (mean ± SEM, L0, n = 2; L1, n = 2; L2, n = 3, *P < 0.05, ***P < 0.001, t‐test).
-
JC16‐BODIPY uptake was measured in L1 GBM line SCCs or FCCs cultured with or without 200 nM FABP7 inhibitor (FABP7i; mean ± SEM, each condition, n = 3, ***P < 0.0001, linear regression).
-
KL1 SCCs and FCCs were separated by FACS and cultured in high glucose or physiological glucose (PG) conditions, with or without 200 nM FABP7 inhibitor. Cell proliferation was assessed using CyQUANT assays 5 days after treatment (mean ± SEM, n = 2–3, *P < 0.05, one‐way ANOVA with Tukey post‐test).
-
LC16‐BODIPY uptake was measured in the L1 wild‐type (WT) and CRISPR‐FABP7 (crFABP7) cell line (mean ± SEM, each condition, n = 3, *P < 0.05, linear regression).
-
M, NCell death of the crFABP7 cell line in the absence or presence of 5 mM 2‐deoxyglucose (2DG) was quantified by flow cytometry 24 h after treatment through propidium iodide (PI) incorporation (n = 3) (M) and cleaved caspase 3 expression (n = 3) (N). Values are mean ± SEM. ***P < 0.001, t‐test.
-
OKaplan–Meier curves showing the survival of animals intracranially implanted with L2 GBM cells and treated with 15 mg/kg i.p. of the FABP7 inhibitor (FABP7i) once a week for 3 weeks, 500 mg/kg of 2‐deoxyglucose (2‐DG) twice a week for 4 weeks, or a combination of both (n = 5 per group, *P ≤ 0.05, log‐rank test).
-
PPredicted survival curves using the Cox frailty model fit and grouping the results for the L0, L1, and L2 xenografts subjected to the different experimental conditions (*P ≤ 0.05, **P ≤ 0.01, log‐rank test).
-
QPredicted survival curves using the Cox frailty model fit and grouping the results for the L0, L1, and L2 xenografts with or without treatment with the FABP7 inhibitor (***P ≤ 0.001, log‐rank test).
Figure EV4. FABPs in glioma and SCCs.
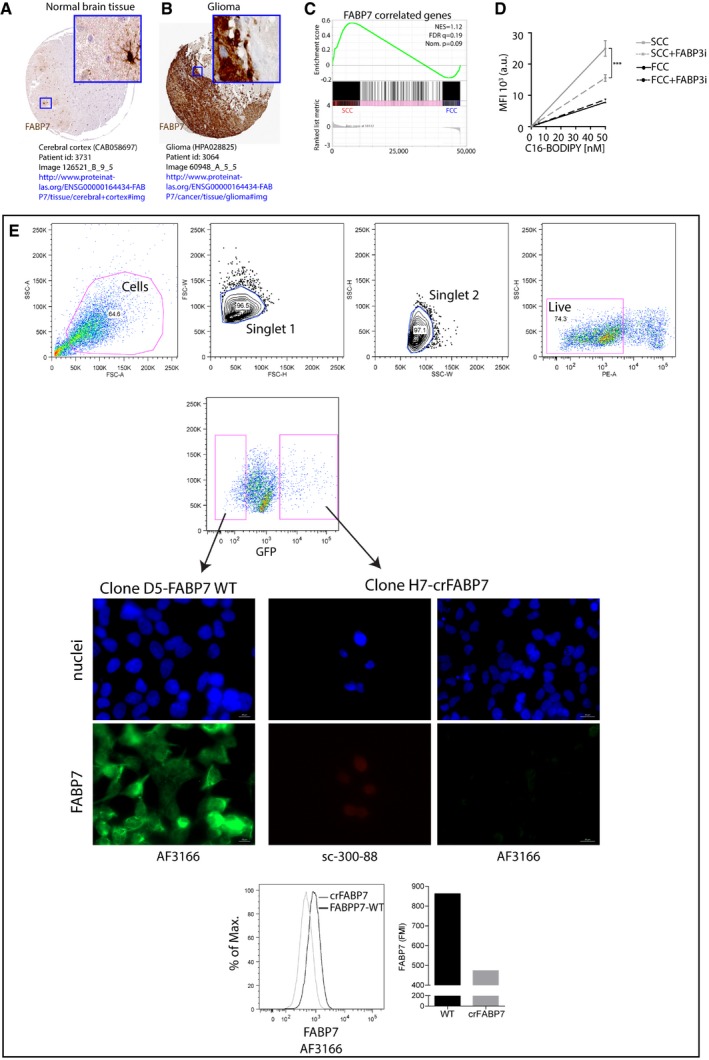
-
A, BFABP7 protein expression in normal brain tissue (A) and glioma (B). Image credit: Human Protein Atlas v16.1, http://www.proteinatlas.org.
-
CGSEA of SCC (n = 3 biological replicates, L0‐1‐2) and FCC (n = 3 biological replicates, L0‐1‐2) RNA seq data sets for enrichment of FABP7‐correlated genes. FDR, false discovery rate; NES, normalized enrichment score; Nom., nominal.
-
DC16‐BODIPY uptake in control conditions or with 250 nM of FABP3 inhibitor (BMS309403; mean ± SEM, n = 3, ***P < 0.0001, linear regression).
-
EFACS of least and most intense GFP‐positive single cells and representative immunofluorescence microscopy images of FABP7 immunoreactivity in wild‐type (clone D5 WT) and crFABP7 (clone H7) clones following CRISPR/Cas9 plasmid transfection for FABP7. FABP7 fluorescence mean intensity (FMI) was higher in wild‐type than crFABP7 clones as measured by flow cytometry. FABP7 expression was assessed using two different antibodies (clone AF3166 and clone sc‐300‐88). Nuclei were labeled with DAPI.
We next investigated the functional roles of FABP7 and FABP3, the level of which was also increased in SCCs compared to FCCs. The uptake of fluorescently labeled fatty acid C16‐BODIPY was significantly decreased after treatment with pharmacological inhibitors of FABP7 (SB‐FI‐26; Kaczocha et al, 2014; Fig 7J) and FABP3 (BMS309403; Fig EV4D) in SCCs, but not in FCCs, suggesting that these fatty acid transporters are essential for lipid uptake, and potentially storage, in SCCs. We then examined whether fatty acid uptake inhibition was associated with an altered ability of GBM cells to adapt and survive in glucose‐restricted conditions. Confirming the results described in Fig 4A and B, reduced glucose levels did not negatively affect the growth of SCCs but significantly decreased the proliferation of FCCs (Fig 7K). However, blockade of FABP7 sensitized SCCs to lower glucose levels. After combining lower glucose conditions and FABP7 inhibition, SCC viability was significantly reduced to a level comparable to that of FCCs when cultured in lower glucose conditions alone. The combination of FABP7 inhibition and decreased glucose levels did not result in any additional growth inhibitory effect on FCCs. Moreover, CRISPR‐/Cas9‐mediated knockout of FABP7 significantly reduced lipid uptake activity (Figs 7L and EV4E) and increased the sensitivity of SCCs to pharmacological inhibition of glycolysis with 2‐DG (Fig 7M and N). Together, these results demonstrate the central role of FABP7 for SCCs' metabolic robustness.
We then tested the effect of FABP7 inhibition on the migration of SCCs. These experiments revealed that FABP7 inhibition caused a significant decrease in cell migration of the total and SCC populations when treated with FABP7i concentrations ranging from 5 to 10 μM (Fig EV5A). In vivo inhibition of FABP7 also resulted in the decrease in cell invasion (Fig EV5B).
Figure EV5. Role of FABP7 in SCCs’ infiltration and resistance to glycolysis inhibition.
