

Abstract
Motivation
A central task of bioinformatics is to develop sensitive and specific means of providing medical prognoses from biomarker patterns. Common methods to predict phenotypes in RNA-Seq datasets utilize machine learning algorithms trained via gene expression. Isoforms, however, generated from alternative splicing, may provide a novel and complementary set of transcripts for phenotype prediction. In contrast to gene expression, the number of isoforms increases significantly due to numerous alternative splicing patterns, resulting in a prioritization problem for many machine learning algorithms. This study identifies the empirically optimal methods of transcript quantification, feature engineering and filtering steps using phenotype prediction accuracy as a metric. At the same time, the complementary nature of gene and isoform data is analyzed and the feasibility of identifying isoforms as biomarker candidates is examined.
Results
Isoform features are complementary to gene features, providing non-redundant information and enhanced predictive power when prioritized and filtered. A univariate filtering algorithm, which selects up to the N highest ranking features for phenotype prediction is described and evaluated in this study. An empirical comparison of pipelines for isoform quantification is reported by performing cross-validation prediction tests with datasets from human non-small cell lung cancer (NSCLC) patients, human patients with chronic obstructive pulmonary disease (COPD) and amyotrophic lateral sclerosis (ALS) transgenic mice, each including samples of diseased and non-diseased phenotypes.
Availability and Implementation
https://github.com/clabuzze/Phenotype-Prediction-Pipeline.git
Contact
clabuzze@iastate.edu, antoniom@bc.edu, watsondk@musc.edu, andersonpe2@cofc.edu
1 Introduction
Comprehensive analysis of high-throughput sequencing data remains a challenging task due to the inherent complexities of genetic transcript analysis from next-generation sequencing data (Kanitz et al., 2015). An especially difficult aspect is the accurate estimation of gene and isoform transcript expression in RNA-Seq data (Liu et al., 2014). Several methods have been developed which claim to approach the problem with an empirically superior algorithm; however, an objective analysis using non-simulated data is often difficult (Leng et al., 2013; Trapnell et al., 2011; Wang and Cairns, 2014).
In RNA-Seq, ‘reads’ represent sequenced transcript fragments. The total number of reads aligning to each transcript is quantified as counts. It is difficult to verify isoform expression because correlation with phenotypes is rarely annotated in genomic databases and comprehensive validation using PCR is unrealistic. An optimal pipeline for isoform expression quantification is necessary in order to apply the full potential of high-throughput sequencing data to biomedical analysis (Kanitz et al., 2015). However, distinct algorithm design and the loss of biological validation make analyses of isoform expression algorithms difficult (Leng et al., 2013; Trapnell et al., 2011; Wang and Cairns, 2014).
Processing samples using RNA-Seq technology captures the expression of genes and isoforms, generated by alternative splicing (Li and Dewey, 2011; Trapnell et al., 2011; Wang and Cairns, 2014). Previous methods of comparing transcript quantification and differential expression techniques have relied on the analysis of false-discovery rates and commonly identified alternative splicing patterns (Liu et al., 2014). An objective method to compare algorithms that quantify transcript expression is to measure the ability of machine learning techniques to predict the phenotype of biological samples processed with each algorithm (Anderson et al., 2014). Prediction accuracy can be used as a metric to determine which tool most reliably quantifies expression.
Two of the most utilized transcript assembly and differential expression pipelines include Tophat/Cufflinks and RSEM/EBSeq (Leng et al., 2013; Trapnell et al., 2011). These approaches utilize varying statistical methods, each claiming to optimally address the challenge. SeqGSEA is another differential expression and alternative splicing platform that has yielded competitive results with respect to the two well-established pipelines (Wang and Cairns, 2014). The state-of-the-art nature of these tools provides a convincing argument to focus a comprehensive analysis on these three methods.
These tools quantify expression for a massive number of transcripts. Feature selection and filtering can reduce the massive number of gene and isoform features to a subset that efficiently represents the original data. Machine learning algorithms such as Sparse Partial Least Squares (Chun and Keleş, 2010), Elastic Net (Zou and Hastie, 2005) and Random Forest (Liaw and Wiener, 2002) may be overwhelmed by noisy datasets due to over-fitting. Therefore, a method to rank these features and select those which best represent the quantitative distance between phenotypes may increase prediction accuracy.
This article describes an empirically optimal method for phenotype prediction, revealing the critical nature of isoforms as features and recommending RSEM as a transcript quantification tool. Our most valuable predictors (MVP) filtering algorithm, increases prediction accuracy and suggests that filtering may allow researchers to focus validation on a relatively small number of transcripts. Our analysis shows that isoforms are complementary to genes, providing non-redundant information and enhanced predictive power.
An analysis of phenotype prediction utilizing both gene and isoform transcripts requires the investigation of distinct transcript quantification methods, a novel investigation of the feature engineering of count-based isoforms into fractional-based isoforms, the MVP univariate filtering method and the evaluation of multiple machine learning algorithms. Comparisons between these pipelines are reported in this paper and recommendations for the optimal pipeline are provided along with a R-based implementation of the pipeline and MVP filtering method.
2 Methods
Three datasets of varying size were used to compare the pipelines generated for this analysis and to empirically develop our MVP filtering method. Included were datasets from human non-small cell lung cancer (NSCLC) patients, human patients with chronic obstructive pulmonary disease (COPD) and samples of pure corticospinal motor neuron populations isolated from amyotrophic lateral sclerosis (ALS) transgenic mice, each including samples of diseased and non-diseased phenotypes. Feature engineering from count-based isoform expression to fractional-based isoform expression is mathematically defined, and we discuss the utility of filtering/feature selection, including the MVP filtering method. We describe the datasets in detail and outline the alignment and transcript quantification processes using the Tuxedo Pipeline, RSEM/EBSeq and SeqGSEA. Finally, the selected machine learning algorithms and differential expression are reviewed.
2.1 MVP filtering algorithm
The MVP method filters gene or isoform expression tables with two phenotypes and sample replicates to prioritize features for phenotype prediction. Before calculating distribution distance, MVP drops features with null or zero expression in any sample. Each feature in the cross-validation test set is guaranteed, therefore, to have non-zero expression, which improves estimates of the sample mean and variance. Filtering the entire dataset for non-zero values does not bias the phenotype distributions as it is permutation invariant. The MVP method only ranks features that have a P-value < α determined by a t-test between phenotypes of the training set samples.
For each significant feature, normal distributions P1 and P2 are modeled for the phenotypes with corresponding means μ1, μ2 and variances . The quantity r ranks features by distance between distributions such that increasing separation between means and decreasing total variance increases r score:
The quantity r orders features similar to P-values generated by the t-test. However, the r quantity is designed to rank features in order of value as predictors by quantifying the distance between phenotype distributions, which may result in a better selection of features compared to P-value alone.
