

Abstract
Significance: Nicotinamide adenine dinucleotide (NAD+) is an essential pyridine nucleotide that serves as an essential cofactor and substrate for a number of critical cellular processes involved in oxidative phosphorylation and ATP production, DNA repair, epigenetically modulated gene expression, intracellular calcium signaling, and immunological functions. NAD+ depletion may occur in response to either excessive DNA damage due to free radical or ultraviolet attack, resulting in significant poly(ADP-ribose) polymerase (PARP) activation and a high turnover and subsequent depletion of NAD+, and/or chronic immune activation and inflammatory cytokine production resulting in accelerated CD38 activity and decline in NAD+ levels. Recent studies have shown that enhancing NAD+ levels can profoundly reduce oxidative cell damage in catabolic tissue, including the brain. Therefore, promotion of intracellular NAD+ anabolism represents a promising therapeutic strategy for age-associated degenerative diseases in general, and is essential to the effective realization of multiple benefits of healthy sirtuin activity. The kynurenine pathway represents the de novo NAD+ synthesis pathway in mammalian cells. NAD+ can also be produced by the NAD+ salvage pathway.
Recent Advances: In this review, we describe and discuss recent insights regarding the efficacy and benefits of the NAD+ precursors, nicotinamide (NAM), nicotinic acid (NA), nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN), in attenuating NAD+ decline in degenerative disease states and physiological aging.
Critical Issues: Results obtained in recent years have shown that NAD+ precursors can play important protective roles in several diseases. However, in some cases, these precursors may vary in their ability to enhance NAD+ synthesis via their location in the NAD+ anabolic pathway. Increased synthesis of NAD+ promotes protective cell responses, further demonstrating that NAD+ is a regulatory molecule associated with several biochemical pathways.
Future Directions: In the next few years, the refinement of personalized therapy for the use of NAD+ precursors and improved detection methodologies allowing the administration of specific NAD+ precursors in the context of patients' NAD+ levels will lead to a better understanding of the therapeutic role of NAD+ precursors in human diseases.
Keywords: NAD+, nicotinamide, sirtuins, oxidative stress, DNA damage
I. Introduction
Pellagra is a syndrome cause by a diet seriously deficient in synthetic precursors for the essential pyridine nucleotide nicotinamide adenine dinucleotide (NAD+), namely niacin (vitamin B3), and tryptophan (75, 117, 255). This lethal disorder can develop within 60 days of maintaining a deficient diet due to the absence of free stores of nicotinic acid (NA) or nicotinamide (NAM) (298). Pellagra is pathologically characterized by a distinct dark pigmented skin rash and the three Ds of dermatitis, diarrhea, and dementia (5). Interestingly, the AIDS dementia complex (ADC) shares some neurological similarities with pellagra in its clinical presentation (55).
In the last century, pellagra was a common disease in rural areas in the poorer southern United States, and was attributed to an unknown infectious pathogen (299). However, it was Dr. Joseph Goldberger, and his associates, of the U.S. Public Health Service, who in 1914 examined the hypothesis that pellagra was due to a dietary deficiency. Subsequently, pellagra was prevented using a diet rich in maize, fresh milk, eggs, and cured meat in these populations (1, 121). Despite these advances, it was not until 1937 that Conrad Elvehjem, a biochemistry Professor, first demonstrated the anti-pellagra genic effect of NAM and NA on the related black tongue disease in malnourished dogs (99, 100).
Individuals diagnosed with pellagra-induced dementia can be successfully treated in the early stages of the disease. However, untreated pellagra results in irreversible neurological damage and eventually death (148). This is primarily due to reduced NAD+ production and availability as NAD+ and its phosphorylated form NADP+ are both essential cofactors and substrates for numerous biological processes (365). A focal reduction in NAD+ availability due to increased turnover or reduced synthesis may also be foundational to the pathology seen in other conditions. It seems to fit the observation of an apparently reversible dementia before frank pathology in patients with ADC. At present, pellagra is a rare condition that has been reported in severe cases of alcoholism and anorexia, or malnourishment in the underdeveloped world (21, 332).
Several biochemical studies have shown that an inefficient production of NAD+, where catabolism exceeds anabolism, may produce cellular dysfunction simply due to dietary lack of niacin (27, 325). It may also be due to the rate-limiting action of cosubstrate-dependent quinolinic acid phosphoribosyltransferase (QPRT) (267, 304). Excess amino acid leucine inhibits QPRT, which prevents the formation of niacin or NA to nicotinic acid mononucleotide (NAMN) (189). Reduced tryptophan availability, particularly after chronic immune activation or in the absence of a tryptophan-rich diet (i.e., soy, meat, fish, eggs, and peanuts), may also be associated with the development of pellagra (217). Essential differences, however, may be observed between ADC and pellagra, as the latter develops as a result of a global bodily deficiency of tryptophan and niacin, while ADC develops as a result of increased tryptophan and NAD+ catabolism at specific, although possibly numerous, sites (Fig. 1). Activation of the tryptophan catabolism may be both positive and negative in ADC. Immune-activated oxidative tryptophan catabolism can positively increase cell viability through increased NAD+ metabolism in brain cells. However, chronic activation of tryptophan catabolism may occur in response to increased NAD+ catabolism. Increased astrocyte and mononuclear phagocyte activation stimulates tryptophan catabolism to maintain NAD+ levels in the early stages of immune activation. However, prolonged immune activation leads to excess macrophage recruitment and activation, reducing the astrocyte-to-neuronal NAD+ supply, resulting in pellagra-like neuronal dysfunction, which may be reversible in the short term (Fig. 1). The characteristic mood disorders and depression of end-stage HIV may be due to increased tryptophan catabolism leading to reduced availability of tryptophan for catabolism via serotonergic pathways (Fig. 1).
FIG. 1.

Putative relationship between changes in tryptophan catabolism and de novo NAD+ synthesis in ADC neuropathology. Immune-activated oxidative l-tryptophan catabolism can contribute positively to the maintenance of cell viability through increased metabolism of NAD+ in astrocytes and mononuclear phagocytes. However, chronic activation of this pathway may also enhance neuronal excitotoxicity through the production of QUIN and possibly 3-HK. 3-HK, 3-hydroxykynurenine; ADC, AIDS dementia complex; IDO, indoleamine 2,3-dioxygenase; IFN-γ, interferon-gamma; NAD+, nicotinamide adenine dinucleotide; PARP, poly(ADP-ribose) polymerase; QUIN, quinolinic acid. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
It is well known that NAD+ concentrations increase under conditions associated with reduced energy loads. These include activities such as fasting, glucose deprivation, caloric restriction (CR), and exercise (68). However, apart from pellagra, NAD+ levels decline in animals on high-fat diets, and during aging and cellular senescence (293). Given that NAD+ levels are elevated under conditions of increased life span or health span, decline under conditions of accelerated aging and/or reduced health span suggests that reduced NAD+ levels may represent a major contributor to the aging process (102). Therefore, supplementation with NAD+ and its precursors may represent a potential therapeutic strategy to mediate protection against accumulation of inflammation and highly volatile reactive oxygen species (ROS) during the aging process.
II. NAD+ Biosynthesis Pathways
Several NAD+ precursors have been identified in our natural diet. These include the amino acid tryptophan, and three forms of vitamin B3—NA, NAM, and nicotinamide riboside (NR) (Fig. 2). Tryptophan catabolism via the kynurenine pathway can lead to de novo NAD+ synthesis (128). When dietary tryptophan is limited, the efficiency of the conversion of tryptophan to NAD+ declines below the well-established conversion ratio of 60:1 (107, 164). NA and NR are precursors found in the basic food chain. NA is produced by plants and algae, while NR is present in milk (338). NAM is formed as a by-product of enzymatic degradation of pyridine nucleotides, and is the main form of vitamin B3 that can be absorbed from animal-based food. The provision of these vitamins to NAD+ is aided by several factors, including the gut microbiome (212, 213). Biosynthetic genes are also regulated by circadian rhythms (243). In addition, the expression levels of a number of enzymes involved in NAD+ anabolism decline with age (236).
FIG. 2.

The NAD+ metabolome. l-TRYP, NA, NAM, NMN, NR, and NAR can be used as precursors for NAD+ synthesis. (A) l-TRYP is catabolized to N-formylkynurenine (N-f-KYN) by IDO or TDO. (B) N-f-KYN is catabolized by arylformidase to form KYN. (C) KATs catabolize KYN to form KA. (D) Kynurenine 3-hydroxylase uses KYN as a substrate to form 3-HK. (E) Kynureninase then forms 3-HAA, which is converted to 2-amino-3-carboxymuconate semialdehyde (not shown) by (F) 3-HAAO. (G) This product is then converted to picolinic acid by picolinic acid carboxylase. (H) Alternatively, the semialdehyde undergoes spontaneous condensation and rearrangement to form QUIN, which forms NAMN by (I) QPRT. (U) NAMN undergoes adenylylation by NMNAT1-3 to form NAAD, which forms NAD+ by (M) glutamine-dependent NAD+ synthetases. NA is used by the Preiss–Handler pathway. (L) NAMN is formed by NAPRT following addition of 5-phosphoribose group from PRPP to NA. (P) NAMPT forms NMN by addition of phosphoribose moiety to NAM. (U) NMN is then converted to NAD+ via the catalytic activity of NMNAT1-3. (N) NAM is also produced as a by-product of NAD-dependent enzymes, for example, PARPs, sirtuins, and CD38. (O) NAM can also be converted to NA by bacterial nicotinamidases. (J) NR is phosphorylated to form NMN by NRK1/NRK2, which is then subsequently converted to NAD+ by NMNAT1-3. (J) NAR can also be used to form NAMN by NRK1/NRK2 or (K) NA by purine nucleoside phosphorylase. (Q) NAM is methylated NNMT to MeNAM and modulates the efficiency of NAD-dependent biological processes. (T) NAD+ can be reduced to form NADH. (R) NAD+ can also undergo phosphorylation to NADP+ (S) and then further reduction to NADPH. 3-HAA, 3-hydroxyanthranilic acid; 3-HAAO, 3-hydroxyanthranilic acid oxygenase; KA, kynurenic acid; KATs, kynurenine aminotransferases; KYN, kynurenine; l-TRYP, l-tryptophan; MeNAM, N-methylnicotinamide; NA, nicotinic acid; NAAD, nicotinic acid adenine dinucleotide; NAM, nicotinamide; NAMN, nicotinic acid mononucleotide; NAMPT, nicotinamide phosphoribosyltransferase; NAPRT, nicotinic acid phosphoribosyltransferase; NAR, nicotinic acid riboside; NMN, nicotinamide mononucleotide; NMNAT, nicotinamide mononucleotide adenylyltransferase; NNMT, nicotinamide N-methyltransferase; NR, nicotinamide riboside; NRK, nicotinamide riboside kinase; PRPP, 5-phosphoribosyl-1-pyrophosphate; QPRT, quinolinic acid phosphoribosyltransferase; TDO, tryptophan 2,3-dioxygenase.
A. Tryptophan catabolism via the kynurenine pathway
Tryptophan is the least abundant amino acid of animal and plant proteins, making up only 1–1.5% of the protein amino acid content (261). Tryptophan was first isolated in 1901 by Sir Frederick Gowland Hopkins and his student S. W. Cole (154), and by 1906 was reported as the first amino acid necessary for growth (261). The kynurenine pathway was first described as a principal route for tryptophan catabolism in 1947 (122). Two major routes for tryptophan catabolism have been identified in mammals that are actively independent of protein anabolism. In the periphery, the kynurenine pathway accounts for up to 95% of tryptophan metabolism, while only about 1% of tryptophan content is converted via the indoleamine pathway to form the neuroactive metabolites, serotonin and melatonin (261).
1. Indoleamine 2,3-dioxygenase-1/2 and tryptophan 2,3-dioxygenase
The kynurenine pathway proceeds with the oxidative cleavage of tryptophan by either indoleamine 2,3-dioxygenase-1 (IDO-1; EC 1.13.11.52) and its isoform IDO2 or tryptophan 2,3-dioxygenase also called tryptophan pyrolase (TDO; EC 1.13.11.11) to produce formylkynurenine (23, 103, 313) (Fig. 2, Step a). Both IDO and TDO are haem-requiring enzymes. IDO is mainly found in extrahepatic tissue, including the brain, placenta, spleen, lung, kidney, alimentary tract, and epididymis. It does not contain activating site for tryptophan analogs and is primarily activated by proinflammatory cytokines, such as interferon-gamma (IFN-γ) (109). Concomitant induction of IDO and free radical production of IFN-γ may at first increase NAD+ biosynthesis to contribute to the regeneration of intracellular NAD+ levels in an environment of increased NAD+ turnover and demand. This suggests a protective role for increased tryptophan catabolism in activated macrophages (Fig. 3). However, TDO is predominantly located in the mammalian liver and can be activated by numerous factors, including fasting, glucocorticoids, hydrocortisone, NA, and l-tryptophan (369).
FIG. 3.
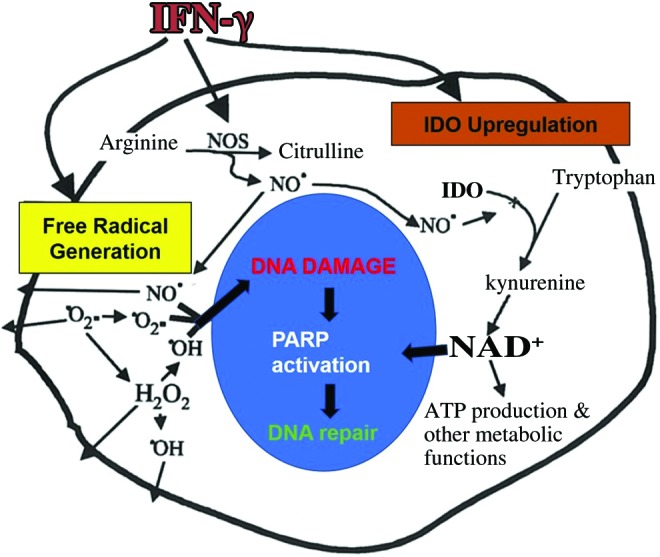
Concomitant induction of IDO and free radical generation by IFN-γ. Chronic immune activation of macrophages and astrocytes will result in increased reactive oxygen and nitrogen species and elevates glutamate levels (in the absence of efficient uptake into astrocytes). A possible relationship exists between IFN-γ-stimulated free radical production and IDO induction, leading to increased de novo synthesis of NAD+. IFN-γ, interferon-gamma. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The metabolic product of IDO-1/2 and TDO activity is the unstable intermediate metabolite, N-formyl kynurenine (N-f-YN) (140), which is rapidly hydrolyzed by kynurenine formylase (EC 3.5.1.9) to form kynurenine (Fig. 2, Step b), the first appreciably stable product of the kynurenine pathway. Kynurenine can cross the blood/brain barrier (BBB) (240) and represents a significant branch point from which three products can be synthesized with the use of three different enzymes, kynureninase (EC 3.7.1.3), kynurenine aminotransferase (KAT; EC 2.6.1.7), and kynurenine 3-hydroxylase (EC 1.14.13.9) (22).
2. Kynureninase
Kynureninase is a cytosolic enzyme that produces anthranilic acid (AA) by the cleavage of the alanine side chain from kynurenine (Fig. 2, Step e). AA can undergo hydroxylation to 5- or 3-hydroxyanthranilic acid (5- or 3-HAA) via nonspecific microsomal hydroxylating enzymes (184, 257, 262). AA can also cross the BBB via passive diffusion. Kynureninase also plays a role in the production of 3-HAA from 3-hydroxykynurenine (3-HK) (Fig. 2, Step e).
The formation of NAD+ from tryptophan is inhibited by inadequate levels of vitamin B6 as kynureninase is dependent on pyridoxyl-5′-phosphate (vitamin B6) as a coenzyme for the conversion of kynurenine to AA, or 3-HK to 3-HAA (238). Low levels of vitamin B6 have been shown to correlate with higher levels of psychological distress (172, 306). The mechanism of B6 involvement in depression is most likely due to the fact that B6 is a cofactor for 5-hydroxytryptophan decarboxylase, the enzyme that catalyzes the last step in serotonin biosynthesis (126, 233).
However, pyridoxine is also a cofactor for several reactions in the brain neurotransmitter pathway, including glutamate decarboxylase and gamma-aminobutyric acid (GABA)-transaminase, the two enzymes required for the synthesis for GABA from glutamate (124, 125, 368). In pyridoxine-dependent epileptic children, inefficient B6 levels resulted in markedly elevated levels of glutamate in the brain (141). Moreover, a decrease in vitamin B6 levels has been associated with a deficiency in both humoral and cell-mediated immune responses, including lymphocyte differentiation and maturation (134).
It has been noted that a decrease in kynureninase activity will reduce further flux through the kynurenine pathway thereby decreasing production of the N-methyl-d-aspartate (NMDA) receptor agonist and excitotoxin, quinolinic acid (QUIN) (111). However, QUIN levels are increased during inflammation, suggesting that kynureninase activity may not be significantly reduced. Vitamin B6 may therefore be used preferentially by the cell for kynureninase activity (e.g., QUIN/NAD production) over GABA transaminase (EC 2.6.1.19; GABA production), 5-hydroxytryptophan decarboxylase (EC 4.1.1.28; serotonin synthesis), and glutamate decarboxylase activity (EC 4.1.1.15), indicating a cell priority for de novo NAD+ biosynthesis under these conditions. The increase in QUIN secretion by activated mononuclear phagocytes during neuroinflammation may indicate an increased demand for NAD+ in these cells, the production of which may be limited under certain conditions by a saturated QUIN ribosylation system (252, 253).
3. Kynurenine aminotransferases
KATs produce kynurenic acid by the transmission of kynurenine (Fig. 2, Step c). Kynurenic acid is a stable compound with nonspecific antagonist action in the brain at the glutamate subtype, NMDA receptor. Both KATs and kynureninase are vitamin B6-dependent enzymes (245).
