

Abstract
Summary
The TRI_tool, a sequence-based web tool for prediction of protein interactions in the human transcriptional regulation, is intended for biomedical investigators who work on understanding the regulation of gene expression. It has an improved predictive performance due to the training on updated, human specific, experimentally validated datasets. The TRI_tool is designed to test up to 100 potential interactions with no time delay and to report both probabilities and binarized predictions.
Availability and Implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Transcriptional regulation (TR) a complex process which controls the cellular gene expression program is among the key deregulated processes in all serious diseases that affect humans including cancer. TR is implemented through non-random combinations of functional dimers or higher order complexes. Therefore, a reliable prediction of intracellular pairs prone to interact will contribute importantly to identifying pharmacologically relevant protein–protein interactions (PPI). The computational PPI predictions rely on extensive resources of known PPI and other types of information such as sequences, protein structures, network associations and coexpression data. However, with the exception of the sequence information, other aforementioned data are time and context dependent. Different proteome-wide prediction methods have demonstrated that the information on amino acid sequences alone may be sufficient to identify novel PPIs (Martin et al., 2005; Shen et al., 2007). In order to enable biomedical investigators to predict PPI and to generate a reliable hypothesis that could be tested experimentally several sequence-based online tools were made available in recent years (Liu and Chen, 2012). Yet, we identified a few possible improvements that will empower those interested in transcriptional control to better leverage the potential of sequence analyses. Firstly, the complexity of TR in humans needs to be considered. Although, the combinatorial nature of the transcriptional control is conserved among prokaryotic and eukaryotic organisms, the level of complexity significantly increases in higher eukaryotes. Secondly, most of the available web tools allow for testing only one interaction at a time or a set of interactions with a time delay due to the job queue systems. In this paper, we present a web-based online tool for automatic prediction of Transcriptional Regulation Interactions (TRI) in humans, the TRI_tool, which addresses the aforementioned issues. The TRI_tool relies on the method based on Chou’s pseudo amino acid composition (PseAAC) model for protein feature representation (Chou, 2001). It was trained on experimentally validated human TRI and consequently, the model entails species specificity. In general, this approach may be extended to different types of interactions for which PPI resources allow for reliable model construction. It has a high predictive performance and capability to test up to 100 potential interactions with no time delay which makes the TRI_tool a unique instrument for biomedical investigators who sought to unravel the molecular mechanisms that direct gene expression.
2 Overview of the TRI_tool
2.1 Implementation
The server side of the software for the sequence encoding calculation and classification was implemented in the JAVA programming language. The random forest model was built using the R language and it was extracted as a plain old java object (POJO) format. The POJO code is imported in the classification module of our software. The HTML pages, with a simple-to-use user interface (Supplementary Fig. S1), are generated using PHP script. The overview of the algorithm of the TRI_tool is presented in Figure 1A.
Fig. 1.
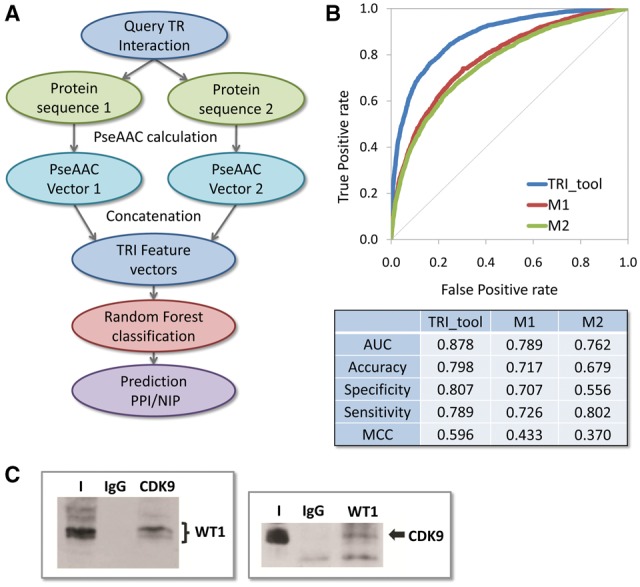
(A) The overview of the algorithm of the TRI_tool; (B) Evaluation of predictive performances of the TRI_tool and two state-of-the-art PPI prediction methods; (C) In vivo binding confirmation. Co-immunoprecipitation of WT1 and CDK9
2.2 TRI_tool predictions and user interface
The TRI_tool interface allows the user to input the protein sequences in a FASTA format and to choose either automatic combination in pairs or to add protein pairs of interest to the input information. The model is trained on 24 488 human TRI and details about datasets are given in the Supplementary Material. Protein sequences are vectorized by PseAAC and coded by the amino acid descriptors relevant for protein interaction properties (Supplementary Table S1). For each protein pair, the output of the TRI_tool is a real value that quantifies the probability of a given pair to form an interaction. The pairs with P > 0.5 are assumed PPI. Additionally, the TRI_tool allows the user to automatically sort results according to P values. The outputs in table format are presented to the user online.