MVP Algorithm:
Input dataset with phenotypes P1 and P2
Retain only features where all samples have non-zero expression
Retain only features where all samples have non-null expression
Perform a t-test on all remaining features between P1 and P2 training set samples
For all features with P-value < α: calculate quantity r using estimates of μ1, μ2 and from the training set phenotype distributions
Select the features with one of the N highest r values as predictors, where N is the number of desired features.
2.2 Feature engineering
In addition to the massive number of isoforms, the robustness of isoform data can be increased by engineering count-based isoform expression to fractional-based isoform expression. Gene expression G, count-based isoform expression C and fractional-based isoform expression F are defined as follows: let Cij be the total read count for isoform where Ij is the set of isoforms of gene , and J is the set of all genes. Read count of gene j is (1). Fractional-based expression of each isoform i of gene j is therefore (2).
| (1) |
| (2) |
Fractional-based isoform expression provides a normalization of isoform expression proportional to the corresponding gene expression. If the expression of all isoforms remained proportional in relation to the gene expression, fractional-based expression can retain the proportionality even in the case of extreme read counts in a sample. This may reduce the impact of samples with outlying read coverage which can impede the accurate estimation of phenotype distributions. Gene data may not be engineered into fractional data and therefore fractional-based isoform features may also be complementary to gene features.
2.3 Datasets
2.3.1 NSCLC
Non-small cell lung cancer RNA samples were taken from 21 patients with clinical outcomes determined by the American College of Surgery Oncology Group (Anderson et al., 2014). Ten of these patients were diagnosed as disease free and 11 were diagnosed with relapse within 3 years of initial surgical resection. A total of 100–200 ng of total RNA was used to prepare libraries using the Illumina protocol for the TruSeq RNA Sample Prep Kit. These RNA-Seq libraries were paired-end sequenced on a HiScanSQ with 2 × 100 cycles and three samples per lane. The quality and adapter content of the paired-end sequences was measured with FASTQC (Patel and Jain, 2012). Trimmomatic 0.33 (Bolger et al., 2014) removed the detected adapter content derived from the TruSeq2 Burnett Adapter Sequences while also trimming the ends of the sequences using the following settings: ILLUMINACLIP:TruSeq2.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:40.
2.3.2 COPD
A 189 sample RNA-Seq COPD dataset of 98 COPD patients and 91 patients with normal lung tissue was discovered in the NCBI GEODatasets Database using the search terms: ‘expression profiling by high throughput sequencing’ [DataSet Type]), 20:1000 [n samples], ‘lung’ (Kim et al., 2015). Ten replicates of each phenotype were randomly selected and the records were obtained using the SRAToolkit v2.5.2 to create a dataset similar in size to the NSCLC dataset (Leionen et al., 2011). This study focuses on the analysis of small/medium size datasets in terms of replicates, even though the COPD dataset offers the opportunity to increase the number of replicates used. These samples had previously been processed as.bam files aligned to the hg19 human genome (UCSC) using Tophat v2.0.0 and as paired end.fastq files for transcriptomic alignment using RSEM v1.2.25.
2.3.3 ALS
UCHL1-eGFP mice were generated to visualize and purify corticospinal motor neurons (CSMN) from the motor cortex, and CSMN identity of eGFP+ neurons was previously confirmed (Yasvoina et al., 2013). hSOD1G93A-UeGFP mice were generated by crossbreeding UCHL1-eGFP with hSOD1G93A mice at Northwestern University. Both healthy (n = 4) and diseased (n = 4) CSMN were isolated from motor cortex upon cortical dissociation and FACS-mediated purification approaches at postnatal day 90, using previously established protocols (Ozdinler and Macklis, 2006). The generated mRNA was converted to a cDNA library using reverse transcription. The samples were sequenced at Iowa State University on an Illumina HiSeq 2500 after cDNA library-prep using Nextera’s DNA Sample Preparation Kit. All eight samples were paired-end sequenced in one lane. The quality and adapter content of the paired-end sequences was measured with FASTQC (Patel and Jain, 2012). BBMap v35.85 was used to remove contaminated sequences (Bushnell, 2015). Trimmomatic 0.33 (Bolger et al., 2014) removed the detected adapter content and the sequences were trimmed using the following settings: ILLUMINACLIP:nextera.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:40.
2.4 Alignment and transcript quantification
The Tuxedo v2.2.1 pipeline begins with Tophat which runs Bowtie2 to align trimmed sequences to a reference genome (Trapnell et al., 2011). The human bowtie indices were created from the hg19 human genome (UCSC) and the mouse bowtie indices were created from the mm10 mouse genome (UCSC). Tophat v2.0.0 was run with the default settings. CuffLinks assembled the transcripts using the corresponding genome GTF file as a reference. CuffMerge condenses each sample’s transcripts into a set which can be compared across all samples and the resulting GTF file was used as a reference for the CuffQuant and CuffDiff steps (Trapnell et al., 2011).
RSEM v1.2.25 ran Bowtie2 to align reads to the transcriptome which was constructed from the human hg19 and mouse mm10 reference genomes and the corresponding GTF file which annotates gene and isoform transcripts. RSEM was run with the default settings, but Bowtie2 was selected for alignment rather than Bowtie. RSEM assembles the transcripts and calculates their abundance using rsem-calculate-expression (Li and Dewey, 2011). The data tables created for each dataset containing the expected counts of genes or isoforms were each piped into EBSeq and normalized (Leng et al., 2013).
SeqGSEA requires.bam files synthesized from genomic alignment (Wang and Cairns, 2014). The files created by Tophat were used to provide input for SeqGSEA. SeqGSEA calculates expression levels on exon counts using a supplemental Python script provided with the SeqGSEA R package. The exon counts were calculated from the Tophat.bam files and the human hg19 and mouse mm10 GTF files. SeqGSEA detects gene expression by totaling the expression of all exons in each gene.
2.5 Differential expression
Due to the variation in differential expression algorithms by the Tuxedo Pipeline, EBSeq and SeqGSEA, a t-test was used to identify differentially expressed transcripts quantified and normalized by each tool. SeqGSEA was particularly problematic because it does not provide P-values for its transcripts, rather ranks the transcripts in order of predicted biological relevance. Therefore, a t-test was used to identify differentially expressed features for input to the machine learning algorithms. The t-test provided consistent differential expression compared to using each tool’s individual differential expression algorithm, which would have otherwise confounded the analysis of each transcript quantification method.
2.6 Machine learning algorithms
The following machine learning algorithms were chosen to perform the phenotype predictions. Random Forest was implemented from the ‘randomForest’ R package (Liaw and Wiener, 2002). The Elastic Net was run using the ‘glmnet’ R package (Friedman et al., 2010; Zou and Hastie, 2005). Each pair of predictions was based on a cross-validated Elastic Net fit which automatically selected the optimal lambda level and feature number. The SPLS algorithm implementation came from the ‘mixOmics’ R package (Chun and Keleş, 2010; Dejean et al., 2011). Each pair of predictions was based on a cross-validation test to select the optimal eta and kappa parameters.
