

Abstract
Introduction:
The ontogeny of drug transport and metabolism is generally studied independently in tissues, yet in the immediate postnatal period the developmental regulation of transporters and metabolizing enzymes must be coordinated. Using the Remote Sensing and Signaling Hypothesis as a framework, we describe how a systems physiology view helps to make sense of how inter-organ communication via hepatic, renal, and intestinal transporters and drug metabolizing (DMEs) enzymes is regulated from the immediate postnatal period through adulthood.
Areas covered:
This review examines patterns of developmental expression and function of transporters and DMEs with a focus on how cross-talk between these proteins in the intestine, liver, and kidney may be coordinated postnatally to optimize levels of metabolites and endogenous signaling molecules as well as gut-microbiome products.
Expert Opinion/Commentary:
Developmental expression is considered in terms of the Remote Sensing and Signaling Hypothesis, which addresses how transporters and DMEs participate in inter-organ and inter-organism small molecule communication in health, development, and disease. This hypothesis, for which there is growing support, is particularly relevant to the “birth transition” and post-natal developmental physiology when organs must deal with critical physiological tasks distinct from the fetal period and where remote inter-organ and possibly inter-organismal (e.g. gut microbiome) communication is likely to be critical to maintain homeostasis.
Keywords: Drug metabolism, transport, ontogeny, development
1. Introduction
Transporters and metabolic enzymes are important determinants of drug absorption, distribution, metabolism, and excretion of drugs. Although they are increasingly recognized to play a major physiological role in maintaining homeostasis by regulating levels of endogenous metabolites, signaling molecules, nutrients, gut microbiome products, and antioxidants in various tissues and body fluids [1]. the developmental regulation of transport and metabolism is an understudied area. In order to improve neonatal health and optimally manage problems associated with premature birth, there is a critical need to better understand the systems physiology of the postnatal period.
It is well established that drug transporters, as well as drug metabolizing enzymes (DMEs), undergo marked increases in expression after birth and through the juvenile period in a variety of tissues, including the intestine, liver, and kidney—sometimes called the gut-liver-kidney axis. These organs are, however, usually discussed individually. A systems physiology view seeks to understand how the intestine, liver, and kidney transporters and metabolic enzymes are regulated from the immediate postnatal period through adulthood. Presumably, this is in the interest of optimizing levels of endogenous metabolites, signaling molecules, and antioxidants as well as gut-derived nutrients and gut microbiome products.
The Remote Sensing and Signaling Hypothesis addresses how multi-specific “drug” transporters (as well as DMEs) participate in inter-organ and inter-organism small molecule communication under steady state physiology as well as when the organism is undergoing dynamic changes, such as after organ injury or during development. This hypothesis, and the growing evidence in support of it, has been detailed elsewhere [1, 2, 3, 4, 5]. Nevertheless, it has been discussed only in passing in the context of the “birth transition” and developmental physiology when multiple organs must communicate remotely to coordinate homeostasis and respond not only to the requirements of tissue growth and maturation, but also to changes in the diet and microbiome, the products of which enter the blood and are subjected to metabolism, distribution and elimination by many of the same DMEs and transporters widely associated with drug disposition.
This is clearly a highly dynamic period where inter-organ and inter-organismal remote communication via small organic molecules is likely to play a crucial role; thus, the Remote Sensing and Signal Hypothesis seems particularly applicable for both understanding systemic developmental physiology involving transporters and DMEs as well as the consequences of drug-metabolite interactions that may perturb normal remote inter-organ and inter-organismal communication, affecting levels of key metabolites, signaling molecules, antioxidants, nutrients and microbiome products in particular tissues and body fluid compartments (e.g. CSF, blood, urine, bile). Using the Remote Sensing and Signaling Hypothesis as a framework, and mainly focusing on the kidney and the liver, we examine existing relevant in vitro, in vivo, and omics data as it pertains to developmental pharmacology and developmental physiology. We argue that the Remote Sensing and Signaling Hypothesis provides a useful framework for a deeper systems-level understanding of the complex interplay between many organs (and other organisms such as those constituting the gut microbiome) during different developmental periods.
2. Transport in Developing and Postnatal Kidney
Membrane transport proteins facilitate the movement of endogenous and exogenous compounds across biological membranes [6, 7, 8]. Multispecific transporters of the solute carrier (SLC) and the ATP-binding cassette (ABC) superfamilies in the renal proximal tubules are involved in sequential uptake and efflux of drugs, metabolites, toxins, nutrients, and signaling molecules from blood into the urine [9]. For many small molecule drugs that bind to plasma proteins, including many antibiotics, antivirals, antifungals, and diuretics, active tubular secretion accounts for a large portion of total clearance[10].
The developmental aspects of renal proximal tubular transport have received considerably less attention than that of DMEs, in part because pharmacokinetic changes mediated by renal transporter activity are generally of a smaller magnitude. For example, strong inhibition of hepatic cytochrome P450 activity is associated with ≥ 5-fold increase in the AUC of a sensitive substrate, whereas inhibition of transporters in the renal proximal tubule generally results in 1 to 3-fold increases in the AUC of substrate drugs [11]. The reasons for this likely include redundant clearance pathways (such as renal filtration and biliary secretion) as well as an upper limit of renal clearance that is approximated by the renal plasma flow. Nevertheless, a quantitative developmental analysis of renal transporters is necessary as a broader view of the endogenous roles of transporters begins to emerge, including the handling of antioxidants, signaling molecules, hormones, nutrients, and neurotransmitters.
Renal transporters are differentiated by membrane localization (basolateral versus apical) as well as substrate specificity. Transporters localized to the basolateral membrane of human tubular epithelial cells recognized as important for the secretion of xenobiotics and endogenous molecules include two members of the organic anion transporter (OAT) family (OAT1 (SLC22A6, originally NKT), OAT3 (SLC22A8)), and the organic cation transporter 2 (OCT2 (SLC22A2)). Clinically relevant apical renal transporters include P-glycoprotein (P-gp or MDR1; ABCB1), multidrug and toxin extrusion protein 1 and 2 (MATE1 (SLC47A1) and MATE2-K (SLC47A2)), and the multi drug resistance-associated protein 2 and 4 (MRP2 (ABCC2) and MRP4 (,ABCC4)) (Figure 1).
Figure 1. Drug Transporters and Metabolic Enzymes in Kidney and Liver.
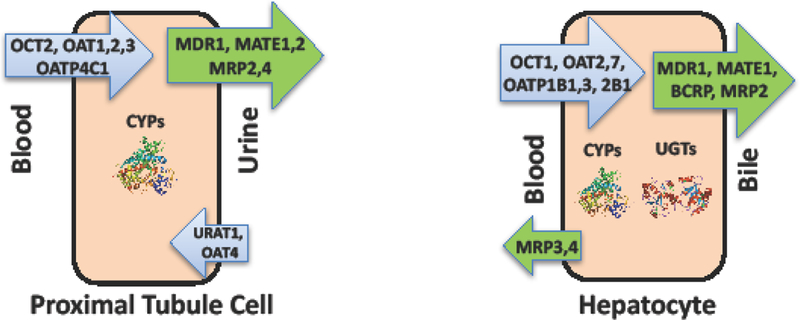
Representative efflux and uptake transporters and metabolic enzymes in human proximal tubule cells and hepatocytes.
