

Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-dependent transcription factor, which is activated by a large group of environmental pollutants including polycyclic aromatic hydrocarbons, dioxins and planar polychlorinated biphenyls. Ligand binding leads to dimerization of the AhR with aryl hydrocarbon receptor nuclear translocator and transcriptional activation of several xenobiotic phase I and phase II metabolizing enzymes, such as cytochrome P4501A1 and glutathione- S -transferase, respectively. Since phase I enzymes convert inert carcinogens to active genotoxins, the AhR plays a key role in tumor initiation. Besides this classical route, the AhR mediates tumor promotion and recent evidence suggests that the AhR also plays a role in tumor progression. To date, no mechanistic link could be established between the canonical pathway involving xenobiotic metabolism and AhR-dependent tumor promotion and progression. A hallmark of tumor promotion is unbalanced proliferation, whereas tumor progression is characterized by dedifferentiation, increased motility and metastasis of tumor cells. Tumor progression and presumably also tumor promotion are triggered by loss of cell–cell contact. Cell–cell contact is known to be a critical regulator of proliferation, differentiation and cell motility in vitro and in vivo . Increasing evidence suggests that activation of the AhR may lead to deregulation of cell–cell contact, thereby inducing unbalanced proliferation, dedifferentiation and enhanced motility. In line with this is the finding of increased AhR expression and malignancy in some animal and human cancers. Here, we summarize our current knowledge on non-canonical AhR-driven pathways being involved in deregulation of cell–cell contact and discuss the data with respect to tumor initiation, promotion and progression.
Introduction
The aryl hydrocarbon receptor (AhR) is a transcription factor belonging to the basic helix-loop-helix/Per/ARNT/Sim (PAS) family ( 1–3 ). Among this group of proteins, the AhR is the only one that is activated by a ligand. It was originally discovered due to its stimulation by a variety of planar aromatic hydrocarbons with benzo[ a ]pyrene (B[ a ]P) as prototype ( 4 ). To date, >400 exogenous ligands have been identified. In addition to polycyclic aromatic hydrocarbons (PAHs), the AhR is activated by dioxins including dibenzofurans and planar polychlorinated biphenyls (PCBs) ( 5 ). One of the most potent ligands known so far is 2,3,7,8-tetrachlorodibenzo- p -dioxin (TCDD). It is generally accepted that the toxic responses of these environmental pollutants are the direct consequence of AhR activation. Interestingly, also naturally occurring compounds, such as indoles and several flavonoids, which are present in food may act as AhR agonists. In search for potential endogenous AhR ligands, diverse compounds such as tryptophan derivatives, arachidonic acid metabolites, equilenin, heme metabolites and indigoids have been characterized ( 6 ). Furthermore, the AhR is activated by UV photoproducts of tryptophan and is regulated by non-ligand signals such as cyclic adenosine monophosphate ( 7 , 8 ). However, the physiological or toxicological consequences of AhR activation by these ligands are mostly unclear.
Biochemical and genetic studies using prototypic AhR agonists, such as B[ a ]P and TCDD, have led to unravel important AhR-dependent pathways and to understand at least some of the toxic effects of these environmental pollutants. A common response after ligand binding to the AhR is induction of gene expression. In the cytosol, the unliganded receptor forms a complex with two heat shock protein 90 (Hsp90) molecules, at least one immunophilin homologous protein and co-chaperones ( Figure 1 ) ( 9–11 ). Binding of the ligand results in nuclear translocation of the AhR, dissociation from the chaperone proteins, heterodimerization with ARNT and subsequent binding of the AhR–ARNT heterodimer to dioxin-responsive elements (DREs) with the consensus core recognition sequence 5′-TNGCGTG-3′, also known as xenobiotic-responsive elements (XREs). This leads to transactivation of several genes encoding phase I and II xenobiotic metabolizing enzymes, such as cytochrome P450s (CYP1A1, CYP1A2 and CYP1B1) and glutathione- S -transferase, NAD(P)H: quinone oxidoreductase 1 and aldehyde dehydrogenase 3, respectively ( Figure 1 ) ( 3 , 12 ). Activation of the AhR pathway by PAHs therefore leads to their detoxication and excretion and, at the same time, to their metabolic activation to genotoxic compounds. Studies in CYP1A1 and CYP1B1 knockout mice indicate that CYP1A1 is predominantly important for detoxication, whereas CYP1B1 is required for metabolic activation of B[ a ]P after oral administration ( 13 , 14 ). B[ a ]P is metabolically activated to the ultimate genotoxic B[ a ]P-7,8-diol-9,10-epoxide and finally binds to DNA forming N 2 -B[ a ]P-7,8-diol-9,10-epoxide-guanine adducts. Since carcinogenicity of PAHs is lost in AhR knockout mice ( 15 , 16 ), it is generally accepted that this ‘canonical’ AhR-dependent pathway is required for tumor initiation by PAHs in animals and very likely in humans as well.
Fig. 1.

Canonical and non-canonical signaling pathways of the AhR. ( A ) Schematic representation of canonical AhR signaling pathway. The cytosolic AhR is complexed by two molecules of Hsp90, XAP2 and the co-chaperone p23. Binding of a ligand, e.g. TCDD, leads to a conformational change, thereby allowing nuclear translocation of the AhR complex. In the nucleus, the AhR dissociates from the complex and dimerizes with ARNT. The AhR–ARNT heterodimer then binds to xenobiotic-responsive elements (XREs) in the promoters of genes encoding for several phase I and phase II metabolizing enzymes but also several other genes, e.g. CYP2S1, COX2 or Slug ( 139 ). Recruitment of additional co-factors and factors of the basal transcription machinery (not shown) finally allows transcription of these genes. GSTM, glutathione- S -transferase M; NQO1, NAD(P)H:quinone oxidoreductase 1; UGT1A, uridine 5′-diphosphate-glucuronosyltransferase 1A; ALDH, aldehyde dehydrogenase ( B – D ) Schematic representation of examples of non-canonical AhR signaling. (B) In pRB-proficient cell lines, activation of the AhR by exogenous ligands may lead to direct interaction with pRB and via several mechanisms to inhibition of the transcription factor E2F ( 99 ). As a consequence, progression from G 1 - to S-phase is blocked. Cell cycle arrest may also be induced by additional mechanisms, such as induction of p27 ( 36 , 140 ). (C) Reciprocal inhibitory effects between the nuclear factor kappa B (NF-κB) and the AhR pathway have been described. One consequence of inhibition of AhR signaling by NF-κB is attenuation of ligand-induced CYP1A1 expression. However, also cooperative effects of the AhR and NF-κB pathways are known ( 37 ). (D) The ligand-activated AhR directly associates with estrogen or androgen receptors (ERα or AR) and modulates their function both positively and negatively. Recently, it was shown that the AhR promotes the proteolysis of ERα/AR through assembling a ubiquitin ligase complex, CUL4B(AhR) ( 38 ).
In contrast to PAHs, TCDD is metabolically inert and hence does not lead to genotoxic metabolites. However, TCDD is known to be one of the most potent tumor promoters in the liver ever studied in animal models ( 17 ). Although still not finally proven in AhR knockout models, it is believed that the tumor-promoting effect of TCDD (and related compounds) is mediated by the AhR. For instance, strains from rats or mice expressing low-affinity AhR show decreased sensitivity in classical tumor initiation/promotion studies ( 18 , 19 ). A role of the AhR in tumor promotion is also suggested by studies in mice expressing a constitutively active AhR ( 20 ). However, the canonical AhR pathway evolving xenobiotic metabolism failed to explain the tumor-promoting effects of TCDD, and no mechanistic link between CYP induction and TCDD toxicity could be established so far ( 21 ). Moreover, in vivo studies in two genetically different rat strains indicate that AhR-driven CYP1A1 induction and tumor promotion can be uncoupled from each other ( 22 ). Although it has been shown that TCDD induces suppression of apoptosis in vitro upon treatment with UV-C ( 23 ) and during tumor promotion in rats ( 24 ), the molecular mechanism of AhR-dependent tumor promotion is unknown to date ( 25 ). However, the fact that mice expressing a constitutively active AhR show an increase in the development of stomach tumors ( 26 ) implies that activation of the AhR leads to deregulation of cell cycle control in vivo . The role of the tumor-promoting effect of the AhR in humans is still unclear. Although epidemiological data indicate that TCDD is a human carcinogen, which causes hematopoietic, lymphatic and possibly breast cancer ( 27 ) and is, therefore, classified as Group 1 carcinogen by the International Agency for Research on Cancer ( 28 ), it does not significantly enhance liver tumor formation in humans. However, this observation does not rule out a role of the AhR in tumor promotion in other organs such as lung, which could occur in response to mixtures of PAHs including weak or non-genotoxic AhR ligands.
Beside its function in tumor initiation and tumor promotion, recent studies suggest that the AhR also plays a role in tumor progression, i.e. in the transition from a benign to a malignant tumor, in animal models and very likely also in humans. For instance, AhR expression is higher in invasive than in non-invasive tumor cells and tissue ( 29 , 30 ), and the level of expression even correlates with malignancy in lung tumors ( 31 ). Down-regulating the AhR function in lung adenocarcinoma cells diminishes anchorage-independent growth in vitro ( 31 ). Up-regulation of nuclear AhR expression in human urothelial tumors is associated with increased invasion and poor prognosis ( 32 ). Importantly, fibroblasts derived from AhR knockout mice show decreased tumorigenicity and migration in a xenograft model due to down-regulation of the proto-oncogene Vav3 leading to a decrease in Rac1 activity ( 33 , 34 ). In support of this, 7,12-dimethylbenz[ a ]anthracene-induced breast tumors in rats are more invasive when the animals are co-exposed to a dioxin-like PCB ( 35 ). In summary, there is strong evidence for a pivotal role of the AhR not only in PAH-dependent tumor initiation but also in tumor promotion and progression. The current data indicate that the AhR induces transcription of genes beyond metabolism. Thus, in addition to the canonical signaling cascade shown in Figure 1A , several non-canonical mechanisms of the AhR have been described recently ( Figure 1B–D ). For instance, activation of the AhR leads to cell cycle arrest in G 1 -phase in several cell lines, which is at least partly due to a direct association of the AhR with the hypophosphorylated retinoblastoma protein (pRB), thereby inhibiting progression into S-phase ( 36 ). It was also found that the transcription factor nuclear factor kappa B modulates AhR signaling. For example, direct interaction of the AhR with nuclear factor kappa B leads to down-regulation of AhR-dependent induction of CYP1A1 in vitro and in vivo ( 37 ). In addition to its well-known function as a transcription factor, the AhR has been shown to possess E3 ubiquitin ligase activity, e.g. leading to degradation of the estrogen receptor α ( 38 ).