4. Kynurenine 3-hydroxylase
Kynurenine 3-hydroxylase is a mitochondrial enzyme that also converts kynurenine to 3-HK by the hydroxylation of the aromatic ring (Fig. 2, Step d). 3-HK is an NADPH-dependent enzyme whose activity appears to be reduced with estrogen and in conditions of hyperthyroidism (28). 3-HK can also cross the BBB, stimulate free radical production, and mediate vasodilation (104).
5. 3-Hydroxyanthranilic acid oxygenase
The catabolism of 3-HAA is mediated by 3-hydroxyanthranilic acid oxygenase (3-HAAO; EC 1.13.11.6), an enzyme found in both cytosol and synaptosomal fractions to produce the intermediate 2-amino-3-carboxymuconic semialdehyde (307) (Fig. 2, Step f).
6. Picolinic acid carboxylase
The enzyme picolinic acid carboxylase (PICAC; EC 4.1.1.45) preferentially converts 2-amino-3-carboxymuconic semialdehyde to 2-aminomuconic semialdehyde with subsequent nonenzymatic conversion to picolinic acid (PIC) (Fig. 2, Step g) (188, 230), a metal chelator (106, 270) and NMDA-receptor antagonist or enzymatic rearrangement leading finally to acetyl CoA (30, 79, 230). The nonenzymatic rearrangement of 2-amino-3-carboxymuconic semialdehyde occurs when PICAC is saturated with substrate to produce QUIN (Fig. 2, Step h). The activity of PICAC has been shown to be inversely proportional to the amount of NAD+ synthesized from tryptophan (305).
7. Quinolinic acid phosphoribosyltransferase
QUIN is converted to NAMN by the enzyme quinolinic acid phosphoribosyltransferase (QPRT; EC 2.4.2.19) (Fig. 2, Step i). QPRT catalyzes the reaction between 5-phosphoribosyl-1-pyrophosphate (PRPP) and QUIN in the presence of Mg2+ to produce NAMN. The maximal enzymatic rate for QPRT is apparently the lowest of all kynurenine pathway enzymes, and is 80 times lower than the preceding enzyme, 3-HAAO. However, the Michaelis–Menton constant (Km) for both 3-HAO and QPRT has been calculated to be the same, and this is likely due to the fact that 3-HAA provides substrate for the production of PIC as well as QUIN. The relative amount of QUIN formed from 3-HAA will therefore by determined by the rate of PICAC activity (168, 176). The behavior of PICAC under inflammatory conditions in the human brain or elsewhere does not appear to have been investigated. However, as IFN-γ appears to only induce IDO, it may cautiously be speculated that PICAC activity is not increased during an inflammatory response. Thus, increased flux through the kynurenine pathway will proportionately increase QUIN production.
QPRT is widely distributed in several tissues, including the liver and brain, and may play an important role in mediating neuroprotection against QUIN-induced toxicity, associated with neurodegenerative diseases, including epilepsy and Huntington's disease (58, 132, 175, 229, 235, 250, 333, 364). The physiological levels of QUIN are thought to be in the low nanomolar range, and QPRT activity increases with increased levels of QUIN. However, at high levels of QUIN (>500 nM), neuronal QPRT activity is saturated (267). This leads to the production of QUIN at a greater rate than the production of NAD+, leading to the accumulation of QUIN- and NMDA-mediated excitotoxicity (254).
PRPP is important in the regulation of QPRT activity (Fig. 4) (33, 161, 162, 168). The rate at which PRPP is synthesized and used determines its steady-state concentration within the cell, which then determines the metabolic progress of pathways competing for PRPP. PRPP is synthesized in the cell in the reaction catalyzed by 5-phosphoribose pyrophosphokinase or PRPP synthetase (EC 2.7.6.1) utilizing a ribose-5-phosphate and ATP. PRPP synthetase has an absolute requirement for inorganic phosphate (Pi) and is elevated in cells undergoing rapid cell division. The activity of PRPP synthetase is competitively inhibited by increased levels of ADP and ATP. The ribose 5-phosphate used in this reaction is generated from glucose 6-phosphate metabolism via the hexose monophosphate shunt or from ribose-1-phosphate (generated by the phosphorolysis of nucleotides) via a phosphoribomutase reaction (73).
FIG. 4.

Cofactors required for QPRT activity and NAD+ synthesis. PRPP is important for the regulation of QPRT activity. PIC, picolinic acid. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Disorders in PRPP-synthetase activity and NAD+ metabolism have been implicated in the development of neurological disorders. PRPP concentrations have been positively correlated with cytosolic NAD+ and ATP levels in whole animals, and the availability of PRPP for NAD+ synthesis may be reduced in the presence of high turnover and de novo synthesis of purine and pyrimidine nucleotides (96). This may occur in ADC and neurodegenerative diseases as a result of free-radical-induced DNA damage and astrogliosis. The increase of QUIN seen in some neuroinflammatory conditions may therefore be a combination of increased flux through the kynurenine pathway coupled with decreased enzyme activity possibly due to the use of PRPP for purine and pyrimidine synthesis in DNA-damaged or mitotic cells.
8. NAD pyrophosphorylase (NAM mononucleotide adenylyltransferase)
Further transformations leading to the synthesis of the parent molecule of the pyridine nucleotides, NAD occurs in the nucleus and possibly the mitochondria. NAMN is catalyzed by NAD pyrophosphorylase or nicotinamide mononucleotide adenylyltransferase (NMNAT; EC 2.7.7.1) in the presence of ATP to produce desamido NAD (193, 296) (Fig. 2, Step u). In the presence of glutamine, desamido-NAD is amidated to the parent pyridine nucleotide, NAD+ (Fig. 2, Step m), the final product of the kynurenine pathway (367). Three isoforms have been identified in humans in several different organelles, namely NMNAT-1 (nucleus), NMNAT-2 (Golgi complex), and NMNAT-3 (mitochondria) (31). The differential localization of these enzymes suggests an organelle-specific function for these proteins, and independent nuclear, mitochondrial, and Golgi-specific NAD+ biosynthetic pathways. Unlike NMNAT-1, which is the preferred enzyme for NAD+ synthesis (157), NMNAT-2 and -3 can also form NADH directly from reduced nicotinamide mononucleotide (NMN) (165). NMNAT activity (and predominantly NMNAT-1) is high and nonrate limiting in catabolic tissue, but not in blood (236).
Apart from NAD+ biosynthesis, some studies have demonstrated that NMNAT isoforms can protect against axonal degeneration both in vitro and in vivo (80, 183, 211). NMNAT has been shown to serve as a stress response protein necessary for thermotolerance and attenuation of oxidative stress-induced shortened life span (11). The same study further showed that NMNAT is transcriptionally regulated by the heat shock factor (HSF) and hypoxia-inducible factor 1α (HIF1α) in vivo. During conditions of heat shock, HSF can bind to the NMNAT promoter, thus inducing NMNAT expression. However, under hypoxic conditions, HIF1α enhances NMNAT levels indirectly via induction of HSF (11). In addition, NMNAT isoforms may exhibit protein chaperone function, exerting neuroprotection in several drosophila and mouse models of neurodegeneration (54, 382). Overexpression of NMNAT-1 has also been shown to partially maintain neuronal function and reduce the levels of biochemical insoluble tau in a mouse model of chronic tauopathy with no significant effect on tau phosphorylation, tau aggregation, or tau-induced inflammation and hippocampal atrophy (12). Furthermore, overexpression of NMNAT-3 mediated axonal protection against tumor necrosis factor-induced and intraocular pressure elevation-induced optic nerve degeneration by reducing the expression of p62 and increasing autophagic flux in a retinal ganglion cell line (182). Taken together, these studies suggest the possibility for new mechanisms of protection for NMNAT enzyme activity in addition to its role as an enzyme for NAD+ biosynthesis.
B. NAD+ production from the vitamin niacin
In addition to its de novo synthesis from tryptophan, NAD+ can also be synthesized from the acid, amide, or riboside form of the vitamin niacin (vitamin B3).
1. NA phosphoribosyltransferase
NA is converted to NAMN by the enzyme nicotinic acid phosphoribosyltransferase (NAPRT) (Fig. 2, Step l) (EC 6.3.4.21) using PRPP as a cosubstrate, in an ATP-dependent manner. As QUIN is converted to NAMN by the enzyme QPRT, the sequence of events leading to NAD+ production is identical after NAMN formation from either substrate (112). NAPRT appears to be expressed in several catabolic tissues, including the colon, heart, kidney, and liver (95). The nondeamidated route of NAD+ synthesis displayed higher relative proportions in blood and small intestine, and higher absolute values in the liver and small intestines compared with the amidated (nicotinamide phosphoribosyltransferase [NAMPT]) route, suggesting the significance of NA as a precursor for NAD+ synthesis in these tissues (236). This has been reaffirmed by several feeding studies that have shown that NA is a more favorable precursor for NAD+ synthesis than NAM in the liver, intestine, and kidney (81). As well, NA has been shown to increase intracellular NAD+ levels in a kidney cell line. In addition, overexpression of NAPRT1 has been shown to mediate protection against oxidative stress-mediated NAD+ depletion (142).
Although tryptophan can be converted to NAM, it cannot be used to produce NA in vertebrate cells expressing the de novo synthesis pathway and NAD+ consuming enzymes, such as poly(ADP-ribose) polymerases (PARPs). NAM can be converted to NA in the intestinal lumen by bacterial nicotinamidase (EC 3.5.1.19) (Fig. 2, Step o). However, one study suggested that sufficient levels of pyrophosphate and NAMN in cells can induce NAPRT to yield NA, thus allowing for the production of NA from tryptophan (212, 214). Further studies are required (and are planned) to test this hypothesis. Bacterial and fungal degradation of NAD+ and direct NA supplementation can also increase NA levels in the alimentary canal for distribution to the rest of the body via vascular blood flow (212).
2. NAM phosphoribosyltransferase
The enzyme NAMPT (EC:2.4.2.12) using PRPP as a cosubstrate converts NAM to NMN (Fig. 2, Step p), and then to NAD+ by the action of NAD pyrophosphorylases in the presence of ATP (Fig. 2, Step u) (351). This amidated route of NAD+ synthesis predominantly displayed the highest rates in the liver and kidney, and lowest in blood (161). The expression of NAMPT is encoded by the pre-B cell colony enhancing factor 1 (PBEF1) gene. NAMPT also known as PBEF or visfatin has been identified as a cytokine that promotes the maturation of B cells when other cytokines, such as IL-7, and stem cell factors are available. It also exhibits insulin mimetic effects (260, 374). The intracellular domain has been shown to activate lymphocytes and function as an NAD+ biosynthetic enzyme (281). However, both the extracellular and intracellular domains exhibit favorable phosphoribosyl activity.
In a cisplatin-induced acute kidney injury (AKI) model, pharmacological manipulation of NAMPT expression via AICAR significantly improved renal function and reduced tubular injury. This effect has been associated with increased messenger RNA (mRNA) expression of SIRT3—a mitochondrial sirtuin—and reduced protein hyperacetylation (237). Inhibition of the NAMPT pathway can impair glucose tolerance and insulin secretion in mice, an effect that can be ameliorated by subsequent supplementation with NMN (275). Despite these findings, inhibition of NAMPT, which anabolizes the substrate for NMNAT in mammalian cells, had no significant effect on NMNAT-1-mediated axonal protection in another study (289).
3. NAM N-methyltransferase
The ability of a cell to salvage NAM into the generation of NAD+ via NAMPT versus methylation of NAM by the enzyme nicotinamide N-methyltransferase (NNMT; EC:2.1.1.1) (Fig. 2, Step q) to N-methylnicotinamide (MeNAM) modulates the efficiency of biological processes dependent on NAD+ (6, 263). N-methylation also regulates the biotransformation and detoxification of certain drugs and other xenobiotic compounds by the liver. The enzymatic activity of NNMT uses S-adenosyl methionine as the methyl donor to form pyridinium ions such as S-adenosyl-l-homocysteine (287). This enzyme is predominantly expressed in the liver. A lower expression has been reported in the kidney, lung, skeletal muscle, placenta, heart, and adipose tissue, although it was not detected in the brain or pancreas (287). Increased activity of NNMT has been shown to facilitate the production of toxic N-methylpyridinium compounds, which have demonstrated neurotoxic properties, and which may be involved in the nigrostriatal degeneration (366).
4. NR kinases
NR or nicotinic acid riboside (NAR) represents newly identified precursors that can be converted to NAD+ via the NR kinase (NRK; EC 2.7.1.173) pathway (Fig. 2, Step j), or by the action of nucleoside phosphorylase and the NAM salvage pathway (38). NRKs are highly conserved in eukaryotic cells, and are encoded by the Nmrk genes. Two NRK enzymes have been identified, NRK1 and NRK2, however, their exact physiological roles remain unclear. While NRK1 is ubiquitously expressed in mammalian tissue, NRK2 is not expressed in the kidney, liver, lung, pancreas, and placenta (272). Using the Nmrk1-deficient mouse model (NRK1KO), it has recently been shown that NRKs are rate limiting for NR/NMN-mediated NAD+ synthesis (272).
5. Purine nucleoside phosphorylase
The second NR salvage pathway is NRK independent, through which NR is broken into a ribosyl product and NAM (Fig. 2, Step k), the latter of which yields NAD+ by NAM salvage. Purine nucleoside phosphorylase (PNP; EC 2.4.2.1) has been shown to convert NAR to NA (Fig. 2, Step k), which is then converted to NAMN by the catalytic action of NAPRT (331). PNP deficiency has been shown to increase the deoxyGTP levels. This in turn inhibits ribonucleotide reductase, which is required for the formation of deoxynucleotides (20, 285, 327). The enzyme deficiency leads to the accumulation of metabolites that can induce toxicity in lymphoid lineage cells (291).
6. Cytosolic 5′-nucleotidases
A recent study showed that NAR can be produced by human cells and forms a critical role in intracellular NAD+ anabolism (190). The study showed that cytosolic 5′-nucleotidases (5′-NTs) can dephosphorylate NAMN and, to a lesser extent, NMN to form NAR. The amount of NAR formed appears sufficient to promote NAD+ synthesis in neighboring cells that are missing the machinery required to utilize non-riboside NAD+ precursors (190).
III. Biological Roles of NAD+
NAD+ is an essential pyridine nucleotide that plays major roles in a number of critical biological processes, including oxidative phosphorylation and ATP production, and synthesis of cholesterol, fatty acids, and steroids (224). The primary function of NAD+ was identified by Warburg and Christian in 1936 (357). NAD+ serves as a hydrogen acceptor allowing the transfer of electrons for oxidation/reduction (i.e., redox) reactions leading to ATP production in the mitochondria. ATP represents the cellular “energy currency,” and a decline in intracellular NAD+ levels leads to reduced levels of ATP, culminating in cell death via energy restriction (373).
Apart from NAD+, its closely related phosphate NADP (Fig. 2, Step r) serves as a cofactor in several anabolic processes, such as fatty acid and cholesterol synthesis (315). The reduced form of NAD+ and NADP are NADH (Fig. 2, Step t) and NADPH (Fig. 2, Step s), respectively. These nucleotides serve as hydride donors, in over 400 enzymatic reactions throughout the body involving dehydrogenases, hydroxylases, and reductases (219). These reduced and phosphorylated forms can interconvert, but do not alter the levels of NAD+.
Importantly, NADPH is an essential coenzyme required for the reduction of ROS (29). Thioredoxin (TXN) is an antioxidant protein that is reduced by thioredoxin reductase in an NADPH-dependent process (61). Glutathione disulfide (GSSG) is also a substrate for glutathione reductase for reduction back to glutathione (GSH) using NADPH. The generation of GSH and TXN is pivotal for the elimination of ROS such as hydrogen peroxide (H2O2) (123). Reduced NADPH production due to decreased NAD+ anabolism (or increased catabolism) can lead to impairments in the cell redox balance leading to perturbations in mitochondrial function and genomic signaling and stability, and subsequently leading to increased vulnerability to necrotic and apoptotic pathways.
Apart from its roles in redox reactions, a large body of evidence has shown that NAD+ is more than a regulator of metabolism, but rather can also participate as the required substrate for several important enzymatic reactions, including DNA repair, epigenetically modulated gene expression, maintenance of intracellular calcium homeostasis, and immunological roles (52, 118, 119) (Fig. 5).
FIG. 5.

Cellular roles of NAD+. The mechanisms of degradation of NAD+, including CD38, PARPs, and sirtuins. NAD+ can be phosphorylated to NADP+. There are also oxireduction reactions of NAD+ to NADH and NADP+ to NADH. CD38 is an NAD-dependent enzyme that leads to the production of cADPR from NAD+ and NADP+, respectively. Cytosolic cADPR target to ryanodine receptors on endoplasmic reticulum, and transient receptor potential mucolipin 1 on lysosomes, regulating intracellular calcium signaling from the endoplasmic reticulum and lysosome-mediated intracellular calcium signaling. cADPR, cyclic-ADP-ribose. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
A. Poly(ADP)-ribosylation and DNA repair
DNA strand breaks are known to occur in response to free radicals, ultraviolet (UV) light, or alkylating chemicals, which activate the enzyme PARP (Fig. 2, Step n) (320). Neuronal and astroglial cells exposed to cytotoxic levels of glutamate and QUIN show both an increase in intracellular oxidative stress and PARP activity (46). PARP-1 (the dominant member of a superfamily of 18 PARP proteins) efficiently detects the presence of DNA breaks by its N-terminal zinc-finger domain (312). The ADP-ribosylation of PARP triggers the recruitment of key proteins that stimulate the repair of DNA damage in less than 15 s (85). Importantly, in order for PARP to carry out its ADP-ribosylating function, it uses the ADP ribose (ADPR) moiety of NAD+ for its supply. Thus, PARP breaks down NAD+ to NAM and an ADP-ribosyl product (Fig. 6A) (145). Possibly as a consequence of DNA strand breaks, recent evidence suggests that the poly(ADP)ribosylation of histones or transcription factors may also be involved in nuclear receptor signaling. Poly(ADP-ribose) metabolism is a dynamic process in which degradation of ADP-ribose polymers occurs relatively rapidly through the action of poly(ADP-ribose) glycohydrolase (295) (Fig. 6A).
FIG. 6.