OAT1 and OAT3 are highly expressed on the basolateral membrane of the renal proximal tubule and facilitate the active uptake of anionic substrates from blood in exchange for intracellular a- ketoglutarate [4, 12]. Few nonsynonymous OAT1 or OAT3 variants have been identified and none are clearly associated with major loss of function [13, 14, 15]. Additional homologs with different substrate specificities have been cloned (OAT2, OAT4, OAT5, OAT6, URAT1) although their importance is not nearly as well understood from the perspective of renal drug handling. OAT1 and OAT3 often have overlapping interactions with common drug substrates [16]. rendering it difficult to quantitate the relative contribution without knocking out individual transporters experimentally. However, recent metabolomics data from the Oatl and Oat3 mouse knockouts suggests that the substrate preference for metabolites may be quite distinct [17, 18, 19]. this at least raises the possibility that Oatl and Oat3 xenobiotic preferences may actually be more different than currently appreciated.
Considerable effort is spent in the drug development setting to characterize the potential of a drug candidate to inhibit and/or undergo transport by these membrane proteins. For example, the U.S. Food and Drug Administration recommends that investigational drugs should be evaluated to determine if they are substrates or inhibitors of OAT1, OAT3, and OCT2 [11] - all of which are involved in the transport of solutes into the kidney tubules. While there has been an increase in research on drug-drug interactions involving SLC and ABC drug transporters in adult cells and tissues, very little work has been done on developing and postnatal tissues, despite the fact that the fetal and postnatal period are periods of particular vulnerability to toxicity [20].
3. Transporter expression in the developing kidney
Despite the contribution of kidney proximal tubule transporters to drug handling and maintenance of homeostasis, relatively little is known about the ontogeny of these transporters. In humans, limited information is available regarding the developmental aspects of mRNA and/or protein expression of renal transporters [21]. Using immunohistochemistry, positive P-gp was observed at the apical side of the tubules at 11 weeks of gestation onward, but not at 7 weeks [22], Quantitation of differentially expressed transport proteins in the human kidney as a function of developmental stage is not currently available; however, a considerable amount of animal data is existing, and these studies have been quite systematic. The expression of various renal drug transporters tends to be low until birth, whereupon there is a rise in both transcript expression as well as function [23, 24, 25]. That said, ever since the discovery of OAT1 as NKT [25]. mid-to- late embryonic expression has been noted in the kidney. By northern blot, expression is detected in the developing murine kidney around embryonic day 16–17, whereas an in situ hybridization signal can be detected around embryonic day 14 [24]. After birth (approximately 18–20 days, depending upon the strain), a rapid increase in Oatl expression occurs with levels approaching those of adult between 1 week and 4 weeks (Figure 2) [23, 26].
Figure 2.
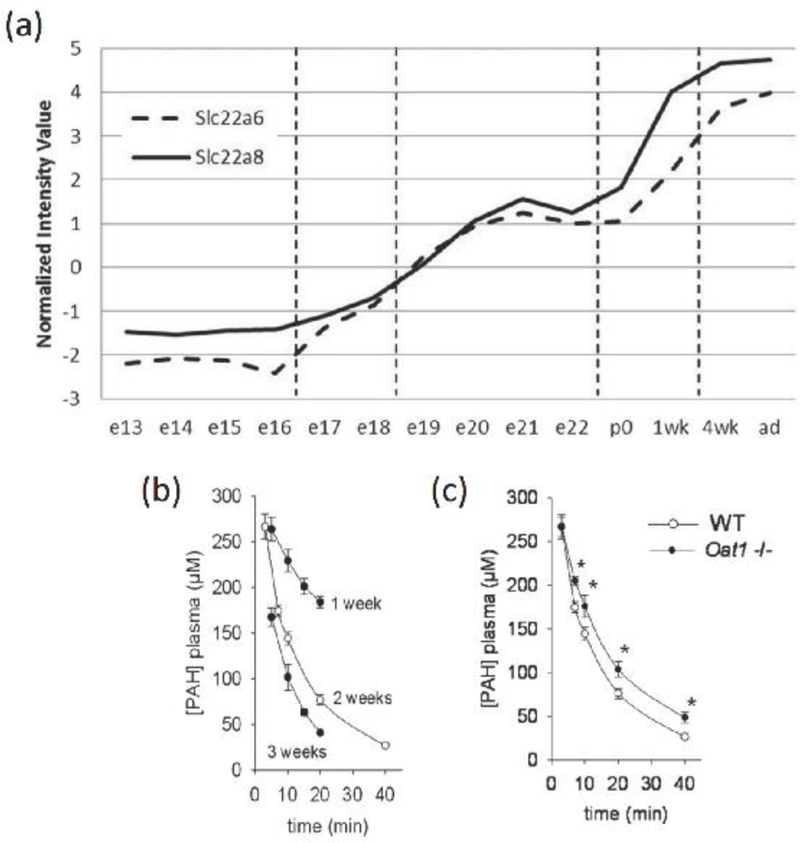
(a) The mRNA expression of Slc22a6 (Oat1, broken line) and Slc22a8 (Oat3, solid line) over the course of kidney development in C57BL/6J mice shows large increases in expression around the postnatal to mature stage; (b) Plasma concentration versus time curves of PAH following 77 umol/kg intravenous dose in wild-type mice demonstrates age-dependent increases in PAH clearance; mean +/− SD shown (n=5 per group) (c) Plasma kinetics of PAH in 2-week-old WT versus Oat1(−/−) mice show PAH clearance is attenuated in Oat1(−/−) mice; mean +/− SD shown (n=5 per group)
The developmental pattern of increased expression in the late fetal period followed by a rapid increase in expression in the immediate postnatal period is similar, but not identical, to that observed for other renal transporters in rodents. For example, the efflux transporters Mrp2 and Mrp4, which are localized to the apical membrane of the proximal tubule, are expressed below adult levels in mice at birth and increase with development [27]. Similarly, mRNA levels of apically expressed MATE1, which mediates the efflux of organic cations, are lower in the proximal tubule during early stages of development as compared with adult. At prenatal day 2, expression was only 12–14% of day 45, whereas MATE expression reached at least half that of 45 day-old mice by day 15 [28], The developmental patterns of mRNA expression in rodents for several other transporters, including Oct2, Octnl, Octn2 and Uratl, resemble that of Oatl and Oat3 [23, 24, 29, 30].
These patterns of developmental expression are consistent with those from non-rodent species. For example, in the ovine renal cortex, OAT1 mRNA abundance is increased at 145 days of gestation (total gestation period of approximately 150 days) and remains elevated in the postnatal period, while OAT3 mRNA abundance was increased postnatally compared to prenatally.[31] Extrapolating animal data to humans in order to predict development and maturation of tubular transport poses a challenge and will require comprehensive time series data and mRNA and protein across species.
More detailed analysis of several SLC22 transporters (including OAT1, OAT3, URAT1 (originally RST in mouse) and OAT2) also reveal early embryonic expression in a variety of non-renal and non-hepatic tissues - including developing neural and vascular tissue [24]. This gave rise to the proposal that these SLC22 “drug” transporters transport endogenous molecules necessary for tissue morphogenesis [24]. indeed, although development is generally normal in knockouts (presumably due to redundancy), these and other so-called drug transporters transport many small molecules that affect signaling events (e.g. metabolite signaling) and which have been shown to affect tissue morphogenesis. For example, cyclic nucleotide-dependent pathways regulate multiple aspects of early kidney development, including Wolffian duct budding [32] and tubulogenesis [33, 34].
