

Abstract
Cell-free biocatalysis systems offer many benefits for chemical manufacturing, but their widespread applicability is hindered by high costs associated with enzyme purification, modification, and immobilization on solid substrates, in addition to the cost of the material substrates themselves. Herein, we report a “bootstrapped” biocatalysis substrate material that is produced directly in bacterial culture and is derived from biofilm matrix proteins, which self-assemble into a nanofibrous mesh. We demonstrate that this material can simultaneously purify and immobilize multiple enzymes site specifically and directly from crude cell lysates by using a panel of genetically programmed, mutually orthogonal conjugation domains. We further demonstrate the utility of the technique in a bienzymatic stereoselective reduction coupled with a cofactor recycling scheme. The domains allow for several cycles of selective removal and replacement of enzymes under mild conditions to regenerate the catalyst system.
Keywords: biocatalysis, biofilms, enzymes, immobilization, surface regeneration
Whole-cell fermentation and immobilized enzymes are competing molecular manufacturing approaches that have complementary advantages and disadvantages.[1] Fermentation processes can be cheap, easily scalable, and can convert abundant feedstocks into a range of higher value molecules through multistep biocatalysis. However, they are not compatible with all transformations, as limited cell permeability of the substrates and products can hinder catalytic efficiency.[2] Furthermore, degradative interactions between native enzymes and molecular intermediates or products can arise unexpectedly, especially for heterologous biosynthetic pathways. Finally, purifications for fermentations can be complicated by the presence of diverse cellular byproducts. In contrast to fermentation, immobilized enzymes offer relatively simpler catalytic processes, streamlined purifications, and the possibility of catalyst recycling.[1b,3] However, this comes at significantly increased costs owing to the need for enzyme purification and immobilization onto a substrate.[4] The immobilization process itself is also a barrier to implementation and may necessitate chemical modification of the enzymes through nonspecific chemical conjugations that can distort enzyme conformation.[5] Crude-cell extracts are also sometimes used in industry, but they are not appropriate for all transformations because of their inherent instability, propensity for undesired side reactions and byproducts, and frequent lack of recyclability.[1b] These are significant concerns in an industry for which cost is a major driving force to stay competitive with chemical synthesis or isolation from natural sources.[6] Furthermore, although great strides have been made in recapitulating multistep pathways with cell-free systems,[7] its industrial viability is highly dependent on the cost and availability of engineered enzymes, and cofactors may need to be added stoichiometrically, again increasing costs. These limitations make the immobilized enzyme approach generally inappropriate for large-scale manufacturing. The complementary advantages and disadvantages of these approaches have also spawned the subfield of cell-free synthetic biology, which depends on the recapitulation of several cell-based processes on versatile inert scaffolds.[4]
Herein, we report a technique to combine the advantages of both of these competing biocatalysis approaches while avoiding some of their pitfalls. Our approach is based on the biofilm integrated nanofiber display (BIND) concept, which we reported on previously.[8] In this technology, which leverages the bio-synthetic potential of microbes to make materials, we engineer the curli amyloid protein, CsgA, which is secreted by E. coli during biofilm formation and forms a durable extracellular network of nanofibers. We repurpose the network by genetically modifying CsgA to serve as a scaffold for the display of heterologous domains that introduce new functions to the nanofibrous material. Indeed, our group and others have reported on modified curli fibers with a wide range of functional properties.[8b,9] In a biocatalytic variation on the BIND concept, the heterologous domain enables the site-specific immobilization of enzymes on the nanofiber surface.[8a]
A distinct advantage of using engineered biosynthetic materials as immobilization substrates is their high degree of programmability, which lends itself to rational design and systematic optimization. In this paper, we demonstrate several new features of the catalytic BIND system, including: one, a suite of mutually orthogonal conjugation domains that enable the simultaneous purification and co-immobilization of multiple enzymes directly from crude cell lysates; two, the use of the system in a novel stereoselective reduction and cofactor recycling scheme involving two enzymes that have not been used in combination before; three, the compatibility of our system with multimeric enzymes; four, the ability to remove and replace one type of enzyme selectively from the material without affecting other immobilized enzyme types.