Cell–cell contact is known to be a critical regulator of cellular proliferation, differentiation and motility. Inhibition of proliferation by cell–cell contact is generally referred to as contact inhibition or contact-dependent inhibition of growth ( 39 ). In adult tissues, contact inhibition is thought to be continuously active, playing a critical role in the repression of somatic cell proliferation. Vice versa, release from contact inhibition in vivo and in vitro is associated with abnormal cellular proliferation ( 40 ). Since tumor promotion is characterized by unbalanced proliferation either due to increased proliferation or decreased level of apoptosis, it is very likely that loss of contact inhibition is a possible event during tumor promotion. Moreover, loss of proper cell–cell adhesion is the sine qua non for tumor progression, which rests on dedifferentiation, migration and invasion of tumor cells.
Recent work from our laboratory and others has shown that the AhR triggers pathways leading to a release from contact inhibition as well as loss of cell–cell adhesion. In this review, we summarize the current knowledge about the action of the AhR on cell–cell contact, thereby inducing loss of contact inhibition, dedifferentiation and migration. (For a recent review on the role of the AhR in cell substratum adhesion, integrins and matrix metabolism, see 41.) In the first section, we will focus on signal transduction pathways of contact inhibition in fibroblasts and in epithelial cells as far it is relevant for understanding the crosstalk between cell–cell contact and the AhR. We will then describe the action of the AhR on contact inhibition. Finally, data will be presented on the involvement of the AhR in disturbing cell–cell adhesion leading to dedifferentiation and migration. The data will be discussed as to tumor promotion and progression.
Contact inhibition and cell–cell adhesion
Contact inhibition
In vitro , contact inhibition becomes apparent by the fact that adherent, non-transformed cells are arrested in G 1 -phase at a critical cell density forming a confluent monolayer. In contrast, transformed cells are characterized by loss of contact inhibition manifested by a higher saturation density and the emergence of multi-layered foci. Despite its importance for cell cycle control, knowledge about the molecular mechanisms mediating contact inhibition and its deregulation during tumorigenesis is still scarce ( 42–46 ). It is important to note that the growth-inhibitory signal of contact inhibition finally leads to a cell cycle arrest in G 0 /G 1 -phase. To date, it is still not fully clear how this inhibitory signal is transduced from the cell membrane to the nucleus and how it is integrated into the cell cycle machinery. In order to understand the impact of contact inhibition on cell cycle regulatory proteins, we shortly describe fundamental issues of the eukaryotic cell cycle (for detailed description of cell cycle regulation, see 47–49 ).
The eukaryotic cell cycle
Cyclin-dependent kinases (Cdks) are known to be master kinases in cell cycle regulation. The activity of Cdks, which belong to the family of serine–threonine kinases, is strictly dependent on association with cyclins. Cyclin protein levels oscillate during the cell cycle in a defined manner, thus—according to the classical view—ensuring proper activation of specific Cdks at the correct time. In G 1 -phase, cyclin D/Cdk4 and downstream cyclin E/Cdk2 phosphorylate members of the pRB family, thus allowing G 1 /S-phase transition. Rb proteins bind to and modulate the activity of transcription factors, such as E2F family members, histone deacetylases and chromatin remodeling complexes, thereby repressing transcription of S-phase-specific genes, such as cyclin A. When pRB becomes phosphorylated by cyclin D/Cdk4 and cyclin A/Cdk2, it dissociates from E2F, thus allowing E2F to function as a transcriptional activator. In early S-phase, Cdk2 then binds to cyclin A, changing substrate specificity of Cdk2. It is generally believed that the critical function of the cyclin A–Cdk2 complex is phosphorylation of substrates that start DNA replication and coordinating the end of S-phase, such as DNA polymerase α, proliferating cell nuclear antigen, replication protein A or cell division cycle 6. S/G 2 - and G 2 /M transitions are regulated by Cdk1, which is sequentially activated by cyclin A and then cyclin B. The activity of Cdks is further modulated by activating and/or inhibiting phosphorylations and dephosphorylations. In addition, the activity of Cdks is regulated by association of small inhibitory proteins, known as p15, p16, 18, p19 (INK family) and p21, p27 and p57 (CIP/KIP family).
Cell cycle and contact inhibition
What happens during contact inhibition? It is now clear that the cell ensures cell cycle arrest by cooperation of several mechanisms ( Figure 2 ). The first evidence that cell cycle regulatory proteins, such as cyclin-dependent kinases, are involved in contact inhibition came from Massagué’s group in 1994. They showed in (Mv1Lu mink) epithelial cells that the protein level of the CIP/KIP inhibitor p27 increases at confluence, which inhibits Cdk2 activity ( 50 ). We and others then showed that p27 is also up-regulated in confluent human fibroblasts ( 51 ), in various epithelial cells and many other cell types ( 52–57 and our unpublished data). Up-regulation of p27 protein levels in contact-inhibited cells is at least partly dependent on sustained activation of p38α mitogen-activated protein kinase (MAPK), which leads to inhibition of the epidermal growth factor receptor signaling ( 58 , 59 ). Although there is no doubt that p27 is a crucial regulator of cell cycle control, it is at least in fibroblasts dispensable for contact inhibition ( 60 ). This indicates further inhibitory mechanisms to be involved such as p16 that was identified as a critical mediator of contact inhibition ( 61 ). As a consequence of inhibition of Cdk4 and Cdk2, pRB remains in its hypophosphorylated state, thus inhibiting progression into S-phase ( 50 , 51 , 61 ). We further provided evidence that upstream of p16, the potential tumor suppressor protein kinase Cδ, is involved in contact inhibition ( 62 ). Although it is very likely that reorganization of the actin cytoskeleton is one major target of protein kinase Cδ, its precise role in contact inhibition and linkage to the cell cycle machinery still has to be resolved.
Fig. 2.

Signaling cascade of contact inhibition in fibroblasts. Cell–cell contacts lead to a rapid activation of protein kinase Cδ (PKCδ) and a persistent activation of p38α MAPK resulting in accumulation of the KIP inhibitor p27, hence inhibiting cyclin E/Cdk2 activity. Additionally, the INK inhibitor p16 is up-regulated, thereby blocking activity of Cdk4. As a result, pRB remains in its hypophosphorylated state and does not allow transcription of S-phase-specific genes, such as cyclin A. If PKCδ plays a role in activation of p38α MAPK or p38α MAPK is involved in p16 up-regulation remains to be elucidated.
Cell membrane proteins
Which cell membrane proteins are responsible for signaling growth inhibition in fibroblasts? Several candidates have been identified, such as contactinhibin and the contactinhibin receptor ( 63 , 64 ), N-cadherin ( 65 ), N-CAM ( 66 ) and others ( 42 ). None of them plays a unique role in contact inhibition and their expression and function entirely depend on the cell type studied.
Cell–cell adhesion in epithelial cells
In epithelial cells, cell–cell adhesion is mediated by Ca 2+ -dependent homophilic interactions of E-cadherin, which does not only induce contact inhibition, i.e. inhibit proliferation, but also maintain the epithelial phenotype and prevent migration ( 67–69 ). Intracellularly, E-cadherin is linked to the actin cytoskeleton by association with α-, β- and/or γ-catenin (plakoglobin). It is generally accepted that one function of E-cadherin is to sequester β-catenin, thereby decreasing the amount of free cytoplasmic β-catenin ( 70 , 71 ). In addition, the cytoplasmic pool of β-catenin is controlled to remain low by proteolytic degradation via the proteasome system ( 72 ). Accumulation of cytoplasmic β-catenin, which occurs physiologically during embryonic development and pathologically during tumorigenesis, leads to increased binding of β-catenin to transcription factors of the TCF/LEF family, thereby inducing transcription of not only proliferative but also mesenchymal genes such as cyclin D1, c-Myc, c-Jun, Id2, Slug or fibronectin ( 73 , 74 ). Although additional roles of β-catenin in mediating contact inhibition have been identified recently ( 75 ), the growth-inhibitory effect of E-cadherin is at least partly mediated by sequestering β-catenin to the plasma membrane, hence preventing transcriptional activation of proliferative genes (76 , 77 and own unpublished observations). By the same mechanism, E-cadherin prevents transcriptional activation of mesenchymal genes, thereby maintaining the epithelial phenotype.
The role of γ-catenin, a close homologue of β-catenin, seems to be different to that of β-catenin. Obviously, its function in cell adhesion is much more important than in intracellular signaling and transcriptional activity ( 78 ). Whereas β-catenin is classified as a proto-oncogene, γ-catenin is considered to be a tumor suppressor. In contrast to β-catenin, γ-catenin is not only associated with E-cadherin but also to desmosomal cadherins ( 79 ). Desmosomes are intercellular junctions probably involved in contact inhibition, epithelial differentiation and inhibition of migration similar to the function of and in cooperation with E-cadherin ( 80 ).