Kidney development begins with the interaction of the metanephric mesenchyme (MM) and the branching ureteric bud (UB) [35, 36]. As a result, the MM undergoes a series of stereotypical steps induced by each UB branch tip, resulting in formation of the proximal convoluted tubule, while the UB undergoes iterations of branching morphogenesis, resulting in the formation of the collecting ducts of the urinary collecting system that funnels into the ureter and bladder. The major renal drug transporters are expressed in the nascent proximal tubule while it develops from the induced MM. Most data derives from rodent studies. Organ cultures of the prenatal embryonic kidney also express these drug transporters and, indeed, the developing proximal tubules of the cultured embryonic kidney are capable of the transport of surrogates of organic anion drugs such as 6-carboxyfluorescein [29]. Moreover, these embryonic kidney organ cultures are capable of interacting with a variety of drug substrates of OAT1 and OAT3, making this a useful ex vivo live assay of intact tissue [37, 38]. This is in contrast to renal slice transport experiments where the tissue is in the process of dying and limitations such as collapsed lumens, damaged tubules, and poor oxygenation may exist. However, the true potential of embryonic organ cultures as ex vivo live tissue assays remains to be exploited.
4. Renal transporter activity as a function of developmental age
The functional activity of renal transporters may be evaluated by characterizing the clearance of probe substrates. For instance, para-amino hippurate (PAH) is the prototypical organic anion substrate and PAH clearance may be used as a diagnostic tool to estimate renal plasma flow. PAH extraction on a single passage through the kidney is approximately 90% due to (i) extensive secretion by the tubules, (ii) free filtration from plasma by the glomerulus, and (iii) non-reabsorption by the tubules. The molecular mechanisms involving PAH tubular secretion have been extensively studied with clear in vitro and in vivo evidence that OAT1 is an efficient PAH transporter [39]. In hOATl expressing Xenopus laevis oocytes, the uptake of 1 μM PAH was 2.7±0.4 pmol/oocyte 30 min with a Km of 3.1 ±0.8 μM and 4.0±1.5 μM in the absence and presence of a chloride buffer, respectively [40]. OAT4 and OAT3 show no or limited PAH transport activity relative to OAT1 [40, 41]. In humans, PAH can be used to measure the effective renal plasma flow (ERPF) and the maximum tubular secretory capacity (TmPAH) (i.e. the difference between the total rate of excretion and quantity filtered by the glomeruli), which is primarily attributable to OAT1 activity.
The clearance of PAH in pediatric patient has been studied prior to the recognition of the role of OATs in tubular secretion and provides the best assessment of postnatal tubular function development to date. In 63 normal neonates between two days and 12 years of age, PAH was used to evaluate maximal tubular secretory capacity and ERPF [42]. The ERPF is lowest in the youngest children and increases gradually, reaching the average adult value around the second year of life. The TmPAH showed higher variability than that of ERPF or GFR. At postnatal age 1–30, the TmPAH was approximately 26% of that during 1 year to < 12 years (Table 1). This data suggests that, as in rodents [23]. postnatal OATl-mediated renal secretion is low in neonates and young infants relative to older children and adults. Further, the tubular secretory capacity may not be predicted on the basis of the GFR alone as the rate of increase in these processes are not identical.
Table 1.
Acquisition of tubular excretory capacity during childhood
| Postnatal age | TmPAH (mg/min/1.73m2) |
|---|---|
| 1–30 days | 19(3.7–30) |
| 31 – 365 days | 51 (35 −64) |
| > 1 year - < 12 years | 72.5 (57.5 −84) |
Data presented as median (interquartile range), n=7 for PNA 1 – 30 days, n=17 for 31–365 days, n=28 for > 1 year – < 12 years. Data abstracted from [35]
TmPAH: maximal tubular excretory capacity for para-amino hippurate
West et al found that in 21 healthy infants aged 0.5 – 110 weeks the maximum tubular secretory capacity was reached in thirty weeks [43]. Again the rates of tubular function and glomerular function differed during the first months of life. Upon linear regression analysis of TmPAH vs body size in infants, a positive intercept is observed equating to zero function at an average of 0.11 square meters body surface area (BSA). One possibility for this finding is the underdevelopment of the OAT system in the newborn kidney.
In addition to PAH studies, the pharmacokinetics of OAT 1/3 drug substrates may also be used to evaluate anionic transport function in pediatric patients. Tenofovir, an antiviral agent indicated for the treatment of HIV-1 infection and chronic hepatitis B, may also be used as a surrogate for organic anion transport capacity. Tenofovir is not metabolized by hepatic CYP enzymes and is renally eliminated by glomerular filtration and tubular secretion with 70–80% of the administered dose recovered in urine. The renal clearance of tenofovir is 2 to 3 times the creatinine clearance demonstrating tubular secretion. Tenofovir tubular secretion involves basolateral uptake by OAT1 and apical efflux out into urine by multi drug resistance protein 4 (MRP4) [44]. In HIV- infected pediatric patients between 8 and 17 years of age, the BSA-normalized renal clearance of tenofovir is similar to that observed in adults [45]. These findings support the view that OAT1 expression and activity reaches adult levels by at least middle childhood.
Ciprofloxacin is a quinolone antibiotic indicated for various infections caused by susceptible isolates. Following intravenous administration, approximately 80% of the dose is eliminated in urine as either an unchanged parent compound or metabolites. Ciprofloxacin renal clearance is nearly 3 times the GFR and is probenecid sensitive (i.e., probenecid reduces renal clearance to a value close to the GFR). InXenopus laevis oocytes and cell monolayers, OAT3 promoted uptake of ciprofloxacin (mOat3; Km = 70 ± 6 μM) with no interaction with either the human or mouse isoforms of OAT1 [46]. This finding was corroborated in Oat3-null mice, in which ciprofloxacin elimination was reduced. Therefore, ciprofloxacin appears to be a more sensitive OAT3 probe substrate than OAT1. Although the pharmacokinetics of ciprofloxacin have been evaluated across the pediatric age continuum, comparatively little is known about renal clearance in children. In one study, the renal clearance of ciprofloxacin in cystic fibrosis patients between 6 and 16 years of age (n=10) was 190 mL/min [47]. This value is lower than that reported in adults (300 mL/min); however, the renal function of the participants of pediatric study was not reported.
DMEs, including CYP450 enzymes, are highly expressed in the proximal tubule [26]. albeit at lower levels than in liver. While this localization of DMEs has clear implications for drug metabolism, the physiological roles are still unfolding. For example, CYP450 enzymes expressed in the kidney are involved in the metabolism of arachidonic acid (AA), whose metabolites, including 14,15-epoxyeicosatrienoic acids (EETs) and 20-hydroxyeicosatetraenoic acid (20-HETE), appear to regulate renal epithelial transport and vascular function [48]. Yet the connection of proximal tubule DMEs to renal “drug” transporters, known to transport prostaglandins and other signaling molecules [4]. remains understudied, and importantly the disposition of compounds that are metabolized and transported in the kidney may change with tubular maturation.
5. Developmental Regulation of Hepatic Drug Metabolizing Enzymes
Hepatic drug clearance is the net result of metabolic enzyme activity and the activity of drug transport proteins responsible for hepatocellular uptake and efflux [49]. Cytochrome P450 (CYP450) enzymes - hemoproteins that catalyze numerous so-called Phase 1 chemical reactions such as hydroxylation, oxidation, and reduction - are responsible for biotransformation of up to 60% to 80% of marketed drugs that are known to be metabolized, and are the primary contributors to hepatic drug metabolism [49, 50, 51]. Further, Phase 2 conjugation pathways, including the UDP-glucuronosyltransferases (UGTs) that catalyze the covalent linkage of glucuronic acid to substrates, may occur either subsequently or in parallel to increase the polarity of the metabolite in order to facilitate excretion from the body [51, 52] (Figure 1).
