

Abstract
Motivation: The advent of next-generation sequencing technologies has increased the accuracy and quantity of sequence data, opening the door to greater opportunities in genomic research.
Results: In this article, we present GNUMAP (Genomic Next-generation Universal MAPper), a program capable of overcoming two major obstacles in the mapping of reads from next-generation sequencing runs. First, we have created an algorithm that probabilistically maps reads to repeat regions in the genome on a quantitative basis. Second, we have developed a probabilistic Needleman–Wunsch algorithm which utilizes _prb.txt and _int.txt files produced in the Solexa/Illumina pipeline to improve the mapping accuracy for lower quality reads and increase the amount of usable data produced in a given experiment.
Availability: The source code for the software can be downloaded from http://dna.cs.byu.edu/gnumap.
Contact: nathanlclement@gmail.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Next-generation sequencing technologies produce fast and accurate results with base pair level resolution. These technologies are currently being applied to a diverse set of biological applications including creating chromatin state maps (Mikkelsen et al., 2007), performing whole-genome evolutionary analyses (McCutcheon and Moran, 2007), determining miRNA expression (Morin et al., 2008), identifying protein–DNA interactions (Johnson et al., 2007), illuminating histone modification sites (Barski et al., 2007) and translocation breakpoint identification (Chen et al., 2008). Speed and quantity of sequenced bases are two areas where next-generation sequencing outperforms traditional sequencing; but the area in which next-generation approaches such as the Solexa platform, developed by Illumina, Inc. (San Diego, CA, USA), provide the greatest benefit are in the additional information included with each run. Not only can they provide millions of bases, but for each of these bases, intensity and accuracy measures are included, which can then be translated into a likelihood score at each base.
For most of the applications that use short-read high-throughput sequencing, tens of millions of DNA or RNA fragments are extracted from a sample. These fragments are hybridized to a slide and sequenced to a specified length. A mapping algorithm is then used to identify the location in a reference genome or transcriptome that was the most probable source for each segment. If a DNA/RNA sequence occurs at multiple locations in the genome, it will be impossible to identify with surety the exact location from which it came. Because of this difficulty, many mapping algorithms discard any sequences that map to multiple locations in the genome. If these reads are discarded, the researcher may miss important genomic features.
As mentioned above, one of the features of next-generation sequencing technology is the assignment of quality scores to each of the four nucleotides on all the positions of the sequence. Variations in chemical processes often result in ambiguous bases, such as assigning nearly equal probabilities to an A and a C. In addition, the first few bases in a Solexa sequencing read are very high in quality, but towards the end of each read, the error rate increases, often dramatically. Many algorithms will only use the nucleotide with the highest probability and ‘call’ that location in the read, ignoring the other three probabilities and the decreasing accuracy near the end of the read. By following this procedure, if there are more than a few bases that have lower probability values, the entire read is discarded. However, if all four probabilities are used in later stages of the mapping process, even the less-confident reads can be mapped, resulting in more usable information from the experiment.
Several applications have attempted to solve the mapping problem. SeqMap (Jiang and Wong, 2008), RMAP (Smith et al., 2008) and ELAND (included as part of the Solexa/Illumina pipeline) all create a hash from the reads. This hash is then used to find matching reads to regions of the genome. Because the genome is not hashed, there is no way to fairly allocate a read across repeat regions. MAQ (Li,H. et al., 2008), SOAP (Li,R. et al., 2008) and Novocraft (unpublished data, http://www.novocraft.com/index.html) also use a hash map, but the reference genome is hashed instead of the reads. MAQ allows up to two mismatches in the first 28 bp. More mismatches are allowed if the Phred-quality score of the entire read is sufficient (Li,H. et al., 2008). Slider (Malhis et al., 2009) gains its speed by lexicographically sorting both the reads and the windowed regions of the reference genome. To account for inexact matches (it does not perform gapped alignments), Slider generates multiple reads with all possible substitutions for each read that has uncertainty in some read positions, leading to a combinatorial increase in the number of reads. Bowtie ‘conducts a quality-aware, greedy, randomized, depth-first search through the space of possible alignments’ (Langmead et al., 2009). Bowtie is extremely fast, but makes compromises to achieve that speed. Most notably, if an exact match does not exist, Bowtie is not guaranteed to map the read.
A rigorous probabilistic approach to mapping repeat regions and reads with lower quality scores can result in a significantly larger number of mapped reads. This can often lead to the identification of regions of interest on the genome that otherwise would have been overlooked—for example, mapping to the large number repetitive genomic elements in mammalian genomes. This article, along with the Genomic Next-generation Universal MAPper (GNUMAP) program, focuses on overcoming the aforementioned inaccuracies for an overall increase in data usage and more accurate read mapping to a reference genome.