To enable the immobilization of multiple enzymes simultaneously, we designed and tested a set of engineered curli fibers displaying conjugation domains and a complementary set of enzyme fusions (Figure 1). In each case, one fragment of a pairwise binding interaction was fused to the C terminus of CsgA and the other fragment to the N terminus of a model enzyme, α-amylase (Amy). We previously reported the immobilization of α-amylase to curli nanofibers by using the SpyTag-SpyCatcher conjugation pair (ST/SC).[8a] The other interactions were chosen to be complementary and orthogonal to ST/SC. InaD and Tip-1 are both derived from PDZ domains and bind to small peptide ligands.[10] In the case of InaD, a disulfide bond is formed with the EFCA tetrapeptide. Tip-1 binds noncovalently to the WRESAI hexapeptide with a dissociation constant (Kd) of approximately 10 nM. ePDZ is an engineered affinity clamp protein composed of a fibronectin type III domain fused to an Erbin PDZ domain through a flexible linker, and it binds with sub-nanomolar affinity to a short peptide called C-tag.[11] Calmodulin (CaM) is a naturally occurring clamp protein that exhibits Ca2+-dependent binding to the M13 peptide.[12] SZ16 and SZ21 are a pair of synthetic α-helical peptides that heterodimerize to form superhelical bundles with each other (Kd < 10 nM).[13] Overall, the panel represented conjugation interactions with a range of stabilities upon exposure to different solvent conditions, enabling removal and replacement of enzymes.
Figure 1.
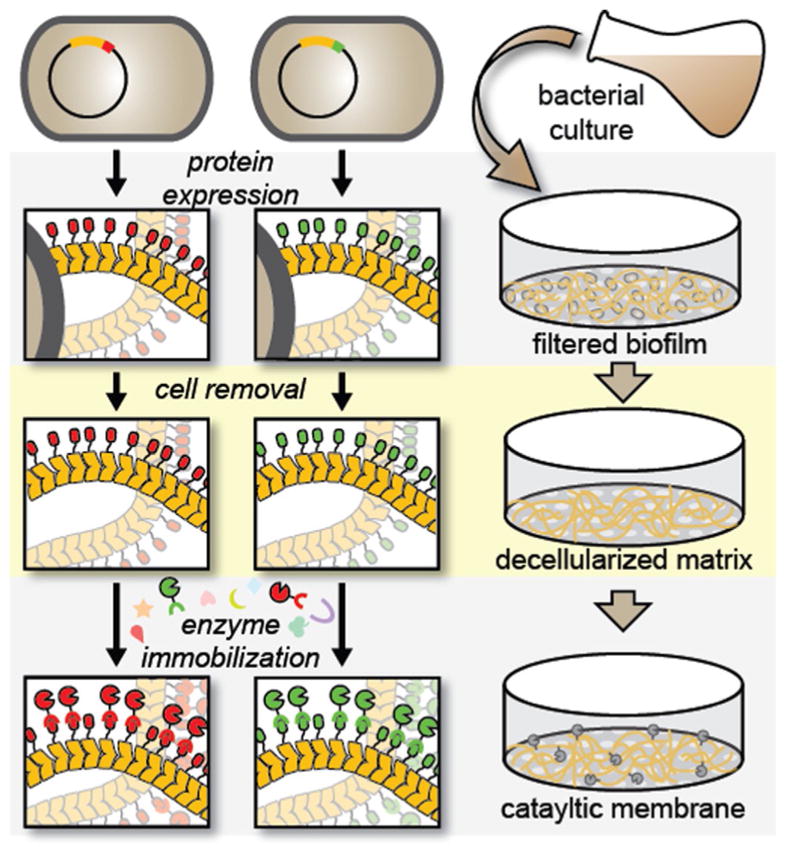
Schematic depicting the preparation of curli fiber mats directly from unprocessed bacterial cultures and subsequent immobilization of enzymes by using a straightforward filtration protocol. The curli fibers are formed by recombinant production of the CsgA protein, which self-assembles into a nanofibrous mesh after secretion into the growth medium. Enzyme immobilization occurs through a specific interaction between genetically programmed conjugation domain pairs and can be done with purified enzyme or directly from crude cell lysate.