Individual Phase I and Phase II hepatic enzymes have unique patterns of expression during development [53, 54, 55, 56]. Significant maturation occurs during the first few years of life, which includes both the size and blood flow to the liver as well as the expression of metabolic enzymes [53]. For the majority of CYP450 enzymes, a gradual increase in expression exists from birth through the first year of life, eventually reaching adult levels, though the rates of increase with time differ and are not necessarily linear as a function of age [53]. On the other hand, some enzymes display a switch during development from one distinct isoform to another - most notably CYP3 A7 - which is highly expressed in fetal liver and during the perinatal period but then rapidly declines, followed by an increase in the hepatic expression of CYP3 A4 and CYP3A5 [57, 58, 59, 60]. While CYP3A isoforms are believed to have relatively similar substrate specificity, the metabolic capability of CYP3 A7 appears to be lower than for CYP3 A4 and CYP3A5 [61].
The mechanisms underlying the expression patterns of drug metabolizing enzymes appear linked to birth, as some enzymes (i.e. CYP1A2) are not expressed during early life [62]. In the case of CYP2C9, protein expression in liver is approximately 1% of adult levels during the first trimester, which then undergoes progressive increases in expression during the second and third trimesters to levels approaching 30% of mature levels [63]. Neonatal CYP2C9 content is significantly higher than during the late fetal period, again indicating that birth is a factor driving development [53]. In animal models, CYP450 enzymes within a genomic cluster show similar developmental patterns, potentially indicating regulation by a common pathway during liver development [64].
From a pharmacokinetic perspective, the consequence of the unique developmental patterns of Phase I and Phase II DMEs is that predictions of clearance for hepatically-metabolized drugs based on scaling by body weight alone will not provide accurate predictions in the youngest children [65]. Thus, while weight-based or body surface area-based dosing normalization may be adequate for older children, such dosing strategies are generally unable to provide safe and effective doses for neonates [66]. Physiologically-based PK (PBPK) modeling is a tool that has been applied to integrate anatomical and physiological changes in the pediatric populations along with drug-specific parameters [65, 67].
6. Developmental Regulation of Hepatic Drug Transporters
Data on the ontogeny of hepatic transporters in humans come primarily from gene expression studies or animal studies, although protein expression data is emerging [68, 69, 70]. Differences appear to exist in the expression of individual transporters with age, yet expression of hepatic transporters seems to be similar between older children, adolescents, and adults. Nonetheless, an understanding of the time course of expression during development will aid in predicting the pharmacokinetics of drug substrates in neonates, infants, and younger children, and will also provide insights into the physiological roles of these transporters [1].
The efflux transporter P-glycoprotein is the most well-studied hepatic transporter in terms of developmental expression. In utero, P-glycoprotein is detectable in bile canaliculi by 14 weeks with gradual increases throughout fetal development but is still 20- to 30-fold below that seen in adults [22]. The mRNA expression of P-glycoprotein increases after birth and during the first months of life until 12 months of age where it approaches adult levels [22]. These findings are corroborated by a more recent study that characterized the protein expression of P-glycoprotein along with several other liver drug transporters. Howveer, in this study P-glycoprotein protein expression in liver appeared to develop later, reaching 50% of adult expression at about 3 years of age [68]. This pattern of developmental expression is similar to other hepatic transporters, including OATP1B3 [68]. The mRNA expression from other transporters, including OATP1B1, BSEP, and MRP2, seem to exhibit a different pattern of delayed maturation with reduced expression after birth and during at least the first six months of life [68]. However, protein expression data for NTCP is conflicting and suggests no age-dependency [68]. The most dramatic changes in developmental protein expression have been shown for OCT1, an uptake transporter expressed on the sinusoidal membrane of hepatocytes which transports thiamine and a variety of clinically relevant drugs including metformin [71]. The OCT1 protein expression in neonates is 5-fold lower than that in adults, which then reaches 50% of adult expression around 5 months [68].
7. Mechanisms of regulation of transporter expression during development
The regulation of drug transporter expression in the late prenatal and early postnatal proximal tubule with respect to particular transcription factors/nuclear receptors has been examined in some detail, although much remains to be worked out. The pregnane X receptor (PXR) is well- recognized in the the transcriptional regulation of a number of overlapping genes that code DMEs and transporters [72]. PXR is activated by ligand binding and the coregulation of DMEs and transporters by PXR led in part to the recognition of “interplay” between these proteins in the disposition of substrates [73, 74]. Moreover, another mechanism that has been investigated is the role of the hepatocyte nuclear factors (HNF4a and HNFla) in regulating the expression of proximal tubule transporters as well as cell fate [26, 75]. From introduction of transcription factors into mouse embryonic fibroblasts, it appears that, while HNF4a and HNFla push cells toward a proximal tubule fate based on “signature marker” expression, other mechanisms may also be important, at least in these types of in vitro experiments. This includes the suppression of a tendency towards a dual proximal tubule-hepatocyte fate dependent on a set of liver transcription factors [75]. In such experiments, HNF4a and/or HNFla are sufficient to increase the expression of a number of SLC and ABC drug and other transporters. ChiP-seq analysis of DNA binding sites for HNF4a generally confirms substantial direct binding to the regulatory regions in the genes of these and other transporters and drug metabolizing enzymes [26].
HNF4a and HNFla also appear to cooperate in the transcriptional initiation and maturation of genes associated with Phase I and Phase II drug metabolism in the developing proximal tubule [26] as well as the liver [76]. This apparent orchestrated expression of both transporters and metabolic enzymes in the developing kidney and liver highlights the potential interplay between DMEs and transporters in handling endogenous and exogenous substrates within the developing proximal tubule and hepatocyte.
Moreover, in addition to experiments in which these transcription factors are introduced into cells, there is data from knockouts of HNF4a in a variety of tissues (but not the kidney) and HNFla in mice [77, 78]. Overall, the knockout data supports the importance of these transcription factors in the expression of SLC and ABC drug transporters and drug metabolizing enzymes [77]. In organ cultures of the embryonic kidney, a reported HNF4a antagonist seems to inhibit proximal tubule differentiation and transporter expression [26]. Interestingly, HNF4a, once viewed as an orphan nuclear receptor, appears to be activated by linoleic acid [79]. raising the possibility that a fatty acid transported into the cell by a drug or other SLC transporter can induce the expression of numerous multispecific and monospecific transporters.
In conclusion, the developmental regulation of renal drug, metabolite and toxin transport, largely in the proximal tubule, is clearly an understudied area. This is true in model animals (e.g. rat, mouse)—and even much more so in humans. While there is some insight into transcriptional mechanisms, particularly those mediated by HNF4a and HNFla, and other nuclear receptors/transcription factors, key questions remain unanswered in the perinatal and postnatal setting regarding the mechanisms of generation and maintenance of vectorial transport by apical- basolateral polarization of SLC and ABC transporters, posttranslational modifications, intracellular transporter pools and recycling as well as degradation mechanisms, among other mechanisms as the proximal tubule matures [33].
Almost entirely unstudied is the potential connection between proximal tubule transporters and DMEs, which are expressed at substantial levels [26]. and this connection may change as tubular maturation occurs. Also unclear is the extent to which crosstalk (remote communication) between the kidney and liver is necessary for proper functional maturation of transport and DME capacity in one or both organs. Given that many OAT and other transporter substrates include key metabolites and signaling molecules derived from the gut microbiome [17, 18, 19, 80]. it seems likely that functional maturation of transport and DME activity will depend on the changing gut microbiome in the postnatal period. Indeed, some of these substrates may induce transporter expression, perhaps by directly activating nuclear receptors and other transcription factors and/or epigenetic modifications [1].