2 APPROACH
Many mapping algorithms discard reads from repeat regions and do not utilize the quality scores once the base has been ‘called’. GNUMAP provides a probabilistic approach that utilizes this additional information to provide more accurate results from fewer costly sequencing runs.
2.1 Unique mapping position
Accurately mapping reads to repetitive genomic elements is essential if next-generation sequencing is to be used to draw valid biological conclusions. For example, a ChIP-seq experiment attempts to accurately identify small DNA regions interacting with a protein of interest. Binding motifs often appear in or near repeat regions (Park et al., 2002; van Helden, 2004), reducing the ability to identify motifs in these regions. Other applications such as transcription mapping, alternative splicing analysis and miRNA identification may also suffer from inaccuracies using such a mapping method. Several programs (such as RMAP, SeqMap and ELAND) have attempted to significantly speed up this mapping process through creating a hash map to efficiently map reads to the genome. Reads are broken into short, 9–15 bp segments and assigned a numerical value in the hash map according to their sequence. The genome is then scanned and the hashing function is used to find corresponding locations for the genomic sequences in the read hash table. The reads at these locations are then aligned with the genome until either a match is found or the alignment is deemed too insignificant to continue. This strategy of hashing the reads poses a problem when there are multiple regions in the reference genome that produce the same alignment score. This approach does not identify all matching regions at the same time so that the read can be fairly allocated to all of these regions. This is a significant problem because genomic sequences contain a large number of repeat regions.
In the human reference genome, <85% of 30 bp sequences are unique (Butler et al., 2008). On a smaller scale, there are no unique 9 bp sequences. Due to this redundancy, when many programs find a sequence that matches multiple locations, they erroneously either discard the sequence or report all matching locations. For example, assume that an organism's DNA were sonicated and sequenced, causing each sequence from the genome to appear exactly once in the final set of reads. If a read had originated from a repeat region which occurred three times in the genome, it would also appear three times in the set of reads. Traditional mapping procedures have attempted to score these regions in one of three ways: (i) discard all repeat regions, (ii) record only one position (first or random) for each read or (iii) record all positions as receiving a hit for each read. Discarding these reads results in the loss of up to half of the data (Harismendy et al., 2009). The second method would cause unequal mapping to some of the repeat regions. The third method would result in each location having three times the correct score. Since several algorithms (RMAP, ELAND and SeqMap) hash the reads and then make a single pass through the genome, there is no way to know how many genomic locations will match in order to add one third of a read to each of these locations. The GNUMAP algorithm described below overcomes this problem by hashing the genome and referencing the reads one at a time to the genome. This approach allows for the simultaneous identification of all genomic matches for each read. GNUMAP then accounts for repetitive elements by assigning a proportion of the read to relevant genomic matches based on the relative likelihood that the read maps to each location. It should be noted that MAQ and SOAP also use an approach that hashes the genome; however, they do not proportionally assign multi-hit reads to the genome.
2.2 Information from probability and intensity files
The second problem with current mapping methods for next-generation sequencing data is the disregard for base-calling variability and the frequent discard of lower quality data. Several algorithms (such as MAQ, Bowtie and SOAP) use a single called base at each position in the read, thus ignoring all uncertainty and allowing for increased mapping error rates. This means that reads with a few low-quality bases can lead to an incorrect mapping of a read. When algorithms apply a quality filter to remove these reads, as many as half of the reads may be discarded (Harismendy et al., 2009).
The GNUMAP algorithm effectively incorporates the base uncertainty of the reads into mapping analysis using a Probabilistic Needleman–Wunsch algorithm. The Probabilistic Needleman–Wunsch was developed to improve upon the common dynamic programming algorithm used for sequence alignment to accurately use reads with lower confidence values. The algorithm is discussed further in Section 3 of this article.
3 METHODS
Care must be taken to develop an algorithm that can accurately map millions of reads to the genome in a reasonable amount of time. In the GNUMAP algorithm, the genome is first hashed and then stored in a lookup table rather than hashing the reads. This allows reads to be accounted for in all of the duplicate genome sites. Next, the reads are efficiently stored as a position-weight matrix (PWM) so that quality scores can be used when aligning the read with genomic data. A Needleman–Wunsch alignment algorithm is modified to use these matrices to score and probabilistically align a read with the reference genome. Figure 1 is a flowchart which shows the major steps of the algorithm.
Fig. 1.