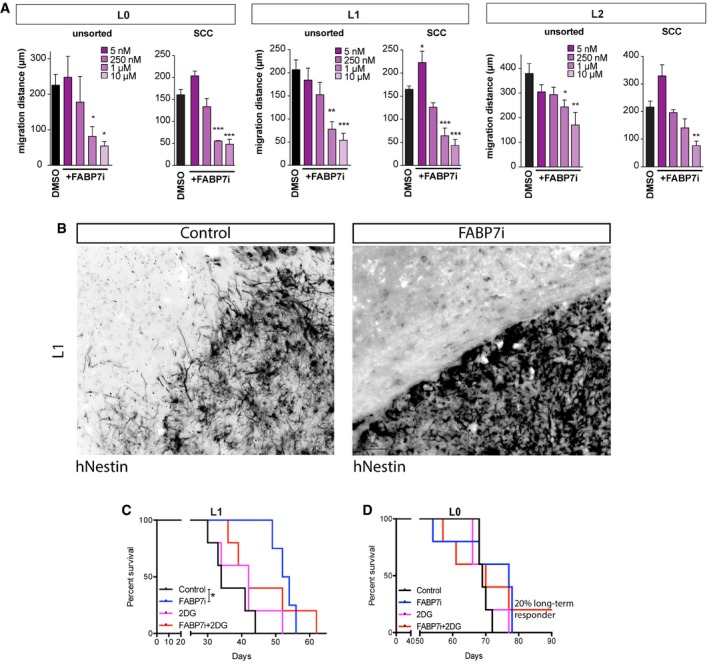
-
ATotal and SCC populations were treated with FABP7i concentrations ranging from 5 nM to 10 μM. In L0 and L1, 1 and 10 μM concentrations of FABP7i significantly reduced the migration distances of overall unsorted and SCC populations, whereas only 10 μM FABP7i was significant for L2 SCC (n = 3, mean ± SEM, *P < 0.05, **P < 0.001, ***P < 0.0005, one‐way ANOVA with Tukey post‐test).
-
BIn vivo inhibition of FABP7 also resulted in decreased tumor cell invasion. Scale bar, 50 μm.
-
C, DThe effects of FABP7 inhibition, alone or in combination with pharmacological targeting of glycolysis with 2‐DG, were conducted on tumors derived from the xenotransplants of L1 (n = 4–5) (C) and L0 (n = 5–6) (D) GBM patient‐derived cells lines. *P < 0.05, log‐rank test.
Finally, we tested the effects of FABP7 inhibition, alone or in combination with 2‐DG, following the xenotransplants of three different GBM lines (Figs 7O–Q, and EV5C and D). Combinatorial treatment showed a synergistic effect in L2 (Fig 7O). Because the results were less robust for L0 and L1 (Fig EV5C and D), we employed the Cox frailty model fit to estimate predicted survival curves for our experimental treatment groups including all three GBM cell lines. The mortality hazard ratio (HR) of the group treated with 2‐DG alone did not differ significantly from the control group's (Fig 7M, HR = 0.84, P = 0.32). Both of the groups treated with FABP7i had significantly lower mortality hazards than the control group, with the combinatorial treatment exhibiting the greatest significance (FABP7i only: HR = 0.43, P = 0.032; FABP7i + 2DG: HR = 0.28, P = 0.002; Fig 7P). The average (main) effect HR for 2‐DG did not differ from 1 (HR = 0.74, P = 0.278), while the average effect HR for FABP7i was significantly < 1 (Fig 7Q, HR = 0.38, P = 0.001). Therefore, survival was significantly improved when tumors were treated with FABP7i, whether alone or in combination with 2‐DG, with the largest significant effect occurring following the combined treatment. By contrast, survival was not improved after 2‐DG treatment alone. These results demonstrate that successful targeting of SCCs via FABP7 inhibition can improve survival.
Together, our results show that blocking lipid metabolism through fatty acid transport inhibition negates the ability of SCCs to survive metabolic stresses such as glucose restriction.
Discussion
The Warburg effect has been widely described in GBM and other tumor types. Here, we show that GBMs are metabolically heterogeneous and contain FCCs that rely on aerobic glycolysis and SCCs that depend on mitochondrial OxPhos for their survival and proliferation. Furthermore, compared to FCCs, SCCs contain increased levels of metabolites and components that are involved in lipid metabolism, storage, and transport and are also found overexpressed in recurrent GBM tumors. These properties provide SCCs with a survival advantage when these cells are exposed to metabolic stresses such as glucose deprivation or glycolysis inhibition. We show that the resistance of SCCs to glucose deprivation or glycolysis inhibition can be prevented by blocking fatty acid uptake, through the pharmacological inhibition or deletion of the fatty acid transporter FABP7 gene, in vitro and in vivo. Importantly, SCCs show increased chemotherapy resistance and migration/invasion compared to the rest of the tumor cell population, further suggesting their central role in tumor recurrence.
Our data show that SCCs possess greater migration and invasion capabilities as well as higher resistance to TMZ than FCCs. Previous studies have reported that an EMT‐like process positively correlates tumor cell invasion and chemoresistance (Qi et al, 2012; Siebzehnrubl et al, 2013; Depner et al, 2016) and that quiescent GBM cells display heightened chemoresistance (Chen et al, 2012; Campos et al, 2014). In addition, we have found that the knockdown or overexpression of the EMT transcription factor ZEB1 affects GBM cell proliferation, with ZEB1 overexpression reducing tumor cell proliferation and enhancing invasion (Siebzehnrubl et al, 2013). Here, our results show that GBM SCCs display increased expression of ZEB1 and greater resistance to chemotherapy, as well as higher migration and invasion capabilities than FCCs, in vitro and in vivo. We also find that ZEB1 knockdown significantly reduces SCC invasion. Together, these findings suggest a positive correlation between tumor cells’ slow proliferation rate, ZEB1 expression, invasion, and chemoresistance.
An increasing number of studies have been exploring tumor metabolism as a targetable vulnerability that is specific to treatment‐resistant, tumor‐propagating cells. Recently, metabolic heterogeneity has been described in animal models of GBM (Vlashi et al, 2011; Marin‐Valencia et al, 2012; Conrad et al, 2014) and other tumors, e.g., melanoma, lymphoma, leukemia, and lung and pancreatic cancer (Caro et al, 2012; Lagadinou et al, 2013; Roesch et al, 2013; Viale et al, 2014; Hensley et al, 2016).Using isotopic labeling, Marin‐Valencia and colleagues have demonstrated that functional mitochondrial OxPhos activity is maintained in GBMs (Marin‐Valencia et al, 2012), suggesting that those tumors might not exclusively rely on aerobic glycolysis, as originally described. SCCs have been shown to display specific metabolic pathways geared toward the utilization of mitochondrial respiration in other cancer types (Caro et al, 2012; Lagadinou et al, 2013; Roesch et al, 2013; Viale et al, 2014). Complementing those studies, our results demonstrate increased mitochondria and OxPhos activities in the GBM SCC population. Interestingly, our bioinformatics analyses reveal an increased expression of genes regulating mitochondrial OxPhos in recurrent GBMs compared to primary tumors, and we have found an increased expression of mitochondria‐specific genes in SCCs that are similar to those found in recurrent tumors. Chemoresistance has also been positively correlated with increased mitochondrial activity in GBM (Oliva et al, 2011; Wolf, 2014). Thus, our findings suggest that chemoresistant SCCs may be a major driver of GBM recurrence, and further studies will be needed to fully understand the mechanisms underlying the role of SCCs in recurrent tumors.
We have recently reported that treating GBM‐bearing animals with a custom high‐fat/low‐carbohydrate diet significantly improved animal survival in an orthotopic patient‐derived xenograft model; however, tumor progression still persisted (Martuscello et al, 2016). Based on the metabolic characterization of SCCs described here, we hypothesized that SCCs might drive disease progression under glucose‐restrictive conditions by bypassing glycolysis and utilizing mitochondrial OxPhos. We have found that SCCs are sensitive to the pharmacological inhibition of the electron transport chain and mitochondrial OxPhos, but resistant to glucose deprivation or glycolysis inhibition with 2‐DG. These treatments have opposite effects on the FCC population. Importantly, targeting both OxPhos and glycolytic pathways in vitro or in vivo has a synergistic inhibitory effect on the total GBM cell population's viability, which is likely due to the restriction of cells' metabolic plasticity and prevention of metabolic compensatory mechanisms. Our results demonstrate the existence of a fundamental metabolic dichotomy between SCCs and FCCs in GBM. Whether these metabolic divergences are the cause or effect for the differential growth patterns between SCCs and FCCs still remains to be determined. In addition, the detailed mechanisms underlying the metabolic specificities of these two cell populations need to be further explored.