2.7 Evaluation
In order to empirically compare multiple pipelines using various methods of transcript quantification, feature engineering, filtering and machine learning algorithms, the prediction accuracy of each pipeline may be compared via receiver operating curve (ROC) analysis. The ROC can be generated from the sensitivity and specificity of phenotype prediction (Robin et al., 2011). For each pipeline comparison, cross-validation is used to create ROC curves and measure AUC. Below is an explanation of the leave-two-out cross-validation setup and summary of the pipeline comparison tests performed.
2.7.1 Cross-validation
To measure the predictive accuracy of each phenotype prediction pipeline, it is important to perform many predictions using the selected machine learning algorithms to quantify the sensitivity and specificity of the predictions. Using the pipelines for each dataset provides further information and comparison for each method. To perform leave-two-out cross-validation tests on each dataset, one sample of each phenotype is dropped iteratively while the remaining samples form the training set. Leave-two-out tests provide a robust estimation of the accuracy of each pipeline for phenotype prediction. The differential expression, filtering and machine learning steps are performed on the training set to select genes or isoforms that best represent the quantitative difference between the phenotypes. Then, the machine learning algorithm predicts the phenotypes of the dropped samples. By iterating through all possible training sets for a dataset, a robust analysis of the predictive accuracy of each pipeline is recorded in a ROC curve. This is generated from the sensitivity and specificity of the leave-two-out cross-validation test predictions. The AUC is a value between 0 and 1 that represents the accuracy of the predictions and is used in this study to compare the effectiveness of phenotype prediction pipelines.
2.7.2 Pipeline comparisons
Possible pipelines were permuted by selecting one option from each of the following steps. For each dataset, the 54 resulting pipelines were compared via leave-two-out cross-validation prediction tests.
Transcript Quantification Method: Cufflinks, RSEM, SeqGSEA
Feature Format: Genes, Count-based Isoforms, Fractional-based Isoforms
Filtering: None, MVP
Machine Learning Method: Random Forest, Elastic Net, SPLS
3 Results and discussion
To incrementally determine the optimal pipeline through analysis of the empirical data, the following questions must be answered:
Which method reliably quantifies transcripts by producing consistently high AUC scores?
Does the MVP filtering method enhance or decrease prediction accuracy?
Which feature produces the most accurate predictions?
Which machine learning algorithm most consistently performs better or equal to the others?
Are isoform features redundant or complementary to gene features?
The answers to these questions will be investigated in the following sections by analyzing the AUC scores generated via leave-two-out cross-validation prediction tests on each dataset.
3.1 Transcript quantification
Transcript expression quantification, the calculation of read abundance for both genes and isoforms, is a difficult task. The optimal statistical technique required for this task is still being explored (Leng et al., 2013; Trapnell et al., 2011; Wang and Cairns, 2014). One goal of this study is to determine whether Cufflinks, RSEM or SeqGSEA consistently produces the highest phenotype prediction accuracy as a result of a superior transcript quantification algorithm.
Our study compares the AUC scores generated by pipelines including each transcript quantification tool with and without the MVP filter method (Fig. 1). Pipelines using RSEM for transcript quantification and the MVP filtering method for feature selection had significantly higher AUC scores than pipelines using the other transcript quantification methods.
Fig. 1.
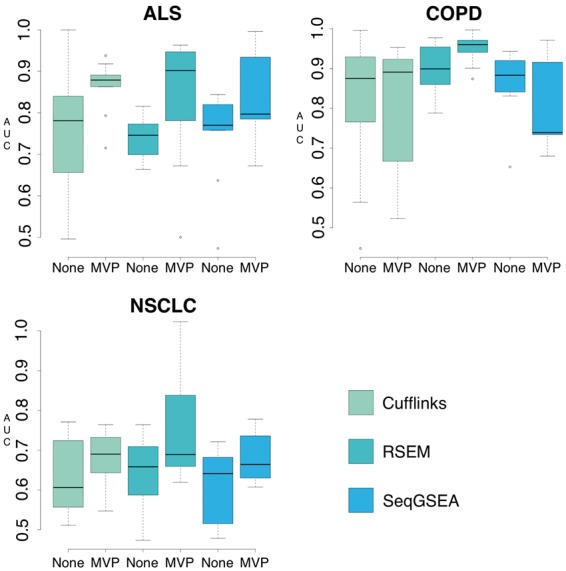
Phenotype prediction by pipelines using variable transcript quantification tools and filtering. AUC values were generated by running each dataset (NSCLC, ALS, COPD) through 54 pipelines that varied in transcript quantification tool, feature type (gene, isoform count, isoform fraction), use of filtering and machine learning algorithm (Random Forest, Elastic Net, SPLS). Predictive results are shown grouped by dataset, transcript quantification tool and use of filtering
We have compiled detailed descriptions of pipelines and corresponding AUC scores (Tables 1–3). For many of the pipelines processed using Cufflinks for transcript quantification, the AUC scores were comparable to those using RSEM when analyzing the ALS and COPD datasets (Tables 1 and 2). RSEM, however, performed better on the NSCLC dataset compared to both Cufflinks and SeqGSEA, especially when predictions were generated from fractional-isoform based data (Table 3). It is likely that AUC scores had large variability when using SeqGSEA due to the large number of exons in any dataset making even more difficult the challenge of selecting reliable features (Fig. 1).
Table 1.
