

Incorporation of multiple enrichment biomarkers into prospective clinical trials is an active area of investigation. This report examines the factors that correlate with successful clinical trial enrollment in a large molecular prescreening program in advanced colorectal cancer.
Keywords: prescreening, molecular, colorectal cancer, targeted, screening
Abstract
Background
Incorporation of multiple enrichment biomarkers into prospective clinical trials is an active area of investigation, but the factors that determine clinical trial enrollment following a molecular prescreening program have not been assessed.
Patients and methods
Patients with 5-fluorouracil-refractory metastatic colorectal cancer at the MD Anderson Cancer Center were offered screening in the Assessment of Targeted Therapies Against Colorectal Cancer (ATTACC) program to identify eligibility for companion phase I or II clinical trials with a therapy targeted to an aberration detected in the patient, based on testing by immunohistochemistry, targeted gene sequencing panels, and CpG island methylation phenotype assays.
Results
Between August 2010 and December 2013, 484 patients were enrolled, 458 (95%) had a biomarker result, and 157 (32%) were enrolled on a clinical trial (92 on biomarker-selected and 65 on nonbiomarker selected). Of the 458 patients with a biomarker result, enrollment on biomarker-selected clinical trials was ninefold higher for predefined ATTACC-companion clinical trials as opposed to nonpredefined biomarker-selected clinical trials, 17.9% versus 2%, P < 0.001. Factors that correlated positively with trial enrollment in multivariate analysis were higher performance status, older age, lack of standard of care therapy, established patient at MD Anderson, and the presence of an eligible biomarker for an ATTACC-companion study. Early molecular screening did result in a higher rate of patients with remaining standard of care therapy enrolling on ATTACC-companion clinical trials, 45.1%, in contrast to nonpredefined clinical trials, 22.7%; odds ratio 3.1, P = 0.002.
Conclusions
Though early molecular prescreening for predefined clinical trials resulted in an increase rate of trial enrollment of nonrefractory patients, the majority of patients enrolled on clinical trials were refractory to standard of care therapy. Within molecular prescreening programs, tailoring screening for preidentified and open clinical trials, temporally linking screening to treatment and optimizing both patient and physician engagement are efforts likely to improve enrollment on biomarker-selected clinical trials.
Clinical Trials Number
The study NCT number is NCT01196130.
introduction
As our understanding of the heterogeneity of cancer has grown, efforts to develop anticancer agents likely to be active in patients with specific genomic alterations have increased. The recent approvals of agents targeting HER2 in breast cancer, ALK fusions in lung cancer, and BRAF V600E in melanoma are examples of the success of such efforts.
As drug development utilizing biomarker enrichment attempts to incorporate earlier phase clinical trials and to engage less prevalent molecular alterations, the challenges of identifying the relevant biomarker-positive clinical trial eligible patients increase. A number of molecular prescreening approaches have been proposed to address these issues [1, 2]. Recently, a limited number of reports from prospective clinical trials incorporating molecular prescreening have been published and have consistently demonstrated low rates of biomarker-selected clinical trial enrollment ranging from 0% to 26% of enrolled patients [3–8].
The Assessment of Targeted Therapies Against Colorectal Cancer (ATTACC) clinical trial was designed to serve as an umbrella molecular screening program for fluoropyrimidine refractory metastatic colorectal cancer (CRC) to facilitate enrollment on companion clinical trials that enriched or selected the biomarker of interest for experimental drug therapy. The companion studies for ATTACC were preselected and designed as early proof of principle phase I or II trials in which clear demonstration of activity based on response rate would provide activity signals within each enriched group. This report describes the initial experience with this umbrella molecular screening program and evaluates factors that correlate with successful clinical trial enrollment.
methods
ATTACC umbrella screening clinical trial and companion clinical trials
Patients with metastatic or unresectable/locally advanced CRC, Karnofsky performance status ≥60, age ≥18 years, and prior treatment with fluoropyrimidine-based chemotherapy (or adjuvant relapse <6 months) were eligible for enrollment on the ATTACC (NCT01196130) umbrella study. All patients provided written consent for this University of Texas MD Anderson Cancer Center (MDACC) institutional review board approved protocol.