Modulation of PARP activity. (A) PARP and PARG enzymatic activity. PARP breaks down NAD+ to NAM and an ADP-ribosyl product degradation of ADP-ribose polymers occurs relatively rapidly through the action of PARG. (B) Relationship between DNA damage, PARP activation, and NAD+ depletion. Under normal physiological conditions, PARP activation leads to repair of damaged DNA. However, increased PARP activity resulting in decreased NAD+ has been shown to decrease ATP as well as cause cell lysis and death (45, 203) (B). PARG, poly(ADP-ribose) glycohydrolase. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
A significant decrease in intracellular NAD+ has been reported in the brain and a variety of other cell types as a result of DNA strand breaks and PARP activation following exposure to H2O2, nitric oxide, HIV infection, or during inflammation (7, 326, 330). Increased PARP activity resulting in decreased NAD+ has been shown to decrease ATP and neurotransmitter levels in the brain as well as cause cell lysis and death (45, 203) (Fig. 6B). Inhibition of PARP activity, following oxidant injury, has been shown to preserve NAD+ and ATP levels preventing cell lysis (14), although damage to the DNA is probably not prevented. In a pancreatic islet cell population lacking expression of PARP, NAD+ depletion does not occur after oxidant injury despite DNA strand breaks occurring to the same degree (68). This demonstrates that activation of PARP is the major cause of NAD+ depletion in these oxidant injury cells. Elevated levels of free radicals, oxidants, and excitotoxins have been reported in inflammatory mediated diseases of the brain, and in some cases, DNA damage has been demonstrated (2, 220, 221, 341, 380). This suggests that NAD+ depletion through PARP activation may play a role in central nervous system (CNS) dysfunction and pathology under these conditions (Fig. 6).
More recently, it has been suggested that PARP activation, rather than NAD+ decline may be responsible for cell death following exposure to genotoxic insult. For example, poly(ADP)ribosylation has been shown to directly inhibit the glycolytic enzyme hexokinase leading to a significant reduction in glycolysis before NAD+ depletion, mitochondrial dysfunction, and neuronal cell death (17). Moreover, direct poly(ADP)ribosylation of glyceraldehyde 3-phosphate dehydrogenase is the primary cause of cell death in kidney tubules following ischemic injury (94). These studies suggest that the beneficial effects of PARP inhibition may be due to altered metabolic effects independent of maintenance of NAD+ levels during pathological conditions.
PARP also appears to play a positive role in the upregulation of the tumor suppressor protein, p53. For example, PARP-deficient cell lines derived from Chinese hamster V79 cells failed to undergo poly(ADP)ribosylation and activate p53 following treatment with etoposide (363). PARP can also activate DNA-dependent protein kinases that regulate p53 activity through phosphorylation (318). Therefore, on the contrary to reported benefits of PARP inhibitors, pharmacological inhibition of PARP activity may contribute to genomic instability with resulting risk of cancer formation.
B. CD38/CD39/CD73/CD157 and secondary messenger signaling
The immune-associated ectoenzymes CD38, CD39, CD73, and CD157 represent another class of NAD+-consuming enzymes (155) (Fig. 2, Step n). These enzymes require NAD+ to produce ADPR and hydrolyze the secondary messenger signaling molecule, cyclic-ADP-ribose (cADPR), which helps mediate intracellular calcium transients (Fig. 7). CD38 has also demonstrated an immunomodulatory role (135). For instance, the presence of CD38 on T lymphocytes influences the ability of antigen-presenting cells to stimulate antigen-specific T cells (256). Upregulation of CD38 expression also signals maturation of dendritic cells during inflammatory cytokine activation and acts as a modulating adhesion and signaling molecule between dendritic cells and lymphocytes (105). In cardiomyocytes, exogenous stimulants may stimulate an increase in intracellular calcium, which leads to activation of CD38 (147). CD38 expression has also been shown to increase with age (65), and this is most likely attributed to an age-related increase in circulating inflammatory cytokines, and reduced CD38 function has been associated with poor immune responses.
FIG. 7.

Stoichiometry of CD38-mediated Ca2+ mobilizing and NADase activities. (A) CD38 requires NAD+ to produce ADPR and hydrolyze the secondary messenger signaling molecule, cADPR, which helps mediate intracellular calcium transients. ADPR, ADP ribose. (B) CD38 also converts NADP+ to NAADP+ via base exchange (NADase activity of CD38). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Given that 100 molecules of NAD+ must be hydrolyzed to generate 1 molecular of cADPR, it is highly likely that CD38 is a major regulator of intracellular NAD+ levels (76). Accordingly, we found a fivefold increase in NAD+ levels in CD38 knockout neuronal cells compared with controls (52). Therefore, CD38 may not only represent an inefficient secondary messenger enzyme but also as an NADase that primarily regulates intracellular levels of NAD+ and its physiological processes (Fig. 8).
FIG. 8.

Schematic representation of CD38-mediated intracellular Ca2+ secondary messenger signaling. CD38 is also an NADase, which primarily regulates intracellular levels of NAD+ and its physiological processes. CD38 also catalyzes a base exchange between NADP and NA, leading to the formation of NAADP, which is also used as a hydrolytic substrate. NAADP, nicotinic acid adenine dinucleotide phosphate. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
CD38 has also been shown to use β-NAD+ as a substrate, but no α-NAD+ or NADH. CD38 can also catalyze a base exchange between NADP and NA, leading to the formation of nicotinic acid adenine dinucleotide phosphate (NAADP), which is also used as a hydrolytic substrate (90). It can also metabolize analogs of NAD+, including nicotinamide guanine dinucleotide (NGD+) and nicotinamide hypoxanthine dinucleotide (NHD+), yielding cyclic compounds (cGDPR and cIDPR, respectively). These compounds exhibit fluorescent properties, but not calcium releasing (383). They represent useful biochemical agents for examining ADP-ribosyl cyclase activity.
Prolonged activation of CD38 following cardiac stress has been shown to induce a sustained Ca2+ release leading to cardiac hypertrophy and arrhythmias (130). Supporting evidence comes from male CD38 knockout mice, which reported improved cardiac function, while treatment with ADPR cyclase inhibitors led to antiarrhythmic effects in multiple in vitro models and cardiac Ca2+ overload studies (131). Similarly, inhibition of CD73 has been shown to mediate protection against renal stressors, and CD39 activity mediated protection against renal ischemic injury (268).
CD38 can also regulate the activity of PARP and other NAD+-dependent enzyme SIRT1 activities by potentially reducing the accessibility of NAD+ to its preferred enzymatic targets (348). NAM, which is generated by the catalytic activity of CD38, also represents an endogenous metabolite of SIRT1 enzyme. Therefore, it has been postulated that CD38 may in fact be an important regulator on intracellular NAD+ levels and SIRT1 activity, thus influencing SIRT1 functions, including maintenance of cellular bioenergetics, obesity, and senescence. Interestingly, one study reported no significant effects on NAD+ levels in CD38 knockout mice compared with wild-type animals (377). Therefore, the amount of benefit due to CD38 inhibition or ablation warrants further investigation.
Novel CD38 inhibitors may also be useful for treatment degenerative disorders where optimal NAD+ and NADPH anabolism remains crucial to attenuate oxidative stress insult, the latter of which serves as the ultimate electron donor supporting glutathione peroxidases, peroxiredoxins, and glutaredoxins. However, inhibition of CD38 may also result in a deleterious impact on immunological function. CD38/cADPR also signals oxytocin release, which regulates many social behaviors, and inhibiting this process may induce several forms of mental impairment. Moreover, niacin deficiency, observed in patients with pellagra, often progresses to dementia similar to schizophrenia (98, 264), and this could be due to impaired cADPR formation.
C. Sirtuin activity
Another important NAD+-dependent function is the activity of the silent information regulators of gene transcription, or sirtuin family of enzymes (Fig. 2, Step n). Sirtuins are a family of class III NAD+-dependent histone deacetylases exhibiting protein lysine deacetylase, and partial ADP-ribose transferase activities. In the reaction mediated by sirtuins, an acetyl-modified lysine is bound to a target protein and NAD+ in specific pockets (346). Deacetylation occurs when the modified lysine side chain is coupled to the cleavage of the glycosidic bonds in NAD+, leading to the generation of the deacetylated lysine, acetylated ADP-ribose, and NAM as by-products (101) (Fig. 9).
FIG. 9.

Sirtuin enzymatic activity. NAM is rendered as a by-product of sirtuin-mediated deactylation. Deacetylation occurs when the modified lysine side chain is coupled to the cleavage of the glycosidic bonds in NAD+, leading to the generation of the deacetylated lysine, acetylated ADP-ribose, and NAM as by-products. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
At present, seven classes of sirtuins (SIRT1–7) have been identified in mammalian cells, each of which are localized in various cellular organelles, and mediate a diverse range of important biological functions (53) (Fig. 10). SIRT1 and SIRT6 are nuclear proteins associated with the maintenance of chromatin structure, DNA repair, and gene expression. It has been suggested that SIRT1 may play a pivotal role in promoting cellular longevity and may hold the key to slowing development of the aging phenotype (290). SIRT1 has been shown to influence the acetylation status of several important transcription factors, including the metabolic regulator, peroxisome proliferator-activated receptor-γ (PPARγ), tumor suppressor protein (p53), and the cell growth-linked FOXO forkhead family of transcription factors (192). However, some evidence suggests that SIRT6 may also contribute to an age-resistant phenotype (300). SIRT2 is predominantly a cytoplasmic protein where it regulates gene expression by deacetylating transcription factors that shuttle from the cytoplasm to the nucleus (282). SIRT3, SIRT4, and SIRT5 are found in the mitochondrion where they respond to changes in mitochondrial redox status by altering the enzymatic activity of specific downstream targets, including manganese superoxide dismutase (MnSOD) (249). SIRT7 is localized in the nucleolus of mammalian cells and has been associated with cellular growth and metabolism (347). The biological relevance of sirtuins in redox processes is discussed further in section IV. Importantly, the beneficial effects of sirtuin activity are only achieved if NAD+ levels are optimal.
FIG. 10.

Functions of NAD-dependent sirtuins and relevant transcription factors. Sirtuin-mediated deacetylation affects numerous target enzymes and transcription factors relevant to aging and disease. Importantly, sirtuin activities stimulate OXPHOS, while yet unknown acetylation mechanisms serve to inhibit anti-OXPHOS. OXPHOS, oxidative phosphorylation. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
D. Principal causes of NAD+ decline
Apart from deficiency within the NAD+ biosynthesis process, there are principally two conditions under which NAD+ depletion may occur: (i) excessive DNA damage due to free radical or UV attack, leading to hyperactivation of PARP. This ultimately leads to a high turnover and subsequent depletion of NAD+. The resulting energy crisis and reduced ATP production can lead to cell death via either an apoptotic or necrotic pathway (ii). A chronic increase in immune activation and inflammatory cytokine production can accelerate CD38 activity and contribute to NAD+ decline. While several clinical disorders and degenerative disorders can meet these criteria, chronic accumulation of oxidative stress and inflammation during advanced age represents a major driver of NAD+ decline (49). Promotion of NAD+ anabolism using NAD+ precursors may represent a clinically relevant therapeutic strategy to ameliorate age-related decline in cellular energy.
IV. Redox Roles of Sirtuins and Transcriptional Regulation
Since the term of their discovery, sirtuins have been associated with life span extension. However, while the longevity enhancing capacity of sirtuins has been established in several small model systems, the modes of action of sirtuins underlying these beneficial effects remain unclear. Chronic accumulation of damage over time represents the main phenotype associated with the aging process. In particular, chronic oxidative stress can induce damage to diverse macromolecules, and perturb mechanisms with which they are repaired. Recent evidence suggests that the beneficial effects of sirtuins may be mediated by their ability to regulate redox processes. In this section, we investigate the association between sirtuins and their redox environment, and review how sirtuin-mediated deacetylation affects target enzymes and transcription factors.
A. SIRT1
As previously mentioned, tumor suppressor p53 represents the first deacetylation substrate of SIRT1. The transcription factor p53 has been shown to activate numerous pro- and antioxidant genes, including sestrins, MnSOD, and glutathione peroxidase 1 (284). The binding and deacetylation of p53 by SIRT1 at Lys382 mediate its transcriptional activity (208). SIRT1 deacetylation of p53 has been shown to influence the cellular localization of p53 in response to oxidative stress, and may serve as a metabolic switch between antioxidant protection and apoptotic cell death. For instance, in murine embryonic stem cells, the absence of antioxidants in cell culture media induced mitochondrial translocation of p53, while in SIRT1 knockout cells, increased oxidative stress induced nuclear translocation of p53 leading to an antioxidant response (139). Similarly, upregulation of SIRT1in mesangial cells attenuated the induction of p53-mediated apoptotic pathway following exposure to pathological concentrations of H2O2. However, at lower concentrations of H2O2, the SIRT1-p53 interaction led to an induction of antioxidant processes (191).
While the adaptive role of SIRT1 against ROS stress has been well established in vitro, studies using live animal have been less convincing. This is due to the high levels of embryonic lethality following the production of SIRT1−/− mice. However, one study using heterozygous SIRT1 knockout mice reported increased vulnerability to renal oxidative stress, and combined SIRT1+/− p53+/− showed greater susceptibility to tumor development compared with p53 haploinsufficiency alone (146).
SIRT1 has also been shown to deacetylate and activate FOXO3a following exposure to oxidative stress (57). FOXO3a appears to be an important transcriptional activator of the SOD2 gene, which encodes for the production of the endogenous antioxidant protein MnSOD. The catalase enzyme, which acts directly on free radicals, is predominantly localized in peroxisomes and represents another target of FOXO3a (185). As per the relationship between SIRT1 and p53, low levels of H2O2 can mediate FOXO3a-mediated induction of catalase, while cytotoxic levels of H2O2 can induce FOXO3a-mediated apoptosis (144). In cardiovascular disease, increased oxidative stress can upregulate SIRT1expression and stimulation of catalase and MnSOD expression. However, higher levels of SIRT1 can lead to cardiac hypertrophy and cell death via apoptotic pathways (9). Taken together, these studies collectively suggest that SIRT1 serves as an ROS sensor, capable of inducing protection at low-level stress, while inducing apoptosis at severe stress levels.
Recently, it has also been shown that mechanisms responsible for the regulation of the intracellular NAD+:NADH ratio can also affect SIRT1 function via AMP-activated kinase (AMPK), an essential regulator of cellular energy homeostasis. Several studies have shown that reduced glucose available in myoblasts induced activation of AMPK and upregulation of NAMPT, leading to increased levels of intracellular NAD+ and activated SIRT1, and culminating in the activation of several transcriptional mediators, including FOXO proteins and PGC-1α, thus enhancing catabolism and mitochondrial biogenesis (110). Furthermore, activation of AMPK stimulated transcriptional activity downstream of SIRT1 in another study (66). SIRT1 can also activate AMPK through positive feedback mechanism. For instance, liver kinase B1 (LKB1), which phosphorylates and activates AMPK under low nutrient levels, can be deacetylated by stimulation or overexpression of SIRT1 (either directly or indirectly). This promotes translocation of LKB1 from the nucleus to the cytosol, which further phosphorylates AMPK (192). As such, there seem to be multiple levels of metabolic regulation occurring through the AMPK–SIRT1 axis, and many of these steps require further elucidation.
Apart from the AMPK-SIRT1 axis, SIRT1 can also interact and deacetylate PGC-1α. PGC-1α is an important transcriptional coactivator that stimulates mitochondrial biogenesis and indirectly also mitochondrial dynamics in a tissue-dependent manner. For instance, increased hepatic SIRT1 due to fasting can deacetylate PGC-1α leading to both inhibition of glycolytic genes and increased expression of genes associated with gluconeogenesis (276). Another study showed that SIRT1 could directly interact with and deacetylate PGC-1α in adrenal PC12 cells leading to reduced PGC-1α transcriptional activity and related mitochondrial oxidative metabolism (246). However, in skeletal muscle, increased SIRT1 activity and PGC-1α deacetylation led to an increase in mitochondrial fatty-acid oxidation (116). Reduced PGC-1α activity associated with reduced expression of the mitochondrial antioxidant protein, MnSOD, providing additional support for the role of SIRT1 on control of redox stressors (207).
Recent studies have shown that calorie restriction and the phytochemical resveratrol, which are known another to activate SIRT1, can enhance endothelial nitric oxide synthase (eNOS) expression and promote mitochondrial biogenesis by upregulating transcription factors such as PGC-1α (78). Similarly, SIRT1 has been shown to deacetylate eNOS in vivo, leading to increased eNOS activity and intracellular NO production (225). Therefore, SIRT1 represents a key regulator of vascular tone dependent on eNOS.
Moreover, it is well established that the transcriptional response to hypoxia is regulated mainly by the HIF family of proteins, of which HIF1α and HIF2α are well characterized [reviewed in Majmundar et al. (216)]. It has been demonstrated that both HIF1α and HIF2α can be deacetylated by SIRT1 by two separate and distinct mechanisms. Under normal physiological conditions, SIRT1 can bind to, and deacetylate, HIF1α, preventing HIF1α from interacting with the transcriptional coactivator p300, inhibiting its transcriptional activity (200). However, under hypoxic conditions, the decline in the NAD+:NADH ratio and available NAD+ for optimal SIRT1 activity due to reduced oxygen levels allows HIF1α to remain acetylated, thus preventing its hypoxic transcriptional activity (200). On the contrary to its effect on HIF1α, SIRT1 can also form a complex with SIRT1 under hypoxic conditions and is deacetylated at three lysine residues (K385, K685, and K741) in the carboxy terminus, leading to increased transcriptional activity of HIF2α and related proteins, and erythropoietin in particular (92).
B. SIRT2
The expression of SIRT2 has also been shown to be upregulated at both the mRNA and protein levels in response to cellular stressors such as oxidative stress. Numerous studies have demonstrated that increased SIRT2 expression following oxidative insult can lead to cellular apoptosis via induction of the proapoptotic protein Bim (350). Overexpression of SIRT2 has also been shown to promote neurodegeneration, although the exact mechanism remains unclear (322). In the absence of the SIRT2 gene, upregulation of the cytosolic chaperone 14-3-3ζ, sequesters the proapoptotic mitochondrial protein BAD in the cytosol and mediates protection against anoxia–reoxygenation-induced cell death (210). SIRT2 inhibitors have been shown to ameliorate α-synuclein-mediated toxicity in a cellular model of Parkinson's disease (PD) (210). However, under low-stress conditions, SIRT2 upregulates mitochondrial MnSOD via FOXO3a deacetylation, leading to a reduction in the levels of ROS.