ALS dataset analysis
| Transcript quantification | Feature type | Filter | Machine learning Alg. | AUC | Confidence Int. | Sens. | Spec. | Mean features* |
|---|---|---|---|---|---|---|---|---|
| Cufflinks | Genes | None | Elastic Net | 0.496 | 0.283–0.709 | 0.625 | 0.625 | 516 |
| Cufflinks | Genes | None | Random Forest | 0.766 | 0.558–0.973 | 1.000 | 0.750 | 516 |
| Cufflinks | Genes | None | SPLS | 0.606 | 0.391–0.820 | 0.688 | 0.688 | 516 |
| Cufflinks | Genes | MVP | Elastic Net | 0.918 | 0.809–1.000 | 1.000 | 0.813 | 38 |
| Cufflinks | Genes | MVP | Random Forest | 0.883 | 0.756–1.000 | 1.000 | 0.750 | 38 |
| Cufflinks | Genes | MVP | SPLS | 0.863 | 0.719–1.000 | 0.750 | 1.000 | 38 |
| Cufflinks | Isoform Count | None | Elastic Net | 0.781 | 0.601–0.962 | 0.875 | 0.813 | 1146 |
| Cufflinks | Isoform Count | None | Random Forest | 0.815 | 0.643–0.986 | 1.000 | 0.750 | 1146 |
| Cufflinks | Isoform Count | None | SPLS | 0.656 | 0.449–0.863 | 0.698 | 0.813 | 1146 |
| Cufflinks | Isoform Count | MVP | Elastic Net | 0.793 | 0.607–0.979 | 0.813 | 0.813 | 50 |
| Cufflinks | Isoform Count | MVP | Random Forest | 0.879 | 0.752–1.000 | 1.000 | 0.750 | 50 |
| Cufflinks | Isoform Count | MVP | SPLS | 0.715 | 0.516–0.913 | 0.750 | 0.813 | 50 |
| Cufflinks | Isoform Fraction | None | Elastic Net | 1.000 | 1.000–1.000 | 1.000 | 1.000 | 93 |
| Cufflinks | Isoform Fraction | None | Random Forest | 0.840 | 0.687–0.993 | 1.000 | 0.750 | 93 |
| Cufflinks | Isoform Fraction | None | SPLS | 0.938 | 0.815–1.000 | 0.938 | 1.000 | 93 |
| Cufflinks | Isoform Fraction | MVP | Elastic Net | 0.891 | 0.776–1.000 | 1.000 | 0.750 | 47 |
| Cufflinks | Isoform Fraction | MVP | Random Forest | 0.871 | 0.739–1.000 | 1.000 | 0.750 | 47 |
| Cufflinks | Isoform Fraction | MVP | SPLS | 0.938 | 0.815–1.000 | 0.938 | 1.000 | 47 |
| RSEM | Genes | None | Elastic Net | 0.699 | 0.489–0.909 | 0.750 | 0.875 | 631 |
| RSEM | Genes | None | Random Forest | 0.758 | 0.545–0.971 | 1.000 | 0.750 | 631 |
| RSEM | Genes | None | SPLS | 0.746 | 0.561–0.932 | 0.750 | 0.750 | 631 |
| RSEM | Genes | MVP | Elastic Net | 0.824 | 0.653–0.995 | 0.938 | 0.750 | 50 |
| RSEM | Genes | MVP | Random Forest | 0.947 | 0.880–1.000 | 1.000 | 0.750 | 50 |
| RSEM | Genes | MVP | SPLS | 0.500 | 0.265–0.735 | 0.438 | 0.938 | 50 |
| RSEM | Isoform Count | None | Elastic Net | 0.699 | 0.501–0.898 | 0.875 | 0.688 | 740 |
| RSEM | Isoform Count | None | Random Forest | 0.773 | 0.572–0.975 | 1.000 | 0.750 | 740 |
| RSEM | Isoform Count | None | SPLS | 0.746 | 0.566–0.926 | 0.813 | 0.750 | 740 |
| RSEM | Isoform Count | MVP | Elastic Net | 0.910 | 0.811–1.000 | 1.000 | 0.750 | 50 |
| RSEM | Isoform Count | MVP | Random Forest | 0.963 | 0.904–1.000 | 1.000 | 0.875 | 50 |
| RSEM | Isoform Count | MVP | SPLS | 0.672 | 0.451–0.892 | 0.625 | 0.938 | 50 |
| RSEM | Isoform Fraction | None | Elastic Net | 0.664 | 0.463–0.866 | 0.875 | 0.563 | 180 |
| RSEM | Isoform Fraction | None | Random Forest | 0.779 | 0.582–0.977 | 1.000 | 0.750 | 180 |
| RSEM | Isoform Fraction | None | SPLS | 0.816 | 0.643–0.990 | 0.875 | 0.813 | 180 |
| RSEM | Isoform Fraction | MVP | Elastic Net | 0.781 | 0.585–0.978 | 1.000 | 0.750 | 41 |
| RSEM | Isoform Fraction | MVP | Random Forest | 0.902 | 0.787–1.000 | 1.000 | 0.750 | 41 |
| RSEM | Isoform Fraction | MVP | SPLS | 0.949 | 0.871–1.000 | 1.000 | 0.875 | 41 |
| SeqGSEA | Genes | None | Elastic Net | 0.637 | 0.435–0.839 | 0.813 | 0.563 | 331 |
| SeqGSEA | Genes | None | Random Forest | 0.770 | 0.565–0.974 | 1.000 | 0.75 | 331 |
| SeqGSEA | Genes | None | SPLS | 0.820 | 0.666–0.975 | 0.813 | 0.750 | 331 |
| SeqGSEA | Genes | MVP | Elastic Net | 0.785 | 0.608–0.963 | 0.813 | 0.750 | 47 |
| SeqGSEA | Genes | MVP | Random Forest | 0.981 | 0.946–1.000 | 0.938 | 0.938 | 47 |
| SeqGSEA | Genes | MVP | SPLS | 0.750 | 0.553–0.947 | 0.813 | 0.750 | 47 |
| SeqGSEA | Exon Count | None | Elastic Net | 0.473 | 0.259–0.687 | 0.438 | 0.750 | 1181 |
| SeqGSEA | Exon Count | None | Random Forest | 0.770 | 0.565–0.974 | 1.000 | 0.750 | 1181 |
| SeqGSEA | Exon Count | None | SPLS | 0.820 | 0.647–0.994 | 0.688 | 1.000 | 1181 |
| SeqGSEA | Exon Count | MVP | Elastic Net | 0.906 | 0.781–1.000 | 0.938 | 0.875 | 50 |
| SeqGSEA | Exon Count | MVP | Random Forest | 0.996 | 0.985–1.000 | 1.000 | 0.938 | 50 |
| SeqGSEA | Exon Count | MVP | SPLS | 0.934 | 0.818–1.000 | 0.875 | 1.000 | 50 |
| SeqGSEA | Exon Fraction | None | Elastic Net | 0.844 | 0.701–0.986 | 0.813 | 0.813 | 2669 |
| SeqGSEA | Exon Fraction | None | Random Forest | 0.758 | 0.545–0.971 | 1.000 | 0.750 | 2669 |
| SeqGSEA | Exon Fraction | None | SPLS | 0.766 | 0.558–0.973 | 1.000 | 0.750 | 2669 |
| SeqGSEA | Exon Fraction | MVP | Elastic Net | 0.672 | 0.466–0.878 | 0.750 | 0.750 | 50 |
| SeqGSEA | Exon Fraction | MVP | Random Forest | 0.797 | 0.612–0.982 | 1.000 | 0.750 | 50 |
| SeqGSEA | Exon Fraction | MVP | SPLS | 0.785 | 0.590–0.980 | 1.000 | 0.750 | 50 |
*Mean number of transcripts selected as features per leave-two-out cross-validation test.
Table 2.