We used an established Congo Red binding assay to test the ability of E. coli (strain PHL628)[14] to produce, secrete, and assemble amyloid fibers composed of the various CsgA fusion proteins (Figure S1 a in the Supporting Information). None of the fusions significantly hampered curli fiber production, and some fusions actually bound to 6.5 times more Congo Red per OD600 than the wildtype CsgA curli fibers. Although we did not investigate this signal enhancement in further detail, it is possible that some cell lysis could have released protein aggregates that bind nonspecifically to Congo Red. In parallel experiments, the α-amylase fusions were tested to confirm that their activity was maintained after domain fusion. We were surprised to find that some of the fusions appeared to enhance enzyme activity significantly (Figure S1 b, c). In all subsequent experiments, we accounted for these discrepancies by adjusting the concentration of the immobilized enzyme to normalize the total enzyme activity across samples.
Our initial attempts to test the activity of various immobilized enzyme variants on filtered biofilm mats gave unpredictable results, which we confirmed with control experiments to be, in part, attributable to cellular consumption of the NAD(P)H cofactors (Figure S2 a). Therefore, to get rid of the cells we adapted a protocol to prepare cell-free curli fiber mats by filtration of induced E. coli cultures, followed by treatment of the filtered biomass with 6M guanidinium chloride (GdmCl) and extensive washing with tris-buffered saline, 0.1% Tween 20 (TBST) buffer.[8a,9a] Scanning electron microscopy (SEM) analysis of the filtered biofilms before and after GdmCl treatment confirmed that the cells could be removed and that the fiber mesh structure of the curli network could be revealed upon treatment, which suggested that nonspecifically bound extracellular material and cellular debris were washed away (Figure 2). Cell viability and NADH consumption dropped significantly after GdmCl treatment (Figure S2 a, b).
Figure 2.

Scanning electron microscopy images of deposited curli biofilm after incubating with a) TBST or b) 6 M GdmCl.
The orthogonality of the conjugation domains was tested on the filtered curli fiber mats combinatorially by exposing each of the CsgA fusions to the entire panel of α-amylase fusions (Figure 3a). Amylase activity was used as a metric for immobilization efficiency. SC-Amy and InaD-Amy, both of which form covalent bonds with their cognate partner domains, showed high selectivity. In contrast, Tip1-Amy and ePDZ-Amy were much less selective and exhibited considerable cross-reactivity with curli fiber mats regardless of the identity of the CsgA fusion. This is perhaps not surprising for ePDZ given the known binding interactions between naturally occurring curli fibers and mammalian ECM proteins such as fibronectin.[15] However, as ePDZ-Amy showed the highest absolute quantities of immobilized enzyme, it may be useful for maximizing the surface density, as long as specificity is not important. As expected, the CsgA-CaM biofilm exhibited high selectivity for M13-Amy in the presence of Ca2+ and could not be conjugated to the curli fiber mats in the absence of Ca2+ (Figure S3). Similarly, SZ16-Amy bound specifically to CsgA-SZ21 but did show some cross-reactivity with CsgA-CaM, even in the absence of Ca2+. This could be explained by the inherent affinity of CaM for α-helical peptide ligands.[16] Overall, the results suggest that at least three conjugation pairs, ST/SC, EFCA/InaD, and CaM/M13, can be used simultaneously for co-immobilization of different enzymes on the basis of their mutual orthogonality.
Figure 3.