Alterations in lipid metabolism have been described in various cancers, including GBM (Bensaad et al, 2014); however, the role of lipids in tumor initiation, maintenance, as well as migration and treatment sensitivity is not fully understood. Here, we find that SCCs exhibit an up‐regulation of lipid‐related metabolic genes. Interestingly, our analyses show that recurrent GBM tumors display an increased expression of the same lipid metabolism pathways when compared with primary GBM tumors, further suggesting the role of SCCs in tumor relapse. Intriguingly, a similar link between stemness and metabolic specificities exists in normal neural and GBM tissue. Both stem‐like GBM SCCs and adult mammalian neural stem cells lack glucose dependency and are able to oxidize diverse sources of fuel, including fatty acids (Stoll et al, 2015). In contrast, similarly to transit‐amplifying neural progenitor cells, GBM FCCs display lower requirements for OxPhos metabolism and depend on glycolysis (Candelario et al, 2013; Li et al, 2014; Stoll et al, 2015). Together, our findings suggest that the slow‐cycling phenotype, lipid metabolism, treatment resistance, and tumor recurrence might be closely connected.
Furthermore, a recent report by Lin et al (2017) has demonstrated that mitochondrial OxPhos activity is dependent on fatty acid oxidation to support the proliferation of GBM cells cultured in serum‐free conditions. Other studies have shown that GBM cells store fatty acids as lipid droplets in response to metabolic stresses such as hypoxia and use a FABP‐dependent mechanism to oxidize fatty acids for energy production and adapt to environmental disruptions (Bensaad et al, 2014). Our results validate these findings and further demonstrate that the SCC population is characterized by a higher expression of FABPs and greater lipid droplet amounts when SCCs are grown in normoxic or nutrient‐replete conditions. We propose that SCCs accumulate energy reserves as lipid droplets in these conditions, through a mechanism controlled by FABP‐associated pathways, as fatty acid transport is prevented by the inhibition of these pathways. In addition, our study shows increased SCC survival and preferential utilization of lipids droplets in response to glucose restriction or glycolysis inhibition. Moreover, the metabolic resistance of SCCs to glucose deprivation or glycolysis inhibition can be prevented by the pharmacological inhibition or genomic deletion using CRISPR/Cas9 of FABP7, both of which prevent the uptake of fatty acids upstream of intracellular lipid metabolic pathways, in vitro and in vivo. Importantly, we have found that FABP7 inhibition leads to overall increased survival, whether alone or in combination with glycolysis inhibition, suggesting that FABP7 is central to lipid metabolism in SCCs and that targeting FABP7‐related metabolic pathways is a viable therapeutic strategy. At high glucose concentrations, FABP7 inhibition did not prevent cell survival and proliferation, which suggests that the fatty acids can be synthesized for metabolism and energy production from other nutrients, including glucose or glutamine. We also found, in SCCs compared to FCCs, a significantly increased presence of autophagosomes and lysosomes and expression of associated pathways, which are known to mediate catabolic pathways that provide energy to cells in response to decreased nutrient availabilities. Future studies will be needed to determine whether lipid reserve utilization is dependent on those autophagosomal and lysosomal pathways.
Chemotherapeutic agents, such as TMZ, induce oxidative stress by increasing ROS (Chandra et al, 2000; Zhang et al, 2010), as well as FABP expression (Bensaad et al, 2014), lipid droplet content (Bensaad et al, 2014), and antioxidant properties through the glutathione pathway (Landriscina et al, 2009), all of which have all been linked to TMZ resistance (Oliva et al, 2011). The increased levels of glutathione‐related metabolites observed in GBM SCCs suggest that these cells’ increased antioxidant activity, which could also involve the FABP/lipid droplet axis, might cause their chemoresistance (Bensaad et al, 2014). Additionally, as autophagy has been linked to resistance to chemotherapy (White et al, 2015; Belounis et al, 2016; Guo & White, 2017), including TMZ (Yan et al, 2016), the autophagosomes that we observed in SCCs may also contribute to TMZ resistance. Therefore, autophagy might represent a pathway that could be therapeutically targeted to sensitize SCCs to TMZ.
We propose that the specific characteristics of GBM SCCs play a critical role in their tolerance to chemotherapy and that interference with lipid metabolism in SCCs may be exploited to target those cells and overcome tumor chemoresistance. Moreover, peptidome analysis of GBM has identified FABP7 as one of the top ten GBM‐specific, HLA molecule‐associated peptide with high immunogenic properties (Dutoit et al, 2012), a characteristic that could be exploited for immunotherapeutic targeting of SCCs. Communication between cancer cells and their microenvironment, including immune cells, has been suggested, and the implication of fatty acids as regulators of this crosstalk has started to gain recognition (Beloribi‐Djefaflia et al, 2016). Further investigations are needed to fully understand the role of fatty acids in this intercellular communication process, especially in SCCs.
SCCs have been garnering increasing attention in the cancer research field, and a better understanding of their specific features and vulnerabilities holds great therapeutic promise, potentially enabling the development of novel targeted treatments to overcome tumor relapse (Graham et al, 2002; Dembinski & Krauss, 2009; Gao et al, 2010; Pece et al, 2010; Roesch et al, 2010, 2013; Caro et al, 2012; Moore et al, 2012; Lagadinou et al, 2013; Campos et al, 2014; Viale et al, 2014; Zeuner et al, 2014; Oshimori et al, 2015). Our characterization of the nature of GBM's metabolic heterogeneity might have a significant impact on the improvement of metabolic therapies, particularly those targeting the Warburg effect. In addition, our study provides a new basis for studying the link between GBM chemoresistance and tumor cell metabolism, particularly lipid metabolic pathways in SCCs. Further identification of the lipid metabolites that are present in GBM SCCs may reveal a unique metabolic marker signature for treatment‐resistant/tumor‐initiating cell populations, as well as potential new targets for cancer stem cell‐specific therapies.
Materials and Methods
Cell culture
The primary cell lines used in this study, Line 0 (L0), Line 1 (L1) and Line 2 (L2) (Deleyrolle et al, 2011; Siebzehnrubl et al, 2013), were isolated from human GBM tumors and cultured as previously described and approved by Institutional committee (IRB; Deleyrolle et al, 2011; Siebzehnrubl et al, 2011, 2013; Sarkisian et al, 2014; Hoang‐Minh et al, 2016). Informed consent was obtained from all subjects, and the experiments conformed to the principles set out in the WMA Declaration of Helsinki and Department of Health and Human Services Belmont Report. The lines were authenticated using STR analysis (University of Arizona Genetics Core). Cells were grown as floating spheres and maintained in Neurocult NS‐A medium (StemCell Technologies) in the presence of 20 ng/ml human EGF. When the spheres reached approximately 150 μm in diameter, they were enzymatically dissociated by digestion with Accumax (Innovative Cell Technologies, Inc.) for 10 min at 37°C. Cells were then washed, counted using Trypan blue to exclude dead cells, and replated in fresh complete medium. To generate TMZ‐resistant cells, cells were initially treated with 500 μM TMZ for one passage and then continuously exposed to 20 μM TMZ.
Isolation of fast‐ and slow‐cycling cells
Populations of slow‐cycling cells (SCCs) and fast‐cycling cells (FCCs) were identified and isolated primary human glioblastoma cell lines based on their capacity to retain CellTrace dyes (carboxyfluorescein succinimidyl ester—CFSE or Cell Trace Violet‐CTV, Invitrogen), as described previously (Deleyrolle et al, 2011), and grouped as CFSE/Violethigh‐ top 10% and CFSE/Violetlow‐ bottom 10%. For the experiments presented in Fig 1D and E, FCCs were isolated as CFSElow‐ bottom 85% (Deleyrolle et al, 2011). Our gating strategy allows for the isolation of functional and phenotypic extremes with similar size population, homogenizing for sorting time and hence overcoming the issue of fluorescence‐activated cell sorting (FACS)‐related metabolic stress. Both SCC and FCC populations are able to expand in vitro and in vivo, demonstrating their viability and expansion capacities (Deleyrolle et al, 2011). Although this strategy does not capture the full spectrum of cellular population contained in GBM, it provides a relevant paradigm to compare defined cellular components or states with distinct proliferation properties. Proliferation was assessed based on CellTrace fluorescence intensity decay rate over time measured by flow cytometry and identified 6–8 days after labeling. This process enabled the separation of rapidly proliferating (FCCs) and slowly proliferating (SCCs) cell fractions (top and bottom 10%) based on CellTrace fluorescence intensity, which is proportional to dye dilution. We used these most extreme fractions of the proliferation spectrum in order to ensure clear and distinct separation of FCCs and SCCs based on cell cycle kinetics. All experiments were performed immediately after FACS of those SCC and FCC populations.