Epithelial–mesenchymal transition
Disruption of cell–cell adhesion (both E-cadherin-mediated cell adhesion and desmosomes) does not only permit the cells to undergo uncontrolled proliferation but also to dedifferentiate to a mesenchymal phenotype and to migrate, a process which is referred to as epithelial–mesenchymal transition (EMT). EMT is not only a physiological process during embryonic development but also a pathological process whereby primary in situ tumors progress toward an invasive or metastatic phenotype. A pivotal initial event in EMT is loss of expression of E-cadherin and γ-catenin. As a consequence, cells lose epithelial markers (i.e. cytokeratine 18), acquire mesenchymal markers such as vimentin, fibronectin and N-cadherin and express proteases that promote cell migration and invasion. Critical mediators of EMT are the zinc finger transcription factors Snail and Slug, which repress transcription of E-cadherin as outlined above ( Figure 3 ) ( 81 ).
Fig. 3.

Schematic illustration of EMT. In epithelial cells, cell–cell adhesion is mediated by homophilic interactions of E-cadherin. One consequence is sequestration of β-catenin, thereby preventing its nuclear translocation. Over-expression of master regulators of EMT, such as Snail and Slug (and others) lead to down-regulation of E-cadherin, hence allowing nuclear translocation of β-catenin. In association with transcription factors (TF) of the TCF/LEF family, β-catenin induces transcription of proliferative and mesenchymal genes (see text for detail). This process is referred to as EMT.
Cell–cell contact as regulator of the AhR
Early work revealed that the AhR is transiently activated by loss of cell substratum adhesion ( 82 , 83 ). Cho et al. then demonstrated that the AhR is regulated by cell density, which rests on the observation that the AhR is activated by loss of cell–cell contact in the absence of exogenous ligands ( 84 ). Thus, in dense cultures of murine C3H10T1/2 fibroblasts, neither nuclear localization of the AhR nor the expression of the AhR target gene CYP1B1 is detectable. However, loss of cell–cell contact either by suspension or seeding the cells at low density provokes nuclear translocation of the AhR with concomitant expression of CYP1B1. In contrast to sustained activation of CYP1B1 by TCDD, transcription of CYP1B1 due to loss of cell–cell contact appears to be transient. Similar findings were obtained with the human keratinocyte cell line HaCaT. Localization of the AhR was found to be predominantly nuclear at sparse cell density and cytoplasmic at confluence ( 85 ). Similar data were found upon down-regulating E-cadherin by calcium depletion and in a wound-healing assay. Accordingly, expression of the AhR target gene CYP1A1 is only detectable at low cell density. Furthermore, nuclear export of the AhR is inhibited in sparse cultures by phosphorylation of Ser-68 in the nuclear export signal of the AhR, which is mediated by p38 MAPK ( 85 ). In accordance with these in vitro findings, the authors demonstrate phosphorylation of the AhR at Ser-68 and nuclear staining of the AhR in the liver of 3-methylcholanthrene-treated mice. Hence, cell–cell contact by E-cadherin provokes a yet unknown signal leading to dephosphorylation of the AhR. It is therefore likely that subcellular distribution of the AhR is regulated by a fine-tuned balance of phosphatases and phosphatase inhibitors in the nucleus, which is dependent on cell density. In contrast to the in vitro data described above, expression of the AhR- and TCDD-dependent CYP1A1 induction increases with differentiation of cells in the human epidermis ( 86 ). The AhR appears to be localized in the nucleus in proliferating and differentiating murine keratinocytes independent on prior exposure to TCDD ( 87 ). Accordingly, exposure of rodent skin to PAHs results in preferential induction of CYP1A1 in differentiating keratinocytes ( 88 ).
The AhR is a deregulator of contact inhibition
The first evidence that ligands of the AhR induce proliferation in confluent cell cultures was provided by Milstone and LaVigne more than two decades ago ( 89 ). When confluent cultures of newborn human foreskin keratinocytes were treated with nanomolar concentrations of TCDD, cell proliferation was significantly stimulated. The group of Birnbaum then demonstrated that TCDD is able to induce a release from contact inhibition in two human squamous carcinoma cell lines ( 90 ). Whereas treatment of exponentially growing cultures with TCDD had no effect on proliferation, treatment of pre-confluent cultures induced a 2-fold increase in saturation density. Later on, similar effects of TCDD on contact inhibition have been observed in the rat liver oval cell line WB-F344 ( 57 , 91 ), Madin-Darby canine kidney and Madin-Darby bovine kidney cells ( 92 : supplementary data, 93 ). Interestingly, exposure of WB-F344 cells to other AhR ligands such as PAHs, planar PCBs and some flavonoids also induces a release from contact inhibition, which supports that release from contact inhibition is mediated by the AhR ( 94–97 ). Although TCDD-dependent release from contact inhibition was observed in several cell lines and appears to be a common phenomenon, this effect is cell type specific since it is not observed in 5L hepatoma, Hepa, primary murine hepatocytes or HaCaT cells (92 and own unpublished observations). Understanding these cell type-specific responses to TCDD would be instrumental to uncover the basis for the known cell type and organ specificity of TCDD poisoning ( 98 , 99 ).
WB-F344 cells ( 100 ) are the best characterized oval cells available so far; when transplanted in vivo , they fully differentiate into hepatocytes ( 101 ). Oval cells, which are liver stem cells, function as a regenerative reservoir in acute liver damage ( 102) and may give rise to liver tumors in rodents and humans ( 103–105 ). Very recently, Hailey et al. ( 106 ) published a 2 year exposure study in which rats had been exposed to TCDD or dioxin-like compounds. This study revealed for the first time that oval cells might be targets of TCDD action during liver carcinogenesis. Since transplantation of chemically transformed WB-F344 cells results in the formation of hepatocarcinomas, cholangiocarcinomas and hepatoblastomas ( 107 ), we focused on WB-F344 cells as a relevant stem cell culture model for mechanistic studies on TCDD-dependent loss of contact inhibition.
When confluent WB-F344 cultures are treated with nanomolar concentrations of TCDD (or other weak or non-genotoxic AhR ligands such as benzo[ b ]fluoranthene or PCB126), cells cease from G 1 arrest and enter S-phase resulting in a 2-fold increase in saturation density and the emergence of multi-layered foci, which are characteristic features of transformed cells ( 57 , 91 , 94 , 96 , 108 ). That neither exponentially growing cells nor serum-deprived cultures respond to the growth stimulatory effect of TCDD indicates that TCDD does not exert a mitogenic effect per se but specifically interferes with the signaling cascade of contact inhibition. The fact that TCDD induces a release from G 1 arrest strongly suggests that cell cycle proteins are targets of TCDD. Indeed, exposure of confluent WB-F344 cells to AhR ligands, such as TCDD, leads to an increase in the S-phase-specific cyclin A and in cyclin A/Cdk2 activity ( 57 , 92 , 109 ). Interestingly, no increase in the phosphorylation level of pRB was observed (own unpublished observation), indicating that S-phase entry occurs downstream of pRB. In line, ectopic expression of cyclin A in confluent WB-F344 cultures is sufficient to overcome G 1 arrest. Although AhR activation may lead to induction of the activator protein-1-transcription factor c-Jun in 5L hepatoma cells ( 110 ), we determined that JunD, but not c-Jun, in association with its partner ATF2 is responsible for cyclin A induction. Activator protein-1 designates a family of dimeric transcription factors consisting of homodimeric Jun family members (c-Jun, JunD and JunB) or heterodimers consisting of Jun with one of the Fos family members (c-Fos, FosB, Fra1 and Fra2) or ATF members (ATF2 and ATFa) ( 111 ). Some of them are activated by kinases, such as c-Jun NH 2 -terminal kinase (JNK), p38 MAPK (p38) or extracellular signal-regulated kinase (ERK). Interestingly, no increase in the activity of any of the three MAPKs (ERK, p38 and JNK) could be observed in WB-F344 cells in response to TCDD (112 and own unpublished observations). Functional interference with AhR and ARNT revealed that induction of JunD, transcriptional activation of cyclin A and release from contact inhibition in response to TCDD are absolutely dependent on the AhR but very likely independent of ARNT ( 92 , 109 ). This novel signaling cascade triggered by the AhR strongly differs from the classical AhR–ARNT pathway ( Figure 4 ). The functional significance of this pathway has yet to be determined.
Fig. 4.