8. Expert Opinion
The appreciation of the importance of interorgan (eg. liver, kidney, intestine) and interorganismal (eg. microbiome-host) communication to functional maturation of postnatal transport and DME activity is consistent with the Remote Sensing and Signaling Hypothesis [1, 3, 4, 5]. Indeed, the transition between the late prenatal and immediate postnatal periods is one of the most profound in terms of renal gene expression [81, 82] as well as organ and systemic physiology [21, 83]). How the transport and other functions (e.g. DME activity) in all these different organs becomes orchestrated to achieve homeostasis of small molecules in various tissues and body fluids is one of the great questions in developmental physiology and pharmacology. It must depend on coordination of small molecule signaling and remote organ sensing (i.e. the mutual sensing of the transport capacity and types of substrates by the kidney, liver, intestine, and other organs) and constant feedback during this particularly dynamic period of life.
The Remote Sensing and Signaling Hypothesis, first proposed a decade ago [1, 2, 3, 4, 5, 84]. attempts to explain how this “remote interorgan and interorganismal communication” might occur in the context of the need to optimize whole organ and systemic homeostasis of small molecules transported by SLC and ABC drug and other transporters [4, 5]—with postnatal organ maturation being just one important context (Figure 3). Such a SLC and ABC transporter network — functioning to optimize metabolites (eg. uric acid, carnitine) and signaling molecules (eg. alpha-ketoglutarate, beta-hydroxybutyrate, short chain fatty acids, bile acids) at the cellular, organ, and system levels — must be closely linked to classical homeostatic systems such as the neuroendocrine system, the growth factor-cytokine system, and autonomic nervous system. Coordination of all these systems would seem to be especially crucial in the immediate postnatal period, and defects in coordination could lead to disease or death.
Figure 3. Small molecule remote communication in the prenatal and postnatal periods.
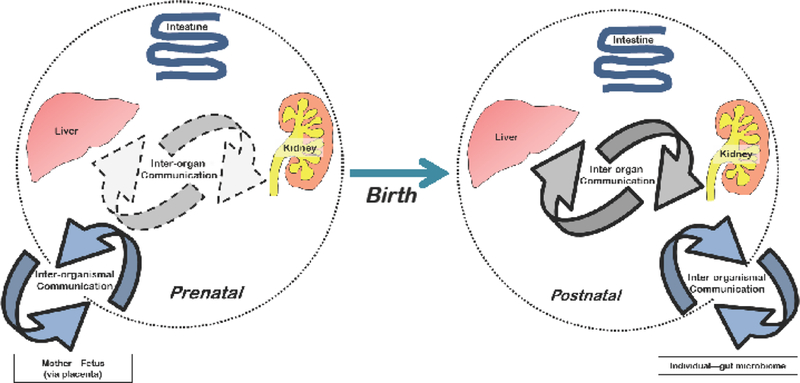
Regulated, transporter-mediated movement of small molecules that reflect (and affect) cell states and/or tissue physiology. Movement across the intestine enables communication of the body with the gut microbiome. Additional inter-organismal communication takes place from maternalfetus and mother-nursing infant. Figure 3. Small molecule remote communication in the prenatal and postnatal periods Page 32 of 32 URL: http://mc.manuscriptcentral.com/eomtE
At different points in postnatal development, for example, immediately after birth compared to the late juvenile period, the whole system may need to optimize for different sets of small molecules. Furthermore, individual organs such as the liver and kidney may need to optimize for entirely different small molecules (compared to the whole system), depending on organ function requirements at that point in development. Finally, the optimal degree of inter-organ communication (e.g. gut-liver-kidney) and, indeed, inter-organismal (e.g. gut microbiome) communication is likely to change during postnatal development (as will the microbiome itself).
It is to be emphasized that, apart from some mechanism of coordinated transcriptional regulation such as that involving HNFla/HNF4a, the Remote Sensing and Signaling Hypothesis leaves open the possibility that different mechanisms—substrate induction, internalization or relocalization to the plasma membrane of transporters (from an internal pool), covalent modification, regulated degradation—are more or less important at distinct postnatal time points. Thus, the individual tissues and groups of remotely communicating tissues may employ different molecular and cellular regulatory mechanisms (upon transporters and DMEs) at distinct time points to achieve local and systemic homeostasis.
This will, however, remain largely a theoretical framework until it is thoroughly investigated in the postnatal developmental context. For there is an acute need to understand the systems physiology of the postnatal period in order to improve neonatal health and, particularly, to optimally manage the problems of premature birth. The Remote Sensing and Signaling Hypothesis provides a framework for beginning to understand the key roles that SLC and ABC transporters (as well as DMEs) in helping to maintain small molecule homeostasis after birth. In order to test this hypothesis in the context of postnatal organ development and organ cross-talk, it will be necessary to obtain a considerable amount of time series “omics” data (e.g. transcriptomics, ChiP-seq, epigenetic, metabolomics, in vivo functional data) over the entire postnatal period through adulthood from multiple organs and body fluid compartments. This will most readily be done in rodent models, but it is important to obtain as much supportive data in humans as possible. An equally great challenge will be the integration of such omics data over time at multiple scales (e.g. from transcriptional regulation through cellular and organ function) and then integrating the multiscale analysis in one developing organ with another in order to identify potential mechanisms of remote organ crosstalk during postnatal development. Success with these or similar studies will provide the foundation of many years of exciting work in neonatal pharmacology and physiology.
Article highlights box.
The transition between the late prenatal and immediate postnatal periods is one of the most profound in terms of gene expression as well as physiology. A systems physiology view, using the Remote Sensing and Signaling Hypothesis as a framework, seeks to understand how the intestine, liver, and kidney transporters and drug metabolizing enzymes (DMEs) are regulated from the immediate postnatal period through adulthood.
The developmental regulation of renal and hepatic drug transporters and DMEs must be coordinated to optimize for different sets of small molecules.
A deeper understanding of changes over developmental time in gene and protein expression (including function and regulation), organ and systemic metabolism, and classical organ physiology—both in the context of individual organs and multiple “remotely communicating” organs—is critical for more appropriate pharmaceutical treatment of neonates and young children.