A flow-chart of the GNUMAP algorithm. First, the algorithm will incrementally find a k-mer piece in the consensus Solexa read. This k-mer is used as an index into the hash table, producing a list of positions in the genome with the exact k-mer sequence. These locations are expanded to align the same l nucleotides from the read to the genomic location. If the alignment score passes the user-defined threshold, the location is considered a hit, and recorded on the genome for future output.
Step 1: Hashing and storing the genome
Hashing a large portion of the data allows for quick data retrieval while still maintaining a reasonable amount of memory use. GNUMAP creates a hash table from the genome instead of the reads, allowing for the computation of a probabilistic scoring scheme.
The entire genome is hashed based upon either a user-supplied hash size or the default hash size of nine. A larger hash size will tolerate fewer mismatches. For example, in a 30 bp read, a hash size of 9 bp will guarantee that the read is matched to every possible location while still allowing for three mismatches. Larger hashes will require more memory, but will also reduce the search space. The amount of memory, B, required based on the number of bases in the genome, s, and the mer-size of the hash, k, can be computed as follows:
| (1) |
For example, for a genome (s) of 200 000 bp and a mer-size (k) of 9, the total memory used (B) will be 4 * (49 + 200000)≈2 Mb of RAM.
Step 2: Processing the reads
One of the novel approaches implemented by GNUMAP lies in the data structure used for storing the reads. Instead of storing the reads as simple sequences, or even sequences with an attached probability as in the FASTQ format, each sequence is stored as a position weight matrix (PWM) (see Table 1 for an example).
Table 1.
Dynamic Programming (DP) matrix for probabilistic Needleman–Wunsch
| j | 0 | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|---|
| PWM | A | 0.059 | 0.000 | 0.172 | 0.271 | 0.300 |
| C | 0.108 | 0.320 | 0.136 | 0.209 | 0.330 | |
| G | 0.305 | 0.317 | 0.317 | 0.164 | 0.045 | |
| T | 0.526 | 0.578 | 0.375 | 0.356 | 0.325 | |
|
| ||||||
| NW | T | T | T | T | C | |
| 0 | −2 | −4 | −6 | −8 | −10 | |
| T | −2 | 0.052 | −1.948 | −3.948 | −5.948 | −1000 |
| T | −4 | −1.844 | 0.208 | −1.792 | −3.792 | −5.792 |
| C | −6 | −3.844 | −1.792 | −0.520 | −2.448 | −4.448 |
| A | −8 | −5.844 | −3.792 | −2.374 | −0.978 | −2.978 |
| C | −10 | −1000 | −5.792 | −4.131 | −2.774 | −1.318 |
Aligning the genomic sequence TTCAC and read TTTTC, with the optimal alignment shown in bold. Also notice the PWM for the sequence, with several fairly ambiguous positions (especially the final position, probably representing a C, even though the probabilities for the C and T are nearly equal). The underlined position is computed in Equation 3.
Raw data from the Solexa/Illumina platform are obtained as either an intensity file or a probability file. From either of these files, it is possible to compute a likelihood score for any nucleotide of any position on any given read. There will often be a lack of distinction between the most probable and another base, such as the ambiguity between G and T seen in position 3 of the PWM in Table 1. Since each read is stored in memory as a PWM, the information included in each base call allows for the correct mapping of a given sequence. Converting these bases to a single probability score will result in the loss of information (Fig. 5c).
Fig. 5.

Comparison of false positive rates in the detection of Solexa/Illumina spikes. For each point on the line corresponding to a particular algorithm, the value on the x-axis indicates the spike number for that algorithm. The y-axis value for that point indicates the number of spikes actually in the benchmark dataset. The difference between the number of spikes found by the algorithm and the diagonal is the number of false positives, i.e. GNUMAP and Novocraft had the lowest false positive rate. GNUMAP was correctly able to identify the top 36 spikes in the test dataset. SeqMap and RMAP performed similarly, as did MAQ and SOAP.
Step 3: Score individual matches
In order to match the reads to the reference genome, the reads are first subjected to a quality filter, removing reads with too many unknown bases. In order to pass GNUMAP's quality filter, a sequence (stored as a PWM) must be able to obtain a positive score when aligned with its own consensus sequence (the sequence created from using only the most probable bases). Using this method, very few reads are discarded by the quality filter (usually only removing reads identified by the Solexa pipeline as having an intensity of zero at each base).
A sliding window of size k is used to create a hash value which can be used to find matching positions in the reference genome. The matching genomic sequence is then aligned to the read using the probabilistic Needleman–Wunsch algorithm (Table 1).