All ATTACC-companion clinical trials required written inclusion in the ATTACC umbrella trial protocol with biomarker eligibility and prioritization clearly defined. Over the 40-month time period studied in this report for the ATTACC umbrella trial, 10 different ATTACC-companion clinical trials existed (supplementary Tables S1 and S2, available at Annals of Oncology online). The predefined ATTACC enrichment biomarkers for the companion clinical trials were CIMP-high, PTEN loss by immunohistochemistry (IHC), BRAF V600E, PI3KCA activating mutations, NRAS activating mutations, and elevated serum fibroblast growth factor (FGF). Until April 2012, a MassARRAY platform was utilized for hotspots in 11 cancer genes and an Ampli-Seq 46 or 50 gene cancer panel using Ion Torrent PGM Sequencer (Life Technologies, CA) was utilized (supplementary Methods, available at Annals of Oncology online).
patients and data collection
Data were collected prospectively regarding demographics, prior chemotherapy treatment, tumor specimen acquisition and testing, molecular results, study allocation, and death. Patients were considered refractory at the time of ATTACC enrollment if they had previously been treated with oxaliplatin, irinotecan, anti-EGFR therapy if KRAS wildtype, or regorafenib after November 2012. RECIST v1.1 radiographic responses and date of progression for clinical trial enrolled patients were collected from each clinical trial. Retrospective review of patient records was done to classify reasons for non-ATTACC enrollment.
As the panel of tested ATTACC biomarkers changed over time depending on open companion clinical trials, biomarker testing success was defined as the ability to obtain results for any one of the predetermined biomarkers of interest. Physician utilization rate of ATTACC was defined as the number of patients enrolled by a single physician in ATTACC over the study time period divided by the number of new CRC consultations determined though physician billing codes over the same time period.
statistical methods
Fisher's exact test and Wilcoxon rank sum test were used to compare patient characteristics between groups, and logistic regression analysis was used to determine the association of clinical trial enrollment with patient characteristics. The Kaplan–Meier method was used to estimate the probability of overall survival (OS) and progression-free survival (PFS). For clinical trial-treated patients, a 1:1 propensity score matching method was applied to balance the potential cofounders, which included age, distance, gender, race, MDACC physician, performance status, number of prior lines, standard-of-care agent remaining, and status of the 7 ATTACC biomarkers. R package MatchIt with nearest neighbor matching method was used for the 1:1 matching. Seventy-four of the 157 patients were matched. Log-rank test and Cox proportional hazards models were applied for the time-to-event outcome analysis. All statistical analyses were two-sided and were carried out using S-Plus software (TIBCO Software, Inc., Palo Alto, CA) and SAS software (SAS Corporate, Cary, NC).
results
patient and biomarker characteristics
Between August 20, 2010 and December 31, 2013, 484 patients were enrolled to the ATTACC umbrella study Figure 1. The characteristics of the overall enrolled population were a median age of 56 years (range 20–81 years), 57% male gender, and 93% KPS ≥ 80. Ethnicity was 73% Caucasian, 12% Hispanic, 9% African-American, and 4% Asian reflecting the ethnic diversity at our institution. The median OS for the entire cohort was 9.4 months (95% confidence interval 8.7–10.3).
Figure 1.
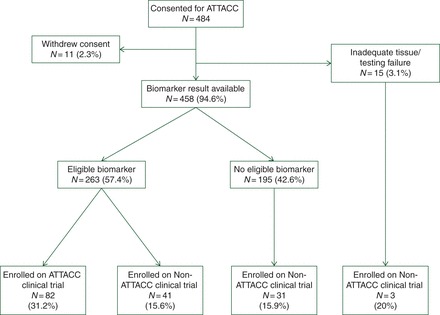
Diagram of patient testing and trial allocation on ATTACC. The eligible biomarker population was defined as a biomarker result that met an ATTACC-companion trial eligibility.
A biomarker result was available for 458 patients (94.6%) and the inability to obtain at least one of the ATTACC-tested biomarkers due to insufficient tumor tissue occurred in 15 cases (3.1%). The median time from ATTACC consent to result for all tested biomarkers was 32 days, with an interquartile range of 25–43. This reflected the slow turn-around time for CIMP testing as PIK3CA and PTEN testing were 21 and 22 days, respectively. In part, this delay reflected the median time of 8 days to obtain outside paraffin tissue, with a longer time to obtain outside tissue statistically associated with longer time to biomarker result, P < 0.001. Forty-five percent of patients were refractory to standard-of-care therapy at the time of ATTACC consent. Tumor tissue analyzed was from a metastatic site in 25.4% cases.