C. SIRT3
Isocitrate dehydrogenase 2 (IDH2) represents another major target of SIRT3, a mitochondrial sirtuin. IDH2 uses NADPH to generate reduced GSH to mediate an antioxidant affect. Schlicker et al. showed SIRT3, but not SIRT5, could deacetylate IDH2 at K211 and K212 residues to promote its activity (294). It has been shown that the GSH:GSSG ratio and the level of NADPH are increased in the liver, brain, and the inner ear following CR in an SIRT3-dependent manner (311). As well, SIRT3 directly deacetylates and inhibits the activity of IDH2, and SIRT3 overexpression increased NADPH levels and reduced oxidative stress-mediated cell death (311). Taken together, these studies suggest that CR, SIRT3, and IDH2 represent important targets for the management and treatment of age-related hearing loss, and that maintenance of intracellular NAD+ levels modulates the cellular response to degeneration.
Like IDH2, SIRT3 has been shown to mediate SOD2 activity by regulating mitochondrial FOXO3a activity, although the exact mechanism remains unclear. One study using overexpression of SIRT3 in mouse embryonic fibroblasts shows that the levels of ROS were dramatically reduced in an SOD2-dependent manner (266). Similarly, hyperacetylation of SOD2 in SIRT3-deficient mice led to reduced SOD2 activity and upregulation of ROS production (329). However, differences in the site-specific regulation of SOD2 by SIRT3 have been reported, and this is likely due to differences in cell type, species, or stress conditions.
Additional mitochondrial targets of SIRT3 and SIRT5 have also been recently identified, which can regulate oxidative stress. SIRT3 has been shown to deacetylate complexes I, II, III, and IV and glutamate dehydrogenase, regulating glutamate oxidative stress yielding NADPH, deacetylated by SIRT3 (and antagonized by SIRT4-mediated ADP-ribosylation) (205). SIRT5 can also deacetylate cytochrome c (294). As well, SIRT3 and SIRT5 can both regulate mitochondrial-localized reactions of the urea cycle. To be more specific, SIRT3 can deacetylate ornithine transcarbamoylase, while SIRT5 acts on carbamoyl-phosphate synthase 1 to enhance urea cycle function and promote the clearance of oxidative stress-promoting ammonium (137).
SIRT3 has been recently shown to be important for the regulation of normal cardiac function and protection against cardiac pathologies. Knockout of SIRT3 has been shown to increase the hyperacetylation of mitochondrial protein, leading to spontaneous cardiac hypertrophy with age and >50% reduction in ATP levels (319). Reduced SIRT3 expression and hyperacetylation of cardiac mitochondrial enzymes have also been reported in mouse models for cardiac disorders, and poor human hearts (156). As well, increased activity of acyl-CoA dehydrogenase and other enzymes involved in fatty acid oxidation (FAO) has also been reported in SIRT3 knockout mice (13). However, another study reported reduced rates of FAO in the hearts of fasted animals (151). These differences may be attributed to variation in the type of stressors that can influence the activity of protein acetylation.
Renal stress has been shown to reduce the expression of SIRT3. For instance, SIRT3 mRNA expression was shown to be decreased in a model of free fatty acid-associated tubulointerstitial inflammation, and this occurred parallel to increased levels of ROS and markers of inflammation compared with age-matched control animals (187). Interestingly, retroviral overexpression of SIRT3 attenuated these changes, suggesting that optimal SIRT3 function is necessary for renal function (370). Similarly, high-glucose levels were shown to decrease the mRNA and protein expression of SIRT3, and supplementation with NAD+ ameliorated high-glucose-induced mesangial hypertrophy and SIRT3 expression at both the genomic and protein levels (390). Taken together, these findings suggest that SIRT3 can protect against renal degeneration in diabetic nephropathy.
D. SIRT4
Like SIRT3, SIRT4 appears to be highly expressed in catabolic tissue such as the brain, heart, liver, and kidney (136). SIRT4 has been shown to protect against hypoxia-induced apoptosis in cardiomyoblast cells (202). However, knockout of SIRT4 protected against angiotensin-II induced cardiac hypertrophy and fibrosis in mice, suggesting that SIRT4 may be directly involved in the pathogenesis of cardiovascular disease (209). While both studies suggest a discrepancy for the exact role of SIRT4 in cardiac function, it appears likely that these effects are due to the modulatory role of SIRT4 on cellular oxidative stress levels.
There also exists a strong correlation between kidney function, SIRT4 levels, and the NAD+ metabolome. For instance, cotreatment with cisplatin and the phytochemical curcumin restored NAD+ levels and attenuated the decline in NAMPT, SIRT1, SIRT3, and SIRT4 expression due to cisplatin-induced nephrotoxicity (342). However, it is unlikely that these effects are directly in response to SIRT4, as the levels of NAMPT, SIRT1, and SIRT3 were also affected. Additional work is necessary to evaluate the role and modes of action of SIRT4 in degenerative disorders of the brain, heart, and kidney, and other age-related conditions associated with NAD+ depletion.
E. SIRT5
The exact roles of SIRT5 in maintaining normal cellular homeostasis is not well understood. One study found no significant differences between the heart weight and rate, and systolic blood pressure in SIRT5 knockout mice exposed to a high-fat diet (379). However, another study showed that protein succinylation is uniquely elevated in SIRT5 knockout mice (248). These proteins include those involved in fatty acid metabolism, amino acid catabolism, the TCA cycle, oxidative phosphorylation, and ketone and pyruvate metabolism (41). In mice exposed to cardiac ischemia, a larger infarct volume and elevated oxidative stress were reported in SIRT5 knockout hearts compared with wild-type controls (41). These changes were accompanied by increased fibrosis, and reduced shortening fraction and ejection fraction compared. Increased activity of succinate dehydrogenase (SDH) was also reported in SIRT5 knockout mice, and SDH inhibitors reduced infarct size to “normal” levels (41). This suggests that the protective effects of SIRT5 may be mediated by desuccinylation of SDH.
Similarly, knockout of SIRT5 also resulted in hypersuccinylation of mitochondrial protein, and post-translational modification of malonylation and glutarylation in the kidney (198). In addition, SIRT5 has been shown to deacetylate carbamoyl-phosphate synthetase 1 (CPS1), leading to increased activity of CPS1 and reduced plasma urea levels (242). Increased blood ammonia levels were reported in SIRT5 knockout mice compared with age-matched wild-type controls. These findings provide a key role for the role of SIRT5 in the regulation of ammonia.
F. SIRT6
While the effect of redox stressors on SIRT6 function remains nascent in current literature, one study has shown that knockdown of SIRT6 can induce accelerated senescence as evidenced by the development of degenerative features, shortened telomere length, and reduced life span (239). Interestingly, HIF1α has been shown to be upregulated in cells lacking SIRT6, leading to an increased glucose uptake and improved glycolysis (384). In normal mice embryonic fibroblast cells, SIRT6 serves as an H3K9 histone deacetylase, inhibiting HIF1α-dependent transcription of multiple glycolytic genes, thus acting as a corepressor of HIF1α.
G. SIRT7
Of the family of sirtuins, SIRT7 remains the least investigated. One study showed that knockdown of SIRT7 enhances acetylation of p53, leading to increased vulnerability to genotoxic insult (241). SIRT7 has also been shown to inhibit cell proliferation following exposure to high oxidative stress levels (344).
H. Activation by NAD+ precursors
A growing body of evidence suggests that upregulation of NAD+ anabolism can influence processes regulated by sirtuins. These pathways may therefore be upregulated with NAD+ or NAD+ precursors, or other means of manipulating NAD+ biosynthesis pathways. It has been shown that the Km for SIRT3 and SIRT5 is significantly lower than the levels of mitochondrial NAD+, suggesting that the activity of these sirtuins is rate limited by the availability of mitochondrial NAD+ levels (150). Current evidence suggests the importance of SIRT1 and SIRT3 in regulating the beneficial effects of NAD+, and the effects of NAD+ supplementation on other sirtuins remain unclear. Examining whether the activity of other sirtuins is affected by NAD+ therapy represents an emerging area of research. It is likely that NAD+ supplementation may activate multiple members of the sirtuin family leading to diverse effects on multiple biological processes, and thus improving brain, cardiac, and renal function under different stressors.
V. Distribution of the NAD+ Metabolome
It is well established that NAD+ (in particular the NAD+/NADH ratio) is a master regulator of cellular bioenergetics. The total intracellular NAD+ content is estimated to be in the range of 0.2–0.5 mM (388). This concentration is within the estimated NAD+ Km value of PARPs (0.02–0.08 mM) (15) and SIRT1 (0.56 mM) for NAD+ (278). This means that the availability of the essential substrate NAD+ is rate limiting for PARPs and SIRT1. For instance, low NAD+ levels due to increased PARP activity lead to reduced SIRT1 activity, whereas higher NAD+ levels enhance PARP and SIRT1 activities. Research from our group has demonstrated that reduced levels of NAD+ due to chronic oxidative stress and hyperactivation of PARPs are associated with significantly reduced sirtuin activity (51, 223).
Metabolomic profiling of the NAD+ metabolome in peripheral blood mononuclear cells (PBMCs), plasma, and urine in an overnight fasting human subject has recently been published (337). The study showed that the phosphorylated NAD+ metabolites—NAMN, nicotinic acid adenine dinucleotide (NAAD), NADP+, NMN, and ADPR—are found exclusively in blood cells, but not in plasma or urine. The levels of NA, NAM, and NR are considerably low in normal fasting blood (337). Very few studies have examined the levels of these NAD+ metabolites due to limitations in accurately measuring them. Using gas chromatography–mass spectrometry, one study reported that the concentration of NAM in fasting blood was about 300 nM, and the level of NA was 30 nM (72). This provides evidence for the physiological importance of NAM as the preferred form of niacin to extrahepatic tissue. The blood levels of both NA and NAM can be significantly increased following supplementation with vitamin B3. These pharmacological doses range between 1 and 3 g of NA or NAM. In comparison, a niacin-rich meal contains about 10 g of vitamin B3 composed of a mixture of NA and NAM, the concentrations of which vary with their content in plant and animal foods (231, 232).
It has been previously shown that small amounts of NA can be converted to NAD+ in the intestine and liver, and NA may not be detected in systemic blood. Moreover, the catalytic activity of NAD+ glycohydrolases or ADP-ribosylation in the small intestine or liver can induce the release of NAM into the blood stream (274). NAM from the diet may also be used to form NAD+ in the small intestine and liver, and may also be released into the blood stream. Expression of hepatic NNMT leads to the formation of MeNAM from NAM, thus maintaining SIRT1 activity in the liver (152).
It remains unclear whether NAM can accumulate in the blood stream following an NAM-rich meal, or can be stored in several tissues for generation of NAD+ as required and later released into the blood stream to maintain threshold levels in the blood stream. However, one study previously showed that up to 60% of the total NAD+ levels are depleted in red blood cells in a rat model of niacin deficiency. The remaining 40% appeared resistant to depletion (274). On the contrary, the levels of NAD+ in the liver were considerably higher and depleted at a slower rate during deficiency (274). Short- or long-term storage of NAD+ may take place in the liver and red blood cells, where it regulates blood NAM levels during periods of niacin deficiency, for example, during fasting. Under normal physiological conditions, high-affinity transporters are required to facilitate the transfer of NA and NAM into extrahepatic tissues, which are present in the blood stream at low- to mid-nanomolar concentrations. Understanding the interactions between these precursors can help us to elucidate appropriate pharmacological doses of NA and NAM.
Recently, NR has been identified as an NAD+ precursor vitamin that is uniquely and orally bioavailable in mice and humans (337). Blood NAD+ levels have been shown to increase by 2.7-fold following a single daily dose of NR (1000 mg) for 7 days, with a concurrent increase in NAAD by up to 45-fold in PBMCs. While it is unclear how an oral dose of NR can raise NAAD levels, it has been suggested that NR may be partially converted to NAM via the NAD+ salvage pathway (337). Such conversion may stimulate bacterial hydrolysis of NAM to NA, culminating in the production of NAD+ using an NAAD intermediate. Another study showed that NMN is metabolized extracellularly to yield NR, which is then converted to NAD+ intracellularly (272). Therefore, NR and NMN represent convergent supplementation strategies to enhance NAD+ anabolism.
VI. Subcellular Compartmentalization of NAD+
Traditionally, it was thought that NAD+ was distributed in the nucleus, as only one form of NMNAT was identified as nuclear in origin (296). Nuclear NAD+ was therefore available to catalyze poly(ADP-ribose) formation but could also equilibrate in the cytosol via nuclear pores (32). Until recently, the significance of mitochondrial NAD+ was unclear, and it was thought that NAD+ could be transported in its intact form into the mitochondria (138). However, it is now understood that there are three intracellular NAD+ compartments—the nucleus, cytosol, and mitochondria (31). Subcellular compartmentalization of NAD+ is thought to play a critical role following niacin depletion. As total intracellular NAD+ levels decline, distinct subcellular stores of NAD+ may influence the outcome of competition between biochemical processes dependent on NAD+ consumption, leading to significant alterations in metabolic pathways that are involved in tissue pathologies.
Recently, three distinct NMNAT enzymes have been discovered, localized to the nucleus (NMNAT-1), mitochondria (NMNAT-2), and the Golgi apparatus (NMNAT-3) (31). While the levels of NMN required for the catalytic activity of NMNAT-1 and NMNAT-2 are very close, a higher amount of NMN is required for NMNAT-2 activity (121). The differential expression of NMNAT enzymes in different intracellular compartments suggests multiple roles for promoting optimal metabolic function in a variety of cells, or an additional mechanism for adaptive response to stress. For example, one study showed that niacin deficiency with normoxia reduced lung NAD+ levels in Fisher-344 rats by 40% (273). Interestingly, exposure to chronic hypoxic conditions induced poly(ADP-ribose) formation in lung tissue, but did not reduce lung NAD+ content, rather NAD+ levels remained at near-control nontreated levels in niacin-deficient lung tissue.
NMNAT-1 plays an important role in mediating NAD+ synthesis to close proximity of the main enzyme responsible for ADP-ribosylation, PARP-1, but also including PARP-2 and 3, tankrases, and sirtuins. While it is likely that nuclear NAD+ may enter the cytosol via specific nuclear pores, there are also additional benefits for the formation of NAD+ in the nucleus. Overexpression of NMNAT-1 has been shown to rescue neurons from axonal degeneration, known as Wallerian degeneration (381). Similarly, inactive mutant forms of NMNAT-1 also demonstrated beneficial effects against neural loss, possibly due to a chaperone effect (54, 382). NMNAT-1 can direct NAD+ synthesis toward the active site of automodified PARP-1 via noncovalent interactions between NMNAT-1 and poly(ADP-ribose) (32).
Moreover, the mitochondrion represents the main site for important redox reactions, including the TCA cycle and oxidative phosphorylation for ATP production. As well, it is also home to mitochondrial poly(ADP-ribose) metabolism and SIRT3–5 activities (247). These fundamental processes need to be maintained if possible even in the presence of NAD+ decline due to increased cellular ADP-ribosylation and niacin deficiency. NAD+ can be released from the mitochondria into the cytosol and nucleus through specific permeability transition pores during conditions of apoptosis or necrosis (89, 149). Therefore, high starting mitochondrial levels of NAD+, which are an order of a magnitude greater than cytosolic levels, are necessary to maintain optimal redox function.
On the other hand, the Golgi apparatus is involved in packaging and transfer of macronutrients to other organelles, and for clearance from the cell. It is likely that the Golgi apparatus may regulate NAD+ levels in other organelles, although this remains uncertain. NAD+ may be excreted from the Golgi apparatus and into the cytosol, or it may be released in the extracellular space to act as a substrate for important ecto-mono(ADP-ribosyl)transferases and/or ADP-ribosyl cyclases, which do not normally have access to significant amounts of NAD+ (31).
The effect of NAD+ precursors in the subcellular distribution remains uncertain and several questions remain unanswered. Will nuclear NAD+ be made more available following treatment with high levels of vitamin B3, since it has the greatest capacity to modulate poly(ADP)ribosylation and repair of DNA damage, will there also be an increase in cytosolic NAD+, given that brain cyclic ADP-ribose levels can increase, and what are the effects of high levels of vitamin B3 on the mitochondrial NAD+ pool? Interestingly, NAPRT, the enzyme responsible for the conversion of NA to NAD+ is found in the cytoplasm (142). Therefore, supplementation with high levels of NA may alter the subcellular contents of NAD+.
VII. Modulation of NAD+ Metabolism by Caloric Restriction
It is well established that CR represents the most efficacious intervention to promote longevity in several short-lived species, including mice and rats, and maintain a healthy and average life span in primates. CR is defined as a 20% reduction in calorie intake compared to ad libitum feeding without incurring malnutrition or reduction in important vitamins and nutrients (222). Although the molecular basis of CR remains unclear, it is thought that CR regulates fat and carbohydrate metabolism, ameliorates oxidative stress and inflammation, activates a stress-induced hormetic response that downregulates insulin and insulin-like signaling (ILS), amino signaling target of rapamycin (TOR)-S6 kinase pathway, and the glucose signaling Ras-protein kinase A (PKA) pathway (36). It is believed that regulation of macromolecule consumption is a direct response to reduced diet, while hormesis and downregulation of TOR and PKA are most likely the molecular aspect of CR.
Several studies have examined the effect of CR in a variety of model organisms. In yeast, exposure to sublethal stress conditions increases expression of nicotinamidases, thus altering NAD+ metabolism and enhancing the activity of Sir2, an yeast homologue of mammalian SIRT2 (16). This is evidenced by repression of age-associated extrachromosomal ribosomal DNA circles (309). Downregulation of TOR and PKA also mediates the beneficial effects of CR on life span as reported in cell survival studies (361). On the contrary, longevity in worms is mediated by inactivation of ILS or forkhead FoxO transcription factor daf-16 (25). While additional mechanisms may be attributed to CR in mammals, alterations in the NAD+ metabolome and increased sirtuin activity may play a prominent role in mediating health benefits reported in the brain and liver following a CR diet.