COPD dataset analysis
| Transcript quantification | Feature type | Filter | Machine learning Alg. | AUC | Confidence Int. | Sens. | Spec. | Mean features* |
|---|---|---|---|---|---|---|---|---|
| Cufflinks | Genes | None | Elastic Net | 0.996 | 0.990–1.000 | 0.980 | 0.990 | 1297 |
| Cufflinks | Genes | None | Random Forest | 0.910 | 0.867–0.953 | 0.930 | 0.810 | 1297 |
| Cufflinks | Genes | None | SPLS | 0.817 | 0.757–0.876 | 0.680 | 0.840 | 1297 |
| Cufflinks | Genes | MVP | Elastic Net | 0.953 | 0.929–0.978 | 0.860 | 0.890 | 50 |
| Cufflinks | Genes | MVP | Random Forest | 0.935 | 0.904–0.967 | 0.890 | 0.860 | 50 |
| Cufflinks | Genes | MVP | SPLS | 0.891 | 0.846–0.935 | 0.720 | 0.950 | 50 |
| Cufflinks | Isoform Count | None | Elastic Net | 0.875 | 0.826–0.924 | 0.820 | 0.820 | 2959 |
| Cufflinks | Isoform Count | None | Random Forest | 0.929 | 0.895–0.963 | 0.910 | 0.810 | 2959 |
| Cufflinks | Isoform Count | None | SPLS | 0.564 | 0.484–0.644 | 0.250 | 0.910 | 2958 |
| Cufflinks | Isoform Count | MVP | Elastic Net | 0.667 | 0.590–0.744 | 0.600 | 0.780 | 50 |
| Cufflinks | Isoform Count | MVP | Random Forest | 0.923 | 0.889–0.957 | 0.880 | 0.800 | 50 |
| Cufflinks | Isoform Count | MVP | SPLS | 0.532 | 0.451–0.613 | 0.820 | 0.330 | 50 |
| Cufflinks | Isoform Fraction | None | Elastic Net | 0.766 | 0.702–0.830 | 0.520 | 0.880 | 2283 |
| Cufflinks | Isoform Fraction | None | Random Forest | 0.941 | 0.912–0.971 | 0.890 | 0.840 | 2283 |
| Cufflinks | Isoform Fraction | None | SPLS | 0.448 | 0.366–0.530 | 0.500 | 0.640 | 2283 |
| Cufflinks | Isoform Fraction | MVP | Elastic Net | 0.728 | 0.648–0.807 | 0.740 | 0.850 | 50 |
| Cufflinks | Isoform Fraction | MVP | Random Forest | 0.892 | 0.845–0.939 | 0.770 | 0.940 | 50 |
| Cufflinks | Isoform Fraction | MVP | SPLS | 0.523 | 0.445–0.611 | 0.560 | 0.690 | 50 |
| RSEM | Genes | None | Elastic Net | 0.860 | 0.808–0.912 | 0.900 | 0.700 | 1982 |
| RSEM | Genes | None | Random Forest | 0.844 | 0.791–0.897 | 0.700 | 0.890 | 1982 |
| RSEM | Genes | None | SPLS | 0.870 | 0.820–0.921 | 0.810 | 0.800 | 1982 |
| RSEM | Genes | MVP | Elastic Net | 0.960 | 0.934–0.986 | 0.900 | 0.940 | 50 |
| RSEM | Genes | MVP | Random Forest | 0.915 | 0.878–0.952 | 0.870 | 0.840 | 50 |
| RSEM | Genes | MVP | SPLS | 0.874 | 0.828–0.920 | 0.750 | 0.840 | 50 |
| RSEM | Isoform Count | None | Elastic Net | 0.788 | 0.724–0.852 | 0.870 | 0.660 | 3435 |
| RSEM | Isoform Count | None | Random Forest | 0.899 | 0.855–0.942 | 0.820 | 0.870 | 3435 |
| RSEM | Isoform Count | None | SPLS | 0.941 | 0.912–0.970 | 0.820 | 0.910 | 3435 |
| RSEM | Isoform Count | MVP | Elastic Net | 0.972 | 0.952–0.991 | 0.900 | 0.940 | 50 |
| RSEM | Isoform Count | MVP | Random Forest | 0.997 | 0.993–1.000 | 1.000 | 0.960 | 50 |
| RSEM | Isoform Count | MVP | SPLS | 0.937 | 0.906–0.967 | 0.890 | 0.820 | 50 |
| RSEM | Isoform Fraction | None | Elastic Net | 0.977 | 0.961–0.993 | 0.890 | 0.970 | 321 |
| RSEM | Isoform Fraction | None | Random Forest | 0.954 | 0.926–0.982 | 0.860 | 0.970 | 321 |
| RSEM | Isoform Fraction | None | SPLS | 0.962 | 0.938–0.986 | 0.890 | 0.980 | 321 |
| RSEM | Isoform Fraction | MVP | Elastic Net | 0.965 | 0.941–0.989 | 0.900 | 0.970 | 50 |
| RSEM | Isoform Fraction | MVP | Random Forest | 0.961 | 0.937–0.985 | 0.870 | 0.990 | 50 |
| RSEM | Isoform Fraction | MVP | SPLS | 0.950 | 0.923–0.976 | 0.870 | 0.900 | 50 |
| SeqGSEA | Genes | None | Elastic Net | 0.908 | 0.870–0.947 | 0.730 | 0.920 | 672 |
| SeqGSEA | Genes | None | Random Forest | 0.851 | 0.797–0.905 | 0.860 | 0.790 | 672 |
| SeqGSEA | Genes | None | SPLS | NA | NA–NA | NA | NA | NA |
| SeqGSEA | Genes | MVP | Elastic Net | 0.734 | 0.663–0.805 | 0.710 | 0.710 | 50 |
| SeqGSEA | Genes | MVP | Random Forest | 0.739 | 0.671–0.807 | 0.600 | 0.810 | 50 |
| SeqGSEA | Genes | MVP | SPLS | 0.680 | 0.604–0.755 | 0.640 | 0.730 | 50 |
| SeqGSEA | Exon Count | None | Elastic Net | 0.653 | 0.578–0.728 | 0.360 | 0.870 | 21862 |
| SeqGSEA | Exon Count | None | Random Forest | 0.858 | 0.800–0.915 | 0.900 | 0.850 | 21862 |
| SeqGSEA | Exon Count | None | SPLS | 0.908 | 0.860–0.955 | 0.830 | 0.950 | 21862 |
| SeqGSEA | Exon Count | MVP | Elastic Net | 0.737 | 0.668–0.807 | 0.770 | 0.660 | 50 |
| SeqGSEA | Exon Count | MVP | Random Forest | 0.706 | 0.633–0.779 | 0.840 | 0.580 | 50 |
| SeqGSEA | Exon Count | MVP | SPLS | 0.801 | 0.738–0.864 | 0.790 | 0.790 | 50 |
| SeqGSEA | Exon Fraction | None | Elastic Net | 0.831 | 0.777–0.885 | 0.800 | 0.810 | 41563 |
| SeqGSEA | Exon Fraction | None | Random Forest | 0.932 | 0.896–0.969 | 0.900 | 0.890 | 41563 |
| SeqGSEA | Exon Fraction | None | SPLS | 0.943 | 0.908–0.978 | 0.990 | 0.890 | 41563 |
| SeqGSEA | Exon Fraction | MVP | Elastic Net | 0.916 | 0.875–0.957 | 0.880 | 0.890 | 50 |
| SeqGSEA | Exon Fraction | MVP | Random Forest | 0.948 | 0.920–0.977 | 0.820 | 1.000 | 50 |
| SeqGSEA | Exon Fraction | MVP | SPLS | 0.971 | 0.951–0.992 | 0.900 | 0.960 | 50 |
*Mean number of transcripts selected as features per leave-two-out cross-validation test.