Biomarker alteration rates for all tested biomarkers are shown in Figure 2. A total of 263 patients, 54.3%, had at least one biomarker alteration making them eligible for one of the 10 predefined ATTACC-companion clinical trials. The majority of patients enrolled on ATTACC-companion studies were selected based on either CIMP-high status or the presence of a BRAF V600E mutation, which were biomarkers and companion trials that were available for the majority, 98% and 79% respectively, of the study time period (supplementary Table S1, available at Annals of Oncology online).
Figure 2.
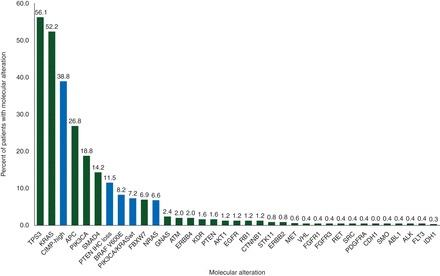
Percent of molecular alterations in all tested cancers with light gray (blue online) representing alterations targeted by ATTACC-companion studies. The number of patients tested is 246 except for KRAS (n = 469), CIMP (n = 415); BRAF V600E (n = 391); PTEN IHC (n = 383); PIK3CA (n = 362); NRAS, AKT1, MET, GNAS, and IDH1 (n = 332). Results represent sequencing of hotspot regions as described in supplementary Methods, available at Annals of Oncology online.
enrollment on to clinical trials
A total of 157 of 484 consented patients (32.4%) enrolled on a clinical trial (Figure 1). The majority of patients, 92 (59%), enrolled on biomarker-selected clinical trials. Enrollment on to biomarker-selected clinical trials outside of the ATTACC-companion clinical trials was rare, with only 10 patients (2.1%) enrolling on non-ATTACC biomarker-selected clinical trials. Thus, enrollment on biomarker-selected clinical trials was ninefold higher for ATTACC-companion clinical trials as opposed to non-ATTACC biomarker-selected clinical trials, 17.9% versus 2.1%, P < 0.001.
The 82 ATTACC-companion clinical trial enrolled patients represented 31.2% of the 263 ATTACC biomarker eligible patients. Interestingly, the frequency of enrollment on non-ATTACC clinical trials was comparable across all populations; biomarker eligible 15.6%, nonbiomarker eligible patients 15.9%, and no biomarker result 20%.
The 75 patients that enrolled on non-ATTACC clinical trials were enrolled across 29 different clinical trials, supplementary Tables S2 and S3, available at Annals of Oncology online. Study enrollment was biomarker based in 10 patients, based in part on the ATTACC-associated testing: ALK mutation (1), C-MET expression (1), PIK3CA mutation (3) NRAS (1), and KRAS mutation (4).
factors correlating with enrollment on clinical trials
In an effort to explore the factors that correlated with clinical trial enrollment within a molecular prescreening study, both univariate and multivariate analyses were conducted, Table 1. In multivariate modeling, factors correlating with enrollment on a clinical trial were a KPS > 80, lack of standard-of-care agents, an ongoing patient–physician relationship as reflected by a longer time from MDACC presentation to ATTACC enrollment, older age, and an eligible biomarker for an ATTACC-companion study, Table 1. Though not significant in multivariate modeling, a shorter travel distance from MDACC correlated with clinical trial enrollment.
Table 1.