Scratch assay
Sorted SCCs and FCCs were plated as described previously (Siebzehnrubl et al, 2013) at 2 million cells per well of a six‐well plate pre‐coated with poly‐D‐lysine and laminin, in medium containing 1% fetal bovine serum. Twenty‐four hours after plating, a scratch was made with a 200‐μl pipette tip. Cells were imaged at the time of lesion, as well as 24 h later, and the distance traveled by the most migratory cells was recorded.
Migration assay
For quantification of cell migration, tumor spheres were plated onto a laminin/poly‐D‐lysine coated surface at low density and in the presence of growth factors, FABP7 inhibitor (SB‐FI‐26), or DMSO as a solvent control. Images were taken from the same spheres 2 and 24 h after plating with a Leica DM IL microscope equipped with a DFC3000G camera and Leica application suite X software. The greatest distance of outgrowing cells was measured using ImageJ, and migration distance was calculated as the difference between the two time points. Only spheres with a diameter greater than 50 μm, 2 h after plating, were used to measure migration distance.
In vivo invasion quantification
To quantify the effects of ZEB1 on the invasion of SCC, GBM cells were stably transfected with shZEB1 or shControl constructs as described (Siebzehnrubl et al, 2013). After selection, transfected cells were loaded with CFSE and separated into SCC and FCC fractions, and each intracranially injected into 5 SCID mice as described (Siebzehnrubl et al, 2013). Mice were transcardially perfused 12 weeks after implantation, their brains harvested and post‐fixed in 4% formalin overnight. Brains were sectioned and stained and analyzed using the Invasion Index as described (Siebzehnrubl et al, 2013).
To assess the intrinsic invasion capacity of SCC and FCC populations, sorted SCC/FCC populations were allowed to recover for 24 h in culture and then transduced with lentiviral vectors encoding for humanized eGFP (SCCs) or humanized RFP (FCCs; both kind gift of Dr Lung‐Ji Chang, University of Florida). In both cases, the transduction efficiency was > 95%. Cells were allowed to expand and then dissociated into single cells and mixed at a ratio of 1:1. 105 cells of this mixture were intracranially implanted into SCID mice. Mice were transcardially perfused 6 weeks after implantation and their brains harvested and post‐fixed in 4% formalin overnight. Brains were sucrose‐protected, frozen, and sectioned. Sections were counterstained with Hoechst 33342, mounted onto slides, and imaged for GFP and RFP using an Olympus BX‐81 DSU spinning‐disk confocal microscope and SlideBook software.
Cell viability/proliferation assays
The methyltetrazolium bromide (MTT) assay was used as an indicator of cell viability and performed as described (Siebzehnrubl et al, 2013). Briefly, 2,000–5,000 cells were plated per well in 96‐well plates, in medium containing 1% fetal bovine serum. The cell populations were treated with TMZ 1 day after plating and analyzed with MTT assay 96 h later. Bar graphs were derived from individual concentration measurements, which were compared to the appropriate controls.
Propidium iodide incorporation and expression of cleaved caspase 3 were used to compare the effects of glucose restriction and/or mitochondrial function inhibition (by rotenone or metformin treatment). Briefly, cells were labeled with CellTrace dye and grown in complete medium for 5–7 days before being placed in high glucose (HG; > 500 mg/dl) or physiological glucose conditions (PG; 90–110 mg/dl) and/or treated with rotenone (0.5–1 μM) or metformin (10–20 mM) for 24 h. Media glucose concentrations were monitored daily and maintained constant throughout the experiments by adding glucose to the cell cultures as needed, which prevented the glucose supply exhaustion that might have occurred due to FCCs’ higher division rate. Propidium iodide and cleaved caspase 3 staining were quantified using flow cytometry. The effects of restricting glucose along with mitochondrial targeting using rotenone or metformin were investigated using the CyQUANT™ assay. Cells were plated at 60,000 cells per well in 96‐well plates and exposed to the treatments alone or in combination (physiological glucose, 0.5 μM rotenone, and 10 mM metformin). CyQUANT binding dye was added to each well and incubated for 30 min at 37°C before being quantified using Biotek™ Cytation™ 3 Cell Imaging Multi‐Mode Reader.
Assessment of mitochondrial function
Cells were seeded at a density of 30,000 cells in 80 μl medium per well in XF96‐well microplates (Seahorse Bioscience; n = 10) pre‐coated with 22.4 μg/ml Cell‐Tak Adhesive (Corning). SCCs and FCCs were incubated for 24 h in standard growth medium in a humidified incubator at 37°C with 5% CO2. After 24 h, the standard medium was exchanged for XF Base Medium pH 7.4 (Seahorse Bioscience) supplemented with 25 mM glucose, 2 mM l‐glutamine, and 1 mM sodium pyruvate. The cells were then incubated for 1 h at 37°C without CO2. OCRs were measured using the XF Cell Mito Stress Assay (Seahorse Bioscience) and prior to and following additions of the following: (i) ATP synthase inhibitor (1 μM oligomycin), (ii) uncoupler [1 μM carbonyl cyanide 4‐(trifluoromethoxy)phenylhydrazone (FCCP)], and (iii) complex I/II inhibitors (0.5 μM rotenone/antimycin A). Data were analyzed using Wave Desktop Software (Seahorse Bioscience), following the manufacturer's instructions, and normalized to protein levels.
ATP level measurement
ATP levels were measured using the luciferase‐based ATP‐lite assay (Perkin Elmer) as per the manufacturer's instructions. Briefly, 10,000 SCCs or FCCs were seeded per well (n = 10) of a black‐walled 96‐well tissue culture plate. Luminescence (indicative of intracellular ATP levels) was measured using a Spectra Max i3x microplate reader (Molecular Devices) and normalized to protein levels for each well.
Electron microscopy
SCCs and FCCs were separated by FACS before being fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) overnight and washed with 0.1 M cacodylate buffer again. Cells were then post‐fixed in 1% osmium tetroxide for 1 h before additional buffer washes. Cells were dehydrated through an ethanol series followed by three additional 100% ethanol. Subsequently, cells were infiltrated with a mixture of 100% ethanol and Eponate 12 resin (Ted Pella Inc., Redding, CA, USA) and then pure Eponate 12 resin overnight. Cells were embedded in Eppendorf tubes and then placed in a 60°C oven for polymerization. Ultrathin 70‐ to 80‐nm‐thick sections were cut on a Leica UltraCut microtome. Sections were then stained with 5% uranyl acetate for 15 min followed by 2% lead citrate for 15 min. Mitochondria were imaged with a JEOL JEM‐1400 transmission electron microscope (Tokyo, Japan) equipped with a Gatan US1000 CCD camera (Pleasanton, CA). The data described here were gathered on the JEOL JEM‐1400 120 kV TEM supported by a National Institutes of Health Grant S10 RR025679.