Acknowledgments
Funding:
This work was funded in part by grants from the NIH (U54HD090259 & DK109392) awarded to S Nigam
Footnotes
Declaration of interest:
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Bibliography
- 1.Brouwer KL, Aleksunes LM, Brandys B, et al. Human Ontogeny of Drug Transporters: Review and Recommendations of the Pediatric Transporter Working Group. Clin Pharmacol Ther. 2015. September;98(3):266–87. doi: 10.1002/cpt.l76. PubMedPMID: ; PubMed Central PMCID: PMCPMC4731327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn SY, Nigam SK. Toward a systems level understanding of organic anion and other multispecific drug transporters: a remote sensing and signaling hypothesis. Mol Pharmacol. 2009. September;76(3):481–90. doi: 10.1124/mol.109.056564. PubMedPMID: ; PubMed Central PMCID: PMCPMC2730381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu W, Dnyanmote AV, Nigam SK. Remote communication through solute carriers and ATP binding cassette drug transporter pathways: an update on the remote sensing and signaling hypothesis. Mol Pharmacol. 2011. May;79(5):795–805. doi: 10.1124/mol.110.070607. PubMed PMID: ; PubMed Central PMCID: PMCPMC3082935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nigam SK, Bush KT, Martovetsky G, et al. The organic anion transporter (OAT) family: a systems biology perspective. Physiol Rev. 2015. January;95(l):83–123. doi: 10.1152/physrev.00025.2013. PubMed PMID: ; PubMed Central PMCID: PMCPMC4281586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nigam SK. What do drug transporters really do? Nat Rev Drug Discov. 2015. January;14(l):29–44. doi: 10.1038/nrd4461. PubMedPMID: ; PubMed Central PMCID: PMCPMC4750486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.International Transporter C, Giacomini KM, Huang SM, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010. March;9(3):215–36. doi: 10.1038/nrd3028. PubMed PMID: ; PubMed Central PMCID: PMCPMC3326076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sai Y Biochemical and molecular pharmacological aspects of transporters as determinants of drug disposition. Drug Metab Pharmacokinet. 2005. April;20(2):91–9. PubMedPMID: [DOI] [PubMed] [Google Scholar]
- 8.Giacomini KM SY. Gilman’s The Pharmacological Basis of Therapeutics. Brunton LL LJ, Parker RL, editor New York: McGraw-Hill; 2006. [Google Scholar]
- 9.Nigam SK, Wu W, Bush KT, et al. Handling of Drugs, Metabolites, and Uremic Toxins by Kidney Proximal Tubule Drug Transporters. Clin J Am Soc Nephrol. 2015. November 06;10(ll):2039–49. doi: 10.2215/CJN.02440314. PubMedPMID: ; PubMed Central PMCID: PMCPMC4633783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bow DA, Perry JL, Simon JD, et al. The impact of plasma protein binding on the renal transport of organic anions. J Pharmacol Exp Ther. 2006. January;316(l):349–55. doi: 10.1124/jpet.105.093070. PubMedPMID: [DOI] [PubMed] [Google Scholar]
- 11.Mulligan N, Schalkwijk S, Best BM, et al. Etravirine Pharmacokinetics in HIV-Infected Pregnant Women. Front Pharmacol. 2016;7:239. doi: 10.3389/fphar.2016.00239. PubMed PMID: ; PubMed Central PMCID: PMCPMC4972814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright SH, Dantzler WH. Molecular and cellular physiology of renal organic cation and anion transport. Physiol Rev. 2004. July;84(3):987–1049. doi: 10.1152/physrev.00040.2003. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 13.Bhatnagar V, Xu G, Hamilton BA, et al. Analyses of 5’ regulatory region polymorphisms in human SLC22A6 (OAT1) and SLC22A8 (OAT3). J Hum Genet. 2006;51(6):575–80. doi: 10.1007/sl0038-006-0398-l. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 14.Fujita T, Brown C, Carlson EJ, et al. Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1). Pharmacogenet Genomics. 2005. April;15(4):201–9. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 15.Xu G, Bhatnagar V, Wen G, et al. Analyses of coding region polymorphisms in apical and basolateral human organic anion transporter (OAT) genes [OAT1 (NKT), OAT2, OAT3, OAT4, URAT (RST)]. Kidney Int. 2005. October;68(4): 1491–9. doi: 10.1111/j.1523-1755.2005.00612.x. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 16.Liu HC, Goldenberg A, Chen Y, et al. Molecular Properties of Drugs Interacting with SLC22 Transporters OAT1, OAT3, OCT1, and OCT2: A Machine-Learning Approach. J Pharmacol Exp Ther. 2016. October;359(l):215–29. doi: 10.1124/jpet.116.232660. PubMed PMID: ; PubMed Central PMCID: PMCPMC5034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bush KT, Wu W, Lun C, et al. The drug transporter OAT3 (SLC22A8) regulates endogenous metabolite flow through the gut-liver-kidney axis. J Biol Chem. 2017. August 01. doi: 10.1074/jbc.Ml17.796516. PubMedPMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wikoff WR, Nagle MA, Kouznetsova VL, et al. Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oatl). J Proteome Res. 2011. June 03;10(6):2842–51. doi: 10.1021/pr200093w. PubMed PMID: ; PubMed Central PMCID: PMCPMC3 201759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu W, Bush KT, Nigam SK. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci Rep. 2017. July 10;7(1):4939. doi: 10.1038/s41598-017-04949-2. PubMedPMID: ; PubMed Central PMCID: PMCPMC5504054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sekine T, Endou H. Children’s toxicology from bench to bed—Drug-induced renal injury (3): Drug transporters and toxic nephropathy in childhood. J Toxicol Sci. 2009;34 Suppl 2:SP259–65. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 21.Nigam SK, Bhatnagar V. How much do we know about drug handling by SLC and ABC drug transporters in children? Clin Pharmacol Ther. 2013. July;94(l):27–9. doi: 10.1038/clpt.2013.82. PubMed PMID: [DOI] [PubMed] [Google Scholar]
- 22.van Kalken CK, Giaccone G, van der Valk P, et al. Multidrug resistance gene (P- glycoprotein) expression in the human fetus. Am J Pathol. 1992. November; 141(5): 1063–72. PubMed PMID: ; PubMed Central PMCID: PMCPMC1886663. [PMC free article] [PubMed] [Google Scholar]
- 23.Sweeney DE, Vallon V, Rieg T, et al. Functional maturation of drug transporters in the developing, neonatal, and postnatal kidney. Mol Pharmacol. 2011. July;80(l): 147–54. doi: 10.1124/mol.110.070680. PubMed PMID: ; PubMed Central PMCID: PMCPMC3127534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavlova A, Sakurai H, Leclercq B, et al. Developmentally regulated expression of organic ion transporters NKT (OAT1), OCT1, NLT (OAT2), and Roct. Am J Physiol Renal Physiol. 2000. April;278(4):F635–43. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Nieto CE, You G, Bush KT, et al. Molecular cloning and characterization of NKT, a gene product related to the organic cation transporter family that is almost exclusively expressed in the kidney. J Biol Chem. 1997. March 07;272(10):6471–8. PubMed PMID [DOI] [PubMed] [Google Scholar]