Table 3.
NSCLC dataset analysis
| Transcript quantification | Feature type | Filter | Machine learning Alg. | AUC | Confidence Int. | Sens. | Spec. | Mean features* |
|---|---|---|---|---|---|---|---|---|
| Cufflinks | Genes | None | Elastic Net | 0.416 | 0.348–0.483 | 1.000 | 0.000 | 1682 |
| Cufflinks | Genes | None | Random Forest | 0.635 | 0.555–0.715 | 0.809 | 0.618 | 1682 |
| Cufflinks | Genes | None | SPLS | 0.613 | 0.534–0.693 | 0.900 | 0.545 | 1682 |
| Cufflinks | Genes | MVP | Elastic Net | 0.543 | 0.465–0.622 | 0.891 | 0.345 | 50 |
| Cufflinks | Genes | MVP | Random Forest | 0.603 | 0.526–0.603 | 0.809 | 0.455 | 50 |
| Cufflinks | Genes | MVP | SPLS | 0.664 | 0.587–0.741 | 0.818 | 0.627 | 50 |
| Cufflinks | Isoform Count | None | Elastic Net | 0.506 | 0.436–0.577 | 0.264 | 0.836 | 3554 |
| Cufflinks | Isoform Count | None | Random Forest | 0.671 | 0.593–0.748 | 0.791 | 0.636 | 3554 |
| Cufflinks | Isoform Count | None | SPLS | NA | NA–NA | NA | NA | NA |
| Cufflinks | Isoform Count | MVP | Elastic Net | 0.541 | 0.465–0.618 | 0.623 | 0.445 | 50 |
| Cufflinks | Isoform Count | MVP | Random Forest | 0.581 | 0.505–0.656 | 0.636 | 0.536 | 50 |
| Cufflinks | Isoform Count | MVP | SPLS | 0.447 | 0.370–0.524 | 0.555 | 0.500 | 50 |
| Cufflinks | Isoform Fraction | None | Elastic Net | 0.411 | 0.358–0.464 | 1.000 | 0.000 | 2623 |
| Cufflinks | Isoform Fraction | None | Random Forest | 0.497 | 0.418–0.575 | 0.645 | 0.473 | 2623 |
| Cufflinks | Isoform Fraction | None | SPLS | NA | NA–NA | NA | NA | NA |
| Cufflinks | Isoform Fraction | MVP | Elastic Net | 0.632 | 0.558–0.707 | 0.718 | 0.709 | 50 |
| Cufflinks | Isoform Fraction | MVP | Random Forest | 0.636 | 0.562–0.710 | 0.764 | 0.518 | 50 |
| Cufflinks | Isoform Fraction | MVP | SPLS | 0.590 | 0.515–0.665 | 0.355 | 0.818 | 50 |
| RSEM | Genes | None | Elastic Net | 0.487 | 0.417–0.558 | 0.864 | 0.182 | 1216 |
| RSEM | Genes | None | Random Forest | 0.629 | 0.550–0.708 | 0.764 | 0.609 | 1216 |
| RSEM | Genes | None | SPLS | 0.558 | 0.479–0.638 | 0.864 | 0.409 | 1216 |
| RSEM | Genes | MVP | Elastic Net | 0.559 | 0.482–0.636 | 0.945 | 0.254 | 50 |
| RSEM | Genes | MVP | Random Forest | 0.529 | 0.449–0.610 | 0.836 | 0.382 | 50 |
| RSEM | Genes | MVP | SPLS | 0.668 | 0.590–0.745 | 0.845 | 0.627 | 50 |
| RSEM | Isoform Count | None | Elastic Net | 0.421 | 0.366–0.476 | 1.000 | 0.000 | 1747 |
| RSEM | Isoform Count | None | Random Forest | 0.609 | 0.530–0.688 | 0.845 | 0.500 | 1747 |
| RSEM | Isoform Count | None | SPLS | 0.535 | 0.453–0.616 | 0.800 | 0.509 | 1747 |
| RSEM | Isoform Count | MVP | Elastic Net | 0.519 | 0.441–0.597 | 0.791 | 0.364 | 50 |
| RSEM | Isoform Count | MVP | Random Forest | 0.581 | 0.500–0.661 | 0.682 | 0.618 | 50 |
| RSEM | Isoform Count | MVP | SPLS | 0.738 | 0.671–0.805 | 0.782 | 0.691 | 50 |
| RSEM | Isoform Fraction | None | Elastic Net | 0.373 | 0.333–0.414 | 1.000 | 0.000 | 880 |
| RSEM | Isoform Fraction | None | Random Forest | 0.586 | 0.509–0.664 | 0.800 | 0.500 | 880 |
| RSEM | Isoform Fraction | None | SPLS | 0.664 | 0.586–0.743 | 0.864 | 0.645 | 880 |
| RSEM | Isoform Fraction | MVP | Elastic Net | 0.923 | 0.886–0.961 | 0.955 | 0.818 | 50 |
| RSEM | Isoform Fraction | MVP | Random Forest | 0.589 | 0.513–0.664 | 0.273 | 0.909 | 50 |
| RSEM | Isoform Fraction | MVP | SPLS | 0.817 | 0.762–0.817 | 0.909 | 0.636 | 50 |
| SeqGSEA | Genes | None | Elastic Net | 0.415 | 0.355–0.474 | 1.000 | 0.009 | 935 |
| SeqGSEA | Genes | None | Random Forest | 0.609 | 0.531–0.686 | 0.709 | 0.600 | 935 |
| SeqGSEA | Genes | None | SPLS | 0.551 | 0.470–0.632 | 0.882 | 0.427 | 935 |
| SeqGSEA | Genes | MVP | Elastic Net | 0.532 | 0.455–0.609 | 0.473 | 0.691 | 50 |
| SeqGSEA | Genes | MVP | Random Forest | 0.530 | 0.453–0.607 | 0.282 | 0.855 | 50 |
| SeqGSEA | Genes | MVP | SPLS | 0.637 | 0.562–0.711 | 0.691 | 0.609 | 50 |
| SeqGSEA | Exon Count | None | Elastic Net | 0.393 | 0.334–0.452 | 0.036 | 0.964 | 8245 |
| SeqGSEA | Exon Count | None | Random Forest | 0.582 | 0.506–0.658 | 0.555 | 0.627 | 8245 |
| SeqGSEA | Exon Count | None | SPLS | 0.541 | 0.460–0.621 | 0.873 | 0.418 | 8245 |
| SeqGSEA | Exon Count | MVP | Elastic Net | 0.507 | 0.429–0.585 | 0.636 | 0.491 | 50 |
| SeqGSEA | Exon Count | MVP | Random Forest | 0.678 | 0.602–0.743 | 0.600 | 0.682 | 50 |
| SeqGSEA | Exon Count | MVP | SPLS | 0.569 | 0.492–0.647 | 0.664 | 0.564 | 50 |
| SeqGSEA | Exon Fraction | None | Elastic Net | 0.378 | 0.313–0.443 | 0.991 | 0.036 | 7524 |
| SeqGSEA | Exon Fraction | None | Random Forest | 0.621 | 0.546–0.697 | 0.673 | 0.591 | 7524 |
| SeqGSEA | Exon Fraction | None | SPLS | 0.528 | 0.450–0.606 | 0.727 | 0.464 | 7524 |
| SeqGSEA | Exon Fraction | MVP | Elastic Net | 0.636 | 0.563–0.555 | 0.563 | 0.691 | 50 |
| SeqGSEA | Exon Fraction | MVP | Random Forest | 0.564 | 0.488–0.640 | 0.536 | 0.591 | 50 |
| SeqGSEA | Exon Fraction | MVP | SPLS | 0.507 | 0.430–0.585 | 0.464 | 0.655 | 50 |
*Mean number of transcripts selected as features per leave-two-out cross-validation test.