In rodents, brain total NAD levels were reportedly increased in CR-treated mice, while NAM levels decreased concurrently (265). These observations occurred in parallel to increased neuronal SIRT1 activity, which lowered Alzheimer's associated-neuropathology. In another study, hepatic total NAD levels increased in fasted mice, and these changes were accompanied by increased SIRT1 activation, PGC1α deacetylation, and increased mitochondrial biogenesis (276) (Fig. 11). Three mechanisms have been developed to explain these changes in the NAD+ metabolome following CR: (i) increased systemic mobilization of NAD+ precursors, NAM and NR, since increased l-tryptophan and NA availability is dependent on dietary availability; (ii) reduced NAD+ catabolism if major NAD-consuming enzymes such as PARPs and CD38 are negatively modulated by CR; and (iii) CR-mediated negative regulation of the NR and/or NAR pathways may increase brain and hepatic NAD+ levels.
FIG. 11.
Modulation of NAD+ and NAD-dependent pathways by caloric restriction in mice and humans. Caloric restriction has been shown to increase neuronal SIRT1 activity in humans. In mice, hepatic total NAD+ levels increased in fasted mice, and these changes were accompanied by increased SIRT1 activation, PGC1α deacetylation, and increased mitochondrial biogenesis. SIRT, sirtuin. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Information obtained from experimental small model organisms has provided insight into the molecular basis of CR and aging. However, there are still discrepancies in studies using larger animals. CR has been investigated in rhesus monkeys, which are the closest experimental model organism to human in a controlled environment (226). One study conducted by the National Institute of Aging (NIA) reported no significant improvement in life span. However, a positive trend to slow down the onset of age-related degenerative diseases was observed (227). In contrast, another study by the Wisconsin National Primate Research Center (WNPRC) showed significant improvements in both life span and health span (82, 83). These discrepancies have been attributed to differences in dietary composition and heterogenic genetic backgrounds of the subjects (Fig. 11).
Nevertheless, the beneficial effects of CR have been documented in the Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy (CALERIE) study conducted by the National Institute of Health (NIH). The study showed that a 2-year 25% CR regimen provided significant health benefits in nonobese humans, including reduced inflammatory markers and cardiometabolic risk factors (280). However, given the results observed in rhesus monkeys, longer studies with a larger sample size need to be conducted to validate the potential effects of CR on human life span and normal physiological function.
VIII. Beneficial Effects of NAD+ Precursors
NAD+ anabolism in mammalian cells is known to occur through two major pathways; the de novo and the salvage pathways. To determine nutritional and therapeutic benefits due to maintenance of NAD+ levels in tissues, organs, and cells, supplementation with either NAD+ and its reduced form NADH or its precursors represents a potential therapeutic strategy to slow down the aging process and/or improve the management of age-related degenerative disease. Oral supplementation with NAD+ and NADH has not shown any significant elevation in plasma or tissue levels of NAD+, potentially due to inefficient metabolism of NAD+ through the gut, thus leading to poor bioavailability (177). In addition, oral NADH may not be oxidized to NAD+ in the body, may not be efficiently absorbed by the gastrointestinal system, or may be converted to a product before absorption that cannot yield NAM (34, 35). At present, intravenous infusion of NAD+ is the only recognized effective means of clinically increasing systemic NAD+ levels. However, it is anticipated that some of the alternative NAD+ precursors, including NA, NAM, NMN, NR, and NAR (Fig. 12), are likely to provide some benefits.
FIG. 12.

Chemical structure of NAD+ precursors.
A. Nicotinic acid
NA represents the acid form of niacin. It is commonly prescribed clinically for the treatment of hyperlipidemia. It has been reported that daily intake of 1–3 g reduces blood triglyceride levels and low-density lipoproteins (LDLs), while increasing the level of high-density lipoprotein (HDL), thus favorably regulating the LDL:HDL ratio (133, 343). Our research group was the first to show that exogenous NA efficiently increased intracellular NAD+ levels in brain cells (127). However, NA therapy induces significant skin flushing in a majority of individuals, thus limiting its clinical uses. A mild skin flush has been reported in patients exposed to 50 mg oral NA, and the upper tolerable limit for NA has been set to 35 mg per day for adults in the United States and Canada (314). The lipid-lowering effects of NA are thought to be mediated by binding of NA to the cell surface of a G-protein-coupled receptor known as HM74A or GPR109A (314). This association in adipocytes suppresses triglyceride lipolysis, culminating in the reduction of circulating fatty acids, and reduced liver very LDL formation and circulating LDL-cholesterol (314) (Fig. 13).
FIG. 13.

Mechanisms of action of NA in dyslipidemia. The lipid-lowering effects of NA are thought to be mediated by binding of NA to the cell surface of a G-protein-coupled receptor known as HM74A or GPR109A. This association in adipocytes suppresses triglyceride lipolysis, culminating in the reduction of circulating fatty acids, and reduced liver very LDL formation and circulating LDL-cholesterol. LDL, low-density lipoprotein. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The uncomfortable side effect occurs because of an NA-mediated stimulation of HM74A in some skin immune cells, which results in the conversion of the omega-6 metabolite arachidonic acid into prostaglandin E2, stimulating vasodilation of skin capillaries, causing skin flush (314) (Fig. 14). RUP25, a receptor that differs from HM74A by only one amino acid, has been identified. RUP25 has been shown to exhibit greater affinity to NA than HM74A, and has been associated with extreme skin flush reactions in some people (314). This often dramatic and unwelcome side effect has therefore restricted NA applications to essentially a treatment-resistant lipid-lowering therapy (170).
FIG. 14.
Schematic representation of the molecular mechanism of skin flushing following treatment with NA. NA-mediated stimulation of HM74A in some skin immune cells results in the conversion of the omega-6 metabolite AA into prostaglandin E2, stimulating vasodilation of skin capillaries, causing skin flush. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
In addition, a ketone body, beta-hydroxybutyrate, is the natural ligand for HM74A, which is produced during fasting (314). While NA demonstrated a greater affinity to HM74A (100 nM required for half-maximal) compared to beta-hydroxybutyrate (700 nM required for half-maximal), endogenous NA levels do not reach the concentrations required to activate this receptor, while ketone bodies circulate at the required levels (314). However, other mechanisms have been prescribed to account for the effects of NA on dyslipidemia. These include but are not limited to inhibition of liver diacylglycerol acetyltransferase, inhibition of pathways associated with the clearance of HDL in the liver (74, 345), and activation of PPAR-mediated cholesterol transport from extrahepatic tissue (84, 169, 171). More recently, SIRT1 has been shown to be a positive regulator of the liver X receptor (LXR). SIRT1-mediated deacetylation of LXR at conserved lysine residues can lead to activation of LXR, which regulates cholesterol levels, HDL biogenesis, and lipid homeostasis (199).
Likewise, elevated levels of NA have been shown to improve genomic integrity by reducing micronucleus frequency, and NA deficiency results in chromosomal instability (178–181). Treatment with NA has been reported to delay carcinogenesis, enhance repair efficiency following γ- and X-irradiation in mouse melanoma cells and human PBMCs, and improved neuronal function following hypoxic insult (244, 362). NA has also been shown to enhance endothelial protection by increasing endothelial levels of NADP+ and GSH (114). However, these studies were performed using concentrations ranging between 250 and 1000 μM in the culture medium, which is beyond the physiological concentrations in humans.
B. Nicotinamide
NAM, the amide form of vitamin B3, is also generated as a by-product of SIRT-mediated deacetylation, PARP-mediated ADP-ribosylation, and CD38 NADase and ADP-ribosylation activities, which can be converted back to NAD+ via the salvage pathway. High levels of NAM have been used to enhance radiotherapy or chemosensitize solid tumors by promoting microvascular flow inside the tumor (3, 4). The clinical regimen describes oral doses (3–6 g) aimed at increasing systemic blood levels to 700 μM or higher, combined with inhalation of 95% oxygen/5% carbon dioxide (358, 359). This leads to improved tumor blood flow and oxygen generation, thus enhancing the effect of radiation by inhibiting myosin light chain kinase (MLCK). Decreased phosphorylation of MLCK disrupts vascular smooth muscle contraction, promoting vasodilation (283). However, the concentrations used in vitro are an order of magnitude higher than clinical systemic levels, and the macrovascular effects appear to be independent of effects on NAD+ production.
NAM has also been shown to prevent or slow down the progression of several types of diabetes in animal models, although this effect was not reproducible using gram amounts of NAM in a randomized control trial (10, 113, 215, 323, 371, 386, 387). NAM has also been used as a potential therapeutic strategy to limit vascular injury and ischemia to the brain and other tissue in response hypoxic and/or chemotoxic insult in several animal models with some success (91, 301, 302, 334, 352).
Topical NAM formulations have also been successfully used for the treatment of inflammatory skin conditions, including rosacea, autoimmune bullous dermatoses, and acne (251). NAM has also been previously used for the maintenance of skin integrity, lowering sebum levels, and reducing hyperpigmentation spots and redness (328). NAM has also been shown to reduce acute and chronic effects of UV-induced skin damage by preventing the expression of inflammatory mediators IL-6 and TNFα, and the DNA damage markers cyclobutane pyrimidine dimers and 8-oxo-7,8-dihydro-2-deoxyguanosine (234). As well, NAM has been shown to improve UV-induced immunosuppression and photocarcinogenesis in rodent models and human studies (115). Similarly, human clinical studies have shown that oral NAM can significantly reduce actinic keratosis compared to a placebo, and may likely to also be useful for the prevention of nonmelanoma skin cancer (321).
However, it is well established that as a by-product of NAD+ catabolism, NAM also serves as a natural feedback inhibitor for NAD-dependent enzymes (Fig. 15). For example, PARP, sirtuin, and CD38 activities are proportionately inhibited as NAM concentrations increase, and this has been postulated as the mechanism for the antidiabetic effects of NAM in humans. While NAD+ levels are still elevated, the important NAD-dependent functions (e.g., SIRT1 activity) are inhibited. Moreover, NAM supplementation worsened liver fat accumulation in a choline-deficient rat model, and this effect was attributed to the accumulation of poly(ADP-ribose), and a reduction in epigenetic methylation due to the use of methyl groups in NAM excretion (19, 173). Therefore, although exogenous NAM can be converted to NAD+ it is again not considered an ideal supplement, particularly in the medium to longer term due to its enzyme inhibiting and methyl depleting potential.
FIG. 15.

Mechanisms of action of NAM and its effect on the NAD+ metabolome. NAM also serves as a natural feedback inhibitor for NAD-dependent enzymes. For example, PARP, sirtuin, and CD38 activities are proportionately inhibited as NAM concentrations increase, and this has been postulated as the mechanism for the antidiabetic effects of NAM in humans. While NAD+ levels are still elevated, the important NAD-dependent functions (e.g., SIRT1 activity) are inhibited. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
C. Nicotinamide mononucleotide
NMN is an important precursor for NAD+ synthesis from NAM. Supplementation with NMN has been shown to have a positive effect on insulin levels most likely through action on pancreatic β cells (316). NMN supplementation has also been shown to reduce obesity and vascular damage in several in vitro and in vivo models (87, 355). NMN has also been reported to improve CNS function by increasing brain mitochondrial respiratory deficits, protecting against amyloid-beta (Aβ) oligomer-induced toxicity and cognitive impairment, and to ameliorate reactive glial-induced motor neuron loss, and maintenance of neural stem and progenitor cells (356). It has also been shown that NMN can protect against cerebral ischemia-induced apoptosis and enhances neurogenesis following cerebral injury (360). NMN treatment also upregulated Nrf2 and HP-1 protein expression and promoted Nrf2 nuclear translocation for its transactivation following postischemic neuroinflammation, to attenuate secondary neurological injury (360). Similarly, overexpression of NAMPT, the enzyme required for NMN anabolism, appears neuroprotective in stroke (353, 354).
A recent study showed that NMN treatment significantly improved major pathological hallmarks of Alzheimer's disease (AD) in APPswe/PS1dE9 AD transgenic (tg) mice, including cognitive impairment, neuroinflammation, Aβ deposition, and synaptic loss (372). Another study also found that NMN treatment inhibited JNK activation and APP-mediated amyloidogenic processing by APP-cleavage secretase in AD-tg mice (349, 356). Accordingly, it is likely that NMN may represent a new therapeutic target for the treatment and management of AD and other age-related degenerative diseases.
In NDUSF4KO mice—a mouse model for cardiac-specific complex I deficiency—mice exhibited a reduced NAD+/NADH ratio, hyperacetylation of mitochondrial protein, including the mitochondrial permeability transition pore (mPTP), impaired oxidative phosphorylation, and increased vulnerability to cardiac stress (174). Treatment with NMN was able to partially restore the intracellular levels of NAD+ and attenuated the hyperphosphorylation of mPTP and improved heart failure following exposure to chronic stress. Moreover, these mice also exhibited increased activation of SIRT3, and its deacetylation of key protein, including mPTP due to enhanced activity following increased NAD+ levels after NMN treatment, may also explain the beneficial effects of NAD+ in NDUFS4KO (174).
Despite these reported benefits of NMN treatment, evidence also suggests that NMN is effectively contained within the cell membranes and is not subject to high diffusion gradients. This has raised the question of whether NMN is able to effectively traffic across most cells. Interestingly, extracellular NMN may be actively produced from direct metabolism of exogenous NAD+ (388). However, further work is required to establish the range of conditions for which NMN may prove beneficial in humans.
D. Nicotinamide riboside
NR is a naturally occurring precursor of NAD+ originally isolated from fresh milk (338). Exogenous treatment with NR has been shown to increase intracellular NAD+ levels in a variety of cell lines. Supplementation with NR protected murine dorsal root ganglion neurons from axonopathy via a mechanism involving the transcriptional induction of NRK2 gene (288). As this effect is not reproducible by NA or NAM, NR represents a major precursor in the CNS when the de novo synthesis of NAD+ by the kynurenine pathway is impaired. NR has been shown to efficiently increase NAD+ levels without causing any adverse skin flushing in contrast to NA, or liver damage in contrast to NAM (337). NR has also been shown to serve as a cholesterol-lowering agent in obese mice (67). Recent studies have shown that NR is the mitochondrially favored NAD+ precursor, and the beneficial in vivo effects of NR have been attributed to modulation of mitochondrial sirtuin activities, as well as nucleocytosolic targets, including PARPs, sirtuins, CD38, NAD-dependent oxidoreductases, and NADPH-dependent ROS detoxification enzymes. Supplementation with NR has also been shown to reduce the acetylation state of several protein targets of SIRT3, including SOD2 and NADH ubiquinone oxidoreductase subunit A9, suggesting that NR may be used to pharmacologically activate SIRT3 (67). Moreover, administration of NR slowed down neurite degeneration after noise exposure by the NAD+-SIRT3 pathway (56).
As yet, the effect on metabolic health of NR as an exclusive source of niacin remains unclear. Supplementation of dietary NR in mice overexpressing the putative human oncogene, unconventional prefoldin RPB5 interactor (URI), reduced dysplastic lesions and prevented tumor development, thus providing evidence for NAD+ supplementation as a novel approach for the treatment and management of hepatocellular carcinoma (340). Overexpression of human URI drives the development of dysplasia in hepatocytes via mechanisms involving the aryl hydrocarbon receptor and estrogen receptor (OR), and impaired kynurenine pathway metabolism (340). End-stage tumors in hURI-overexpressing mice regressed with increased apoptosis in mice supplemented with NR (320, 340).
Despite the beneficial effects of NR supplementation, the doses often used to produce beneficial effects are remarkably high (400 mg NR/kg body weight/day) compared with current commercially available supplements (6–500 mg/kg body weight/day). One study showed that NR (300 mg/kg body weight/day) reduced exercise performance in rats, and previously reported ergogenic effects of NR could not be confirmed (186). Two hypotheses have been postulated to explain this observation: (i) based on similar effects of NA and NAM, it is likely that NR may also reduce FAO during exercise, leading to earlier fatigue; and/or (ii) NR may also alter the redox properties of NAD+ and NADP+, leading to a nonoptimal reductive state (186). Additional studies are warranted to examine the effects of NR on exercise performance.
Recently, the effect of a wide range of dietary NR concentrations on metabolic flexibility and gene expression in epididymal white adipose tissue was examined in mice exposed to a mildly obesogenic (40% fat) diet (303). The study showed that 30 mg NR/kg diet was most beneficial for improving metabolic health, with regard to metabolic flexibility and increased expression of PPARγ, a master regulator of adipogenesis, and SOD2 and PRDX3, two antioxidant genes (303). The study concluded that 30 mg NR/kg diet represented the optimal concentration to potentiate metabolic health.
E. Nicotinic acid riboside
The least examined of the NAD+ precursors, NAR has been shown to be produced in human cells through NMN and NAMN dephosphorylation by cytosolic 5′-NTs (38, 190). It is anticipated that this metabolite will represent an important precursor for NAD+ generation. Low micromolar concentrations of NAR have already been demonstrated to produce sufficient amounts of NAD+ to maintain cell viability. One study showed that NAR can be produced and delivered by cells at physiologically sufficient levels (190). It is likely that other cell types may use NAR and transport it between each other.
IX. Pharmacokinetics of NAD+ Precursors
Boosting the NAD+ pool by utilizing precursor molecules may have multiple health benefits and a diverse range of therapeutic implications. NA, NAM, NMN, and NR have been publicized as potent NAD+ boosters. NMN and NR may also be used as a general supplement in patients who have adverse responses to NA and NAM. The pharmacokinetics of NA and NAM has been extensively investigated, while the pharmacokinetic properties of NR have only recently been determined in mice and a middle-aged human subject (337). However, pharmacokinetic effects of NMN and, more so, NAR have not yet been fully investigated in either the human or murine model.