- 26.Martovetsky G, Tee JB, Nigam SK. Hepatocyte nuclear factors 4alpha and lalpha regulate kidney developmental expression of drug-metabolizing enzymes and drug transporters. Mol Pharmacol. 2013. December;84(6):808–23. doi: 10.1124/mol.113.088229. PubMed PMID: ; PubMed Central PMCID: PMCPMC3834141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maher JM, Slitt AL, Cherrington NJ, et al. Tissue distribution and hepatic and renal ontogeny of the multidrug resistance-associated protein (Mrp) family in mice. Drug Metab Dispos. 2005. July;33(7):947–55. doi: 10.1124/dmd.105.003780. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 28.Lickteig AJ, Cheng X, Augustine LM, et al. Tissue distribution, ontogeny and induction of the transporters Multidrug and toxin extrusion (MATE) 1 and MATE2 mRNA expression levels in mice. Life Sci. 2008. July 4;83(l-2):59–64. doi: 10.1016/j.lfs.2008.05.004. PubMed PMID: ; PubMed Central PMCID: PMCPMC2494953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sweet DH, Eraly SA, Vaughn DA, et al. Organic anion and cation transporter expression and function during embryonic kidney development and in organ culture models. Kidney Int. 2006. March;69(5):837–45. doi: 10.1038/sj.ki.5000170. PubMed PMID: ; PubMed Central PMCID: PMCPMC2825705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmadimoghaddam D, Zemankova L, Nachtigal P, et al. Organic cation transporter 3 (OCT3/SLC22A3) and multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter in the placenta and fetal tissues: expression profile and fetus protective role at different stages of gestation. Biol Reprod. 2013. March;88(3):55. doi: 10.1095/biolreprod.112.105064. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 31.Wood CE, Cousins R, Zhang D, et al. Ontogeny of expression of organic anion transporters 1 and 3 in ovine fetal and neonatal kidney. Exp Biol Med (Maywood). 2005. October;230(9):668–73. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 32.Tee JB, Choi Y, Shah MM, et al. Protein kinase A regulates GDNF/RET-dependent but not GDNF/Ret-independent ureteric bud outgrowth from the Wolffian duct. Dev Biol. 2010. November 15;347(2):337–47. doi: 10.1016/j.ydbio.2010.08.029. PubMed PMID: ; PubMed Central PMCID: PMCPMC2981800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gallegos TF, Martovetsky G, Kouznetsova V, et al. Organic anion and cation SLC22 “drug” transporter (Oatl, Oat3, and Octl) regulation during development and maturation of the kidney proximal tubule. PLoS One. 2012;7(7):e40796. doi: 10.1371/journal.pone.0040796. PubMed PMID: ; PubMed Central PMCID: PMCPMC3396597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santos OF, Moura LA, Rosen EM, et al. Modulation of HGF-induced tubulogenesis and branching by multiple phosphorylation mechanisms. Dev Biol. 1993. October;159(2):535–48. doi: 10.1006/dbio.l993.1262. PubMed PMID: [DOI] [PubMed] [Google Scholar]
- 35.Nigam SK, Shah MM. How does the ureteric bud branch? J Am Soc Nephrol. 2009. July;20(7): 1465–9. doi: 10.1681/ASN.2008020132. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 36.Steer DL, Shah MM, Bush KT, et al. Regulation of ureteric bud branching morphogenesis by sulfated proteoglycans in the developing kidney. Dev Biol. 2004. August 15;272(2):310–27. doi: 10.1016/j.ydbio.2004.04.029. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 37.Nagle MA, Truong DM, Dnyanmote AV, et al. Analysis of three-dimensional systems for developing and mature kidneys clarifies the role of OAT1 and OAT3 in antiviral handling. J Biol Chem. 2011. January 07;286(1):243–51. doi: 10.1074/jbc.Ml10.139949. PubMed PMID: ; PubMed Central PMCID: PMCPMC3012981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Truong DM, Kaler G, Khandelwal A, et al. Multi-level analysis of organic anion transporters 1,3, and 6 reveals major differences in structural determinants of antiviral discrimination. JBiol Chem. 2008. March 28;283(13):8654–63. doi: 10.1074/jbc.M708615200. PubMed PMID: ; PubMed Central PMCID: PMCPMC2417182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eraly SA, Vallon V, Vaughn DA, et al. Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. J Biol Chem. 2006. February 24;281(8):5072–83. doi: 10.1074/jbc.M508050200. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 40.Rizwan AN, Krick W, Burckhardt G. The chloride dependence of the human organic anion transporter 1 (hOATl) is blunted by mutation of a single amino acid. J Biol Chem. 2007. May 04;282(18): 13402–9. doi: 10.1074/jbc.M609849200. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 41.Burckhardt G, Bahn A, Wolff NA. Molecular physiology of renal p-aminohippurate secretion. News Physiol Sci. 2001. June; 16:114–8. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 42.Rubin MI, Bruck E, Rapoport M, et al. Maturation of Renal Function in Childhood: Clearance Studies. J Clin Invest. 1949. September;28(5 Pt 2): 1144–62. doi: 10.1172/JCI102149. PubMed PMID: ; PubMed Central PMCID: PMCPMC439672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.West JR, Smith HW, Chasis H. Glomerular filtration rate, effective renal blood flow, and maximal tubular excretory capacity in infancy. J Pediatr. 1948. January;32(l):10–8. PubMed PMID: [DOI] [PubMed] [Google Scholar]
- 44.Imaoka T, Kusuhara H, Adachi M, et al. Functional involvement of multidrug resistance- associated protein 4 (MRP4/ABCC4) in the renal elimination of the antiviral drugs adefovir and tenofovir. Mol Pharmacol. 2007. February;71(2):619–27. doi: 10.1124/mol.106.028233. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 45.King JR, Yogev R, Jean-Philippe P, et al. Steady-state pharmacokinetics of tenofovir- based regimens in HIV-infected pediatric patients. Antimicrob Agents Chemother. 2011. September;55(9):4290–4. doi: 10.1128/AAC.01334-10. PubMed PMID: ; PubMed Central PMCID: PMCPMC3165337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vanwert AL, Srimaroeng C, Sweet DH. Organic anion transporter 3 (oat3/slc22a8) interacts with carboxyfluoroquinolones, and deletion increases systemic exposure to ciprofloxacin. Mol Pharmacol. 2008. July;74(l): 122–31. doi: 10.1124/mol.107.042853. PubMed PMID: ; PubMed Central PMCID: PMCPMC2822873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schaefer HG, Stass H, Wedgwood J, et al. Pharmacokinetics of ciprofloxacin in pediatric cystic fibrosis patients. Antimicrob Agents Chemother. 1996. January;40(l):29–34. PubMed PMID: ; PubMed Central PMCID: PMCPMC163051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao X, Imig JD. Kidney CYP450 enzymes: biological actions beyond drug metabolism. CurrDrugMetab. 2003. February;4(l):73–84. PubMedPMID: [DOI] [PubMed] [Google Scholar]
- 49.Momper JD, Venkataramanan R, Nolin TD. Nonrenal drug clearance in CKD: Searching for the path less traveled. Adv Chronic Kidney Dis. 2010. September;17(5):384–91. doi: 10.1053/j.ackd.2010.05.009. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 50.Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013. April; 138(1): 103–41. doi: 10.1016/j.pharmthera.2012.12.007. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 51.Zamek-Gliszczynski MJ, Hoffmaster KA, Nezasa K, et al. Integration of hepatic drug transporters and phase II metabolizing enzymes: mechanisms of hepatic excretion of sulfate, glucuronide, and glutathione metabolites. Eur J Pharm Sci. 2006. April;27(5):447–86. doi: 10.1016/jejps.2005.12.007. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 52.Almazroo OA, Miah MK, Venkataramanan R. Drug Metabolism in the Liver. Clin Liver Dis. 2017. February;21(l):l–20. doi: 10.1016/jcld.2016.08.001. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 53.Hines RN. Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol. 2007;21(4): 169–75. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 54.Ince I, Knibbe CA, Danhof M, et al. Developmental changes in the expression and function of cytochrome P450 3 A isoforms: evidence from in vitro and in vivo investigations. Clin Pharmacokinet. 2013. May;52(5):333–45. doi: 10.1007/s40262-013-0041-1. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 55.Krekels EH, Danhof M, Tibboel D, et al. Ontogeny of hepatic glucuronidation; methods and results. Curr Drug Metab. 2012. July;13(6):728–43. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 56.McCarver DG, Hines RN. The ontogeny of human drug-metabolizing enzymes: phase II conjugation enzymes and regulatory mechanisms. J Pharmacol Exp Ther. 2002. February;300(2):361–6. PubMedPMID: [DOI] [PubMed] [Google Scholar]