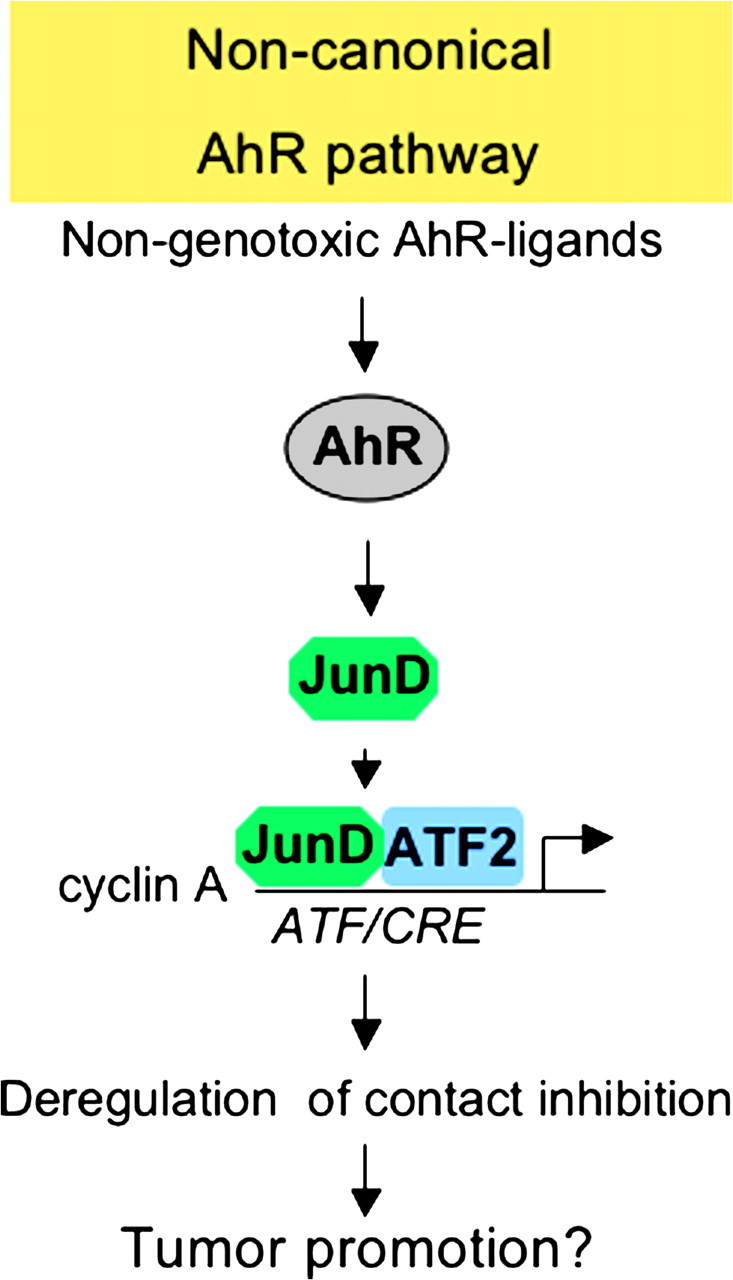
Proposed novel non-canonical pathway of the AhR. According to the proposed novel non-canonical pathway, non-genotoxic ligands, such as TCDD, lead to activation of the AhR, and very likely independent from ARNT, to induction of JunD, which after heterodimerization with ATF2 results in transcriptional activation of cyclin A finally leading to a release from contact inhibition. If genotoxic ligands may also induce this novel pathway is currently not known.
In line with a direct effect of TCDD on cell–cell contact, no secretion of the soluble factor transforming growth factor-β was observed ( 113 ). This implies that additional targets of TCDD might be cell adhesion molecules. Interestingly, γ-catenin is significantly down-regulated in response to TCDD, whereas no decrease in E-cadherin, α- or β-catenin could be detected ( 108 ). Regulation of desmosomal proteins by TCDD has not been analyzed so far. It is not clear if down-regulation of γ-catenin impairs the function of E-cadherin-mediated cell adhesion and/or of desmosomal cell–cell contacts. The precise mechanism of γ-catenin down-regulation also remains to be elucidated. Since (i) inhibition of γ-catenin degradation by a proteasome inhibitor does not reverse γ-catenin down-regulation and (ii) γ-catenin messenger RNA is down-regulated, it is reasonable to conclude that transcription of γ-catenin is blocked by TCDD, hypothetically by promoter methylation as it has been observed for TCDD-dependent silencing of p16 and p53 ( 114 ) or, alternatively, by binding of the AhR to an inhibitory XRE. Such inhibitory XREs have been identified in the promoter region of c-Fos, COX2 and cathepsin D gene ( 115–117 ). It is supposed that the AhR binds to these DNA sequences, thereby attenuating the activity of other transcription factors such as Sp1 ( 118 ). Interestingly, a GC-rich Sp1-binding region is located closely upstream of a putative inhibitory XRE in the human γ-catenin promoter ( 119 ).
The AhR is a deregulator of cell–cell adhesion
Increasing evidence is provided that the AhR can stimulate migration and EMT in several cell lines and in vivo . Thus, exposure of the human breast cancer epithelial cell line MCF-7 to TCDD or 3-methylcholanthrene leads to down-regulation of E-cadherin, loss of cell–cell adhesion and increased mobility of the cells ( 120 ). In contrast to the signaling cascade described in WB-F344 cells, where no increase was observed in the activity of any of the MAPKs in response to TCDD, TCDD leads to a late and persistent activation of JNK in MCF-7 cells. This again indicates cell type and species specificity of AhR function. A gain-of-function analysis revealed that expression of a constitutively active AhR mimicks the effect of TCDD on JNK, i.e. scattering and migration, which supports that the AhR plays a pivotal role in migration of MCF-7 cells. Pharmacological inhibition of JNK blocks the effect of constitutively active AhR and TCDD arguing for JNK as a central mediator of AhR-induced motility. Possible downstream targets of JNK, such as c-Jun, have not been identified in this study. Interestingly, the JNK/c-Jun pathway has recently been shown to induce DNA methyltransferase 1, thereby leading to promoter methylation on CpG islands and decreased transcription of E-cadherin ( 121 ). Whether a similar mechanism is involved in AhR-triggered down-regulation of E-cadherin in MCF-7 cells is not known. In search for an AhR target gene in MCF-7 cells, the authors further identified Nedd9/Hef1/Cas-L as the most consistently induced gene in response to 3-methylcholanthrene. They also showed for the human liver hepatoma cell line HepG2 that Nedd9/Hef1/Cas-L is a target gene of the AhR, containing two xenobiotic-responsive elements in its promoter, which mediates the effects on JNK activation and E-cadherin down-regulation ( 122 ).
Another important AhR target regulating migration might be the nuclear factor of activated T-cells (NFAT) c1/autotaxin signaling pathway ( Figure 5 ). Very recently, Seifert et al. ( 123 ) discovered that TCDD activates the transcription factor NFAT in MCF-7 cells leading to increased migration. Activation of NFAT resulted in enhanced expression of its target gene autotaxin , which is known to exhibit lysophospholipase D activity, thereby generating lysophosphatidic acid (LPA). Although the authors did not investigate the effect of LPA on E-cadherin expression in MCF-7 cells, LPA is known to induce breakdown of E-cadherin-mediated junctions in other cell lines ( 124 ).
Fig. 5.
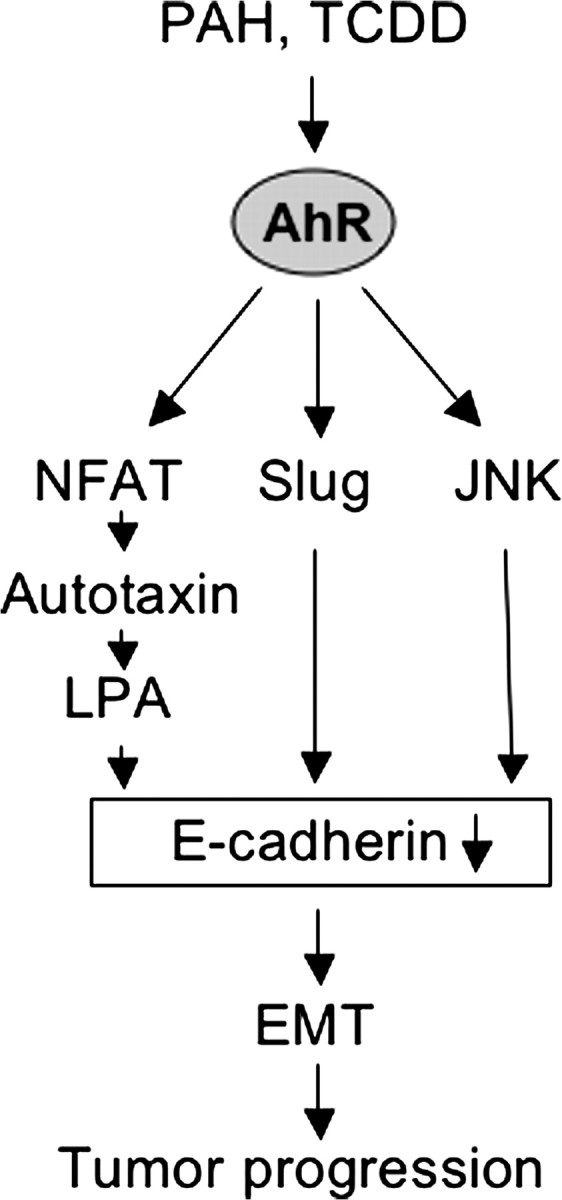
Potential role of the AhR in EMT and tumor progression. Tumor progression is generally characterized by dedifferentiation of a cell from an epithelial to a mesenchymal phenotype and increased motility. A central process is down-regulation of E-cadherin. Transcriptional inhibition of E-cadherin may either be mediated by AhR-dependent activation of the transcriptional repressor Slug and/or activation of JNK. Breakdown of E-cadherin may also be achieved through activation of the NFAT/autotaxin/LPA pathway (see text for detail).
Ikuta and Kawajiri ( 125 ) detected the zinc finger transcription factor Slug as direct target gene of the AhR. Activation of the AhR either by 3-methylcholanthrene or calcium depletion in MCF-7 or HaCaT cells induces transcriptional activation of Slug by binding of the AhR–ARNT complex to an identified XRE in the Slug promoter. Expression of Slug results in down-regulation of the epithelial marker cytokeratine-18 and up-regulation of the mesenchymal marker vimentin. Whether there is a mechanistic link between JNK and Slug, as suggested by observations in lens epithelial cells ( 126 ), remains to be determined.