The probabilistic Needleman–Wunsch score (PNWScore) for read r and genomic sequence S at position i, j in the dynamic programming matrix NW can be calculated as:
| (2) |
given that costk,j is the cost of aligning the character at position rj with the character k. For example, using the PWM in Table 1, the calculation of the score for position 3, 3 in the dynamic programming matrix would be:
| (3) |
(For this example, a match yields a cost of 1, a mismatch yields a cost of −1 and the cost for a gap is −2. This results in a cost of −0.520 which is stored at position 3, 3 in the dynamic programming matrix NW.
Step 4: Processing scores
Once the read has been scored against all plausible matches in the genome, a proportional share of this read will be added to all the matching genomic locations. In order to compute the hit score at a position in the reference genome, a posterior probability for each read is computed. For a read r, the algorithm first finds the n most plausible match locations on the genome, M1,…, Mn. These matches are scored using the probabilistic Needleman–Wunch algorithm, to obtain the scores Q1,…, Qn. The value added to the genome G for each read, r, obtained from each significant match location Mj, signified by GMj, will then be
| (4) |
where nMk is the number of times the sequence located at position Mk appears in the genome.
When using this scoring method, the total score for each sequence at a particular site in the genome is weighted by its number of occurrences in the genome. If a given sequence occurs frequently, the value added to a particular matching site in the final output is down-weighted, removing the bias that would occur if the match was added to all repetitive regions in the genome. If, however, there are the same number of duplicate reads as the number of times the sequence is duplicated in the genome then a whole read will be added to each of the duplicate locations in the final output.
This scoring technique requires the hashing and storing of the genome instead of the set of reads. Because the score for a given read is not only calculated from its alignment score but also by the number of occurrences of similar regions in the genome, the genome must be scanned for each read to fairly allocate the read across all matching sites.
Step 5: Create output
After all the reads have been matched and scored on the genome, two output files are created. The first file identifies the highest scoring match for each read, and the second file contains the genome in .sgr format for viewing in the UCSC Genome Browser (http://genome.ucsc.edu/) or Affymetrix's Integrated Genome Browser (IGB) (http://www.affymetrix.com).
4 RESULTS
GNUMAP was tested using four datasets—two real and two simulated. The first dataset is a human ChIP-seq experiment attempting to identify the in vivo binding sites of the ETS1 transcription factor. The second dataset is a human small RNA sequencing experiment. In these two examples, we illustrate the ability for GNUMAP to find biologically relevant features while maintaining a low false discovery rate as compared with other mapping algorithms. In addition, in order to justify GNUMAP's probabilistic approach, we analyze the overall read quality and estimate sequencing error rates. We show that GNUMAP is much less prone to mapping errors than other algorithms based on these metrics.
In addition, we generated two spike-in datasets that illustrate GNUMAP's accuracy in a setting where the correct answer is known. For the first test dataset, we sampled random simulated reads from promoter regions from the Caenorhabditis elegans genome. Thirty-five ‘spikes’ were used as the data and the accuracy of several mapping algorithms was compared. We also generated a dataset simulating the specific case where many of the bases are miscalled by the sequencing method and show that GNUMAP can still accurately map these reads. For our performance benchmark, GNUMAP processed 100 000 reads in 47 s and correctly mapped 71 262 of them. As a comparison, MAQ correctly mapped 62 208 in 46.5 s. The performance study shown in Table 3 indicates that GNUMAP achieves reasonable performance when compared with other applications, but is able to map significantly more reads.
Table 3.
Performance comparison
| Benchmark data |
Human genome |
|||
|---|---|---|---|---|
| Program | Time | # mapped | Time | # mapped |
| GNUMap | 47.9 s | 71 262 | 985 m 14 s | 7 739 321 |
| Bowtie | 7.0 s | 62 298 | 14 m 43 s | 6 699 526 |
| SOAP | 11.7 s | 62 208 | 32 m 20 s | 6 764 050 |
| MAQ | 46.5 s | 62 208 | *3488 m 28 s | 6 764 054 |
| Slider | 16 m 31 s* | 58 551 | Crashed | Crashed |
| SeqMap | 81.2 s | 56 326 | 1703 m 04 s | 5 455 538 |
| Novocraft | 24.4 s | 56 238 | *920 m 25 s | 5 306 782 |
| RMAP | 9.2 s | 1202 | *295 m 54 s | 3 447 086 |
Bold values show that GNUMAP achieves the best performance.