- 57.de Wildt SN, Kearns GL, Leeder JS, et al. Cytochrome P450 3A: ontogeny and drug disposition. Clin Pharmacokinet. 1999. December;37(6):485–505. doi: 10.2165/00003088-199937060-00004. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 58.Riffel AK, Schuenemann E, Vyhlidal CA. Regulation of the CYP3A4 and CYP3A7 promoters by members of the nuclear factor I transcription factor family. Mol Pharmacol. 2009. November;76(5):1104–14. doi: 10.1124/mol.109.055699 PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 59.Strougo A, Yassen A, Monnereau C, et al. Predicting the “First dose in children” of CYP3 A-metabolized drugs: Evaluation of scaling approaches and insights into the CYP3A7-CYP3A4 switch at young ages. J Clin Pharmacol. 2014. September;54(9): 1006–15. doi: 10.1002/jcph.294. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 60.Vyhlidal CA, Pearce RE, Gaedigk R, et al. Variability in Expression of CYP3A5 in Human Fetal Liver. Drug Metab Dispos. 2015. August;43(8): 1286–93. doi: 10.1124/dmd.ll5.064998. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams JA, Ring BJ, Cantrell VE, et al. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. DrugMetab Dispos. 2002. August;30(8):883–91. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 62.Song G, Sun X, Hines RN, et al. Determination of Human Hepatic CYP2C8 and CYP1A2 Age-Dependent Expression to Support Human Health Risk Assessment for Early Ages. Drug Metab Dispos. 2017. May;45(5):468–475. doi: 10.1124/dmd.l16.074583. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 63.Johansson M, Strahm E, Rane A, et al. CYP2C8 and CYP2C9 mRNA expression profile in the human fetus. Front Genet. 2014;5:58. doi: 10.3389/fgene.2014.00058. PubMed PMID: ; PubMed Central PMCID: PMCPMC3971157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cui JY, Renaud HJ, Klaassen CD. Ontogeny of novel cytochrome P450 gene isoforms during postnatal liver maturation in mice. Drug Metab Dispos. 2012. June;40(6): 1226–37. doi: 10.1124/dmd.l_11.042697. PubMedPMID: ; PubMed Central PMCID: PMCPMC3362787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maharaj AR, Edginton AN. Physiologically Based Pharmacokinetic Modeling and Simulation in Pediatric Drug Development. CPT Pharmacometrics Syst Pharmacol. 2014. November;3(ll):l–13. doi: 10.1038/psp.2014.45. PubMedPMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edginton AN, Shah B, Sevestre M, et al. The integration of allometry and virtual populations to predict clearance and clearance variability in pediatric populations over the age of 6 years. Clin Pharmacokinet. 2013. August;52(8):693–703. doi: 10.1007/s40262-013-0065-6. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 67.Miyagi SJ, Long-Boyle JR. Predicting Pediatric Drug Disposition-Present and Future Directions of Pediatric Physiologically-Based Pharmacokinetics. Drug Metab Lett. 2015;9(2):80–7. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 68.Prasad B, Gaedigk A, Vrana M, et al. Ontogeny of Hepatic Drug Transporters as Quantified by LC-MS/MS Proteomics. Clin Pharmacol Ther. 2016. 0ct;100(4):362–70. doi: 10.1002/cpt.409. PubMedPMID: ; PubMed Central PMCID: PMCPMC5017908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mooij MG, Nies AT, Knibbe CA, et al. Development of Human Membrane Transporters: Drug Disposition and Pharmacogenetics. Clin Pharmacokinet. 2016. May;55(5):507–24. doi: 10.1007/s40262-015-0328-5. PubMedPMID: ; PubMed Central PMCID: PMCPMC4823323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodieux F, Gotta V, Pfister M, et al. Causes and Consequences of Variability in Drug Transporter Activity in Pediatric Drug Therapy. J Clin Pharmacol. 2016. July;56 Suppl 7:S173–92. doi: 10.1002/jcph.721. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 71.Chen L, Shu Y, Liang X, et al. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc Natl Acad Sci USA. 2014. July 08;111(27):9983–8. doi: 10.1073/pnas.1314939111. PubMed PMID: ; PubMed Central PMCID: PMCPMC4103324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tolson AH, Wang H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. AdvDrugDeliv Rev. 2010. October 30;62(13): 1238–49. doi: 10.1016/j.addr.2010.08.006. PubMed PMID: ; PubMed Central PMCID: PMCPMC2991607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Christians U, Schmitz V, Haschke M. Functional interactions between P-glycoprotein and CYP3A in drug metabolism. Expert Opin Drug Metab Toxicol. 2005. December;l(4):641–54 doi: 10.1517/17425255.1.4.641. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 74.Wacher VJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome P450 3 A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog. 1995. July;13(3):129–34. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 75.Martovetsky G, Bush KT, Nigam SK. Kidney versus Liver Specification of SLC and ABC Drug Transporters, Tight Junction Molecules, and Biomarkers. Drug Metab Dispos. 2016. July;44(7): 1050–60. doi: 10.1124/dmd.ll5.068254. PubMedPMID: ; PubMed Central PMCID: PMCPMC4931883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell. 2010. February 16; 18(2): 175–89. doi: 10.1016/j.devcel.2010.01.011. PubMedPMID: . [DOI] [PubMed] [Google Scholar]
- 77.Bonzo JA, Ferry CH, Matsubara T, et al. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J Biol Chem. 2012. March 02;287(10):7345–56. doi: 10.1074/jbc.Ml_11.334599. PubMedPMID: ; PubMed Central PMCID: PMCPMC3 293558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Servitja JM, Pignatelli M, Maestro MA, et al. Hnflalpha (MODY3) controls tissue- specific transcriptional programs and exerts opposed effects on cell growth in pancreatic islets and liver. Mol Cell Biol. 2009. June;29(ll):2945–59. doi: 10.1128/MCB.01389-08. PubMed PMID: ; PubMed Central PMCID: PMCPMC2682018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yuan X, Ta TC, Lin M, et al. Identification of an endogenous ligand bound to a native orphan nuclear receptor. PLoS One. 2009;4(5):e5609. doi: 10.1371/journal.pone.0005609. PubMed PMID: ; PubMed Central PMCID: PMCPMC2680617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu W, Jamshidi N, Eraly SA, et al. Multispecific drug transporter Slc22a8 (Oat3) regulates multiple metabolic and signaling pathways. Drug Metab Dispos. 2013. 0ct;41(10): 1825–34. doi: 10.1124/dmd.113.052647. PubMedPMID: ; PubMed Central PMCID: PMCPMC3781372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsigelny IF, Kouznetsova VL, Sweeney DE, et al. Analysis of metagene portraits reveals distinct transitions during kidney organogenesis. Sci Signal. 2008. December 09;l(49):ral6. doi: 10.1126/scisignal.1163630. PubMedPMID: ; PubMed Central PMCID: PMCPMC3 016920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stuart RO, Bush KT, Nigam SK. Changes in global gene expression patterns during development and maturation of the rat kidney. Proc Natl Acad Sci USA. 2001. May 08;98(10):5649–54. doi: 10.1073/pnas.091110798. PubMed PMID: ; PubMed Central PMCID: PMCPMC33267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ahn SY, Nigam SK. Organic Anion Transport in the Developing Kidney. 5th ed. 2017. (Fetal and Neonatal Physiology). [Google Scholar]
- 84.Kaler G, Truong DM, Khandelwal A, et al. Structural variation governs substrate specificity for organic anion transporter (OAT) homologs. Potential remote sensing by OAT family members. JBiol Chem. 2007. August 17;282(33):23841–53. doi: 10.1074/jbc.M703467200. PubMed PMID: ; PubMed Central PMCID: PMCPMC3 812435. [DOI] [PMC free article] [PubMed] [Google Scholar]