A. Nicotinic acid
The pharmacokinetics of NA has been previously examined using pharmacological doses of NA and several extended release formulations. An open-label, dose-rate escalation, crossover study administered 12 human subjects with 2000 mg NA in solution form at slow (25 mg niacin aqueous solution administered every 10 min for 80 doses), intermediate (50 mg niacin aqueous solution administered every 10 min for 40 doses), or fast (100 mg niacin aqueous solution administered every 10 min for 20 doses) (232). Peak NA levels varied between 10 μM (slow release) and 240 μM (fast release). Interestingly, the area under the curve (AUC) for the slow release formulation was 25-fold lower than the fast release counterpart. NA in the slow release preparation is taken up by the intestine and liver, forming NAD+, and released into the circulation as NAM (232). Importantly, a concentration of 10 μM is estimated to be about 30-fold higher than physiological levels of NAM in the blood. Fast release formulation of NA not only yields higher peak levels and AUC, it also elevates peak NAM levels to 16 μM. NA is preferentially removed from circulation at high levels with a half-life of 1 h, compared with a half-life of 4 h for NAM (231, 232). Therefore, high doses of NA also elevated the levels of NAM to supraphysiological levels. Therefore, the effects of NA on lipids may also be due to the protective effects of NAM and increased NAD+ anabolism.
In a recent study comparing the efficiency of NAD+ precursors to generate NAD+ in mice following oral gavage, NA produced the lowest levels of NAD+ (337). However, the kinetics of hepatic NAD+ accumulation was 4–6 h faster than either NAM or NR. Oral administration of NA doubled hepatic NAD+ (from 1 to 2 mM) by increasing the level of NAAD (an intermediate), and enhanced NAD+ catabolism as reported by increased levels of MeNAM (337). The liver promoted NAD+ anabolism as long as enough NA is available, while increasing the activity of NAD-dependent processes, some of which generate NAM as a by-product (337). Increased NNMT expression due to increased levels of MeNAM stabilizes hepatic SIRT1 protein and regulates lipid levels in mice and humans (152, 194, 336).
Another study reported significant changes in the levels of NAD+ following oral supplementation over a 2–3-week feeding interval (26). It was previously thought that NAD+ levels will continue to increase in response to time before reaching a plateau. In rats supplemented with NA (30 and 4000 mg/kg), bone marrow NAD+ significantly increased in animal fed with 4000 mg/kg (26). However, NAD+ levels were downregulated as consumption became chronic, and it was unclear whether this effect was due to either NA uptake alone, associated with the conversion of NA to NAM, or altered NAD+ catabolism in bone marrow. Therefore, it has been postulated that pharmacological responses to long-term supplementation with NAD+ precursors may change over time (26). This also raises the important question of whether higher NAD+ levels have the potential to induce a deleterious impact on cellular function, thus stimulating an adaptive response.
B. Nicotinamide
The pharmacokinetics of 3–6 g of oral NAM in humans has been previously investigated (93). Higher doses are prone to produce adverse reactions, including nausea and vomiting. The peak blood levels of NAM were between 1 and 2 mM. This is estimated to be more than 3000-fold higher than circulating levels (93). This figure is also well above the minimum concentration associated with radiation sensitivity. The half-life of NAM is 4–5 h, which provides sufficient time to facilitate carbogen breathing and radiation therapy. It has been suggested that if MLCK represents the likely target of NAM, then it is required to accumulate inhibitory concentrations inside a cell. While intracellular transporters for NAM have been previously identified, mechanisms for cellular responses to high concentrations of NAM remain unclear. One study showed that radiation sensitivity remained after a decline in circulating levels of NAM, suggesting that the beneficial effects of NAM may be related to additional downstream effects, including increased NAD+ generation (279), and/or inhibition of poly(ADP-ribose) and DNA damage-induced apoptosis (153).
While high doses of NA have been shown to increase NAM levels, the effect of increased NAM on NA levels remains unclear. At present, enzymatic conversion of NAM to NA has not been identified. However, the deamidation of NAM via oral and intestinal microflora is possible (212). One study showed that significant amounts of salivary NAM were converted to NA, although circulating levels of NA following large oral dosing of NAM remained undetected (317). It is likely that the quantification methods used in this study were not sensitive enough to clearly delineate changes in the NAD+ metabolome. Skin flushing, a common adverse effect following NA treatment, has also been reported with NAM, suggesting that NA may be increased following administration of high doses of NAM (167).
A recent study comparing three NAD+ precursor vitamins provided in bolus at equivalent oral doses also demonstrated increased hepatic NMN, NAAD, NAD+, and NADP+ levels, and NAD+ catabolic activity as evidenced by elevations in MeNAM and ADP-ribose (337). However, while the AUC of increased NAD+ due to NAM showed a 50% benefit compared with NA, the study demonstrated a 50% deficit in NAM-mediated accumulation of ADP-ribose compared with NA (337). These studies also suggest that NAAD may represent a biomarker for increased NAD+ synthesis, and is independent of traditional NAAD precursors such as l-tryptophan and NA (337). Unlike NA, NAM is not a potent cholesterol-lowering agent, and high levels of NAM may inhibit PARP and sirtuin activities.
Evidence for potential adaptive responses following high doses of NAM has been previously reported. Rats supplemented with high doses of NAM (4 g/kg) exhibited elevated levels of NAD+ and ADP-ribose in the brain. Interestingly, these changes were accompanied by impaired cognition as reported by impaired performance in a hippocampal-dependent spatial learning test (197). Increased NAD+ can ultimately lead to increased activity of a diverse range of enzymes, including PARPs, sirtuins, and CD38/CD157. This in turn may have dynamic effects in cellular function.
C. Nicotinamide mononucleotide
The detection of NMN in blood remains challenging. While the concentration of NMN has been reported to be around 50 μM in plasma (275), NMN levels were undetectable in another study (272). These differences can be attributed to different detection techniques for NMN. For instance, using an high-performance liquid chromatography (HPLC)-based method, intracellular concentrations of NMN and NAD+ were reported to increase up to 500 pmol/mg and 50 pmol/mg of white adipose and pancreatic tissue 15 min after intraperitoneal injection of 500 mg/kg of NMN (376). However, hepatic NMN and NAD+ levels were reported to reach 10 and 4000 pmol/mg, respectively, 6 h after oral gavage of 185 mg/kg (337). Similarly, the level of NMN has been reported to be around 1.5 pmol/mg tissue in tumors, and 80 nM in ascite fluid (310).
NMN appears to be stable in plasma and cell media supplemented with 10% fetal bovine serum (FBS), and no increases in NAM levels were reported after a 1-h incubation (272). However, NMN injections led to significant increases in NAM levels in plasma, which suggests that NMN may be partially converted to NAM following intraperitoneal injection. The presence of NAM in mice plasma following NMN injection suggests that NMN may be initially converted to NR (272).
A recent study demonstrated that dephosphorylation of NMN into NR, which is required to produce NAD+ in yeast, represents a major step as an exogenous NAD+ precursor in mammalian cells (272). It is thought that the extracellular receptor CD73 may act as an NR-releasing enzyme. CD73 has both pyrophosphatase and 5′NT activity, facilitating the conversion of extracellular NAD+ and NMN to NR, which in turn can be used to stimulate further NAD+ synthesis. This is supported by another study which showed that gene silencing of CD73 inhibits the use of NMN as a potential NAD+ precursor (310). In addition, NRK1 has recently been identified as an important rate-limiting enzyme for the conversion of NMN to NR for NAD+ synthesis (272), and provides a reliable explanation to account for overlapping effects reported for NMN and NR.
D. Nicotinamide riboside
Evaluating the pharmacokinetics of NR in mammalian tissue has been limited by poor sensitivity and detection of NR in biological samples. As well, results of a clinical trial aimed at investigating the pharmacokinetics of NR in healthy human subjects are not yet available. However, NR was degraded rapidly after incubation in murine plasma (∼10% NR was degraded after 10 min, and 66% was degraded after 1 h), leading to comparable increases in NAM. These results hint at the presence of plasma factors that can degrade NR to NAM (272). NR degradation followed by detection of NAM has also been observed in cell media containing 10% FBS (272). Importantly, NR is stable in protein factions in milk with a potential lifetime of 1 week (338). NR may also be circulated in a cell-bound form for several hours.
One study showed that NR is safe and orally bioavailable in mice and humans with no adverse effects reported (337). However, a future study will need to incorporate a validated flushing symptom questionnaire to assess whether NR may be associated with any flushing episodes. Higher levels of NMN, NAMN, NAM, NAAD, NAD+, and NADP+ were produced following oral gavage of NR compared with oral NAM (337). In addition, ADP-ribose levels were more significantly elevated compared with NA and NAM, suggesting that NR can enhance the activity of NAD+-consuming enzymes more than mole-equivalent doses of NAM and NA (337). More recently, a randomized, double-blinded, placebo-controlled study showed that NR in combination with pterostilbene, a naturally occurring phytochemical found in blueberries, can increase NAD+ in a dose-dependent manner in whole blood lysates throughout the entire 8-week trial (88). Taken together, this suggests that NR is a more potent precursor of NAD+ synthesis and NAD-dependent activities than amidated and acidic forms of niacin.
E. Nicotinic acid riboside
To our knowledge, the pharmacokinetic properties of NAR have not been reported in the literature. Understanding the pharmacology of NAR has remained difficult due to its low physiological submicromolar concentration. The 1H-nuclear magnetic resonance (NMR) method has shown some success in detecting NAR in cell culture medium because of the observed chemical shifts (190). Also, low sensitivity of detection methodologies requires acquisition of spectra over extensive periods of time. Overexpression of NAPRT in HEK293 cells led to the detection of NAR in the cell culture medium, and this effect is due to increased NA catabolism (190). In HeLa cells, NAR, but not NR, is released in amounts that are sufficient to maintain NAD+ biosynthesis and cell survival (190).
F. Nicotinic acid adenine dinucleotide—a biomarker of elevated NAD+ metabolism
As mentioned above, NA, NAM, and NR have been shown to increase the levels of the intermediate, NAAD. However, the increase in the levels of NAAD was lowest following ingestion of NA (337). This finding suggests that increased NAD+ anabolism by supplementation with NAD+ precursors not only increases the accumulation of by-products of NAD+ catabolism (such as ADP-ribose and MeNAM) but also stimulates retrograde synthesis of NAAD and NAMN. As the rate of NAD+ anabolism increases, NAD+ is deamidated to form NAAD by a yet unknown mechanism. Similarly, it is also possible that deamidation of NMN can lead to increased levels of NAMN and NAAD. While the deamidation reaction is yet to be verified, it is possible that NAAD may also be formed from NAADP, although the mechanism responsible for this elusive biochemical reaction is yet to be identified.
X. Effects of NAD+ Precursors on NAD-Dependent Processes
Apart from the beneficial effects of NAD+ precursors on normal cellular function, including NAD+ and NADP-dependent reactions and ADP-ribosylation, these substrates for NAD+ anabolism share a common effect of increasing intracellular NAD+ levels in multiple cellular compartments. It has been considered that normal metabolic processes may be fulfilled following recommended daily intake of vitamin B3, and that higher doses may induce alternate mechanisms. However, it is now clear that physiological roles of NAD+ precursors may be different than the doses present naturally in the diet.
A. NAM and PARPs
Whether NAM can inhibit PARP-1 activity remains controversial. The Ki value for NAM-mediated inhibition of PARP-1 ranges between 30 and 200 μM in a cell-free system. The Ki value in cultured cells was threefold greater than in a cell-free system (271). This suggests that the uptake and/or conversion of NAM to NAD+ may be limited in cell cultures. In mammals, oral NAM is metabolized by the small intestine and liver before it enters the blood stream. NAM is taken up by extrahepatic tissue in small amounts where it is immediately converted to NAD+. Moreover, blood volumes are significantly lower than tissue volumes. Therefore, it is less likely that tissue NAM levels will reach the concentrations needed to inhibit PARP-1 activity.
On the contrary, the conversion of NAM to NAD+ due to increased substrate has been shown to promote poly(ADP-ribose) levels. For instance, supplementation with NAM (1 g/kg) increased hepatic NAD+ levels by 50%, while basal poly(ADP-ribose) levels increased by twofold (163). This suggests that NAM was more effective at enhancing substrate pools than mediating PARP-1 inhibition. However, poly(ADP-ribose) content was the same when the same animals were exposed to a hepatocarcinogen to enhance PARP-1 activity. NA also promoted higher levels of poly(ADP-ribose) formation (163). Taken together, it is likely that basal PARP-1 activity may be regulated differently than DNA damage-induced PARP-1 activity, and NAM may be more effective at inhibiting the latter form of PARP-1 activity.
In another study, increased PARP-1 activity was reported in extrahepatic tissue in response to oral dosing of NAM (4 g/kg). In that study, bone marrow NAD+ levels increased by 2.5-fold, basal poly(ADP-ribose) levels increased by fivefold, while DNA damage-induced poly(ADP-ribose) increased by twofold (42, 43). Similarly, studies in radiation sensitization models showed that radiation sensitivity due to NAM was due to mechanism(s) independent of inhibition of PARP activity and DNA repair processes (279).
B. NAM and sirtuins
Mammalian sirtuins have developed low NAD+ binding affinities, which ensured that their deacetylase activities can be efficiently regulated by minor changes in the intracellular concentrations of NAD+, thus serving as potent NAD+ sensors. Reduced intracellular levels of NAD+ during aging can downregulate sirtuin activity and SIRT1-mediated deacetylation of p53 (51). On the contrary, increased intracellular NAD+ levels, either due to CR or NAD+ supplementation, can upregulate sirtuin activity. While resveratrol, a plant-derived stilbene putatively allosterically activates SIRT1 only, NAD+ supplementation can activate almost all seven forms of mammalian sirtuins. For example, regulation of SIRT3 by intracellular NAD+ levels has been demonstrated to be the major determinant of cellular resilience against apoptosis (143).
If the increase in NAM in biological systems is capable of inhibiting PARP-1 activity, then it may also inhibit other NAD+-dependent processes such as sirtuins. High levels of NAM may inhibit NAM cleavage reactions or mediate competitive inhibition at NAD+-binding sites leading to altered function or sirtuin enzymes, to ultimately enhance the levels of NAD+. Physiological levels of NAM are within the same range as the IC50 of several sirtuins (159), therefore suggesting that sirtuins may act as NAM sensors as well as NAD+ sensors.
NAM has been shown to bind to a specific conserved region in the catalytic site of sirtuins, inducing a reverse base-exchange reaction with an intermediate, rather than deacetylation, thus inhibiting sirtuin deacetylase activity (129). The base-exchange equilibrium constant has been estimated to be about 20 for SIRT1 (37). This means that the maximum possible activation of SIRT1 by full inhibition of the base exchange reaction at any NAM concentration is greater than Sir2 in yeast. Recently, isonicotinamide (isoNAM), a synthetic analog of NAM, has been shown to compete with NAM for binding at the catalytic site (228). However, unlike NAM, isoNAM does not substantially react with the intermediate, leading to increased Sir2 activity.
NAM represents a physiological inhibitor of sirtuins. The IC50 values for inhibition of bacterial Sir2, yeast Sir2, mouse Sir2, SIRT1, SIRT2, SIRT3, and SIRT5 were measured to be 26, 120, 160, 50, 100, 36.7 μM, and 1.6 mM, respectively (129). The concentration of NAM in yeast nuclei has been estimated to be 10–150 μM, which suggests that NAM is a regulator of Sir2 activity in vivo (292). Yeast and bacterial sirtuins have lower Kms and Kds for NAD+, compared with mammalian sirtuins, and therefore may be less sensitive to changes in intracellular NAD+ concentrations than their mammalian counterpart (68). Therefore, increased NAD+ levels are more likely to result in activation of mammalian sirtuins. In mammalian cells, low levels of NAM have been reported in several rat tissues (142). This is likely due to rapid catabolism of NAM for the production of NAD+ and related pyridine nucleotides. However, high concentrations of NAM (up to 300 μM) have been reported in the brain of Tg2576 mice (265), providing additional evidence for NAM as a regulator of sirtuin activity in mammalian tissue. Moreover, the ratio of NAD+ to NAM in subcellular subcompartments can decline with age, therefore lowering sirtuin activities (223). This effect may be due to increased utilization of NAD+ by PARPs and reduced NAD+ anabolism from NAM by NAMPT-mediated salvage pathway (160).
Depending on their physiological roles, several other mechanisms have been attributed to account for the regulation of sirtuins by NAM. For instance, NAM can only partially inhibit Sir2AF2 (SIRT2 homologue from Archaeon Archaeglobus fulgidus), whereas it is a full inhibitor of SIRT1 (129). As well, NAM is a competitive inhibitor of SIRT3, in contrast to noncompetitive inhibition reported for other sirtuins (129). Mammalian SIRT3 is a mitochondrial sirtuin that has demonstrated tumor suppressive effects and regulates glycolytic metabolism (18). Inhibition of SIRT3 by NAM may increase glycolytic metabolism and inhibit aerobic glycolysis and thus reducing cancer cell biomass and improving chronic tissue damage.
Apart from base exchange, which is known to increase the rate of forward reaction for sirtuin activity, direct competition has been postulated as another mechanism to explain the greater degree of competition between NAM and NAD+ in the inhibition kinetics of SIRT3 compared with that of SIRT1 (129). This involves NAD+ binding to the catalytic site in the presence of NAM (129). Elucidating the mechanisms of action of NAM against SIRT3 and other sirtuins can help to develop more efficient inhibitors and activators of sirtuins that can be translated to the clinic.
C. CD38-mediated processes
It has been proposed by our group and others that CD38 can regulate SIRT1 activity by modulating the availability of the essential substrate NAD+, and NAM to the SIRT1 enzyme (52). This can have a profound effect on modulating obesity, metabolic disorders, cellular energy homeostasis and cellular senescence, and aging. By promoting intracellular NAD+ anabolism while reducing NAM levels, inhibition of CD38 can increase SIRT1 activity.
Several CD38 inhibitors have been identified. These include NAM and NA, NAD+ analogs such as arabiono-NAD, and reducing agents including dithiothreitol (76). CD38, due to its effect on calcium generation, also serves as an important mediator of smooth muscle contraction, cell death and apoptosis, neural and hormonal signaling, and egg fertilization (76). Therefore, CD38 inhibition may be useful under pathological conditions where calcium homeostasis is impaired, including hypertension, cardiac ischemia, asthma, and dysfunctional labor.