4.1 Quality score analysis
In order to justify the need for a probabilistic approach to read mapping, we conducted a quality score analysis on the _prb.txt files from the ChIP and small RNA datasets [for a similar quality analysis, see Ossowski et al., (2008) where the authors evaluated an algorithm that produced spliced alignments of short sequence reads]. The _prb.txt file contains qualities Q that can be transformed into the probability P of each position being a particular nucleotide using the formula P=1−1/(10Q/10+1). We observed that only 71.6% (ChIP) and 75.9% (small RNA) of the base probabilities were >90%. This indicates that there are a large number of ambiguous bases in these data.
We also considered the number of reads that contain only high probability bases. Only 14.5% (ChIP) and 13.1% (small RNA) of the reads consisted of base probabilities that were all >90%. Additionally, only 47.2% (ChIP) and 54.1% (small RNA) reads contained only probabilities >50%. Therefore, the majority of probes contain at least one ambiguous base position. The profile of a random read that represents the typical profile of a read from these experiments is given in the Supplementary Material. This justifies the need for a probabilistic alignment algorithm to account for these ambiguities in an unbiased way. Furthermore, methods that rely solely on ‘called’ reads will give equal weight to each position regardless of the unequal uncertainty associated with the positions, potentially leading to less accurate results.
4.2 Sequencing error analysis
In a Solexa/Illumina sequencing experiment, 5′ and 3′ adapters are ligated to DNA/RNA fragments as part of the sequencing procedure. We have observed that multiple 5′ and 3′ adapters often join to each other without a DNA/RNA fragment between them. In this case, the sequencing reactions will return the 5′ adapter. A typical sequencing run will contain hundreds of thousands of such reads. These reads are ideal for evaluating the sequencing error rate for the experiment. For genomic DNA, the 5′ adapter sequence is 33 bases long, and for small RNA experiments the adapter is 20 bases long. These adapters are constructed so they do not perfectly match any genomic fragment. For example, the best human match and the small RNA adapter have only 10 bases in common. For this reason, we hypothesize that if we see a read that contains a close match to the adapter sequence, then the read is directly sequencing the adapter.
For our error analysis, we searched for any read (or part of the read) whose called sequence matched the adapter with three or less mismatches. We fit a logistic regression model to estimate the sequencing error rate for each read position, while also accounting for adapter synthesis and base composition error rates (model definition and details are given in the Supplementary Materials). Table 2 contains the estimates for α (transformed back to probabilities) for several read mapping methods. From the table, note that the methods that call the reads based on the most probable base (SeqMap, SOAP, MAQ, Bowtie) have the highest base-calling errors. This means that these algorithms are already at a disadvantage (higher error rate) before they even begin mapping the reads. Other methods first filter out the reads they deem ‘low-quality’ before mapping. For example, RMAP recommends filtering any read that does not have at least 10 consecutive bases with Q ≥ 8. In the case of the data presented here, RMAP filters out 30.2% (ChIP) and 35.8% (RNA) of the reads. It is interesting to note that GNUMAP's probabilistic representation of the read results in a lower error rate without removing any of the data. However, if one insists on filtering low-quality reads, GNUMAP's performance is further improved.
Table 2.
Sequencing error rates for the ChIP and RNA experiments
| Read mapping method | ChIP error rate | RNA error rate |
|---|---|---|
| (Algorithms) | (% data used) | (% data used) |
| Highest intensity base | — | 2.26 |
| (None) | (100) | |
| Most probable base (MPB) | 2.29 | 4.17 |
| (SeqMap, SOAP, MAQ, Bowtie) | (100) | (100) |
| MPB + chastity filtering | — | 4.45 |
| (Solexa Pipeline) | (97.8) | |
| MPB + quality filtering | 0.83 | 2.44 |
| (RMAP) | (69.8) | (64.2) |
| Probability mapping | 0.67 | 1.64 |
| (GNUMAP, Novo, Slider) | (100) | (100) |
| Probability + quality | 0.27 | 1.11 |
| (GNUMAP) | (69.8) | (64.2) |
Notice that the probabilistic mapping (GNUMAP) error rates are lower than those for other methods. In particular, the error rates for GNUMAP (using all the data) are lower using than those that incorporate base-calling and filtering (deleting 30–35% of the data).
Figure 2 shows these values (transformed to probabilities). At the beginning of the read, the GNUMAP error rate consistently outperforms all other methods for both datasets. Later in the read, GNUMAP's good performance is reduced because the base qualities are reduced. However, we consider this an advantage, as this means that the latter bases in the read are given less weight in the read mapping. Based on Figure 2b most of the other methods have higher error rates at the beginning of the read, meaning that the latter bases will have more weight in the mapping (because there are fewer mismatches), even though the read quality is lower for these bases.