Generation and maintenance of FABP7‐depleted cell lines
To generate FABP7‐depleted patient‐derived GBM cell lines, we screened and identified CRISPR‐/Cas9‐encoding plasmids containing a GFP reporter gene that could target human FABP7 [Sigma‐Aldrich; CRISPR/Cas‐GFP vector (pU6‐gRNA‐CMV‐Cas9:2a:GFP); primer pair ID: HS0000240647; FABP7 gRNA target sequence: CTTGACTGATAATTACCGT]. For transfection experiments, GBM cells were grown on 10‐cm2 plates and transfected (Lipofectamine 2000; Life Technologies) at 60–70% confluence with 0.5 μg/ml of the CRISPR‐/Cas9‐encoding plasmid DNA. Twenty‐four to 48 h after transfection, GFP‐positive cells were sorted as individual clones into 96‐well plates containing 250 μl of complete medium supplemented with hEGF using a BD FACS Aria II Cell Sorter (BD Biosciences, San Jose, CA), excluding cell debris and dead cells from the analysis by forward‐ and side‐scatter gating and PI exclusion. Stable cell lines from each GFP‐positive clone were then expanded and screened for the presence of FABP7 by immunofluorescence microscopy analysis as well as flow cytometry. GFP‐positive clones with undetectable FABP7 levels were designated CRISPR FABP7 (crFABP7, L1 clone H7). For immunostaining, once cells formed spheres greater than 100 μm in diameter in each well, the spheres were mechanically dissociated, replated, and expanded into Lab‐Tek chambered slides in 5% FBS‐supplemented complete medium. After 2–3 days, cells were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (4% PFA) for immunohistochemical analysis as described below.
Animal experiments
Adult male NOD‐SCID mice (7–15 weeks old) were used for in vivo tumor implants following NIH and institutional (IACUC) guidelines and regulations for animal care and handling. The mice colonies were maintained at the University of Florida's animal facility. Animals were randomized to cages following implantation. For in vivo tumor invasion assay, FACS cells were intracranially implanted as previously described (Deleyrolle et al, 2011; Siebzehnrubl et al, 2013; Hoang‐Minh et al, 2016) and invasion assay was performed 10 weeks after implant. For in vivo TMZ treatment, animals were implanted with 100,000 cells immediately after cell sorting. Tumor‐bearing animals were intraperitoneally treated with five injections of 20 mg/kg TMZ over 5 days at 3 (hGBM L0) or 4 (hGBM L2) weeks after implantation. For in vivo restricted glucose and mitochondria targeting experiments, animals were xenografted with SCCs or FCCs and subjected to either a high‐carbohydrate control diet or a custom‐supplemented high‐fat/low‐carbohydrate dietary regimen (sHFLC; Martuscello et al, 2016). Each group received vehicle or rotenone treatment (0.5 mg/kg i.p, once a week for 6 weeks).
Mass spectrometry‐based metabolite screening
CellTrace‐labeled cells were cultured in gliomasphere growth conditions for 5–7 days before being separated into SCCs and FCCs using FACS. Upon isolation, cells were placed into 10 mM ammonium acetate for metabolic fingerprinting using UHPLC/HRQMS. Detected metabolites were identified based on both retention time and mass accuracy using major metabolite databases, including the Human Metabolome DataBase (HMDB), Madison Metabolomics Consortium Database (MMCD), Metlin, LIPID MAPS, and our 700 compound internal library (from the Southeast Center for Integrated Metabolomics). For final identification, tandem MS was performed to confirm assignment. Statistical analyses were performed using JMP 11 and Metaboanalyst (http://www.metaboanalyst.ca), a free R‐based metabolomic statistical analysis package. In addition, multivariate statistics including principal components analysis (PCA) and partial least squares‐discriminant analysis (PLS‐DA) were used to identify metabolites that might differentiate the cell lines or cellular subtypes.
Lipid uptake
Each cell line was labeled with CellTrace dye and grown for 5–7 days. Cells were then dissociated and treated with different C16‐BODIPY (BODIPY®FLC16, Molecular probes) concentrations and incubation times described below. Fatty acids conjugated to BODIPY fluorophore undergo nativelike metabolism and transport. Dose–response was performed with 0, 0.5, 2.5, 5, 10, 25, and 50 nM BODIPY, and the time course was done at a concentration of 5 nM for 1, 4, 5, 10, and 15 min. The cells were then washed and fixed with 4% paraformaldehyde. To determine the amount of fatty acid uptake, cells were analyzed by flow cytometry on a BD LSRII Flow Cytometer. Fatty acid transporter inhibitors SB‐FI‐26 (Cayman Chemicals, #14191) and BMS309403 (Millipore, #34310) were used to inhibit FABP7 (Kaczocha et al, 2014) and FABP3 (Furuhashi et al, 2007), respectively.
Dyes and antibodies
Dyes and primary antibodies used for flow cytometry or immunocytochemistry included CellTrace™ Violet and CFSE Cell Proliferation Kit (Molecular Probes), DAPI (Molecular Probes), Hoechst (Thermo Scientific), DRAQ5 (Thermo Scientific), propidium iodide (Molecular Probes), LipidTox (ThermoFisher Scientific), MitoTracker Green and Orange (Molecular Probes), MitoProbe DiIC1(5) (Molecular Probes), FABP7 (Santa Cruz Biotechnology, #sc‐30088), FABP7 (R&D Systems, #AF3166), human Nestin (Millipore, #MAB5326), N‐cadherin (Millipore, #04‐1126), beta‐catenin (Sigma, #C2206), ZEB1 (Sigma, #HPA027524), VDAC1 (Abcam, #ab15895), cleaved caspase‐3 (Cell Signaling Technology, #9661S), NDUFA4 (Abcam, #ab129752), ATP synthase (BD Biosciences #612518), LC3 (Cell Signaling Technology, #3868S), and LAMP2 (Abcam, # 25631) antibodies.
Flow cytometry
Six to eight days post‐CellTrace load, labeling was performed using the antibodies and dyes that are listed above and according to the manufacturer's protocol. Staining was quantified by flow cytometry (BD LSRII), and percent of immunoreactive cells or mean fluorescence intensity (MFI) were reported.
Image acquisition and invasion measurement
Tumor invasion was measured using human‐specific nestin labeling (Millipore, MAB5326). Full images of brain sections were obtained by multiple gray scale imaging acquired using Spot Advanced software (Spot Imaging Solutions), merged into full images, and inverted into black‐and‐white images using Photoshop CS6 (Adobe Systems). Staining threshold levels were adjusted in ImageJ software to distinguish tumor from background, as previously described (Siebzehnrubl et al, 2013). Invasion index was obtained by calculating the ratio of the squared‐perimeter distance over the area (P2/A). Dissociated tumors are associated with higher invasion indices compared to more spherical tumors characterized by lower invasion indices. High‐power images of stained tissues were taken using an IX81‐DSU spinning‐disk confocal microscope (Olympus) fitted with a 60× water immersion objective, and all images were captured as z‐stacks (0.5 μm steps). For 3D imaging, pictures were acquired using a UPLSAPO 60× water objective and Hamamatsu ORCA‐AG Camera. Images were captured as z‐stacks (0.5 μm steps). All image analyses and 3D surface reconstructions utilized the 3i SlideBook v4.2 Software (with Deconvolution Module). Image capture settings were standardized across samples. 3D surface reconstruction rendering cutoff values were also standardized in the 3i SlideBook software.
RNA sequencing and GSEA
The autophagosome–lysosome gene set was compiled by combining the list of genes from The Human Lysosome Gene Database (http://lysosome.unipg.it/index.php) and the GO_Autophagosome gene set (http://software.broadinstitute.org/gsea/msigdb/cards/GO_AUTOPHAGOSOME.html). Enrichment of the gene signature was assessed using GSEA (http://www.broadinstitute.org/gsea/index.jsp), and P‐values were obtained by permuting the phenotypes (1,000 permutations). To broaden the validity of the gene signature enrichment, additional gene sets were used based on previously published autophagosome–lysosome gene signatures (Jegga et al, 2011; Perera et al, 2015). The stem cell signature was derived from Wong et al (2008). GBM single‐cell RNA sequencing data were generated from (Venteicher et al, 2017). Differentially expressed genes were extracted from groups by nonparametric t‐test (P < 0.05). Gene set enrichment analysis was performed using GenePattern ssGSEA.
Bioinformatics analysis
Using information from the TCGA dataset (Cancer Genome Atlas Research Network, 2008), we analyzed the RNA expression of 155 primary (de novo) and 14 recurrent patients’ GBM tumors. All 20,530 identified genes were normalized by log2 transformation and centered by mean value. Results were presented using a volcano plot comparing recurrent and primary GBM gene expression using a threshold of a twofold change with P < 0.05 (Mann–Whitney U‐test, Subio platform) as significant difference. For pathway analyses, we used the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database platform (Szklarczyk et al, 2015; http://string-db.org) to identify functional networks differentially activated between primary and recurrent tumors. The datasets that were utilized are indicated in the text, Figure legends, and on the Figures. TCGA data were accessed at https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp.