3 Discussion
The TRI_tool performance was evaluated by comparison with state-of-the-art sequence-based PPI prediction tools M1 (Guo et al., 2008) and M2 (Pitre et al., 2008). All three methods were assessed by 3-fold cross-validation and the evaluation shows that the TRI_tool significantly outperforms other methods (Fig. 1B, Supplementary Fig. S2 and Table S4). Furthermore, to demonstrate the effectiveness of the TRI_tool, we applied it to the prediction of interactions of Wilm’s tumor protein (WT1), a transcription factor and regulatory molecule which plays a critical role in development and hematopoiesis. WT1 is overexpressed in numerous human malignancies (Huff, 2011) acting either as an oncogene or tumor suppressor and therefore, mapping its interactions will enhance the understanding of functional complexity. Towards this goal, we assessed prospective interactions of WT1 with: (1) a set of protein kinases which participate in TR (Supplementary Table S5) and (2) its known interactor. Kinases are prime targets for the development of selective inhibitors of cancer signaling pathways and we wanted to pinpoint TRI relevant for drug discovery. Based on the user-defined input sequence pairs, the TRI_tool predicted interaction between WT1 and validated anti-cancer target cyclin-dependent kinase, CDK9. The candidate interaction was tested and confirmed by immunoprecipitation in human leukemia cell line K562 (Fig. 1C). The capability of the TRI_tool to predict known interactions was confirmed by correctly predicting the WT1 interaction with TFIIB which was described in the literature, but not included in the training set (Supplementary Table S6).
4 Conclusion
The TRI_tool is intended for biomedical investigators who work on understanding the regulation of gene expression and search for new treatments for complex diseases and cancer. The TRI_tool allows for fast and efficient testing of up to 100 sequence pairs and provides output as binarized predictions and probability values which enable users to prioritize candidate interactions.
Supplementary Material
Acknowledgements
We are grateful to Jude Fitzgibbon from the Barts Cancer Institute for insightful discussions and to Yungki Park from the Hunter James Kelly Research Institute for method M2 implementation.
Funding
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia [ON173001].
Conflict of Interest: none declared.
References
- Chou K.C. (2001) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins, 43, 246–255. [DOI] [PubMed] [Google Scholar]
- Guo Y. et al. (2008) Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res., 36, 3025–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff V. (2011) Wilms’ tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat. Rev. Cancer, 11, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.P., Chen L. (2012) Proteome-wide prediction of protein–protein interactions from high-throughput data. Protein Cell, 3, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S. et al. (2005) Predicting protein–protein interactions using signature products. Bioinformatics, 21, 218–226. [DOI] [PubMed] [Google Scholar]
- Pitre S. et al. (2008) Global investigation of protein–protein interactions in yeast Saccharomyces cerevisiae using re-occurring short polypeptide sequences. Nucleic Acids Res., 36, 4286–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. et al. (2007) Predicting protein–protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA, 104, 4337–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.