Catalytic activity of curli fiber mats functionalized with α-amylase containing different conjugation domain pairs and their stability. A) α-Amylase activity measurements for curli fiber mats functionalized with various conjugation domain pairs. Orthogonality was assessed by measuring immobilized enzyme activity after exposure of the mats to α-amylase bearing a panel of mismatched conjugation domains. Complementary conjugation domains are coded with axis labels of matching colors. B) Schematic depicting the cycle of enzyme immobilization, removal, and re-immobilization. C) Enzyme activity measured before and after exposure of functionalized mats to enzyme-removal conditions and after re-immobilization of freshly purified α-amylase fusions. Various enzyme-removal conditions were tested, including harsh ones (6 M GdmCl) and milder ones. Enzyme activity measured after initial immobilization, removal, and after re-immobilization. Bars show mean ± standard deviation. D) Summary of conjugation domain stability derived from panel c.
Several of the conjugation domain pairs (WRESAI/Tip1, C-tag/ePDZ, CaM/M13, and SZ16/SZ21) make use of noncovalent bonds only. Given the extreme stability of curli fibers under conditions that denature most proteins,[8a,9a] we reasoned that these interactions could be disrupted by chaotropes to remove the enzymes while leaving the curli fiber mats intact for refunctionalization. Indeed, 6 M GdmCl removed 70–90 % of the noncovalently bound enzymes from the surface (Figure 3c). After re-exposure to the purified soluble enzymes, the curli fiber mats regained at least 90 % of their original activity. In the cases of WRESAI/Tip1 [(117 ± 8) %] and SZ16/SZ21 [(154 ± 4) %], the higher enzyme activity upon regeneration may have been owing to the exposure of more conjugation domains that had previously been blocked by nonspecific protein adsorption from cell lysates. In contrast, enzymes immobilized with covalent conjugation pairs (ST/SC and EFCA/InaD) remained attached to the biofilms as expected. However, InaD-Amy could be removed from curli fiber mats by exposure to urea and dithiothreitol (DTT) (Figure 3c). Some of the interactions could be disrupted with milder conditions. The SZ21/ SZ16 interaction could be disrupted by exposure of the functionalized curli fiber mats to pH 5 aqueous buffer, albeit with less efficiency than with GdmCl. Similarly, M13-Amy could be removed from CsgA-CaM fiber mats with exposure to the Ca2+ chelator ethylene glycol-bis-(beta-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA).[17] As with the noncovalent interactions, the curli fiber mats displaying EFCA, SZ21, and CaM could be refunctionalized by exposure to the appropriate purified enzyme fusions and regained their original catalytic activity. Importantly, these milder conditions worked for a specific conjugation pair and did not affect the activity of mats functionalized with other Amy fusions (Figure 3d).
To demonstrate the regeneration potential of the curli fiber mats with repeated use, we exposed some of them to several cycles of enzyme functionalization, removal, and re-immobilization. InaD-Amy-functionalized mats maintained 60–85 % of their activity over four such cycles with urea and DTT as the removal agents (Figure 4a). M13-Amy-functionalized mats similarly maintained their full activity through four cycles, although the removal efficiency dropped over the course of the experiment (Figure 4b).
Figure 4.