Quantitative RT–PCR
SCCs and FCCs were isolated from cell lines L0, L1, and L2 as described above. RNA was extracted using TRIzol. After treatment with RNase‐free DNase I, cells from each group were purified using the RNeasy Mini Kit (Qiagen). RNA quantity and purity were determined using a NanoDrop ND‐1000, and RNA integrity was assessed by determining the RNA integrity number and 28S/18S ratio using a Bioanalyzer 2100 (Agilent Technologies). A quantity of 500 ng of high‐quality RNA (260/280 ratios slightly higher than 2.0 and 260/230 ratios higher than 1.7) for each group was converted into cDNA using the RT2 First Strand cDNA Kit (SABiosciences). All qPCRs used the RT2 SYBR Green qPCR Master Mix (SABiosciences). Fatty acid metabolism gene expression was determined using the Fatty Acid Metabolism PCR Array (PAHS‐007Z, SABiosciences), and the C100 Touch Thermal Cycler CFX96 Real‐Time System (Bio‐Rad) according to the manufacturer's protocol. FABP7 expression levels were assessed using the primers listed in Table EV8. All significant changes in gene expression levels are reported in the article; the complete list of genes assayed on the array can be found at the manufacturer's website (http://www.sabioscience.com/rt_pcr_product/HTML/PAHS-007Z.html). For additional glucose metabolism gene analyses, LDH‐A, B, and C expression levels were detected using the C100 Touch Thermal Cycler CFX96 Real‐Time System (Bio‐Rad) with Actb as control. Primers were purchased from ThermoFisher Scientific. The qRT–PCR primers designed for this study are listed in Table EV8.
Statistics
Values reported in the results are mean values ± SEM, and statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software). Statistical tests are indicated in the text. Comparisons between groups were performed appropriately using either a one‐way ANOVA or Student's t‐test (95% confidence intervals). Groups that showed significant differences with ANOVA were further subjected to Tukey's post hoc analysis. In vivo survival analyses were calculated using log‐rank analyses. Flow cytometry analysis was performed using FlowJo software. We used generalized linear models (GLM) with log‐normal errors (McCullagh & Nelder, 1989) to analyze the effect of experimental factors on mean responses. All experiments analyzed in this way involved at least two experimental factors. Corresponding models included design variables representing the main effects and interactions among factors. Survival time responses were converted to “pseudo‐observations” that more accurately represented the contributions of observed and right‐censored survival times to unbiased survival time mean estimates (Klein et al, 2008). GLM models incorporating a robust “sandwich” estimator for the covariance matrix (equivalent to generalized estimating equation models) were fitted to pseudo‐observation survival times. Residuals from model fits were evaluated graphically to assess model fit assumptions. F tests were used to test the significance of interactions and main effects. Means and 95% confidence intervals (CI) were estimated for various experimental conditions. Percent differences between means, and percent differences between the effects of experimental factors represented by interactions, were estimated and tested for significant difference from zero using t statistic contrasts within the framework of fitted models. Via repeated simulation of responses within our various experimental designs, we determined that we had 80% power to detect 49–55% percent differences between effects within interactions at a 2‐sided significance level of 0.05. Model‐fitting and estimation was carried out using SAS Version 9.4 (SAS Institute, Cary, NC, USA). Data simulation and retrospective power calculations were carried out using R Version 3.5.0 (R Foundation for Statistical Computing, Vienna, Austria).
Study approval
All procedures involving mice were performed according to NIH and institutional guidelines for animal care and handling.
Data accessibility
The RNA sequencing (EGAS00001003251) and metabolomics (Metabolomics Workbench: ST000248) datasets have been deposited.
Author contributions
LBH‐M, FAS, MRS, CAP, BAR, and LPD conceived and designed the research studies; LBH‐M, FAS, CY, KD, TL, NA, MSM, JP, KA, AV, AJ‐P, SS‐H, CM, and LPD collected and/or assembled the data; LBH‐M, FAS, CY, JH, EJS, TJG, DAM, SS‐H, CAP, PK, MRS, BAR, and LPD analyzed and interpreted the data; LBH‐M, FAS, EJS, and, LPD wrote the manuscript. FAS, DAM, MRS, BAR, and LPD provided financial support. All authors gave final approval of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Table EV9
Table EV10
Table EV11
Review Process File
Acknowledgements
Financial support was provided by American Brain Tumor Association Basic Research Fellowship Grant (L.H.M.), Tenovus Cancer Care TIG2015/L19 and the Cancer Research UK Cardiff Centre (F.A.S.), Preston A. Wells, Jr. Endowment 00107592 (J.H.), 1K08CA199224‐01A1 (E.J.S.), American Cancer Society Research Scholar Grant RSG‐13‐031‐01‐DDC (M.R.S.), Preston A. Wells, Jr. Brain Tumor Research Fund (D.A.M.), NINDS R24 NS086554‐01 (B.A.R.), American Cancer Society Chris DiMarco Institutional Research Grant, Accelerate Brain Cancer Cure, American Brain Tumor Association Research Grant DG1800014, and Preston A. Wells, Jr. Brain Tumor Research Fund (L.P.D.). This study was supported by the Robert P. Apkarian Integrated Electron Microscopy Core (RPAIEMC), which is subsidized by the Emory College of Arts and Sciences and the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. The authors would like to thank the University of Florida Interdisciplinary Center for Biotechnology Research Cytometry Core (N. Benson and A. Doty), the Cell & Tissue Analysis Core (D. Smith, NIH Grant # 1S10OD020026), M. Dajac and M. Andrews for technical assistance, the Florida Center for Brain Tumor Research (B. Frentzen, R. McTiernan, and J. Pittman) for access to brain tumor samples, and B. Tugertimur for his assistance in setting up the MTT assays.
The EMBO Journal (2018) 37: e98772
Contributor Information
Brent A Reynolds, Email: brent.reynolds@neurosurgery.ufl.edu.
Loic P Deleyrolle, Email: loic.deleyrolle@neurosurgery.ufl.edu.