Fig. 2.
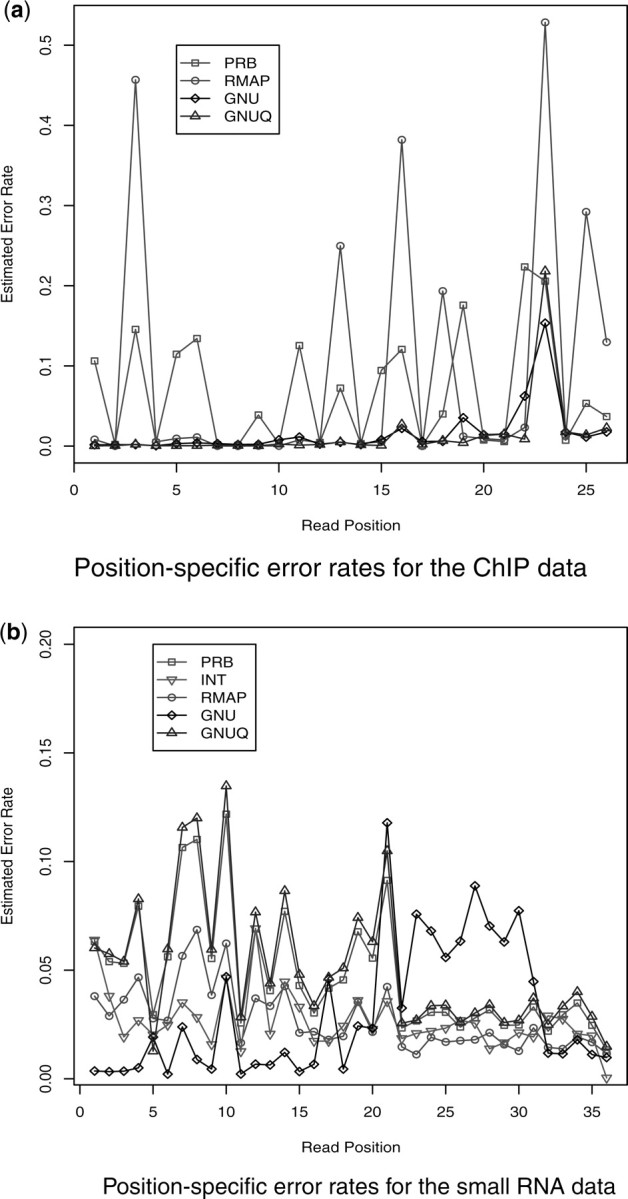
Position-specific base-calling error rates for the various methods (PRB = Most probable base (MPB), INT = Highest intensity base, RMAP = MBP+quality, GER = MPB+chastity, GNU = Probability, GNUQ = Probability+quality). Notice that GNU seems to have the best error rate for the beginning of the read in both cases. In (b) GNUMAP's performance worsens but the overall error rate is still smaller. Plus this means that GNUMAP will rely more on the beginning of the read while mapping. (a) Indicates that the RMAPQ filtering may not be highly effective against base-calling errors.
Figure 2a also indicates a potential problem for read filtering based on quality scores. It is clear that some of the error spikes from the _prb.txt file are removed, but others are amplified.
4.3 ChIP data
A useful test of a mapping algorithm occurs when real data are processed with unknown spike regions. For this example we use the ChIP-seq experiment attempting to identify the in vivo binding sites of the ETS1 transcription factor. We processed the reads using many of the algorithms to determine the sensitivity on real data. Figure 3 shows a spike found by GNUMAP that was not found by any of the other programs. This spike lies in the RALGPS2 gene promoter. The spike occurs in a highly repetitive region that is rich in ‘GGAA’ motifs that can potentially be bound by ETS1. This is an example of a repeat region that holds biological significance that would be missed by most methods.
Fig. 3.

The spike found by GNUMAP in the promoter region of the RALGPS2 gene was not found by any other program. This spike is located in a repeat region and has a ‘GGAA’ motif, indicating that this region may bound by ETS1. Note: The option for SOAP to report every match to a particular location was used here. This may not give an accurate representation of the method, but does show the noise that would occur if each matching location was reported.