The significant increase in prediction accuracy when using RSEM for transcript quantification and the MVP filter for feature selection suggests that an optimal pipeline for phenotype prediction should include these options.
3.2 MVP filtering
This analysis confidently shows MVP filtering consistently enhances the prediction accuracy of each feature type across all datasets when using RSEM for transcript quantification. The mean AUC score for phenotype predictions in each dataset increased when using the MVP filter (Fig. 2). Reducing the feature size per iteration from greater than 1000 on average to 50, with a general increase in accuracy, is more efficient and a significant improvement in feature selection. This is further evidence for the inclusion of a filtering method such as MVP in the development of an optimal phenotype prediction pipeline.
Fig. 2.
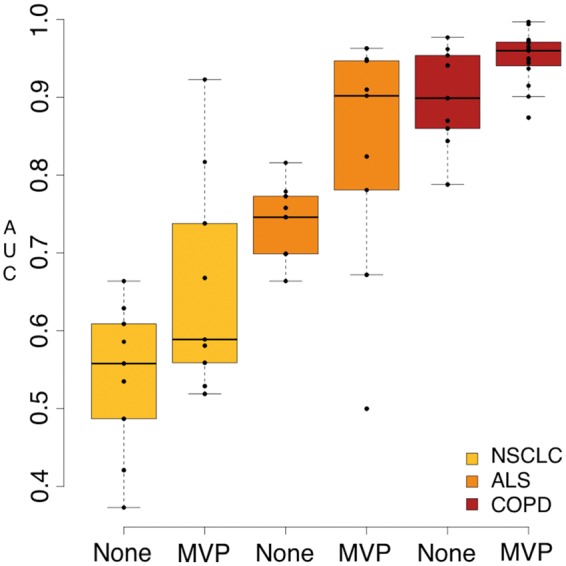
Predictive power of pipelines that use RSEM with varying input datasets and filtering. AUC values were generated by running all input datasets through nine pipelines that all performed transcript quantification with RSEM, but varied in feature type (gene, isoform count, isoform fraction), use of filtering and machine learning algorithm (Random Forest, Elastic Net, SPLS). Predictive results for these pipelines are shown grouped by dataset and use of filtering
The selection of the top 50 features ranked by the MVP filtering method may provide important biomarker candidates to biomedical researchers. Following the identification of consistently selected features in cross-validation tests, these features can be tested using corrected t-tests and other differential expression methods to identify viable biomarker candidates. It may be reasonable to correlate the list of MVP features with features identified as differentially expressed by EBSeq or CuffDiff. The promising genes and isoforms may be further processed using qPCR to validate biological importance.
3.3 Features and feature engineering
Isoform data has not traditionally been included in phenotype prediction. This analysis shows that both count-based isoform data and fractional-based isoform data are competitive in regard to gene expression data and even have enhanced prediction accuracy. The AUC scores of pipelines based on each feature when using RSEM for transcript quantification were compared (Fig. 3).
Fig. 3.
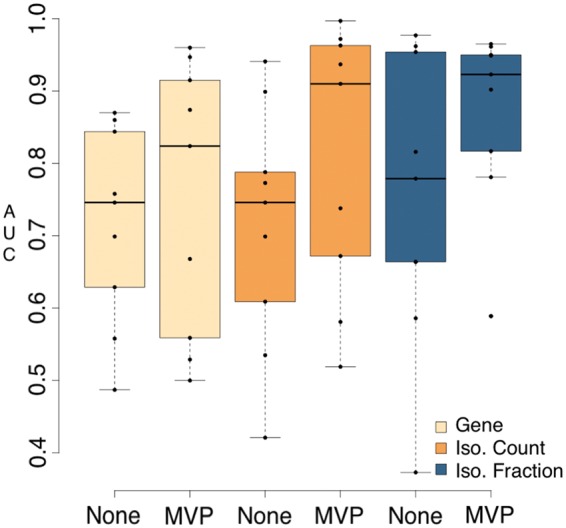
Predictive power of pipelines that use RSEM with varying feature types and filtering. AUC values were generated by running all input datasets (NSCLC, ALS, COPD) through nine pipelines that all performed transcript quantification with RSEM, but varied in feature type (gene, isoform count, isoform fraction), use of filtering and machine learning algorithm (Random Forest, Elastic Net, SPLS). Predictive values are shown grouped by feature type and whether filtering was applied
Count-based isoform expression data using the MVP filtering method generated many of the highest AUC’s in this analysis (Tables 1–3). The enhancement compared to gene expression data may result from the increased number of features that exist compared to genes alone. This result may also implicate the largely overlooked active isoforms that are involved in phenotype expression.
Fractional-based isoform expression produced AUC scores comparable to those of count-based isoform data (Fig. 3). Fractional-based AUC’s seem to be superior when analyzing datasets with features that have outlier expression counts, such as the NSCLC dataset where many outliers were detected using iLOO (George et al., 2015) (Table 3). Fractional-based isoform data quantified by RSEM with MVP filtering produced the only viable AUC scores in the NSCLC dataset (Table 3). The proportional normalization of each isoform in regard to corresponding gene expression may reduce the impact of extreme read counts and outliers. This may make differential expression tests more reliable in such cases and supports the case for performing phenotype prediction from fractional-based isoform data.