However, CD38 is also involved in the release of hormones such as oxytocin and Adrenocorticotropic hormone, which regulate maternal and social behavior (166). Inhibition of CD38 in these conditions may have significant negative effects on psychological function. In addition, CD38 plays an important role in the immune system, and knockout of CD38 has been shown to increase susceptibility to bacterial infection (201). Therefore, the effect of NAD+ precursors and changes to the NAD+ metabolome may have previously unknown effects of CD38 activity and NAD-dependent processes, and may serve as important therapeutic strategies for the treatment of metabolic and inflammatory conditions if appropriate dosage regimens are devised and adapted to meet individual patient requirements.
D. Redox reactions
In eukaryotic cells, the generation of ATP is achieved predominantly by mitochondrial oxidative phosphorylation. In this process, free energy released following the breakdown of carbon substrates is captured by exchanges between electron donors and electron acceptors via the electron transport chain (ETC) leading to ATP production (308). NAD+ serves as an electron acceptor, and its reduction leads to the generation of NADH, which can be subsequently oxidized by complex I of the ETC to produce NAD+. The NAD+/NADH ratio serves as an important indicator of several oxidoreductase enzymes. Elevated levels of NADH can inhibit NAD-dependent processes. A metabolic imbalance in oxidative phosphorylation has been associated with several cardiac, neurological, and renal pathologies (206). Alterations to the ETC can lead to a significant decline in ATP production, increased intracellular Ca2+ influx and free radical production, and lowered NAD+/NADH ratio. A switch between oxidative to anaerobic metabolism in response to several cardiac stressors has been shown to reduce oxidative damage and maintain ATP levels. However, this compensatory mechanism impairs oxidative phosphorylation while limiting the mitochondrial NAD+ pool (97).
Similarly, the NAD+/NADH ratio appears to play a crucial role in the heart and kidney and supplementation with NAD+ precursors has been shown to protect against impairments in oxidative phosphorylation due to cardiac stressors and AKI-induced renal damage (150). In PGC1α-deficient mice, treatment with NAM increased FAO, ATP generation, and the NAD+/NADH ratio to protect against AKI toxicity (339). Therefore, under degenerative conditions associated with impaired oxidative phosphorylation, or other abnormality leading to a decline in NAD+, upregulation of NAD+ anabolism through NAD+ precursors may improve redox function.
Human in vivo studies regarding the effect of NAD+/NADH ratio remain nascent. However, using two-photon microscopy for the quantification of NADH and NADPH in epidermal skin layers, one study reported a significant increase in NADH fluorescence following arterial occlusion, suggesting that there is a reduction in oxidative phosphorylation due to a decline in the need for electron donation for the oxidation of NADH to NAD+ (24). Similarly, reduced NADPH fluorescence emission has been reported in the facial skin of older aged females compared with younger subjects (286). Taken together, these studies provide supportive evidence for the role of NAD+ in regulating cellular bioenergetics.
Impaired poly(ADP)ribosylation has been associated with increased sensitivity to DNA damage underlying skin lesions reported in the human disease of niacin deficiency better known as pellagra (274). In addition, impairment in the formation of cyclic ADP-ribose, which regulates intracellular calcium levels, may contribute to neuronal loss observed in pellagrous dementia (378). However, redox reactions corresponding to the ratio of NAD+/NADH are less prone to be affected by altered NAD+ levels as ADP-ribosylation reactions. NAD+/NADP+ serve as soluble cofactors in a multitude of oxidation/reduction reactions. The catalytic enzymes utilize riboflavin-based nucleotides as a source of prosthetic groups. Others contain iron to facilitate electron transfer. Unlike poly- and mono(ADP-ribosyl)ation reactions, iron and riboflavin deficiencies are not known to induce sun sensitivity of the skin, and dementia, which are two main characteristics of pellagra.
One study investigated the effect of NADH, the reduced form of NAD+, on proliferation, cytokine release, and cell redox status of lymphocytes collected from healthy aged subjects (40). Cells exposed to NADH (500 μM/L) showed increased levels of GSH, and catalase activities, while malondialdehyde and carbonyl proteins are markedly decreased (40), suggesting a decline in oxidative stress. Recently, it has been shown that treatment with 1 mM NADH increased the expression of nuclear Nrf2, catalase activity, and total GSH by increasing SIRT2 function (69).
As well, the effect of niacin deficiency on endogenous antioxidant defence mechanisms, NADPH:NADP+, and GSH:GSSG redox couples remains unclear. Two studies showed that niacin deficiency increased markers of oxidative stress, but did not induce either NADPH or GSH decline (27, 325). This suggests that niacin deficiency impaired poly(ADP-ribose) accumulation but did not stimulate further tissue damage, while maintaining GSH defences. Several mechanisms have been postulated to account for the maintenance of redox reactions during periods of niacin deficiency. These include variations in substrate affinity for NAD+, subcellular localization of enzymes and cofactors, and direct modulation of enzyme activity/expression levels.
XI. Do NAD+ and Related Precursors Display Hormesis?
There is a growing body of evidence which suggests that NAD+ decline is a major contributor to the aging process and may be involved in the pathogenesis of several age-related degenerative diseases affecting the heart, brain, liver, kidney, and skin. These results collectively highlight the potential for NAD+ supplementation, whether using NAD+ alone or NAD+ precursors to protect against aging and associated pathologies. While such prospects are of major clinical significance, the role of NAD+ and its modulation in human aging remains only partially understood. In particular, little is understood regarding the impact of having “very high” NAD+ levels. We suggest that modulation of NAD+ levels may induce a hormetic dose/response that may confound numerous clinical outcomes.
The term hormesis was first incorporated into the biomedical context by Southam and Ehrlich (314a) in 1943 to account for the effects of red cedar tree extracts on wood-rotting fungi (62). The study showed that various species of fungi exhibited low-dose stimulation and a high-dose inhibitory effect on cellular metabolism. By the 21st century, hormesis is now used to define the biphasic dose/response that occurs following exposure to a chemical or physical agent, or as an overcompensatory response to cytotoxic insult (63). Resveratrol, an activator of sirtuins, has recently been shown to induce a hormetic dose/response in a variety of biological models, including breast, prostate, colon, lung, uterine, and leukemia tumor cell lines (64). In these studies, lower concentrations of resveratrol enhanced tumor cell proliferation. However, at higher concentrations, resveratrol induced an inhibitory effect. For instance, resveratrol increased the activity of the vitamin D receptor and promoted proliferation of T47D breast cancer cells up to 4 μM, above which led to reduced proliferation of the tumor cell line (64).
Other studies have shown that resveratrol can protect cultured hippocampal neurons against oxidative stress at concentrations between 5 and 25 μM (108). Resveratrol could also ameliorate inflammation and oxidative stress in cultured tumor cells by inhibition of COX-2 (391). However, when these cells are under conditions of reduced oxygen and glucose availability, resveratrol can induce apoptotic cell death (64).
In light of these findings, it is likely that upregulation of NAD+ anabolism may also conform to a hormesis biphasic dose/response. For example, where neuronal cells are exposed to cellular stress, as may occur due to ischemic insult, or cytotoxins such as glutamate, and Aβ aggregates, increasing NAD+ levels may provide both beneficial and deleterious effects that may be dependent on the dosage and duration of administration relative to the cytotoxic stimulant. For instance, in vitro incubation of naive T cells with NAD+ induced apoptosis, while activated T cells incubated with NAD+ showed no signs of apoptosis (204). It was suggested that ecto-NAD, as substrate of ADP ribosylation, acts on naive, but not on activated T cells (297). This indicates that many effects of NAD+ are dependent on environmental factors that would seem to produce a favorable response.
Competition between NAD+-consuming enzymes also displays hormesis. For example, as previously mentioned, PARP1 activity increases with age due to accumulation of oxidative DNA damage, and in response to high energy intake. Since the Km for NAD+, PARP1, and SIRT1 is relatively similar, the decline in NAD+ levels following PARP1 activation can also induce a decline in SIRT1 activity. Therefore, while low levels of PARP1 activity can repair DNA damage following exposure to mild oxidative stress levels, increased PARP1 activity can lead to cell death via reduced SIRT1 activity and energy restriction, therefore exacerbating disease progression (269). Therefore, rigorous double-blind and placebo-controlled clinical trials are needed to assess the nature of the dose/response effect of NAD+ in humans. Further work will be required to gain further understanding of the role of NAD+ anabolism against aging and age-related diseases.
XII. Limitation of Using In Vitro and In Vivo Studies
Despite the importance of NAD+ metabolism to human health and diseases, determining the levels of NAD+ remains a challenge. As well, while there is clinical significance for supplementation with NAD+ precursor in the clinic, evidence showing increased NAD+ levels on such supplementation is limited. While cell culture and animal models are commonly used in research studies, they are not a true representation of human physiology. Moreover, biochemical assays or analytical methods that are currently used to analyze tissue samples or cell homogenates are vulnerable to changes in pH, temperature, light, and chemical agent or buffer solution. Therefore, more accurate and reliable quantification and extrapolation to in vivo conditions are warranted.
A. Cell culture systems
Accumulating evidence suggests the involvement of oxidative stress, inflammation, and increased l-tryptophan catabolism in several degenerative disorders. This has paved the way for investigation of basic mechanisms of free-radical damage and modulation of the NAD+ metabolome as a therapeutic strategy to protect against it. For example, primary murine and human brain cell cultures, and immortalized cell lines, remain highly useful as models for examining the effect of oxidative damage and adaptive cellular responses. The most common approach to modeling CNS oxidative stress and altered kynurenine pathway metabolism is through exposure of primary glial and neuronal cells to deleterious conditions, and addition of exogenous pro-oxidants and neuroprotective agents (44, 46, 48, 50, 71, 195, 333). Cell culture models have also been used to examine the effects of niacin deficiency, and inhibition of NAD-dependent processes (such as PARP, sirtuin, and CD38 inhibition) on cellular function in several in vitro disease models (46, 47, 52, 119, 267). However, most cell culture components, which are fundamental to these studies, are aimed at maximizing cell growth and survival in culture and do not fully recapitulate natural in vivo biological processes. There is also strong evidence that the beneficial effects of NAD+ precursors in culture systems may be incurred via nonphysiological mechanisms.
Vitamin B3 is present in cell culture in both its amide and acidic form. However, NAM is present at highest concentrations, and this reflects the significance of NAM as the main form of niacin in the blood stream. Commonly used cell culture media (e.g., MEM, Williams, RPMI, BME, L-15, and Dulbecco's) contain between 1 and 4 mg/L of NAM, although more specialized media (MCDB 131 and BGjB) may contain between 6 and 20 mg/L. The equivalent molar concentration for 4 mg/L is ∼33 μM. This amount is about 300-fold greater than the average levels of NAM in plasma. It is likely that these concentrations may have a profound effect on intracellular NAD+ storage, cyclic-ADP- and monoribosylation, and inhibitor studies. Other cell culture media contain equal contents of NAM and NA at concentrations of up to 4 μM. Similarly, the concentration is well above the physiological concentration of NA in systemic circulation. Moreover, these levels are significantly greater than the amount required to activate HM74A receptors if these are expressed in cells in culture.
B. In vivo models
The human life span is much longer than smaller mammalian species, making it difficult to fully characterize the influence of NAD+ metabolism during normal human aging. Therefore, traditional in vivo studies have been performed using animals with phenotypically accelerated aging or prolonged longevity, transgenic, mutant, and knockout models that focus on a single gene's role, to generate reproducible results. Due to their short life span, inbred laboratory rodents, particularly rats and mice (e.g., senescence-accelerated mice), are used as models to investigate the effects of intrinsic and extrinsic factors on life span (324). However, this is quite limited since these inbred models do not provide significant genetic diversity to be compared to humans and correlate poorly with human conditions. To date, more than 150 clinical trial candidates to attenuate inflammation in critically ill patients have failed due to over-reliance on inadequate animal models.
We have addressed the conceptual translation of biochemical data collected from aging female Wistar rats to further enlighten our understanding of the role of NAD+ metabolism and other molecular changes occurring as part of “normal” human aging (51–53). Our physiologically aged Wistar rats were an outbred model, which displays significant genetic diversity within a small number of individuals. This diversity-outcrossed rat model is more representative of a natural population and is therefore a powerful tool in identifying the genetic basis for assessing the efficacy of these pharmacological strategies, and to identify adverse effects in first-line therapeutic tests, which are otherwise nascent in previously inbred animals. Additional effects of aging previously demonstrated in this animal model include a marked decrease in the astrocyte/neuronal ratio; altered pericyte/endothelial relationship affecting “vessel stability”; marked inflammatory cascade, CNS neovascularization and breakdown of the blood/retinal barrier, and a decline in defence-related Fos expression (158, 218).
C. Methods of detection
NAD+ and its related metabolites have been previously measured using a variety of methods. For instance, enzymatic and colorimetric assays, which provide indirect measurements, have inherent difficulties that can affect reliability and are susceptible to significant variation in metabolite levels due to minor differences in temperature and pH, and cannot detect low picomolar levels. Moreover, reverse-phase HPLC, which relies on mobile phases containing buffer salts and ion pairing agents, has been used to increase sensitivity, but is still limited to low micromolar detection levels (70).
In contrast, liquid chromatography/tandem mass spectrometry allows more robust quantification of trace levels of NAD+ metabolites in different biological samples with high specificity and sensitivity. It represents the gold standard in NAD+ metabolomics. However, unlike NMR, complex sampling processes are required (335). The diversity of NAD+ metabolites (i.e., free bases, mono and dinucleotides) makes their simultaneous differential analysis a major challenge (335). We recently developed an improved method to quantify the NAD+ metabolome and adenosine phosphates across biological samples, including the brain and reproductive cells. Its principal features are enhanced resolution and simultaneous quantification of 17 analytes on an amino-phase column, avoiding the need for 2 separate gradients (i.e., alkaline and acidic chromatographic gradients) (60).
Development of nondestructive detection and quantification of the NAD+ metabolome is desirable to elucidate intracellular NAD+ levels and redox state in the intact human and animal body. Recently, a novel magnetic resonance imaging (MRI) has been developed to determine the endogenous 31P MR signals of the NAD+ molecules in live animal brains (389). This technique can resolve the MRI signal of NADH from that of NAD+ by utilizing specific spectroscopic characteristics at a given magnetic field strength. This approach requires ultrahigh fields of 9.4 and 16.4 T. This noninvasive technique has been further used to measure intracellular NAD+ and NADH contents and NAD+/NADH redox state in healthy human brains using a 7-T human MR scanner (389). We were the first to show that intracellular NAD+ levels, the essential substrate for sirtuin activity, decline with age in humans and physiologically aged rats (51, 52, 223). MRI was used to reaffirm these age-dependent increases of intracellular NADH and age-dependent reductions in NAD+, total NAD contents, and NAD+/NADH redox potential of the healthy human brains.
It is anticipated that improvement in methods to quantify the NAD+ metabolome will be developed to help standardize NAD+ research across different laboratories, and to overcome challenges associated with translation of preclinical studies toward clinical practice.
XIII. Prospects of Using NAD+ Precursors in the Clinic
Pellagra, a syndrome caused by a diet deficient in either NA or l-tryptophan, can lead to psychotic symptoms leading to presenile dementia likely due to upregulation of IDO, which can deplete neurons of the essential amino acid, l-tryptophan, causing neurodegeneration. Administration of the NAD+ precursors, NA or NAM, previously improved the neurological state of dementia patients in the 1930s. Pharmacological doses of either NA or NAM have also provided dramatic therapeutic benefits for other diseases, including lipid dyshomeostasis, rheumatoid arthritis, type I diabetes, colitis, multiple sclerosis (MS), and schizophrenia in both animal models and in the clinical setting. Among these precursors, NA appears to specifically activate the G-protein-coupled receptor, GPR109, leading to the release of prostaglandins, PGE2 and PGD2 (314). These prostaglandins exert potent anti-inflammatory effects through endogenous signaling mechanisms. While NAM can prevent MS in animal models, it is also an inhibitor of sirtuins, and may therefore prove detrimental on long-term cell survival and longevity (258, 259).
There is growing evidence suggesting that NAD+ administration may also reduce cellular injury in multiple oxidative stress-induced degenerative diseases. NAD+ treatment has been shown to reduce PARP1-induced astrocyte death (7). PARP1 has been implicated in the pathogenesis of several diseases, including diabetes, AD, and PD (196, 221). Since supplementation with NAD+ can protect against PARP1-mediated cell death, NAD+ administration may improve cell viability in these diseases by at least partially ameliorating PARP1 toxicity. In vitro studies have shown that NAD+ remains protective even when administered at 3–4 h following PARP1 activation, suggesting that NAD+ administration has a long window period for reducing cellular injury (8). In addition, NAD+ may also improve cell viability by enhancing sirtuin activities and/or improving energy metabolism.
While the potential involvement of NAD+ metabolic pathways in energy metabolism and mitochondrial function has been known for quite some time, suggestions of the involvement of NAD+ in DNA repair and longevity have grown at a rapid rate in the last decade. Characterization of the NAD+ synthetic pathways has not only made these advancements possible but also contributed extensively to the understanding of the diverse roles of pyridine nucleotides in cellular biology. Despite this, information regarding the fundamental roles of NAD+ in neurodegeneration and aging remains limited. Further investigations are necessary in this increasingly relevant field.
While the current review herein focused on PARP1 in cellular degeneration, the role of other PARPs such as tankyrases in cellular function remains largely unknown. Since NAD-dependent tankyrases are primary mediators of telomerase activity, it is highly likely that NAD+ may also affect the aging process through regulation of tankyrase activity (385). It would therefore be intriguing to study the effects of NAD+ precursors on tankyrases and telomerases on certain biological functions, including neurogenesis, which might be relevant in the aging brain.