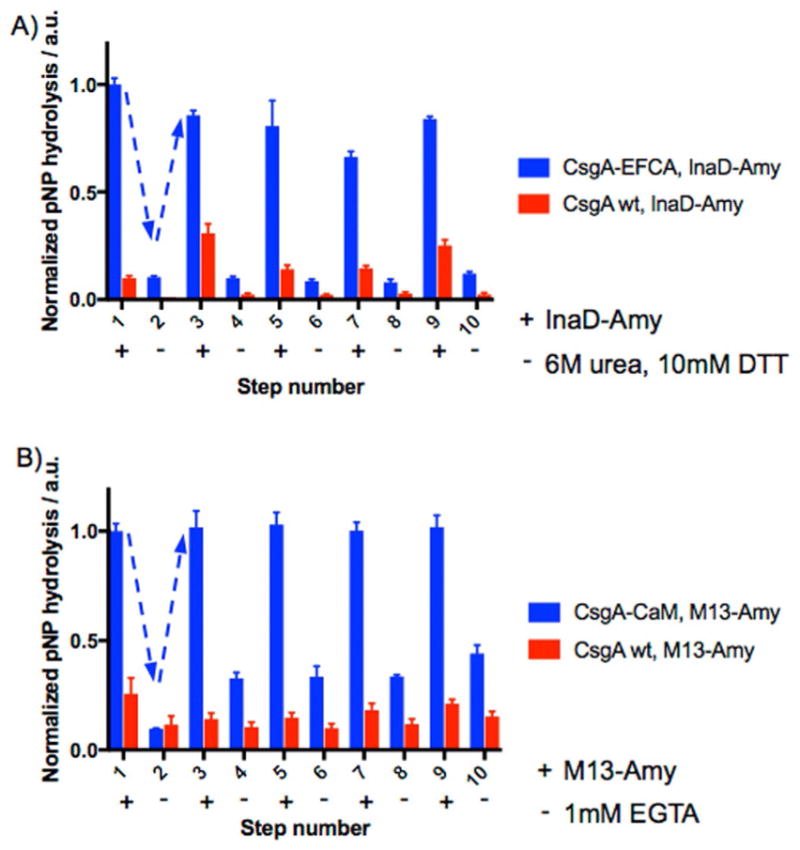
Regeneration of enzymes on curli fiber mats. a) α-Amylase immobilized on mats through the InaD/EFCA interaction could be removed (even step numbers) by exposure to urea and DTT and re-immobilized (odd step numbers) through five cycles while maintaining (84 ± 2) % of its activity from the first immobilization. b) Enzyme regeneration could also be achieved through five cycles with the M13/CaM conjugation pair by using EGTA as the removal agent, while maintaining close to 100 % of the original enzyme activity. Points show mean ± standard deviation. Dashed arrows are provided to guide the eye.
Whereas α-amylase served as a useful and hardy model enzyme, we also explored the compatibility of our system with stereoselective ketone reduction transformations by ketoreductases (KREDS). RADH is a homotetrameric NADPH-dependent alcohol dehydrogenase derived from Ralstonia sp. that has been used previously in various cascade reactions.[18] It exhibits rare compatibility with sterically hindered ketones, which makes it a potentially useful biocatalyst. To avoid the use of stoichiometric amounts of the cofactor, which is prohibitively expensive for most fine-chemical synthesis, we paired RADH with the homodimeric phosphite dehydrogenase (PTDH) from Pseudomonas stutzeri.[19] PTDH converts NAD(P)+ back into NAD(P)H by using phosphite as a cheaper electron donor. Accordingly, we cloned chimeras of the two enzymes (M13-RADH and PTDH-InaD) with flexible linkers separating the conjugation domain from the surface-accessible terminus of each enzyme (Figure 5b, Figures S4–S6).
Figure 5.

A) Schematic of the bienzymatic system for stereospecific ketone reduction by using RADH coupled to NADPH cofactor recycling by PTDH. B) Crystal structure of the homotetrameric RADH (PDB: 4BMS) and the homodimeric PTDH (PDB: 4E5N) with NADPH and NAD+ (blue) bound, respectively. C) MPP production as a function of initial concentration of the cofactor. In one case (circles), only NADP+ was supplied, along with 50 mM phosphite. In another case (squares), only NADPH was supplied without phosphite. D) Enzyme activity on a curli fiber mat functionalized with both M13-RADH and PTDH-InaD. The activity of each enzyme was measured individually, independent of the other by controlling the solution and substrate conditions. M13-RADH only (red), PTDH-InaD only (blue), and their combined activity (green) were measured through various steps of removal and re-immobilization and were normalized to their initial activity. Step 1: initial immobilization; step 2: EGTA to remove M13-RADH; step 3: M13-RADH re-immobilization; step 4: 6 M urea and 10 mM DTT to remove all bound enzymes; step 5: re-immobilization of M13-RADH and PTDH-InaD. Points show mean ± standard deviation.