References
- Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, Sommergruber W, Schweifer N, Wernitznig A, Beug H, Foisner R, Eger A (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26: 6979–6988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloribi‐Djefaflia S, Vasseur S, Guillaumond F (2016) Lipid metabolic reprogramming in cancer cells. Oncogenesis 5: e189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belounis A, Nyalendo C, Le Gall R, Imbriglio TV, Mahma M, Teira P, Beaunoyer M, Cournoyer S, Haddad E, Vassal G, Sartelet H (2016) Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 16: 891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJ, Karpe F, Schulze A, Harris AL (2014) Fatty acid uptake and lipid storage induced by HIF‐1alpha contribute to cell growth and survival after hypoxia‐reoxygenation. Cell Rep 9: 349–365 [DOI] [PubMed] [Google Scholar]
- Blachly‐Dyson E, Forte M (2001) VDAC channels. IUBMB Life 52: 113–118 [DOI] [PubMed] [Google Scholar]
- Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI (2005) High‐resolution genome‐wide mapping of genetic alterations in human glial brain tumors. Cancer Res 65: 4088–4096 [DOI] [PubMed] [Google Scholar]
- Campos B, Gal Z, Baader A, Schneider T, Sliwinski C, Gassel K, Bageritz J, Grabe N, von Deimling A, Beckhove P, Mogler C, Goidts V, Unterberg A, Eckstein V, Herold‐Mende C (2014) Aberrant self‐renewal and quiescence contribute to the aggressiveness of glioblastoma. J Pathol 234: 23–33 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455: 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario KM, Shuttleworth CW, Cunningham LA (2013) Neural stem/progenitor cells display a low requirement for oxidative metabolism independent of hypoxia inducible factor‐1alpha expression. J Neurochem 125: 420–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, Polak K, Tondera D, Gounarides J, Yin H, Zhou F, Green MR, Chen L, Monti S, Marto JA, Shipp MA, Danial NN (2012) Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 22: 547–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D'Alessio AC, Young RA, Weinberg RA (2013) Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154: 61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra J, Samali A, Orrenius S (2000) Triggering and modulation of apoptosis by oxidative stress. Free Radic Biol Med 29: 323–333 [DOI] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF (2012) A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488: 522–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Goldman JG, Kelly L, He Y, Waliczek T, Kordower JH (2014) Abnormal alpha‐synuclein reduces nigral voltage‐dependent anion channel 1 in sporadic and experimental Parkinson's disease. Neurobiol Dis 69: 1–14 [DOI] [PubMed] [Google Scholar]
- Conrad CA, Fueyo J, Gomez‐Manzano C (2014) Intratumoral heterogeneity and intraclonal plasticity: from Warburg to oxygen and back again. Neuro Oncol 16: 1025–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe B (2012) Mitochondrial markers for cancer: relevance to diagnosis, therapy, and prognosis and general understanding of malignant disease mechanisms. ISRN Pathol 2012: 15 [Google Scholar]
- De Rosa A, Pellegatta S, Rossi M, Tunici P, Magnoni L, Speranza MC, Malusa F, Miragliotta V, Mori E, Finocchiaro G, Bakker A (2012) A radial glia gene marker, fatty acid binding protein 7 (FABP7), is involved in proliferation and invasion of glioblastoma cells. PLoS One 7: e52113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleyrolle LP, Harding A, Cato K, Siebzehnrubl FA, Rahman M, Azari H, Olson S, Gabrielli B, Osborne G, Vescovi A, Reynolds BA (2011) Evidence for label‐retaining tumour‐initiating cells in human glioblastoma. Brain 134: 1331–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleyrolle LP, Rohaus MR, Fortin JM, Reynolds BA, Azari H (2012) Identification and isolation of slow‐dividing cells in human glioblastoma using carboxy fluorescein succinimidyl ester (CFSE). J Vis Exp 62: 3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembinski JL, Krauss S (2009) Characterization and functional analysis of a slow cycling stem cell‐like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis 26: 611–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depner C, Zum Buttel H, Bogurcu N, Cuesta AM, Aburto MR, Seidel S, Finkelmeier F, Foss F, Hofmann J, Kaulich K, Barbus S, Segarra M, Reifenberger G, Garvalov BK, Acker T, Acker‐Palmer A (2016) EphrinB2 repression through ZEB2 mediates tumour invasion and anti‐angiogenic resistance. Nat Commun 7: 12329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Czaja MJ (2011) Regulation of lipid droplets by autophagy. Trends Endocrinol Metab 22: 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutoit V, Herold‐Mende C, Hilf N, Schoor O, Beckhove P, Bucher J, Dorsch K, Flohr S, Fritsche J, Lewandrowski P, Lohr J, Rammensee HG, Stevanovic S, Trautwein C, Vass V, Walter S, Walker PR, Weinschenk T, Singh‐Jasuja H, Dietrich PY (2012) Exploiting the glioblastoma peptidome to discover novel tumour‐associated antigens for immunotherapy. Brain 135: 1042–1054 [DOI] [PubMed] [Google Scholar]
- Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, Kono K, Babaev VR, Fazio S, Linton MF, Sulsky R, Robl JA, Parker RA, Hotamisligil GS (2007) Treatment of diabetes and atherosclerosis by inhibiting fatty‐acid‐binding protein aP2. Nature 447: 959–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao MQ, Choi YP, Kang S, Youn JH, Cho NH (2010) CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 29: 2672–2680 [DOI] [PubMed] [Google Scholar]
- Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, Holyoake TL (2002) Primitive, quiescent, Philadelphia‐positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro . Blood 99: 319–325 [DOI] [PubMed] [Google Scholar]
- Guo JY, White E (2017) Autophagy, metabolism, and cancer. Cold Spring Harb Symp Quant Biol 81: 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev‐Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, Wodzak M, Klimko C, McMillan E, Butt Y, Ni M, Oliver D, Torrealba J, Malloy CR, Kernstine K, Lenkinski RE et al (2016) Metabolic heterogeneity in human lung tumors. Cell 164: 681–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang‐Minh LB, Deleyrolle LP, Siebzehnrubl D, Ugartemendia G, Futch H, Griffith B, Breunig JJ, De Leon G, Mitchell DA, Semple‐Rowland S, Reynolds BA, Sarkisian MR (2016) Disruption of KIF3A in patient‐derived glioblastoma cells: effects on ciliogenesis, hedgehog sensitivity, and tumorigenesis. Oncotarget 7: 7029–7043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenboom BW, Suda K, Engel A, Fotiadis D (2007) The supramolecular assemblies of voltage‐dependent anion channels in the native membrane. J Mol Biol 370: 246–255 [DOI] [PubMed] [Google Scholar]
- Jegga AG, Schneider L, Ouyang X, Zhang J (2011) Systems biology of the autophagy‐lysosomal pathway. Autophagy 7: 477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Rebecchi MJ, Ralph BP, Teng YH, Berger WT, Galbavy W, Elmes MW, Glaser ST, Wang L, Rizzo RC, Deutsch DG, Ojima I (2014) Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS One 9: e94200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JP, Gerster M, Andersen PK, Tarima S, Perme MP (2008) SAS and R functions to compute pseudo‐values for censored data regression. Comput Methods Programs Biomed 89: 289–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 11: 325–337 [DOI] [PubMed] [Google Scholar]
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O'Dwyer KM, Liesveld JL, Brookes PS, Becker MW, Jordan CT (2013) BCL‐2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 12: 329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan XY, Jorg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, Guilhamon P, Lee L, Kushida MM, Pellacani D, Park NI, Coutinho FJ, Whetstone H, Selvadurai HJ, Che C, Luu B, Carles A, Moksa M, Rastegar N, Head R et al (2017) Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 549: 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landriscina M, Maddalena F, Laudiero G, Esposito F (2009) Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid Redox Signal 11: 2701–2716 [DOI] [PubMed] [Google Scholar]
- Li L, Candelario KM, Thomas K, Wang R, Wright K, Messier A, Cunningham LA (2014) Hypoxia inducible factor‐1alpha (HIF‐1alpha) is required for neural stem cell maintenance and vascular stability in the adult mouse SVZ. J Neurosci 34: 16713–16719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Condello S, Thomes‐Pepin J, Ma XX, Xia Y, Hurley TD, Matei D, Cheng JX (2017) Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 20: 303–314 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Bollen AW, Aldape KD, Gupta N (2006) Nuclear FABP7 immunoreactivity is preferentially expressed in infiltrative glioma and is associated with poor prognosis in EGFR‐overexpressing glioblastoma. BMC Cancer 6: 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Patel S, Affleck VS, Wilson I, Turnbull DM, Joshi AR, Maxwell R, Stoll EA (2017) Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol 19: 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, Cai L, Good L, Tu BP, Hatanpaa KJ, Mickey BE, Mates JM, Pascual JM, Maher EA, Malloy CR, Deberardinis RJ et al (2012) Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo . Cell Metab 15: 827–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martuscello RT, Vedam‐Mai V, McCarthy DJ, Schmoll ME, Jundi MA, Louviere CD, Griffith BG, Skinner CL, Suslov O, Deleyrolle LP, Reynolds BA (2016) A supplemented high‐fat low‐carbohydrate diet for the treatment of glioblastoma. Clin Cancer Res 22: 2482–2495 [DOI] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM (2014) Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159: 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh P, Nelder JA (1989) Generalized linear models. London; New York, NY: Chapman and Hall; [Google Scholar]
- Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX (2011) Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther 339: 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore N, Houghton J, Lyle S (2012) Slow‐cycling therapy‐resistant cancer cells. Stem Cells Dev 21: 1822–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihiro Y, Yasumoto Y, Vaidyan LK, Sadahiro H, Uchida T, Inamura A, Sharifi K, Ideguchi M, Nomura S, Tokuda N, Kashiwabara S, Ishii A, Ikeda E, Owada Y, Suzuki M (2013) Fatty acid binding protein 7 as a marker of glioma stem cells. Pathol Int 63: 546–553 [DOI] [PubMed] [Google Scholar]
- Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, Sorrentino G, Chianese G, Taglialatela‐Scafati O, Trisciuoglio D, Del Bufalo D, Di Martile M, Di Napoli A, Ruco L, Costantini S, Jakopin Z, Budillon A, Melino G, Del Sal G, Ciliberto G et al (2017) Stearoyl‐CoA‐desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ (vol 36, pg 4573, 2017). Oncogene 36: 4671–4672 [DOI] [PubMed] [Google Scholar]
- Oliva CR, Moellering DR, Gillespie GY, Griguer CE (2011) Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS One 6: e24665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimori N, Oristian D, Fuchs E (2015) TGF‐beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160: 963–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M, Huang L, Ratliff M, Karimian Jazi K, Kurz FT, Schmenger T, Lemke D, Gommel M, Pauli M, Liao Y, Haring P, et al (2015) Brain tumour cells interconnect to a functional and resistant network. Nature 528: 93–98 [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt‐Rosen O, Suva ML, Regev A, Bernstein BE (2014) Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344: 1396–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, Di Fiore PP (2010) Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell 140: 62–73 [DOI] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, Settleman J, Stephanopoulos G, Dyson NJ, Zoncu R, Ramaswamy S, Haas W, Bardeesy N (2015) Transcriptional control of autophagy‐lysosome function drives pancreatic cancer metabolism. Nature 524: 361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi S, Song Y, Peng Y, Wang H, Long H, Yu X, Li Z, Fang L, Wu A, Luo W, Zhen Y, Zhou Y, Chen Y, Mai C, Liu Z, Fang W (2012) ZEB2 mediates multiple pathways regulating cell proliferation, migration, invasion, and apoptosis in glioma. PLoS One 7: e38842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM (2004) ONCOMINE: a cancer microarray database and integrated data‐mining platform. Neoplasia 6: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Fukunaga‐Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M (2010) A temporarily distinct subpopulation of slow‐cycling melanoma cells is required for continuous tumor growth. Cell 141: 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga‐Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow‐cycling JARID1B(high) cells. Cancer Cell 23: 811–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar‐Ramiro A, Ramirez‐Ortega D, Perez de la Cruz V, Hernandez‐Pedro NY, Gonzalez‐Esquivel DF, Sotelo J, Pineda B (2016) Role of redox status in development of glioblastoma. Front Immunol 7: 156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian MR, Siebzehnrubl D, Hoang‐Minh L, Deleyrolle L, Silver DJ, Siebzehnrubl FA, Guadiana SM, Srivinasan G, Semple‐Rowland S, Harrison JK, Steindler DA, Reynolds BA (2014) Detection of primary cilia in human glioblastoma. J Neurooncol 117: 15–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF (2009) Downregulation of miRNA‐200c links breast cancer stem cells with normal stem cells. Cell 138: 592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshan‐Barmatz V, Gincel D (2003) The voltage‐dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem Biophys 39: 279–292 [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Vedam‐Mai V, Azari H, Reynolds BA, Deleyrolle LP (2011) Isolation and characterization of adult neural stem cells. Methods Mol Biol 750: 61–77 [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Silver DJ, Tugertimur B, Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT, Kupper MD, Neal D, Nabilsi NH, Kladde MP, Suslov O, Brabletz S, Brabletz T, Reynolds BA, Steindler DA (2013) The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med 5: 1196–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Settleman J (2010) EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 29: 4741–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh DK, Kollipara RK, Vemireddy V, Yang XL, Sun Y, Regmi N, Klingler S, Hatanpaa KJ, Raisanen J, Cho SK, Sirasanagandla S, Nannepaga S, Piccirillo S, Mashimo T, Wang S, Humphries CG, Mickey B, Maher EA, Zheng H, Kim RS et al (2017) Oncogenes activate an autonomous transcriptional regulatory circuit that drives glioblastoma. Cell Rep 18: 961–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll EA, Makin R, Sweet IR, Trevelyan AJ, Miwa S, Horner PJ, Turnbull DM (2015) Neural stem cells in the adult subventricular zone oxidize fatty acids to produce energy and support neurogenic activity. Stem Cells 33: 2306–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352: 987–996 [DOI] [PubMed] [Google Scholar]
- Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA, Taylor LP, Lieberman F, Silvani A, Fink KL, Barnett GH, Zhu JJ, Henson JW, Engelhard HH, Chen TC, Tran DD, Sroubek J, Tran ND, Hottinger AF, Landolfi J et al (2015) Maintenance therapy with tumor‐treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA 314: 2535–2543 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta‐Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C (2015) STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirinato L, Liberale C, Di Franco S, Candeloro P, Benfante A, La Rocca R, Potze L, Marotta R, Ruffilli R, Rajamanickam VP, Malerba M, De Angelis F, Falqui A, Carbone E, Todaro M, Medema JP, Stassi G, Di Fabrizio E (2015) Lipid droplets: a new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 33: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH II, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi‐Sichani M, Dutton‐Regester K, Lin JR, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CG, Kazer SW et al (2016a) Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science 352: 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, Neftel C, Desai N, Nyman J, Izar B, Luo CC, Francis JM, Patel AA, Onozato ML, Riggi N, Livak KJ et al (2016b) Single‐cell RNA‐seq supports a developmental hierarchy in human oligodendroglioma. Nature 539: 309–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, Kushida M, Head R, Morrissy S, Zhu XM, Aviv T, Voisin V, Clarke ID, Li YS, Mungall AJ, Moore RA, Ma Y, Jones SJM, Marra MA, Malkin D et al (2014) Quiescent Sox(2+) cells drive hierarchical growth and relapse in Sonic Hedgehog Subgroup medulloblastoma. Cancer Cell 26: 33–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez AP, Graef M (2016) Autophagy regulation depends on ER homeostasis controlled by lipid droplets. Autophagy 12: 1409–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, Gillespie SM, Dionne D, Luo CC, Ravichandran H, Mylvaganam R, Mount C, Onozato ML, Nahed BV, Wakimoto H, Curry WT et al (2017) Decoupling genetics, lineages, and microenvironment in IDH‐mutant gliomas by single‐cell RNA‐seq. Science 355: eaai8478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost‐Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y et al (2014) Oncogene ablation‐resistant pancreatic cancer cells depend on mitochondrial function. Nature 514: 628–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian C, Reue K, Christofk H, Mischel PS, Pajonk F (2011) Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci USA 108: 16062–16067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg OH (1926) Ueber den stoffwechsel der tumoren: arbeiten aus dem Kaiser Wilhelm‐institut für biologie, Berlin‐Dahlem. JAMA 87: 1671 [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T (2009) The EMT‐activator ZEB1 promotes tumorigenicity by repressing stemness‐inhibiting microRNAs. Nat Cell Biol 11: 1487–1495 [DOI] [PubMed] [Google Scholar]
- White E, Mehnert JM, Chan CS (2015) Autophagy, metabolism, and cancer. Clin Cancer Res 21: 5037–5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DA (2014) Is reliance on mitochondrial respiration a “chink in the armor” of therapy‐resistant cancer? Cancer Cell 26: 788–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY (2008) Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell 2: 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Xu Z, Dai S, Qian L, Sun L, Gong Z (2016) Targeting autophagy to sensitive glioma to temozolomide treatment. J Exp Clin Cancer Res 35: 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuner A, Francescangeli F, Contavalli P, Zapparelli G, Apuzzo T, Eramo A, Baiocchi M, De Angelis ML, Biffoni M, Sette G, Todaro M, Stassi G, De Maria R (2014) Elimination of quiescent/slow‐proliferating cancer stem cells by Bcl‐XL inhibition in non‐small cell lung cancer. Cell Death Differ 21: 1877–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WB, Wang Z, Shu F, Jin YH, Liu HY, Wang QJ, Yang Y (2010) Activation of AMP‐activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem 285: 40461–40471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Guo P, Kruh GD, Vicini P, Wang X, Gallo JM (2007) Predicting human tumor drug concentrations from a preclinical pharmacokinetic model of temozolomide brain disposition. Clin Cancer Res 13: 4271–4279 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Table EV8
Table EV9
Table EV10
Table EV11
Review Process File
Data Availability Statement
The RNA sequencing (EGAS00001003251) and metabolomics (Metabolomics Workbench: ST000248) datasets have been deposited.