Further evidence for a pivotal role of the AhR in EMT in mammary tumors comes from studies in mouse mammary tumor virus—c-Rel × CK 2a bitransgenic ( 127 ) mice. Mammary tumors and cell lines derived from these bitransgenic mice show a highly invasive phenotype with loss of E-cadherin and γ-catenin expression but expression of fibronectin and vimentin. Interestingly, the level of AhR expression seems to correlate with malignancy ( 128 ). Human mammary tumors also show aberrant expression of the AhR ( 129 ). Co-expression of c-Rel and CK 2 in non-transformed mammary epithelial cells induces the expression of the AhR and Slug and promotes EMT ( 128 ). The c-Rel subunit belongs to the family of nuclear factor kappa B transcription factors ( 130 ), and CK 2 is a ubiquitously expressed serine–threonine kinase ( 131 ). Similar results were obtained in 7,12-dimethylbenz[ a ]anthracene-treated, c-Rel-transformed mammary epithelial cells. Interestingly, the malignant phenotype can be prevented and reversed in vitro by co-treatment with the polyphenol epigallocatechin-3 gallate (EGCG) as—even more importantly—EGCG prevents the invasive phenotype of rat mammary cancer induced by 7,12-dimethylbenz[ a ]anthracene in vivo ( 128 ). The precise mechanism of this protective effect of EGCG is not known so far. However, EGCG has been shown to block AhR-dependent transcription indirectly by binding to Hsp90 ( 132 ). It was found that EGCG binds at or near to a C-terminal ATP-binding site in Hsp90 leading to stabilization of the AhR–Hsp90–XAP2 complex, which still translocates to the nucleus but is unable to dimerize with ARNT ( 133 ). Thereby, AhR-dependent gene transcription cannot occur ( 133 ).
The findings show that ligand-dependent activation of the AhR may induce EMT, a key process during tumor progression. A central mediator of EMT is down-regulation of the cell adhesion molecule E-cadherin. Down-regulation of E-cadherin allows β-catenin to translocate to the nucleus, thereby, in association with transcription factors of the TCF/LEF family, inducing transcription of proliferative and mesenchymal genes. Transcriptional inhibition of E-cadherin may therefore be the result of AhR-dependent activation of the transcriptional repressor Slug and/or activation of JNK. As outlined above, down-regulation of E-cadherin may also be mediated by activation of the transcription factor NFAT, which in turn induces transcription of autotaxin, an enzyme generating LPA. Taken together, at least three pathways seem to become triggered in response to AhR activation ( Figure 5 ) causing E-cadherin breakdown or lack of its expression, thus stimulating EMT.
The AhR: canonical and non-canonical pathways in concert trigger tumor formation and growth
The critical role of the AhR in tumor initiation rests on the canonical pathway in which activation of the AhR results in the expression of phase I and II enzymes that catalyze detoxication but at the same time ‘activation’ of procarcinogens causing DNA adduct formation ( Figure 1 ). The factors regulating the balance between activation of procarcinogens into DNA-reactive metabolites and their detoxication have not been clearly determined yet. However, it is suggested that CYP1A1 is predominantly involved in detoxication, whereas CYP1B1 is required for metabolic activation ( 13 , 14 ). Irrespective of this, it is clear that metabolic activation mediated by AhR-regulated functions is a conditio sine qua non for tumor initiation ( 15 , 16 ). Since tumor initiation is based on fixation of DNA damage by mutations in critical target genes (oncogenes and tumor suppressor genes) and since mutagenesis by base mispairing and translesion synthesis is bound on DNA replication ( 134 ), it is reasonable to posit that any proliferation stimulus during and immediately after the induction of DNA damage and prior to its repair drives the initiation process. Therefore, AhR-triggered functions that end up in stimulation of DNA replication may support DNA damage fixation and, thus, tumor initiation.
An important process counteracting tumor initiation is DNA repair. Data have been reported indicating that ARNT interacts with breast cancer 1 ( 135 ) and TCDD may stimulate homologous recombination ( 136 , 137 ), which suggests a possible involvement of the AhR in modulation of DNA repair processes. It would be highly important to elucidate in more detail the role of the AhR in the regulation and the enzymology of DNA repair processes together with their impact on cell cycle regulation. We should note that an important aspect in genotoxin-driven processes is cell death that may counteract mutagenesis by elimination of heavily damaged cells. Early work in vivo and in vitro ( 23 , 24 ) and the recent finding that the AhR binds to E2F1 and thus is able to inhibit E2F1-mediated apoptosis ( 138 ) may be taken to indicate the complex network of AhR-driven functions that in concert support tumor formation and promotion.
Given the role of proliferation in DNA damage fixation and tumor promotion, it is tempting to speculate that loss of contact inhibition in the initiation and post-initiated stage may support pre-initiated cells to expand. Although still not shown in vivo , a possible mechanism involved might be AhR-dependent up-regulation of the expression of proto-oncogenes, such as cyclin A by a non-canonical pathway. Breakdown of cell–cell adhesion notably by down-regulation of E-cadherin will lead to EMT with induction of proliferative, mesenchymal and invasive genes, thus stimulating the process of tumor progression, in which at the same time further mutations will be accumulating from fixation of spontaneous DNA damage. AhR agonists affect several key players in EMT, such as JNK, Slug and NFAT, thereby leading to loss of E-cadherin function, which supports a role of the AhR in tumor progression. Based on this hypothetical model ( Figure 6 ), AhR-dependent tumor promoters might be powerful tumorigenic agents because they have the capability to enhance fixation of any DNA damage, expansion of initiated cells, i.e. tumor promotion and finally to drive progression. We should note that in the non-experimental situation, individuals are exposed to mixtures of deleterious compounds such as PAHs that may act both as initiator and promoter, which may at the same time activate both the canonical and the non-canonical functions of the AhR.
Fig. 6.

Hypothetical model of the role of AhR in tumor initiation, promotion and progression. Procarcinogens such as PAHs are known to activate the canonical xenobiotic-responsive element-dependent pathway, thereby leading to their conversion to genotoxic metabolites forming DNA adducts. Mutations are fixed by clonal expansion of initiated cells. Non-genotoxic AhR agonists, such as TCDD, are known to increase cell number, either by inhibition of apoptosis (not included in the figure) ( 24 ) or possibly by enhanced proliferation due to loss of contact inhibition providing a mechanistic basis for their tumor-promoting effects. AhR ligands may further lead to breakdown of E-cadherin function by regulating several key players of EMT, thereby driving the process of tumor progression.
Outlook
Both in vitro and in vivo studies suggest that the AhR deregulates cell–cell contact, thereby inducing release from contact inhibition as well as disruption of cell–cell adhesion, hence leading to EMT. Although some important signaling pathways and key players could be identified so far, it is clear that we are still at the beginning of understanding the role of the AhR in these processes. Therefore, intensive research is still needed to unravel the complex signaling cascade of the AhR. Moreover, in vivo studies using appropriate genetic mouse models are needed to finally prove the functional significance of the findings. The fact that EGCG, a component of green tea and inhibitor of the AhR signaling, was able to prevent EMT and thereby the invasive phenotype of breast cancer in an in vivo model is at least promising and might help to better understand the process of invasion and metastasis. Importantly, a number of dietary AhR ligands are present in food, some of them known as agonists and some of them acting as antagonists. To date, very little is known about their adverse or beneficial properties and their potential role in tumor promotion, progression or prevention. The novel findings of the non-canonical pathways triggered by the AhR provide a solid basis of new hypotheses that may serve as a ground for further elucidating the molecular events driven by AhR agonists. They will hopefully also help to understand better clinical observations showing that the AhR expression correlates with malignancy in some human cancers.
Funding
Deutsche Forschungsgemeinschaft (DI793, KA724).