To immobilize M13-RADH and PTDH-InaD simultaneously onto a single curli fiber mat, we first expressed CsgA-CaM and CsgA-EFCA separately and mixed them together prior to deposition on filter plates. The hybrid curli fiber mats were functionalized simultaneously with both enzymes by exposure to crude cell lysates from separate recombinant expressions of M13-RADH and PTDH-InaD (Figure 5a). After the bifunctionalized mat was incubated with all the necessary reaction components – 2-hydroxy-2-methylpropiophenone (2-HPP), sodium phosphite, NADP+, and CaCl2 in pH 7.5 Tris buffer, the product, (R)-2-methyl-1-phenyl-1,2-propanediol (MPP), was isolated with >99 % ee, as confirmed by LC–MS and chiral-phase HPLC (Figures S7–S9). Importantly, this was possible even though only the oxidized cofactor was initially present, which indicated that both enzymes were active. Furthermore, the average volumetric productivity of the mats over the course of the 8 h reaction period was (150 ± 8) mg L−1 h−1 with a conversion of (64 ± 1) % after 24 h (Figure S10). Complete conversion was presumably hindered by the well-known reversibility of the RADH-catalyzed reaction, but the conversion was comparable to those reported previously.[18b,20]
To confirm further the recycling of NADP+ to NADPH by PTDH, we ran two parallel experiments on dual functionalized curli fiber mats (Figure 5c). In one, we varied the concentration of NADPH (0.2–2.0 mM) while keeping the substrate concentration constant (10 mM) in the absence of phosphite. Product formation scaled stoichiometrically with cofactor equivalents, as expected, which confirmed that NADPH regeneration was not possible without phosphite. In a second experiment, we varied the NADPH concentration in the presence of 50 mM phosphite. In this experiment, product was formed at a concentration of approximately 2.6 mM, despite the fact that the concentration of NADP+ was 0.25 mM, which indicated that the cofactor was regenerated in situ at least 10 times in this reaction.
Having demonstrated the compatibility of the conjugation domains in the context of a dual-enzyme system for cofactor recycling, we sought to explore the possibility of the selective removal and re-immobilization of a single enzyme in the presence of another. Therefore, we measured the activity of each enzyme individually and the combined activity of both enzymes on bifunctional curli fiber mats through various cycles of enzyme removal and re-immobilization (Figure 5d, Figure S11). Notably, we performed all (re-)immobilization steps for this experiment by using unpurified cell lysate, demonstrating the simultaneous purification and immobilization concept. We found that M13-RADH could be removed selectively from the mats with EGTA and that fresh M13-RADH could be re-immobilized without disrupting the activity of the co-immobilized PTDH-InaD (Figure 5d, steps 2–3). Furthermore, the combined activity of the bienzymatic system could be restored to its original value after this process. Both enzymes could be removed by exposure to urea and DTT, and re-immobilization of both enzymes from crude cell lysates could be performed siultaneously to restore the full activity of the system (Figure 5d, steps 4–5).
In summary, the biofilm integrated nanofiber display (BIND) technology is a versatile and robust scaffold material for performing biocatalytic transformations. It is highly programmable on the basis of straightforward plasmid-based genetic engineering and can easily be customized to enable multienzyme display. Several enzymes can be immobilized on the scaffold in a site-specific manner, directly from crude cell lysates, and the enzyme-modified material can be used over many cycles without deterioration of the activity. Even if some activity is lost, one type of immobilized enzyme can be refreshed without disrupting the activity of the others, and this enables on-the-fly adjustments to maximize reaction cycles per unit of catalyst. Although the novel coupling of RADH to phosphite dehydrogenase displayed only modest total turnover numbers for NADPH recycling, the BIND system could easily be adapted to more efficient coupled enzyme systems with established compatibility with manufacturing processes.[21] Ongoing work in the laboratory is based on exploring further modifications to render the BIND material production to be more scalable and to fabricate particular morphologies that would be compatible with established fermentation workflows. We envision that this platform would have broad relevance to a range of biocatalysis applications, including chemical and pharmaceutical synthesis and water treatment.