4.4 Spike-in data
One of the most important end results of the mapping process is to identify ‘spike'regions in the reference genome with a significant number of read matches. To compare the accuracy of GNUMAP and other methods, we created a test dataset with a large number of known spikes and evaluated the ability of the applications to map the sequences to these spiked-in regions. Promoter regions (which often contain repetitive elements) from the 15 Mb C.elegans genome were used in the comparison dataset. Fifty base-pair windows from these promoter regions were selected as spiked-in regions, and a random 35 bp sequence within them was inserted into the benchmark dataset. To correct for errors in the benchmark dataset, these regions were mapped back to the genome, adding all repeat regions to the list of expected spikes. Probability and intensity files were created with the probability of 70% for each called base, simulating quality scores commonly seen in Solexa/Illumina data.
4.4.1 Visual analysis
The sequences were matched with several different applications available for comparison (RMAP, SOAP, SeqMap and Novocraft). After the sequences were matched, the resulting output files were parsed and converted into .sgr format capable for viewing and comparison using the Affymetrix IGB. The .sgr files were further processed, creating bins 100 bp in size to show density as well as magnitude. These files were visually compared with that of the original and that of GNUMAP (Fig. 4).
Fig. 4.

Benchmark real spikes (bottom) compared with SeqMap (Jiang and Wong, 2008), SOAP (Li,R. et al., 2008), RMAP (Smith et al., 2008), Novocraft (unpublished data) and GNUMAP. Benchmark data were constructed by sampling from 1000 promoter regions in the C.elegans genome. In (b) [an enlargement of the first boxed region in (a)], SeqMap incremented every location for reads from identical regions, producing a significant false positive spike. Attempting to remove false positives by discarding these reads, such as was done by RMAP, results in missing important information, as can be seen in (c) [an enlargement of the second boxed region in (a)]. Note: The intention of this figure is not to discuss the relative mapping capabilities of all currently implemented programs specifically, but to show the trend that would occur if each of these read-placement techniques were used.
Because the majority of these reads came from promoter regions with a significant number of repetitive regions, differences are apparent among the several algorithms. As discussed previously, the two classical options for aligning sequences from repetitive sequences is to either discard them or count them multiple times. As shown in Figure 4c, when discarding these sequences (default for most programs, and included with RMAP's run), repetitive regions are missed. In the test dataset, this region consists of two highly similar sequences which cannot be identified when discarding reads from repetitive regions.
The second option is to report all locations that show a significant alignment. As shown in Figure 4b, the abnormal spike in the data occurs as a result of SeqMap reporting all these reads. The mapping process of GNUMAP has also identified these repetitive locations, but because of the proportional nature of the scoring algorithm (step 4 of the algorithm), the spike is significantly reduced. As can be seen in the figure, reporting all the locations adds significant noise to the final output.
Instead of discarding repetitive reads or adding them to all matching locations in the genome, the posterior probability of the read matching a reference genome location should be proportionally added to all hit locations (the method employed by both Novocraft and GNUMAP). Using a probabilistic method allows for important regions to be expressed while not confusing the analysis with the overexpression of insignificant regions. Using this method, there is a relation between the amplitude at a given location and the number of reads in the original data that matched that location.
4.4.2 Quantitative analysis
In addition to a visual comparison, a procedure was developed for quantitatively comparing the differences in the mapping methods (Fig. 5). Each algorithm produced a set of spikes that were ordered according to the number of reads that mapped to that location. The top 50 spikes from each application were compared with the known top 50 spikes (from the benchmark dataset). For each ordered spike index, i, in an application, the number of top i spikes occurring in the top 50 spikes of the real dataset was plotted on the y-axis. If the plot followed the diagonal line, the algorithm would have an accuracy of 100%, identifying all correct spikes with no false positives. Falling too far below the diagonal would imply the occurrence of too many false positives which washed out the identification of true positives. RMAP and SeqMap were capable of identifying the highest two spikes (as evidenced by the fact that their lines follow the diagonal for the first two points); however, GNUMAP outperformed all other programs in correctly identifying fewer false positives after all 50 spiked-in regions were processed (Fig. 5a).
These results show that either recording or discarding all ambiguous locations results in a drastic decrease in positive versus false positive rates. For the SeqMap repeat-including algorithm, the high number of false positives washed out nearly every significant alignment, causing a very poor detection rate. RMAP's repeat-discarding algorithm reported so few locations that the true positive rate was significantly low. The only other algorithm that was capable of approaching the accuracy of statistical mapping software was SOAP, with a detection rate nearly equal to that of Novocraft and GNUMAP. However, when reviewing locations such as in Figure 4c, it becomes clear that, while the method employed by the SOAP algorithm is capable of recognizing true positive sequence spikes, the amplitude at these locations is often incorrect.