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- AhR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor nuclear translocator
- B[ a ]P
benzo[ a ]pyrene
- Cdks
cyclin-dependent kinases
- CYP
cytochrome P450
- EGCG
epigallocatechin-3 gallate
- EMT
epithelial–mesenchymal transition
- Hsp90
heat shock protein 90
- JNK
c-Jun NH 2 -terminal kinase
- LPA
lysophosphatidic acid
- MAPK
mitogen-activated protein kinase
- NFAT
nuclear factor of activated T-cells
- PAH
polycyclic aromatic hydrocarbon
- PCB
polychlorinated biphenyl
- pRB
retinoblastoma protein
- TCDD
2,3,7,8-tetrachlorodibenzo- p -dioxin
- XRE
xenobiotic-responsive element
References
- 1.Whitlock JP., Jr. Mechanistic aspects of dioxin action. Chem. Res. Toxicol. 1993;6:754–763. doi: 10.1021/tx00036a003. [DOI] [PubMed] [Google Scholar]
- 2.Hankinson O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 3.Rowlands JC, et al. Aryl hydrocarbon receptor-mediated signal transduction. Crit. Rev. Toxicol. 1997;27:109–134. doi: 10.3109/10408449709021615. [DOI] [PubMed] [Google Scholar]
- 4.Swanson HI, et al. The AH-receptor: genetics, structure and function. Pharmacogenetics. 1993;3:213–230. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Denison MS, et al. Ligand binding and activation of the Ah receptor. Chem. Biol. Interact. 2002;141:2–24. doi: 10.1016/s0009-2797(02)00063-7. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen LP, et al. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fritsche E, et al. Lightening up the UV response by identification of the aryl hydrocarbon receptor as a cytoplasmic target for ultraviolet B radiation. Proc. Natl Acad. Sci. USA. 2007;104:8851–8856. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oesch-Bartlomovicz B, et al. Aryl hydrocarbon receptor activation by cAMP versus dioxin: divergent signaling pathways. Proc. Natl Acad. Sci. USA. 2005;102:9218–9223. doi: 10.1073/pnas.0503488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen HS, et al. Subunit composition of the heterodimeric cytosolic aryl hydrocarbon receptor complex. J. Biol. Chem. 1994;269:27554–27558. [PubMed] [Google Scholar]
- 10.Ma Q, et al. A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 1997;272:8878–8884. [PubMed] [Google Scholar]
- 11.Carver LA, et al. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J. Biol. Chem. 1997;272:11452–11456. doi: 10.1074/jbc.272.17.11452. [DOI] [PubMed] [Google Scholar]
- 12.Nebert DW, et al. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004;279:23847–23850. doi: 10.1074/jbc.R400004200. [DOI] [PubMed] [Google Scholar]
- 13.Uno S, et al. Oral exposure to benzo[a]pyrene in the mouse: detoxication by inducible cytochrome P450 is more important than metabolic activation. Mol. Pharmacol. 2004;65:1225–1237. doi: 10.1124/mol.65.5.1225. [DOI] [PubMed] [Google Scholar]
- 14.Uno S, et al. Oral benzo[a]pyrene in Cyp1 knockout mouse lines: Cyp1A1 important in detoxication, Cyp1B1 metabolism required for immune damage independent of total-body burden and clearance rate. Mol. Pharmacol. 2006;69:1103–1114. doi: 10.1124/mol.105.021501. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu Y, et al. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc. Natl Acad. Sci. USA. 2000;97:779–783. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakatsuru Y, et al. Dibenzo[a,l]pyrene-induced genotoxic and carcinogenic responses are dramatically suppressed in aryl hydrocarbon receptor-deficient mice. Int. J. Cancer. 2004;112:179–183. doi: 10.1002/ijc.20365. [DOI] [PubMed] [Google Scholar]
- 17.Pitot HC, et al. Quantitative evaluation of the promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin of hepatocarcinogenesis from diethyl-nitrosamine. Cancer Res. 1980;40:3616–3620. [PubMed] [Google Scholar]
- 18.Beebe LE, et al. Promotion of N-nitrosodiethylamine-initiated hepatocellular tumors and hepatoblastomas by 2,3,7,8-tetrachloro-dibenzo-p-dioxin or aroclor 1254 in C57BL/6, DBA/2, and B6D2F1 mice. Cancer Res. 1995;55:4875–4880. [PubMed] [Google Scholar]
- 19.Viluksela M, et al. Liver tumor-promoting activity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in TCDD-sensitive and TCDD-resistant rat strains. Cancer Res. 2000;60:6911–6920. [PubMed] [Google Scholar]
- 20.Moennikes O, et al. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004;64:4707–4710. doi: 10.1158/0008-5472.CAN-03-0875. [DOI] [PubMed] [Google Scholar]
- 21.Nukaya M, et al. The role of the dioxin-responsive element cluster between the Cyp1a1 and Cyp1a2 loci in aryl hydrocarbon receptor biology. Proc. Natl Acad. Sci. USA. 2009;106:4923–4928. doi: 10.1073/pnas.0809613106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuomisto J. Does mechanistic understanding help in risk assessment—the example of dioxin. Toxicol. Appl. Pharmacol. 2005;207:S2–S10. doi: 10.1016/j.taap.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 23.Chopra M, et al. Inhibition of UV-C light-induced apoptosis in liver cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2009;111:49–63. doi: 10.1093/toxsci/kfp128. [DOI] [PubMed] [Google Scholar]
- 24.Stinchcombe S, et al. Inhibition of apoptosis during 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated tumor promotion in rat liver. Carcinogenesis. 1995;16:1271–1275. doi: 10.1093/carcin/16.6.1271. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz M, et al. Carcinogenic risks of dioxin: mechanistic considerations. Regul. Pharmacol. Toxicol. 2005;43:19–34. doi: 10.1016/j.yrtph.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 26.Andersson P, et al. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc. Natl Acad. Sci. USA. 2002;99:9990–9995. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pesatori AC, et al. Cancer incidence in the population exposed to dioxin after the “Seveso accident”: twenty years of follow-up. Environ. Health. 2009;8:39. doi: 10.1186/1476-069X-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.IARC. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 69. Lyon: International Agency for Research on Cancer; 1997. IARC working group on the evaluation of carcinogenic risks to humans. Polychlorinated dibenzo-para-dioxins and polychlorinated dibenzofurans. [Google Scholar]
- 29.Yang X, et al. The aryl hydrocarbon receptor constitutively represses c-myc transcription in human mammary tumor cells. Oncogene. 2005;24:7869–7881. doi: 10.1038/sj.onc.1208938. [DOI] [PubMed] [Google Scholar]
- 30.Villano CM, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) expression and invasion in 2058 melanoma cells. Toxicol. Appl. Pharmacol. 2006;210:212–224. doi: 10.1016/j.taap.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Chang JT, et al. Requirement of aryl hydrocarbon receptor overexpression for Cyp1B1 up-regulation and cell growth in human lung adenocarcinomas. Clin. Cancer Res. 2007;13:38–45. doi: 10.1158/1078-0432.CCR-06-1166. [DOI] [PubMed] [Google Scholar]
- 32.Ishida M, et al. Activation of the aryl hydrocarbon receptor pathway enhances cancer cell invasion by up-regulating the MMP expression and is associated with poor prognosis in upper urinary tract urothelial cancer. Carcinogenesis. 2010;31:287–296. doi: 10.1093/carcin/bgp222. [DOI] [PubMed] [Google Scholar]
- 33.Mulero-Navarro S, et al. Immortalized mouse mammary fibroblasts lacking dioxin receptor have impaired tumorigenicity in a subcutaneous mouse xenograft model. J. Biol. Chem. 2005;31:28731–28741. doi: 10.1074/jbc.M504538200. [DOI] [PubMed] [Google Scholar]
- 34.Carvajal-Gonzalez JM, et al. The dioxin receptor regulates the constitutive expression of the Vav3-proto-oncogene and modulates cell shape and adhesion. Mol. Biol. Cell. 2009;20:1715–1727. doi: 10.1091/mbc.E08-05-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nesaretnam K, et al. 3,3’,4,4’-Tetrachlorobiphenyl (TCB) can enhance DMBA-induced mammary carcinogenesis in rats. Eur. J. Cancer. 1999;34:389–393. doi: 10.1016/s0959-8049(97)10026-0. [DOI] [PubMed] [Google Scholar]
- 36.Puga A, et al. The aryl hydrocarbon receptor cross-talks with multiple signal transduction pathways. Biochem. Pharmacol. 2009;77:713–722. doi: 10.1016/j.bcp.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian Y. Ah receptor and NF-kB interplay on the stage of epigenome. Biochem. Pharmacol. 2009;77:670–680. doi: 10.1016/j.bcp.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 38.Ohtake F, et al. AhR acts as an E3 ubiquitin ligase to modulate steroid receptor functions. Biochem. Pharmacol. 2009;77:474–484. doi: 10.1016/j.bcp.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 39.Eagle H, et al. Growth regulatory effects of cellular interaction. Nature. 1967;213:1102–1106. doi: 10.1038/2131102a0. [DOI] [PubMed] [Google Scholar]
- 40.Abercrombie M. Contact inhibition and malignancy. Nature. 1979;281:259–262. doi: 10.1038/281259a0. [DOI] [PubMed] [Google Scholar]
- 41.Kung T, et al. The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism. Biochem. Pharmacol. 2009;77:536–546. doi: 10.1016/j.bcp.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson PJ, et al. Emerging targets: molecular mechanisms of cell contact-mediated growth control. Kidney Int. 2002;61:S99–S105. doi: 10.1046/j.1523-1755.2002.0610s1099.x. [DOI] [PubMed] [Google Scholar]
- 43.Curto M, et al. Nf2/Merlin: a coordinator of receptor signaling and intercellular contact. Br. J. Cancer. 2008;98:256–262. doi: 10.1038/sj.bjc.6604002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallez Y, et al. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta. 2008;1778:794–809. doi: 10.1016/j.bbamem.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Zeng Q, et al. The emerging role of the Hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell. 2008;13:188–192. doi: 10.1016/j.ccr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 46.Takai Y, et al. Nectins and nectin-like molecules: roles in contact inhibition of cell movement and proliferation. Nat. Rev. Mol. Cell Biol. 2008;9:603–615. doi: 10.1038/nrm2457. [DOI] [PubMed] [Google Scholar]