Supplementary Material
Acknowledgments
We gratefully acknowledge funding from the National Science Foundation (NSF) Grant 1410751 (DMR) and the Wyss Institute for Biologically Inspired Engineering. M.G.N. gratefully acknowledges an Early Postdoc Mobility postdoctoral research fellowship (P2BSP3 158817) from the Swiss National Science Foundation (SNSF). This work was supported by the Wyss Institute for Biologically Inspired Engineering.
Footnotes
Supporting Information and the ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/cctc.201701221.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.a) Ricca E, Brucher B, Schrittwieser JH. Adv Synth Catal. 2011;353:2239. [Google Scholar]; b) Sheldon RA, van Pelt S. Chem Soc Rev. 2013;42:6223. doi: 10.1039/c3cs60075k. [DOI] [PubMed] [Google Scholar]
- 2.a) Goldberg K, Schroer K, Lütz S, Liese A. Appl Microbiol Biotechnol. 2007;76:237. doi: 10.1007/s00253-007-1002-0. [DOI] [PubMed] [Google Scholar]; b) Goldberg K, Schroer K, Lütz S, Liese A. Appl Microbiol Biotechnol. 2007;76:249. doi: 10.1007/s00253-007-1005-x. [DOI] [PubMed] [Google Scholar]; c) Xue R, Woodley JM. Bioresour Technol. 2012;115:183. doi: 10.1016/j.biortech.2012.03.033. [DOI] [PubMed] [Google Scholar]; d) Johannes T, Simurdiak MR, Zhao H. Encyclopedia of Chemical Processing. New York: 2006. p. 101. [Google Scholar]
- 3.a) Jia F, Narasimhan B, Mallapragada S. Biotechnol Bioeng. 2014;111:209. doi: 10.1002/bit.25136. [DOI] [PubMed] [Google Scholar]; b) Zhang Y, Ge J, Liu Z. ACS Catal. 2015;5:4503. [Google Scholar]
- 4.You C, Zhang YHP. In: Future Trends in Biotechnology. Zhong J-J, editor. Vol. 131. Springer; Berlin Heidelberg: 2013. p. 89. [Google Scholar]
- 5.Hermanson GT. Bioconjugate Techniques. 2. 2008. [Google Scholar]
- 6.Fu H, Dencic I, Tibhe J, Sanchez Pedraza CA, Wang Q, Noel T, Meuldijk J, de Croon M, Hessel V, Weizenmann N, Oeser T, Kinkeade T, Hyatt D, Van Roy S, Dejonghe W, Diels L. Chem Eng J. 2012;207–208:564. [Google Scholar]
- 7.Rollin JA, Tam TK, Zhang YHP. Green Chem. 2013;15:1708. [Google Scholar]
- 8.a) Botyanszki Z, Tay PKR, Nguyen PQ, Nussbaumer MG, Joshi NS. Biotechnol Bioeng. 2015;112:2016. doi: 10.1002/bit.25638. [DOI] [PubMed] [Google Scholar]; b) Nguyen PQ, Botyanszki Z, Tay PKR, Joshi NS. Nat Commun. 2014;5:4945. doi: 10.1038/ncomms5945. [DOI] [PubMed] [Google Scholar]
- 9.a) Dorval Courchesne N-M, Duraj-Thatte A, Tay PKR, Nguyen PQ, Joshi NS. ACS Biomater Sci Eng. 2017;3:733–741. doi: 10.1021/acsbiomaterials.6b00437. [DOI] [PubMed] [Google Scholar]; b) Chen AY, Zhong C, Lu TK. ACS Synth Biol. 2015;4:8. doi: 10.1021/sb500113b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhong C, Gurry T, Cheng AA, Downey J, Deng Z, Stultz CM, Lu TK. Nat Nanotechnol. 2014;9:858. doi: 10.1038/nnano.2014.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Yan X, Zhou H, Zhang J, Shi C, Xie X, Wu Y, Tian C, Shen Y, Long J. J Mol Biol. 2009;392:967. doi: 10.1016/j.jmb.2009.07.060. [DOI] [PubMed] [Google Scholar]; b) Lu Y, Huang F, Wang J, Xia J. Bio-conjugate Chem. 2014;25:989. doi: 10.1021/bc500134w. [DOI] [PubMed] [Google Scholar]; c) Kimple ME, Sondek J. Biotechniques. 2003;33:578. doi: 10.2144/02333rr01. [DOI] [PubMed] [Google Scholar]