Factors correlating with enrollment on a therapeutic clinical trial
| Enrolled in a clinical trial |
Multivariate analysis |
|||||
|---|---|---|---|---|---|---|
| No, N = 304 (%) |
Yes, N = 154 (%) |
P value | OR | 95% CI | P value | |
| Median age, years (range)a | 54 (20–81) | 59 (26–76) | 0.007 | 1.92 | 1.2–3.1 | 0.007 |
| Median distance from MDACC, miles (range)a | 278.5 (2–7890) | 200 (2–7890) | 0.02 | |||
| Median duration of metastatic disease, months (range) | 20.7 (0–112.4) | 20.6 (0.4–115) | 0.46 | |||
| Gender | ||||||
| F | 137 (45.1) | 58 (37.7) | 0.13 | |||
| M | 167 (54.9) | 96 (62.3) | ||||
| KPS | 0.02 | 2.03 | 1.28–3.24 | 0.003 | ||
| >80 | 118 (39.3) | 78 (51.7) | ||||
| 60–80 | 182 (60.7) | 73 (48.3) | ||||
| Race | 0.82 | |||||
| White | 223 (73.4) | 111 (72.1) | ||||
| Non-white | 81 (26.6) | 43 (27.9) | ||||
| Number of prior lines of therapy | 0.018 | |||||
| ≤2 | 163 (53.6) | 64 (41.6) | ||||
| >2 | 141 (46.4) | 90 (58.4) | ||||
| Standard-of-care agent remaining | <0.001 | 3.31 | 2.08–5.27 | <0.001 | ||
| No | 115 (37.8) | 101 (65.6) | ||||
| Yes | 189 (62.2) | 53 (34.4) | ||||
| Physician ATTACC enrollment rate | 0.3 | |||||
| <20% | 108 (39.6) | 68 (45) | ||||
| ≥20% | 165 (60.4) | 83 (55) | ||||
| Time from first medical oncology consultation to ATTACC consent | <0.001 | 2.96 | 1.64–5.21 | <0.001 | ||
| ≥7 days | 231 (70.6) | 136 (86.6) | ||||
| <7 days | 96 (29.4) | 21 (13.4) | ||||
| Eligible ATTACC biomarker | <0.001 | 4.36 | 2.61–7.25 | <0.001 | ||
| Yes | 140 (46.1) | 123 (79.9) | ||||
| No | 164 (53.9) | 31 (20.1) | ||||
| ATTACC-companion study open | <0.001 | |||||
| Yes | 113 (37.2) | 85 (55.2) | ||||
| No | 191 (62.8) | 69 (44.8) | ||||
aIn multivariate models, value dichotomized by median age (≤60 versus >60) and median miles (≤265 versus >265), respectively.
We retrospectively explored the reasons for nonclinical trial enrollment within the 304 patients with a biomarker result who did not enroll on a clinical trial, Figure 3A. Within this cohort, the primary reason for nonenrollment was the lack of an eligible biomarker for one of the 10 predefined ATTACC-companion clinical trials, 164 patients (54%). Within the remaining 140 patients, the main reasons for nonenrollment were the treating physician opted for standard of care therapy in 26%, the lack of an opening in the ATTACC-companion studies in 18%, and patients opted for treatment elsewhere or were lost to further follow-up in 27%.
Figure 3.
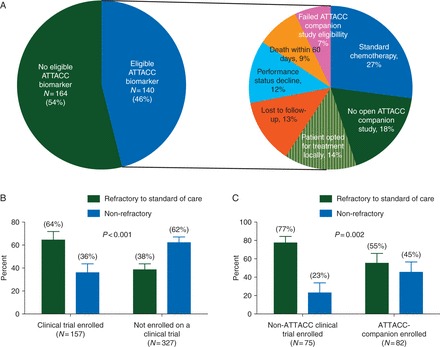
(A) Reasons for nonenrollment on a clinical trial for patients with an eligible biomarker (N = 301). (B) Clinical trial enrollment stratified by the availability of remaining standard-of-care agent for all patients with a biomarker result (N = 458). (C) ATTACC-companion and non-ATTACC clinical trial enrolled patients stratified by the availability of remaining standard-of-care agent (N = 157). Error bars represent 95% confidence intervals.
Of the patients who were enrolled on clinical trials, we sought to explore the clinical factors that correlated with enrollment on ATTACC-companion clinical trials as opposed to non-ATTACC trials, Figure 3B–C. Interestingly, patients and physicians were more willing to enroll into ATTACC-companion studies when compared with non-ATTACC studies before exhausting all available standard-of-care therapies, 45.1% versus 22.7%, respectively [odds ratio (OR) 3.1, P = 0.002]. Also physicians who had a higher ATTACC utilization rate, defined as enrolling >20% of their CRC patients on ATTACC, enrolled more patients on ATTACC-companion, 62.2%, as opposed to non-ATTACC trials, 36.6% (OR 2.0, P = 0.02).
outcome of ATTACC-companion and non-ATTACC clinical trial-treated patients
When comparing the 157 patients treated on clinical trials with the 327 patients not treated on a clinical trial, there was no significant difference in the median OS, 10.2 versus 8.9 m, respectively, P = 0.49. Within the 157 clinical trial-treated patients, partial responses were seen in 5 patients on ATTACC-companion studies and in 1 patient on non-ATTACC studies, P = 0.21. All the responses were seen in patients that were enrolled on biomarker-selected clinical trials, P = 0.08. The median OS and PFS for patients enrolled to ATTACC-companion and non-ATTACC clinical trials was 10.5 versus 9.4, P = 0.018 and 3.3 versus 2.1, P = 0.018, respectively. Due to the confounding prognostic effect of biomarker selection between the two groups, we carried out propensity matching to further explore OS and PFS. While OS did not differ between patients enrolled on ATTACC-companion and non-ATTACC clinical trials, 9.4 versus 8.3 months, P = 0.57, an improvement for PFS was seen, 3.5 versus 2.1 months, P = 0.04.