- 47.Malumbres M, et al. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005;30:630–641. doi: 10.1016/j.tibs.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 48.Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24:2796–2809. doi: 10.1038/sj.onc.1208619. [DOI] [PubMed] [Google Scholar]
- 49.Hochegger H, et al. Cyclin-dependent kinases and cell-cycle transitions: does one fit all? Nat. Rev. Mol. Cell Biol. 2008;11:910–916. doi: 10.1038/nrm2510. [DOI] [PubMed] [Google Scholar]
- 50.Polyak K, et al. p27KIP1, a cyclin-cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22. doi: 10.1101/gad.8.1.9. [DOI] [PubMed] [Google Scholar]
- 51.Dietrich C, et al. Differences in the mechanisms of growth control in contact-inhibited and serum-deprived human fibroblasts. Oncogene. 1997;15:2743–2747. doi: 10.1038/sj.onc.1201439. [DOI] [PubMed] [Google Scholar]
- 52.Reynisdóttir I, et al. Kip/Cip and INK4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-β. Genes Dev. 1995;9:1831–1845. doi: 10.1101/gad.9.15.1831. [DOI] [PubMed] [Google Scholar]
- 53.St. Croix B, et al. E-cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27 KIP1. J. Cell Biol. 1998;142:557–571. doi: 10.1083/jcb.142.2.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki E, et al. Reentry into the cell cycle of contact-inhibited vascular endothelial cells by a phosphatase inhibitor. J. Biol. Chem. 2000;275:3637–3644. doi: 10.1074/jbc.275.5.3637. [DOI] [PubMed] [Google Scholar]
- 55.Nakatsuji Y, et al. Density dependent modulation of cell cycle protein expression in astrocytes. J. Neurosci. Res. 2001;66:487–496. doi: 10.1002/jnr.1240. [DOI] [PubMed] [Google Scholar]
- 56.Charrasse S, et al. N-cadherin-dependent cell-cell contact regulates Rho GTPases and β-catenin localization in mouse C2C12 myoblasts. J. Cell Biol. 2002;158:953–965. doi: 10.1083/jcb.200202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dietrich C, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-dependent release from contact inhibition in WB-F344 cells: involvement of cyclin A. Toxicol. Appl. Pharmacol. 2002;183:117–126. [PubMed] [Google Scholar]
- 58.Faust D, et al. p38α MAPK is required for contact inhibition. Oncogene. 2005;24:7941–7945. doi: 10.1038/sj.onc.1208948. [DOI] [PubMed] [Google Scholar]
- 59.Swat A, et al. Cell density-dependent inhibition of epidermal growth factor receptor signaling by p38α mitogen-activated protein kinase via sprouty2 downregulation. Mol. Cell. Biol. 2009;29:3332–3343. doi: 10.1128/MCB.01955-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakayama K, et al. Mice lacking p27 Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors . Cell. 1996;85:707–720. doi: 10.1016/s0092-8674(00)81237-4. [DOI] [PubMed] [Google Scholar]
- 61.Wieser R, et al. p16 INK4 mediates contact inhibition of growth . Oncogene. 1999;18:277–281. doi: 10.1038/sj.onc.1202270. [DOI] [PubMed] [Google Scholar]
- 62.Heit I, et al. Involvement of PKCδ in contact-dependent inhibition of growth in human and murine fibroblasts. Oncogene. 2001;20:5143–5154. doi: 10.1038/sj.onc.1204657. [DOI] [PubMed] [Google Scholar]
- 63.Wieser R, et al. Isolation and characterization of a 60-70 kD plasma membrane protein involved in the contact-dependent inhibition of growth. J. Cell Biol. 1990;111:2681–2692. doi: 10.1083/jcb.111.6.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gradl G, et al. Density-dependent regulation of growth by contactinhibin and the contactinhibin-receptor. Curr. Biol. 1995;148:399–403. doi: 10.1016/s0960-9822(95)00105-9. [DOI] [PubMed] [Google Scholar]
- 65.Levenberg S, et al. p27 is involved in N-cadherin-mediated contactinhibition of cell growth and S-phase entry. Oncogene. 1999;18:869–876. doi: 10.1038/sj.onc.1202396. [DOI] [PubMed] [Google Scholar]
- 66.Aoki J, et al. Neural cell adhesion molecule mediates contact-dependent inhibition of growth of near-diploid mouse fibroblast cell line m5S/1M. J. Cell Biol. 1991;115:1751–1761. doi: 10.1083/jcb.115.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Takeichi M. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 1995;7:619–627. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- 68.Gumbiner B. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 69.Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem. Soc. Trans. 2008;36:149–155. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gottardi C, et al. E-cadherin suppresses cellular transformation by inhibiting β-catenin signaling in an adhesion-independent manner. J. Cell Biol. 2001;153:1049–1059. doi: 10.1083/jcb.153.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stockinger A, et al. E-cadherin regulates cell growth by modulating proliferation-dependent β-catenin transcriptional activity. J. Cell Biol. 2001;154:1185–1196. doi: 10.1083/jcb.200104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aberle H, et al. β-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Noort M, et al. TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev. Biol. 2002;244:1–8. doi: 10.1006/dbio.2001.0566. [DOI] [PubMed] [Google Scholar]
- 74.Brabletz T, et al. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and β-catenin. Cells Tissues Organs. 2005;179:56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- 75.Perrais M, et al. E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol. Biol. Cell. 2007;18:2013–2025. doi: 10.1091/mbc.E06-04-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dietrich C, et al. Subcellular localization of β-catenin is dependent on cell density. Biochem. Biophys. Res. Commun. 2002;292:196–199. doi: 10.1006/bbrc.2002.6625. [DOI] [PubMed] [Google Scholar]
- 77.Conacci-Sorell M, et al. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J. Cell Biol. 2003;163:847–857. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Simcha I, et al. Differential nuclear translocation and transactivation potential of β-catenin and plakoglobin. J. Cell Biol. 1998;141:1433–1448. doi: 10.1083/jcb.141.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cowin P, et al. Plakoglobin: a protein common to different kinds of intercellular adhering junctions. Cell. 1986;46:1063–1073. doi: 10.1016/0092-8674(86)90706-3. [DOI] [PubMed] [Google Scholar]
- 80.Chidgey M, et al. Desmosomes: a role in cancer? Br. J. Cancer. 2007;96:1783–1787. doi: 10.1038/sj.bjc.6603808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang Y, et al. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 82.Sadek CM, et al. Cytochrome P450IA1 is rapidly induced in normal keratinocytes in the absence of xenobiotics. J. Biol. Chem. 1994;269:16067–16074. [PubMed] [Google Scholar]
- 83.Sadek CM, et al. Suspension-mediated induction of Hepa 1c1c7 Cyp1A1 expression is dependent on the Ah receptor signal transduction pathway. J. Biol. Chem. 1994;269:31505–31509. [PubMed] [Google Scholar]
- 84.Cho YC, et al. Disruption of cell-cell contact maximally but transiently activates AhR-mediated transcription in 10T1/2 fibroblasts. Toxicol. Appl. Pharmacol. 2004;199:220–238. doi: 10.1016/j.taap.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 85.Ikuta T, et al. Cell density regulates intracellular localization of aryl hydrocarbon receptor. J. Biol. Chem. 2004;279:19209–19216. doi: 10.1074/jbc.M310492200. [DOI] [PubMed] [Google Scholar]
- 86.Wanner R, et al. Retinoic acid affects the expression rate of the differentiation-related genes aryl hydrocarbon receptor, ARNT and keratin 4 in proliferative keratinocytes only. Biochim. Biophys. Acta. 1996;1317:105–111. doi: 10.1016/s0925-4439(96)00038-5. [DOI] [PubMed] [Google Scholar]
- 87.Jones CL, et al. Differentiation status of cultured murine keratinocytes modulates induction of genes responsive to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Arch. Biochem. Biophys. 1997;347:163–173. doi: 10.1006/abbi.1997.0350. [DOI] [PubMed] [Google Scholar]
- 88.Reiners JJ, Jr., et al. Differential expression of basal and hydrocarbon-induced cytochrome P-450 monooxygenase and quinone reductase activities in subpopulations of murine epidermal cells differing in their stages of differentiation. Drug Metab. Dispos. 1992;20:360–366. [PubMed] [Google Scholar]
- 89.Milstone L, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces hyperplasia in confluent cultures of human keratinocytes. J. Invest. Dermatol. 1984;82:532–534. doi: 10.1111/1523-1747.ep12261149. [DOI] [PubMed] [Google Scholar]
- 90.Hébert C, et al. Inhibition of high-density growth arrest in human squamous carcinoma cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Carcinogenesis. 1990;11:1335–1342. doi: 10.1093/carcin/11.8.1335. [DOI] [PubMed] [Google Scholar]
- 91.Münzel P, et al. Growth modulation of hepatocytes and rat liver epithelial cells (WB-F344) by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Carcinogenesis. 1996;17:197–202. doi: 10.1093/carcin/17.2.197. [DOI] [PubMed] [Google Scholar]
- 92.Weiss C, et al. TCDD deregulates contact inhibition in rat liver oval cells via Ah receptor, JunD and cyclin A. Oncogene. 2008;27:2198–2207. doi: 10.1038/sj.onc.1210859. [DOI] [PubMed] [Google Scholar]
- 93.Fiorito F, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases bovine herpesvirus type-1 (BHV-1) replication in Madin-Darby Bovine Kidney (MDBK) cells in vitro. J. Cell. Biochem. 2008;103:221–233. doi: 10.1002/jcb.21398. [DOI] [PubMed] [Google Scholar]
- 94.Chramostova K, et al. Polycyclic aromatic hydrocarbons modulate cell proliferation in rat hepatic epithelial stem-like WB-F344 cells. Toxicol. Appl. Pharmacol. 2004;196:136–148. doi: 10.1016/j.taap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 95.Vondrácek J, et al. Induction of aryl hydrocarbon receptor-mediated and estrogen receptor-mediated activities, and modulation of proliferation by dinaphthofurans. Environ. Toxicol. Chem. 2004;23:2214–2220. doi: 10.1897/03-620. [DOI] [PubMed] [Google Scholar]