- 11.a) Huang J, Koide A, Makabe K, Koide S. Proc Natl Acad Sci USA. 2008;105:6578–6583. doi: 10.1073/pnas.0801097105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang J, Makabe K, Biancalana M, Koide A, Koide S. J Mol Biol. 2009;392:1221. doi: 10.1016/j.jmb.2009.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang J, Nagy SS, Koide A, Rock RS, Koide S. Biochemistry. 2009;48:11834. doi: 10.1021/bi901756n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blumenthal DK, Takio K, Edelman AM, Charbonneau H, Titani K, Walsh KA, Krebs EG. Proc Natl Acad Sci USA. 1985;82:3187. doi: 10.1073/pnas.82.10.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Thompson KE, Bashor CJ, Lim WA, Keating AE. ACS Synth Biol. 2012;1:118. doi: 10.1021/sb200015u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reinke AW, Grant RA, Keating AE. J Am Chem Soc. 2010;132:6025. doi: 10.1021/ja907617a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hidalgo G, Chen X, Hay AG, Lion LW. Appl Environ Microbiol. 2010;76:6939. doi: 10.1128/AEM.01254-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olsén A, Herwald H, Wikström M, Persson K, Mattsson E, Björck L. J Biol Chem. 2002;277:34568. doi: 10.1074/jbc.M206353200. [DOI] [PubMed] [Google Scholar]
- 16.a) Meister GE, Joshi NS. ChemBioChem. 2013;14:1460. doi: 10.1002/cbic.201300168. [DOI] [PubMed] [Google Scholar]; b) Shifman JM, Mayo SL. J Mol Biol. 2002;323:417. doi: 10.1016/s0022-2836(02)00881-1. [DOI] [PubMed] [Google Scholar]
- 17.Hennessey JP, Manavalan P, Johnson WC, Malencik DA, Anderson SR, Schimerlik MI, Shalitin Y. Biopolymers. 1987;26:561. doi: 10.1002/bip.360260409. [DOI] [PubMed] [Google Scholar]
- 18.a) Kulig J, Simon RC, Rose CA, Husain SM, Hackh M, Ludeke S, Zeitler K, Kroutil W, Pohl M, Rother D. Catal Sci Technol. 2012;2:1580. [Google Scholar]; b) Wachtmeister J, Jakoblinnert A, Kulig J, Offermann H, Rother D. ChemCatChem. 2014;6:1051. [Google Scholar]; c) Sehl T, Hailes HC, Ward JM, Menyes U, Pohl M, Rother D. Green Chem. 2014;16:3341. [Google Scholar]
- 19.Costas AMG, White AK, Metcalf WW. J Biol Chem. 2001;276:17429. doi: 10.1074/jbc.M011764200. [DOI] [PubMed] [Google Scholar]
- 20.Johannes TW, Woodyer RD, Zhao H. Biotechnol Bioeng. 2007;96:18. doi: 10.1002/bit.21168. [DOI] [PubMed] [Google Scholar]
- 21.Angelastro A, Dawson WM, Luk LYP, Allemann RK. ACS Catal. 2017;7:1025–1029. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.