In this spike-in comparison, Novocraft and GNUMAP performed similarly because they both utilize a posterior probabilistic scoring method (as described in step 4 in Section 3). For this reason, both algorithms are correctly able to discard spurious match locations and include genuine spikes. However, GNUMAP's implementation of a probabilistic Needleman–Wunsch alignment, incorporating the probabilities of every base at each position, is able to out-perform Novocraft in several occasions (see Supplementary Material for further analysis).
5 PERFORMANCE ANALYSIS
Although accuracy is probably the most important feature of a mapping program, the speed of an algorithm is also important. Table 3 presents the results from a benchmark test of several mapping programs. The ‘Benchmark Data’ columns in the table show both the time to perform the mapping of 100K reads on a single chromosome as well as the number of reads correctly mapped. The reads were sampled from both the promoter and genomic regions of C.elegans chromosome I, with bias given to promoter regions to introduce spikes in the data. Various base probabilities (up to three on each read) were changed in order to simulate miscalled bases. We included the ‘*’ for the Slider time to indicate that the result was obtained using different hardware with similar performance since Slider could not run in the same 64-bit environment as the other tests did. The ‘*’ on MAQ, Novocraft and RMAP indicates that we were not able to test using multiple processors. The other programs were able to use the two quad-core 2.5 GHz Xeon processors available on the machine. The ‘Human Genome’ columns show the time to map an actual Solexa lane (11 000 000 reads of input DNA) and the corresponding number of reads mapped to the whole human genome.
In this dataset, GNUMAP was able to correctly map ∼9000 more reads (∼15% increase) than the next best methods (Bowtie, MAQ and SOAP). However, GNUMAP's increased accuracy does come at a cost. While GNUMAP is faster than SeqMap and Slider and comparable with MAQ, GNUMAP runs at 1/2–1/8 the speed of Bowtie, SOAP, NovoAlign and RMAP. Performance optimizations are currently being implemented that should significantly improve GNUMAP performance. We should also note that, as a result of Slider's combinatorial expansion of the reads to account for mismatches, their algorithm produced more that 500 000 reads to map, but still only identifying mappings for 58 551 reads in its histogram.
6 CONCLUSION
Next-Generation sequencing promises to revolutionize biological research by providing millions of bases of sequenced data per experiment. The more accurate approach used by GNUMAP can create a more accurate identification of spike regions in the reference genome.
Mapping algorithms that discard a large number of next-generation sequencing reads will bias the results to discriminate against repeat regions. By utilizing quality information from the raw reads and proportionally sharing the score for the read across matching regions of the genome, a better mapping can be performed.
When dealing with the identification of short motifs in promoter regions, it becomes even more necessary to use all available information in performing a mapping. With smaller datasets (mapping to a single chromosome), it is possible to discard reads in repeat regions while still finding short promoter motifs. However, when mapping a full Solexa/Illumina run of 10 M reads to a whole human genome of 4 Gb, the number of repeat regions found for a short promoter motif will increase. This will cause more reads to be discarded in the motif region, washing out the motif signal when compared with surrounding regions.
GNUMAP has been shown to provide comparatively more accurate mappings of spike regions to a reference genome. It is able to proportionally share a read that maps to a repeat region among match locations. By using methods such as a probabilistic Needleman–Wunsch and statistical mapping algorithms found in GNUMAP, available data can be more fully utilized, creating more accurate, cost-efficient results.
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Butler J, et al. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 2008;18:810–820. doi: 10.1101/gr.7337908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, et al. Mapping translocation breakpoints by next-generation sequencing. Genome Res. 2008;18:1143–1149. doi: 10.1101/gr.076166.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wong W. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24:2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Langmead B, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Malhis N, et al. Slider maximum use of probability information for alignment of short sequence reads and SNP detection. Bioinformatics. 2009;25:6–13. doi: 10.1093/bioinformatics/btn565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon J, Moran N. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc. Natl Acad. Sci. USA. 2007;104:19392–19397. doi: 10.1073/pnas.0708855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen T, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin R, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18:610–621. doi: 10.1101/gr.7179508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, et al. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008;18:2024–2033. doi: 10.1101/gr.080200.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P, et al. Comparing expression profiles of genes with similar promoter regions. Bioinformatics. 2002;18:1576–1584. doi: 10.1093/bioinformatics/18.12.1576. [DOI] [PubMed] [Google Scholar]
- Smith A, et al. Using quality scores and longer reads improves accuracy of Solexa read mapping. BMC Bioinformatics. 2008;9:128–138. doi: 10.1186/1471-2105-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Helden J. Metrics for comparing regulatory sequences on the basis of pattern counts. Bioinformatics. 2004;20:399–406. doi: 10.1093/bioinformatics/btg425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.