discussion
This study reports the results for a large single-center experience with a prospective molecular prescreening umbrella protocol. Through this effort, 32.4% of patients were enrolled on clinical trials, of which 59% enrolled on biomarker-selected clinical trials. Enrollment on biomarker-selected clinical trials was almost entirely dependent on the predefined ATTACC-companion trials, as enrollment on non-ATTACC biomarker-selected studies occurred only in 2.1% of patients. These results strongly support the use of molecular screening within the context of a clinical trial program where predefined, open, and genomically relevant clinical trials are identified before molecular prescreening.
The design of the ATTACC prescreening program allowed for early screening once a CRC patient was fluoropyrimidine refractory. This design was intended to allow for early identification of patients for clinical trials. This approach resulted in early trial engagement with patients as ATTACC-enrolled patients, in comparison with non-ATTACC-enrolled patients, were significantly more likely to have standard-of-care agents remaining at the time of clinical trial enrollment. Inherent within this approach is the ability for patients to continue other therapy while awaiting biomarker results. This allowance represented the major clinical reason for biomarker eligible patients not enrolling on ATTACC, as the retrospective reason for nonenrollment was continuation of standard chemotherapy, continuation of treatment locally, or lost to follow-up in 54% of patients. In addition, the most important factor in clinical trial enrollment aside for an eligible ATTACC biomarker, OR 4.4, was the lack of standard-of-care therapy, OR 3.3. Nearly half of the patients, 45%, enrolled in ATTACC once all standard-of-care agents were exhausted. Therefore, our results suggest that early molecular screening of patients will increase enrollment of nonrefractory patients but results in a reduced overall likelihood that a biomarker-positive patient will enroll on a clinical trial in comparison with screening conducted in a refractory setting. This suggests that molecular prescreening programs should attempt to improve the temporal link of screening to therapeutic clinical trial enrollment. Improved linkage would also address the second most common cause for non-trial enrollment within the biomarker eligible population; the lack of an open ATTACC-companion clinical trial in 18% of patients.
The finding of increased biomarker-selected enrollment within ATTACC-companion studies when compared with non-ATTACC studies was not surprising given the inherent design of the ATTACC screening program; however, the magnitude of this effect was surprising. Enrollment on biomarker-selected clinical trials outside of the ATTACC-companion studies was rare: 10 patients. This occurred despite 246 of our patients, 50.8%, undergoing testing with a 46- or 50-gene cancer panel that enabled the acquisition of not only the predefined ATTACC biomarkers but also additional potentially actionable mutations. Potential explanations for this observation are likely the limited availability of genomically relevant clinical trials, off-protocol use of targeted therapy, and the use of a moderate size mutation panel that was not enriched for CRC targets.
Within the biomarker eligible population, the 31.2% (82 of 263) enrollment rate to the ATTACC-companion clinical trials compares favorably to other recently reported molecular prescreening clinical trials [3–8]. As reported by other studies, the key determinant for nonenrollment was the lack of an eligible biomarker for the pre-specified ATTACC-companion studies. Indeed, trials were available for a minority of the abnormalities detected (Figure 2). It is notable that the rate of actionable DNA mutations defined by ATTACC was only 22%, despite the higher prevalence of traditional ‘un-targetable’ mutations in APC, TP53, and KRAS. Incorporating multiple methods of genomic alteration enabled a 57% eligible biomarker rate within ATTACC, which is similar to other reported prescreening clinical trials in which that rate of actionable genomic alterations has ranged from 65% to 31% [3–8].
Limitation of the ATTACC design was the use of previously collected paraffin tissue for biomarker analysis, which may not account for clonal evolution over time, though neither the site of tissue collection (primary tumor versus metastatic site) nor the proximity of the sample collection to enrollment impacted the biomarker frequencies (supplementary Table S4, available at Annals of Oncology online). In addition, the reliance upon single common molecular alterations may not be sufficient to capture the underlying biological complexities of CRC. Though the time to biomarker result was long in ATTACC, a shorter time to biomarker result did not statistically correlate with an increase in ATTACC-companion clinical trial enrollment. Future efforts should minimize this time to biomarker result to a more clinically acceptable timeframe, which may reduce clinical trial nonenrollment. In addition, the rate of enrollment on ATTACC-companion clinical trials may be underestimated as additional patients may enroll with increasing follow-up time. Time-to-event analyses are potentially biased by both the differing biomarker distributions within compared groups and the potential for both predictive and prognostic effects from the biomarkers.