- 96.Vondrácek J, et al. Aryl hydrocarbon receptor-activating polychlorinated biphenyls and their hydroxylated metabolites induce cell proliferation in contact-inhibited rat liver epithelial cells. Toxicol. Sci. 2005;83:53–63. doi: 10.1093/toxsci/kfi009. [DOI] [PubMed] [Google Scholar]
- 97.Zatloukalová J, et al. β-Naphthoflavone and 3’-methoxy-4’-nitroflavone exert ambiguous effects on Ah receptor-dependent cell proliferation and gene expression in rat liver, stem-like’ cells. Biochem. Pharmacol. 2007;73:1622–1634. doi: 10.1016/j.bcp.2007.01.032. [DOI] [PubMed] [Google Scholar]
- 98.Bock K-W, et al. Ah receptor- and TCDD-mediated liver tumor promotion: clonal selection and expansion of cells evading growth arrest and apoptosis. Biochem. Pharmacol. 2005;69:1403–1408. doi: 10.1016/j.bcp.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 99.Marlowe J, et al. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell. Biochem. 2005;96:1174–1184. doi: 10.1002/jcb.20656. [DOI] [PubMed] [Google Scholar]
- 100.Tsao M-S, et al. A diploid epithelial cell line from normal adult rat liver with phenotypic properties of ‘oval’ cells. Exp. Cell Res. 1984;154:38–52. doi: 10.1016/0014-4827(84)90666-9. [DOI] [PubMed] [Google Scholar]
- 101.Coleman W, et al. Evaluation of the differentiation potential of WB-F344 rat liver epithelial stem-like cells in vivo . Differentiation to hepatocytes after transplantation into dipeptidylpeptidase-IV-deficient rat liver . Am. J. Pathol. 1997;151:353–359. [PMC free article] [PubMed] [Google Scholar]
- 102.Shafritz DA, et al. Liver stem cells and model systems for liver repopulation. J. Hepatol. 2002;36:552–564. doi: 10.1016/s0168-8278(02)00013-2. [DOI] [PubMed] [Google Scholar]
- 103.Steinberg P, et al. Oval cell lines OC/CDE 6 and OC/CDE 22 give rise to cholangio-cellular and undifferentiated carcinomas after transformation. Lab. Invest. 1994;71:700–709. [PubMed] [Google Scholar]
- 104.Libbrecht L, et al. The immunohistochemical phenotype of dysplastic foci in human liver: correlation with putative progenitor cells. J. Hepatol. 2000;33:76–84. doi: 10.1016/s0168-8278(00)80162-2. [DOI] [PubMed] [Google Scholar]
- 105.Dumble ML, et al. Generation and characterization of p53 null transformed hepatic progenitor cells: oval cells give rise to hepatocellular carcinoma. Carcinogenesis. 2002;23:435–445. doi: 10.1093/carcin/23.3.435. [DOI] [PubMed] [Google Scholar]
- 106.Hailey JR, et al. Classification of proliferative hepatocellular lesions in harlan sprague-dawley rats chronically exposed to dioxin-like compounds. Toxicol. Pathol. 2005;33:165–174. doi: 10.1080/01926230590888324. [DOI] [PubMed] [Google Scholar]
- 107.Tsao M-S, et al. Hepatocarcinomas, cholangiocarcinomas, and hepatoblastomas produced by chemically transformed cultured rat liver epithelial cells. A light- and electron-microscopic analysis. Am. J. Pathol. 1987;127:168–181. [PMC free article] [PubMed] [Google Scholar]
- 108.Dietrich C, et al. TCDD-dependent downregulation of gamma-catenin in rat liver epithelial cells (WB-F344) Int. J. Cancer. 2003;103:435–439. doi: 10.1002/ijc.10830. [DOI] [PubMed] [Google Scholar]
- 109.Andrysik Z, et al. The aryl hydrocarbon receptor-dependent deregulation of cell cycle control induced by polycyclic aromatic hydrocarbons in rat liver epithelial cells. Mutat. Res. 2007;615:87–97. doi: 10.1016/j.mrfmmm.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 110.Weiss C, et al. TCDD induces c-jun expression via a novel Ah (dioxin) receptor-mediated p38-MAPK-dependent pathway. Oncogene. 2005;24:4975–4983. doi: 10.1038/sj.onc.1208679. [DOI] [PubMed] [Google Scholar]
- 111.Shaulian E, et al. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 112.Hölper P, et al. Evaluation of the role of c-scr and MAPK in TCDD-dependent release from contact inhibition in WB-F344 cell. Arch. Toxicol. 2005;79:201–207. doi: 10.1007/s00204-004-0624-6. [DOI] [PubMed] [Google Scholar]
- 113.Hölper P, et al. Transforming growth factor-β1 is not involved in 2,3,7,8-tetrachlorodibenzo-p-dioxin-dependent release from contact inhibition in WB-F344 cells. Arch. Toxicol. 2004;78:643–648. doi: 10.1007/s00204-004-0591-y. [DOI] [PubMed] [Google Scholar]
- 114.Ray SS, et al. Dioxin-induced immortalization of normal human keratinocytes and silencing of p53 and p16 INK4a. J. Biol. Chem. 2004;279:27187–27193. doi: 10.1074/jbc.M402771200. [DOI] [PubMed] [Google Scholar]
- 115.Duan R, et al. Transcriptional activation of the c-fos protooncogene by 17beta-estradiol: mechanism of aryl hydrocarbon receptor-mediated inhibition. Mol. Endocrinol. 1999;13:1511–1521. doi: 10.1210/mend.13.9.0338. [DOI] [PubMed] [Google Scholar]
- 116.Olnes MJ, et al. 2,3,7,8-Tetrachlorodibenzo- p -dioxin modulates expression of the prostaglandin G/H synthase-2 gene in rat thymocytes . J. Pharmacol. Exp. Ther. 1996;279:1566–1573. [PubMed] [Google Scholar]
- 117.Krishnan V, et al. Molecular mechanism of inhibition of estrogen-induced cathepsin D gene expression by 2,3,7,8-tetrachlorodibenzo- p -dioxin . Mol. Cell. Biol. 1995;15:6710–6719. doi: 10.1128/mcb.15.12.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Safe S. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol. Lett. 2001;120:1–7. doi: 10.1016/s0378-4274(01)00301-0. [DOI] [PubMed] [Google Scholar]
- 119.Pötter E, et al. Molecular cloning of a functional promoter of the human plakoglobin gene. Eur. J. Endocrinol. 2001;145:625–633. doi: 10.1530/eje.0.1450625. [DOI] [PubMed] [Google Scholar]
- 120.Diry M, et al. Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene. 2006;25:5570–5574. doi: 10.1038/sj.onc.1209553. [DOI] [PubMed] [Google Scholar]
- 121.Tsai C-L, et al. Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH 2 -terminal kinase signaling . Cancer Res. 2006;66:11668–11676. doi: 10.1158/0008-5472.CAN-06-2194. [DOI] [PubMed] [Google Scholar]
- 122.Bui L-C, et al. Nedd9/Hef1/Cas-L mediates the effects of environmental pollutants on cell migration and plasticity. Oncogene. 2009;28:3642–3651. doi: 10.1038/onc.2009.224. [DOI] [PubMed] [Google Scholar]
- 123.Seifert A, et al. TCDD induces cell migration via NFATc1/ATX-signaling in MCF-7 cells. Toxicol. Lett. 2009;184:26–32. doi: 10.1016/j.toxlet.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 124.Jourquin J, et al. Dispersal of epithelial cancer cell colonies by lysophosphatidic acid (LPA) J. Cell Physiol. 2006;206:337–346. doi: 10.1002/jcp.20470. [DOI] [PubMed] [Google Scholar]
- 125.Ikuta T, et al. Zinc finger transcription factor slug is a novel target gene of aryl hydrocarbon receptor. Exp. Cell Res. 2006;312:3585–3594. doi: 10.1016/j.yexcr.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 126.Choi J, et al. Transforming growth factor-β1 represses E-cadherin production via slug expression in lens epithelial cells. Invest. Ophthalmol. Vis. Sci. 2007;48:2708–2718. doi: 10.1167/iovs.06-0639. [DOI] [PubMed] [Google Scholar]
- 127.Eddy SF, et al. Inducible IkB kinase/IkB kinase expression is induced by CK2 and promotes aberrant nuclear factor-kB activation in breast cancer cells. Cancer Res. 2005;65:11375–11383. doi: 10.1158/0008-5472.CAN-05-1602. [DOI] [PubMed] [Google Scholar]
- 128.Belguise K, et al. Green tea polyphenols reverse cooperation between c-Rel and CK2 that induces the aryl hydrocarbon receptor, Slug, and an invasive phenotype. Cancer Res. 2007;67:11742–11750. doi: 10.1158/0008-5472.CAN-07-2730. [DOI] [PubMed] [Google Scholar]
- 129.Yang X, et al. Constitutive regulation of Cyp1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J. Cell. Biochem. 2008;104:402–417. doi: 10.1002/jcb.21630. [DOI] [PubMed] [Google Scholar]
- 130.Karin M. Nuclear factor–κB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 131.Duncan JS, et al. Too much of a good thing: the role of protein kinase CK2 in tumorigenesis and prospects of therapeutic inhibition. Biochim. Biophys. Acta. 2008;1784:33–47. doi: 10.1016/j.bbapap.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 132.Palermo CM, et al. Epigallocatechin gallate inhibits aryl hydrocarbon receptor gene transcription through an indirect mechanism involving binding to a 90 kDa heat shock protein. Biochemistry. 2005;44:5041–5052. doi: 10.1021/bi047433p. [DOI] [PubMed] [Google Scholar]
- 133.Yin Z, et al. Epigallocatechin-3-gallate is a novel Hsp90 inhibitor. Biochemistry. 2009;48:336–345. doi: 10.1021/bi801637q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Christmann M, et al. Mechanisms of human DNA repair: an update. Toxicology. 2003;193:3–34. doi: 10.1016/s0300-483x(03)00287-7. [DOI] [PubMed] [Google Scholar]
- 135.Kang HJ, et al. BRCA1 modulates xenobiotic stress-inducible gene expression by interacting with ARNT in human breast cancer cells. J. Biol. Chem. 2006;281:14654–14662. doi: 10.1074/jbc.M601613200. [DOI] [PubMed] [Google Scholar]
- 136.Chan CYY, et al. TCDD affects DNA double strand-break repair. Toxicol. Sci. 2004;81:133–138. doi: 10.1093/toxsci/kfh200. [DOI] [PubMed] [Google Scholar]
- 137.Lin PH, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces oxidative stress, DNA strand breaks, and poly(ADP-ribose) polymerase-1 activation in human breast carcinoma cell lines. Toxicol. Lett. 2007;172:146–158. doi: 10.1016/j.toxlet.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 138.Marlowe JL, et al. The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis. Mol. Biol. Cell. 2008;19:3263–3271. doi: 10.1091/mbc.E08-04-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gasiewicz TA, et al. Expression and activity of aryl hydrocarbon receptors in development and cancer. Crit. Rev. Eukaryot. Gene Expr. 2008;18:279–321. doi: 10.1615/critreveukargeneexpr.v18.i4.10. [DOI] [PubMed] [Google Scholar]
- 140.Kolluri SK, et al. p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev. 1999;13:1742–1753. doi: 10.1101/gad.13.13.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]