3.4 Machine learning algorithms
Both the Random Forest and Elastic Net machine learning algorithms produced promising results across the datasets, especially after MVP filtering (Fig. 4). SPLS was more variable than both Random Forest and Elastic Net, and produced several NA results when SPLS failed due to low variance features. Random Forest generated the highest observed AUC scores after MVP filtering. The Elastic Net consistently produced results within range or superior to Random Forest, and produced much greater scores in several datasets where Random Forest and SPLS did not generate accurate predictions (Fig. 4).
Fig. 4.
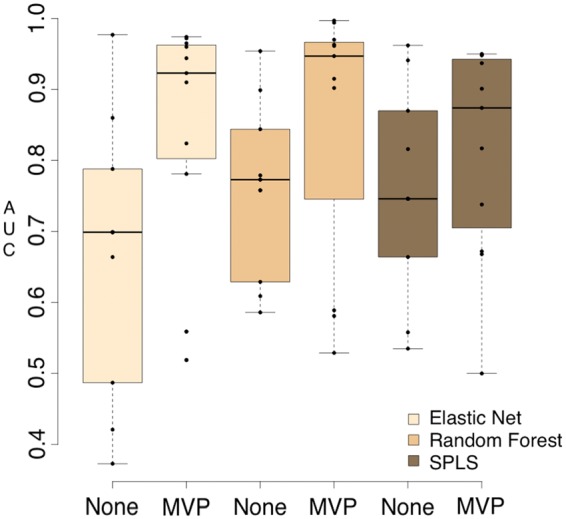
Predictive power of pipelines that use RSEM with varying machine learning algorithms and filtering. AUC values were generated by running all input datasets (NSCLC, ALS, COPD) through nine pipelines that all performed transcript quantification with RSEM, but varied in feature type (gene, isoform count, isoform fraction), use of filtering and machine learning algorithm (Random Forest, Elastic Net, SPLS). Predictive values are shown grouped by machine learning algorithm and whether filtering was applied
Due to the fact that the Elastic Net performed more consistently on all datasets (Tables 1–3) and generated AUC scores comparable in accuracy to Random Forest (Fig. 4), it seems optimal to include the Elastic Net in the phenotype prediction pipeline.
3.5 Complementary features
Isoforms offer a complementary and non-redundant set of features for phenotype prediction. When selected fractional-based isoform features were converted to the respective gene name and compared to the list of gene features for the optimal RSEM-based pipeline, little overlap between the two lists of features was found. There was 4.49% overlap in the ALS dataset, 1.93% overlap in the COPD dataset and 0% overlap in the NSCLC dataset. This reinforces the importance of including isoform expression data in phenotype prediction analyses.
4 Conclusion
The results of this study support two major conclusions. First, we have identified an optimal pipeline for phenotype prediction by answering the previously discussed questions on the construction of such a tool. RSEM generally generated the highest isoform based AUC scores with MVP filtering compared to other transcript quantification tools. We have shown that feature selection via a filtering method, such as the MVP filtering algorithm, consistently increases AUC score. Overall, fractional-based isoform features can be analyzed using the Elastic Net to yield the most consistently accurate phenotype predictions. These methods have been built into an open source pipeline available via GitHub. Second, we have identified the complementary nature of isoform expression data. Isoform features provide non-redundant information and enhanced predictive power compared to gene features. Our paper addresses the necessity of including isoform expression data in phenotype prediction and biomedical data analysis.
4.1 Pipeline
The Phenotype Prediction Pipeline is implemented in R. Extensive documentation and the full source code are available at: https://github.com/clabuzze/Phenotype-Prediction-Pipeline.git
Acknowledgements
The authors would like to thank the College of Charleston for hosting the NSF Omics REU which is supervised by the National Science Foundation DBI Award 1359301, the Medical University of South Carolina for providing the NSCLC Dataset, Northwestern University and Iowa State University for providing the ALS Dataset and Google Cloud Platform for their generous donation and facilitation of supplementary computational power for this and other Omics projects. We also acknowledge support from the Genomics Shared Resource, Hollings Cancer Center, Medical University of South Carolina.
Funding
This study was supported by the ALS Association and the National Science Foundation DBI Award 1359301 and supported in part by the Hollings Cancer Center, Medical University of South Carolina Support Grant (P30 CA 138313).
Conflict of Interest: none declared.
References
- Anderson P. et al. (2014). Predictive modeling of lung cancer recurrence using alternative splicing events versus differential expression data. In: IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology, Niagara Falls, Canada, pp. 1–8.
- Bolger A.M. et al. (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B. (2015). Bbmap. http://sourceforge.net/projects/bbmap/.
- Chun H., Keleş S. (2010) Sparse partial least squares regression for simultaneous dimension reduction and variable selection. J.R. Stat. Soc. Ser. B Stat. Methodol., 72, 3–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejean S., et al. (2011) mixOmics: Omics Data Integration Project http://CRAN.R-project.org/package=mixOmics. R package version 2.9-6..
- Friedman J. et al. (2010) Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw., 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- George N.I. et al. (2015) An iterative leave-one-out approach to outlier detection in rna-seq data. PLoS One, 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanitz A. et al. (2015) Comparative assessment of methods for the computational inference of transcript isoform abundance from RNA-seq data. Genome Biol., 16, 150.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.J. et al. (2015) Comprehensive analysis of transcriptome sequencing data in the lung tissues of COPD subjects. Int. J. Genomics, 2015, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leionen R., et al. (2011) The sequence read archive. Nucleic Acids Res., 39, D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N. et al. (2013) EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics, 29, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A., Wiener M. (2002) Classification and regression by random forest. R News, 2, 18–22. [Google Scholar]
- Liu R. et al. (2014) Comparisons of computational methods for differential alternative splicing detection using RNA-seq in plant systems. BMC Bioinformatics, 15, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdinler P.H., Macklis J.D. (2006) IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat. Neurosci., 9, 1371–1381. [DOI] [PubMed] [Google Scholar]
- Patel R.K., Jain M. (2012) NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One, 7, e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X. et al. (2011) Proc: an open-source package for r and s+ to analyze and compare roc curves. BMC Bioinformatics, 12, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C. et al. (2011) Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol., 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Cairns M.J. (2014) SeqGSEA: a bioconductor package for gene set enrichment analysis of RNA-Seq data integrating differential expression and splicing. Bioinformatics, 30, 1777–1779. [DOI] [PubMed] [Google Scholar]
- Yasvoina M.V. et al. (2013) eGFP expression under UCHL1 promoter genetically labels corticospinal motor neurons and a subpopulation of degeneration-resistant spinal motor neurons in an ALS mouse model. J. Neurosci., 33, 7890–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H., Hastie T. (2005) Regularization and variable selection via the elastic-net. J. R. Stat. Soc. Ser. B, 67, 301–320. [Google Scholar]