Our findings are similar to the prior reports describing performance status, physician engagement, and available trials as important factors correlating with clinical trial enrollment [9–11]. Physician engagement as reflected by both a longer patient–physician relationship and higher physician utilization of the ATTACC prescreening program significantly correlated with clinical trial enrollment. Such engagement may reflect the improved incorporation of upfront discussions with patients related to the implications of the uncertain benefit from phase I and II clinical trial participation, travel, and cost.
Though molecular prescreening represents an inherently inefficient method of clinical trial enrollment, the work presented here demonstrates the successful implementation of a program that prescreens for predefined clinical trials. This work demonstrates that molecular selection does not alleviate standard barriers to clinical trial enrollment. Future efforts within prescreening programs should focus upon temporally linking molecular screening and therapeutic clinical trail enrollment, increasing the number of companion studies, optimizing the time studies are available and open, and improve patient communication regarding the implications of a prescreening program aimed at clinical trial enrollment. While novel therapies are exploring low-prevalence mutations, amplifications, or fusions in CRC without engagement upon more common molecular alterations, these efforts are unlikely to substantially improve the efficiency of future biomarker enrichment strategies in CRC.
funding
This work was supported in part by the Sheikh Khalifa Al Nahyan Ben Zayed Institute for Personalized Cancer Therapy, Cancer Prevention Research Institute of Texas, and the MD Anderson Cancer Center Support grant (P30 CA016672).
disclosure
GM has research support funding from AstraZeneca and GlaxoSmithKline and is on the scientific advisory board for AstraZeneca. MO has research funding from Celgene and Bristol Meyers Squibb. All remaining authors have declared no conflicts of interest.
Supplementary Material
acknowledgements
The authors thank Elena Vess for secretarial assistance. They also thank Laurel Deaton, Shanequa Manuel, and Constance Dennis for assistance in data acquisition.
references
- 1. Andre F, Delaloge S, Soria JC. Biology-driven phase II trials: what is the optimal model for molecular selection? J Clin Oncol 2011; 29: 1236–1238. [DOI] [PubMed] [Google Scholar]
- 2. Rodon J, Saura C, Dienstmann R et al. . Molecular prescreening to select patient population in early clinical trials. Nat Rev Clin Oncol 2012; 9: 359–366. [DOI] [PubMed] [Google Scholar]
- 3. Meric-Bernstam F, Brusco L, Shaw K et al. . Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol 2015; 33: 2753–2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andre F, Bachelot T, Commo F et al. . Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER). Lancet Oncol 2014; 15: 267–274. [DOI] [PubMed] [Google Scholar]
- 5. Kris MG, Johnson BE, Berry LD et al. . Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014; 311: 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chantrill LA, Nagrial AM, Watson C et al. . Precision medicine for advanced pancreas cancer: the Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clin Cancer Res 2015; 21: 2029–2037. [DOI] [PubMed] [Google Scholar]
- 7. Le Tourneau C, Delord JP, Goncalves A et al. . Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 2015; 16: 1324–1334. [DOI] [PubMed] [Google Scholar]
- 8. Lopez-Chavez A, Thomas A, Rajan A et al. . Molecular profiling and targeted therapy for advanced thoracic malignancies: a biomarker-derived, multiarm, multihistology phase II basket trial. J Clin Oncol 2015; 33: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lara PN Jr, Higdon R, Lim N et al. . Prospective evaluation of cancer clinical trial accrual patterns: identifying potential barriers to enrollment. J Clin Oncol 2001; 19: 1728–1733. [DOI] [PubMed] [Google Scholar]
- 10. Go RS, Frisby KA, Lee JA et al. . Clinical trial accrual among new cancer patients at a community-based cancer center. Cancer 2006; 106: 426–433. [DOI] [PubMed] [Google Scholar]
- 11. Kaplan CP, Napoles AM, Dohan D et al. . Clinical trial discussion, referral, and recruitment: physician, patient, and system factors. Cancer Causes Control 2013; 24: 979–988. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.