In addition, NAD+ regulates diverse pathways that may control life span. The importance of NAD+ is further underscored by recent work providing genetic evidence for the existence of several pathways necessary for NAD+ synthesis. For example, the newly identified NAD+ precursor, NR, has been shown to contribute to NAD+ synthesis by at least two unique pathways in the yeast Saccharomyces cerevisiae, and can upregulate intracellular NAD+ levels in mice and humans (338). Both pathways require the NAM ring for entry into the previously established pathways for NAD+ synthesis. Future studies are required to address the importance of NR in human health and disease, and whether it can be effectively used to replenish lowered NAD+ levels in age-related diseases, such as AD. Given the adverse effects associated with high-dose use of NA and NAM, NR may represent an alternative precursor to enhance NAD+ levels.
As well, changes in the NADH level, NAD+ redox potential, and NAD+ levels are likely to be present in other pathological conditions and may be associated with disease progression. In particular, increased oxidative stress and immune activation in AD, PD, ADC, and amyotrophic lateral sclerosis (ALS) may influence the available concentration of these molecules. Future investigation into the metabolism and biological function of NAD+ in these and other degenerative diseases may expose fundamental properties that may involve the use of NAD+ precursors as adjunct therapy for treatment in these diseases, and perhaps may help in slowing down the age-related disease process.
Resveratrol is a polyphenol with major health benefits that is thought to operate through direct activation of the “antiaging” enzyme SIRT1. However, recent reports have challenged this “direct activation” hypothesis, suggesting that the mechanism by which resveratrol increases SIRT1 function is still unknown (39, 86). Previous work from our group has shown for the first time that resveratrol induces a dose-dependent increase in NMNAT-1 activity. As SIRT1 requires NAD+ as a substrate to perform its gene silencing function, higher NAD+ levels will enhance SIRT1 activity. This finding suggests that resveratrol may promote SIRT1 function by enhancing NAD+ synthesis in whole cell systems without requiring direct activation. Our observation that resveratrol increases NAD+ levels in primary human brain cells by acting on NMNAT, together with the neuroprotective effects of green tea polyphenols against QUIN-mediated excitotoxicity (47), supports the view that polyphenols have considerable therapeutic potential, particularly for the treatment of neurodegenerative diseases. As NMNAT can accelerate NAD+ synthesis from all six substrates, QUIN, NR, NMN, NA, NAR, and NAM, NMNAT activation by resveratrol may represent an ideal natural therapeutic to replenish NAD+ levels. Maintenance of higher cellular NAD+ will enhance SIRT1 activity and other NAD+-dependent pathways, impacting positively on cell viability and longevity.
Finally, increased NAMPT has been reported in a mice model of collagen-induced arthritis both in the serum and in the arthritic paw (59). NAMPT inhibition reduced arthritic severity comparable to etanercept, significantly lowered the levels of cytokine release in affected joints, and reduced intracellular NAD+ concentrations in inflammatory cells (59). Therefore, NAMPT may play an important role during inflammatory diseases associated with cytokine secretion from leukocytes. Therefore, increasing NAD+ levels may be deleterious in inflammatory conditions and may exacerbate the disease due to increased NAMPT activity and NAD+ use in immune cells. This represents an additional potential negative to “one-size-fits-all” use of NAD+.
While most of the evidence reviewed in this article strongly supports the current enthusiasm to investigate and develop strategies for increasing NAD+ levels in conditions where NAD+ turnover is high and/or concentrations are reduced, the use of NAD+ enhancing therapeutics in circumstances where cellular NAD+ levels are already adequate may be unwise. Given the complexity of the biochemical systems affected by NAD+ and its associated metabolites a simplistic, one-size-fits-all approach to NAD+ therapeutics will likely limit the true potential of NAD+ treatment and may in fact cause harm under some circumstances. As well, while several NAD+ precursors have been recently identified and examined in several models, a side-by-side comparison of these precursors is nascent in current literature. It is anticipated that these precursors may exhibit important differences in their effect in various pathological disorders (375).
To circumvent this, in addition to the many studies focused on identifying efficient ways of increasing NAD+, additional effort must be applied to the development of cost-effective methods of measuring and correlating NAD+ levels in both tissue and extracellular fluids to cellular and organ health in an effort to establish a clear understanding of what a “healthy” NAD+ level actually is. Armed with this knowledge the clinician may confidently apply NAD+ therapy after an appropriate assessment of NAD+ levels to determine whether the treatment is likely to be effective in each client's case.
XIV. Concluding Remarks
NAD+ research has generated multiple discoveries in the last two decades. Identification of the important role of NAD+ as a cofactor in cellular respiration and energy production was followed by discoveries of numerous NAD+ biosynthesis pathways. In recent years, NAD+ has been shown to play a unique role in DNA repair and epigenetic control through protein deacetylation. Elucidation of the pivotal roles played by NAD+ in linking the key biochemical and cellular processes of oxidative stress and immune activation, energy metabolism, epigenetic control, and cell viability in degenerative disorders and aging will likely prove seminal to the advancement of effective therapeutics in degenerative diseases. NAD+ remains the central molecule in the metabolism and functions of NAD+, NADH, NADP+, and NADPH. Of these four molecules, only NAD+ can be synthesized de novo via the kynurenine pathway, while the generation of NADH, NADP+, and NADPH requires NAD+ as the original precursor. Maintenance of intracellular NAD+ levels is pivotal for the regulation of DNA repair, stress resistance, and cell death, suggesting that NAD+ synthesis through the kynurenine pathway and/or salvage pathway is an attractive target for therapeutic intervention in age-associated degenerative disorders. Agents such as NR, and to a lesser degree, NA and NAM, have been shown to protect severed axons from degeneration in animal models for Wallerian degeneration, and extend life span in small organisms. However, further studies are necessary to clarify the conditions under which specific NAD+ precursors should be used to efficiently promote intracellular NAD+ anabolism. This involves evaluating the pharmacokinetics, safety, and efficacy in healthy and disease models to develop targeted therapies that ameliorate degenerative processes and help maintain and improve health span and longevity.
While it will almost certainly be proved true that NAD+ therapy alone is not the mythical “elixir of life,” its foundational role in cellular energetics, nuclear signaling, and viability suggests it just may be a key ingredient.
Acknowledgment
N.B. received the Australian Research Council DECRA Fellowship.
Abbreviations Used
- 3-HAA
3-hydroxyanthranilic acid
- 3-HAAO
3-hydroxyanthranilic acid oxygenase
- 3-HK
3-hydroxykynurenine
- 5′-NTs
5′-nucleotidases
- AA
anthranilic acid
- Aβ
amyloid-beta
- AD
Alzheimer's disease
- ADC
AIDS dementia complex
- ADPR
ADP ribose
- AKI
acute kidney injury
- AMPK
AMP-activated kinase
- AUC
area under the curve
- BBB
blood/brain barrier
- cADPR
cyclic-ADP-ribose
- CNS
central nervous system
- CPS1
carbamoyl-phosphate synthetase 1
- CR
caloric restriction
- eNOS
endothelial nitric oxide synthase
- ETC
electron transport chain
- FAO
fatty acid oxidation
- FBS
fetal bovine serum
- GABA
gamma-aminobutyric acid
- GSH
glutathione
- GSSG
glutathione disulfide
- H2O2
hydrogen peroxide
- HDL
high-density lipoprotein
- HIF1α
hypoxia-inducible factor 1α
- HPLC
high-performance liquid chromatography
- HSF
heat shock factor
- IDH2
isocitrate dehydrogenase 2
- IDO
indoleamine 2,3-dioxygenase
- IFN-γ
interferon-gamma
- ILS
insulin-like signaling
- isoNAM
isonicotinamide
- KAT
kynurenine aminotransferase
- LDL
low-density lipoprotein
- LKB1
liver kinase B1
- LXR
liver X receptor
- MeNAM
N-methylnicotinamide
- MLCK
myosin light chain kinase
- MnSOD
manganese superoxide dismutase
- mPTP
mitochondrial permeability transition pore
- MRI
magnetic resonance imaging
- mRNA
messenger RNA
- MS
multiple sclerosis
- NA
nicotinic acid
- NAAD
nicotinic acid adenine dinucleotide
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NAD+
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- NAMN
nicotinic acid mononucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- NAPRT
nicotinic acid phosphoribosyltransferase
- NAR
nicotinic acid riboside
- NMDA
N-methyl-d-aspartate
- NMN
nicotinamide mononucleotide
- NMNAT
nicotinamide mononucleotide adenylyltransferase
- NMNAT-1
isoform of NMNAT localized to the nucleus
- NMNAT-2
Golgi complex isoform of NMNAT
- NMNAT-3
isoform of NMNAT localized to the mitochondria
- NMR
nuclear magnetic resonance
- NNMT
nicotinamide N-methyltransferase
- NR
nicotinamide riboside
- NRK
nicotinamide riboside kinase
- PARP
poly(ADP-ribose) polymerase
- PBEF
pre-B cell colony enhancing factor
- PBMC
peripheral blood mononuclear cell
- PD
Parkinson's disease
- PIC
picolinic acid
- PICAC
picolinic acid carboxylase
- PKA
protein kinase A
- PNP
purine nucleoside phosphorylase
- PPAR
peroxisome proliferator-activated receptor
- PRPP
5-phosphoribosyl-1-pyrophosphate
- QPRT
quinolinic acid phosphoribosyltransferase
- QUIN
quinolinic acid
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SIRT
sirtuin
- TDO
tryptophan 2,3-dioxygenase
- tg
transgenic
- TOR
target of rapamycin
- TXN
thioredoxin
- URI
unconventional prefoldin RPB5 interactor
- UV
ultraviolet
References
- 1. Public Health Reports, June 26, 1914. The etiology of pellagra. The significance of certain epidemiological observations with respect thereto. Public Health Rep 90: 373–375, 1975 [PMC free article] [PubMed] [Google Scholar]
- 2. Abeti R. and Duchen MR. Activation of PARP by oxidative stress induced by beta-amyloid: implications for Alzheimer's disease. Neurochem Res 37: 2589–2596, 2012 [DOI] [PubMed] [Google Scholar]
- 3. Agote M, Viaggi M, Kreimann E, Krawiec L, Dagrosa , Juvenal GJ, Pisarev Influence of nicotinamide on the radiosensitivity of normal and goitrous thyroid in the rat. Thyroid 11: 1003–1007, 2001 [DOI] [PubMed] [Google Scholar]
- 4. Agote Robertson M, Finochietto P, Gamba CA, Dagrosa MA, Viaggi ME, Franco MC, Poderoso JJ, Juvenal GJ, and Pisarev MA. Nicotinamide increases thyroid radiosensitivity by stimulating nitric oxide synthase expression and the generation of organic peroxides. Horm Metab Res 38: 12–15, 2006 [DOI] [PubMed] [Google Scholar]
- 5. Aikawa H. and Suzuki K. Lesions in the skin, intestine, and central nervous system induced by an antimetabolite of niacin. Am J Pathol 122: 335–342, 1986 [PMC free article] [PubMed] [Google Scholar]
- 6. Aksoy S, Brandriff BF, Ward A, Little PF, and Weinshilboum RM. Human nicotinamide N-methyltransferase gene: molecular cloning, structural characterization and chromosomal localization. Genomics 29: 555–561, 1995 [DOI] [PubMed] [Google Scholar]
- 7. Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, and Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci 30: 2967–2978, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alano CC, Ying W, and Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem 279: 18895–18902, 2004 [DOI] [PubMed] [Google Scholar]
- 9. Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, and Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res 100: 1512–1521, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Alenzi FQ. Effect of nicotinamide on experimental induced diabetes. Iran J Allergy Asthma Immunol 8: 11–18, 2009 [PubMed] [Google Scholar]
- 11. Ali YO, McCormack R, Darr A, and Zhai RG. Nicotinamide mononucleotide adenylyltransferase is a stress response protein regulated by the heat shock factor/hypoxia-inducible factor 1alpha pathway. J Biol Chem 286: 19089–19099, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ali YO, Ruan K, and Zhai RG. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum Mol Genet 21: 237–250, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, and Lopaschuk GD. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res 103: 485–497, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Altmeyer M. and Hottiger MO. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging (Albany NY) 1: 458–469, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Altmeyer M, Messner S, Hassa PO, Fey M, and Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res 37: 3723–3738, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Anderson RM, Bitterman KJ, Wood JG, Medvedik O, and Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423: 181–185, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Andrabi SA, Umanah GKE, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, Poirier GG, Dawson VL, and Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A 111: 10209–10214, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ansari A, Rahman MS, Saha SK, Saikot FK, Deep A, and Kim KH. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 16: 4–16, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. ApSimon MM, Rawling JM, and Kirkland JB. Nicotinamide megadosing increases hepatic poly(ADP-ribose) levels in choline-deficient rats. J Nutr 125: 1826–1832, 1995 [DOI] [PubMed] [Google Scholar]
- 20. Arpaia E, Benveniste P, Di Cristofano A, Gu Y, Dalal I, Kelly S, Hershfield M, Pandolfi PP, Roifman CM, and Cohen A. Mitochondrial basis for immune deficiency. Evidence from purine nucleoside phosphorylase-deficient mice. J Exp Med 191: 2197–2208, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Badawy AA. Pellagra and alcoholism: a biochemical perspective. Alcohol Alcohol 49: 238–250, 2014 [DOI] [PubMed] [Google Scholar]
- 22. Badawy AA. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res 10: 1178646917691938, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ball HJ, Yuasa HJ, Austin CJ, Weiser S, and Hunt NH. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int J Biochem Cell Biol 41: 467–471, 2009 [DOI] [PubMed] [Google Scholar]
- 24. Balu M, Mazhar A, Hayakawa CK, Mittal R, Krasieva TB, Konig K, Venugopalan V, and Tromberg BJ. In vivo multiphoton NADH fluorescence reveals depth-dependent keratinocyte metabolism in human skin. Biophys J 104: 258–267, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bansal A, Kwon ES, Conte D, Jr., Liu H, Gilchrist MJ, MacNeil LT, and Tissenbaum HA. Transcriptional regulation of Caenorhabditis elegans FOXO/DAF-16 modulates lifespan. Longev Healthspan 3: 5, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartleman AP, Jacobs R, and Kirkland JB. Niacin supplementation decreases the incidence of alkylation-induced nonlymphocytic leukemia in Long-Evans rats. Nutr Cancer 60: 251–258, 2008 [DOI] [PubMed] [Google Scholar]
- 27. Benavente CA. and Jacobson EL. Niacin restriction upregulates NADPH oxidase and reactive oxygen species (ROS) in human keratinocytes. Free Radic Biol Med 44: 527–537, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bender DA. and McCreanor GM. Kynurenine hydroxylase: a potential rate-limiting enzyme in tryptophan metabolism. Biochem Soc Trans 13: 441–443, 1985 [DOI] [PubMed] [Google Scholar]
- 29. Benfeitas R, Uhlen M, Nielsen J, and Mardinoglu A. New challenges to study heterogeneity in cancer redox metabolism. Front Cell Dev Biol 5: 65, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Beninger RJ, Colton AM, Ingles JL, Jhamandas K, and Boegman RJ. Picolinic acid blocks the neurotoxic but not the neuroexcitant properties of quinolinic acid in the rat brain: evidence from turning behaviour and tyrosine hydroxylase immunohistochemistry. Neuroscience 61: 603–612, 1994 [DOI] [PubMed] [Google Scholar]
- 31. Berger F, Lau C, Dahlmann M, and Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280: 36334–36341, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Berger F, Lau C, and Ziegler M. Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci U S A 104: 3765–3770, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhatia R. and Calvo KC. The sequencing expression, purification, and steady-state kinetic analysis of quinolinate phosphoribosyl transferase from Escherichia coli. Arch Biochem Biophys 325: 270–278, 1996 [DOI] [PubMed] [Google Scholar]
- 34. Birkmayer JG. and Nadlinger K. Safety of stabilized, orally absorbable, reduced nicotinamide adenine dinucleotide (NADH): a 26-week oral tablet administration of ENADA/NADH for chronic toxicity study in rats. Drugs Exp Clin Res 28: 185–192, 2002 [PubMed] [Google Scholar]
- 35. Birkmayer JG, Vrecko C, Volc D, and Birkmayer W. Nicotinamide adenine dinucleotide (NADH)—a new therapeutic approach to Parkinson's disease. Comparison of oral and parenteral application. Acta Neurol Scand Suppl 146: 32–35, 1993 [PubMed] [Google Scholar]
- 36. Blagosklonny MV. Hormesis does not make sense except in the light of TOR-driven aging. Aging (Albany NY) 3: 1051–1062, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blum CA, Ellis JL, Loh C, Ng PY, Perni RB, and Stein RL. SIRT1 modulation as a novel approach to the treatment of diseases of aging. J Med Chem 54: 417–432, 2011 [DOI] [PubMed] [Google Scholar]
- 38. Bogan KL. and Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr 28: 115–130, 2008 [DOI] [PubMed] [Google Scholar]
- 39. Borra MT, Smith BC, and Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 280: 17187–17195, 2005 [DOI] [PubMed] [Google Scholar]
- 40. Bouamama S, Merzouk H, Medjdoub A, Merzouk-Saidi A, and Merzouk SA. Effects of exogenous vitamins A, C, and E and NADH supplementation on proliferation, cytokines release, and cell redox status of lymphocytes from healthy aged subjects. Appl Physiol Nutr Metab 42: 579–587, 2017 [DOI] [PubMed] [Google Scholar]
- 41. Boylston JA, Sun J, Chen Y, Gucek M, Sack MN, and Murphy E. Characterization of the cardiac succinylome and its role in ischemia-reperfusion injury. J Mol Cell Cardiol 88: 73–81, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Boyonoski AC, Spronck JC, Gallacher LM, Jacobs RM, Shah GM, Poirier GG, and Kirkland JB. Niacin deficiency decreases bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr 132: 108–114, 2002 [DOI] [PubMed] [Google Scholar]
- 43. Boyonoski AC, Spronck JC, Jacobs RM, Shah GM, Poirier GG, and Kirkland JB. Pharmacological intakes of niacin increase bone marrow poly(ADP-ribose) and the latency of ethylnitrosourea-induced carcinogenesis in rats. J Nutr 132: 115–120, 2002 [DOI] [PubMed] [Google Scholar]
- 44. Braidy N, Brew BJ, Inestrosa NC, Chung R, Sachdev P, and Guillemin GJ. Changes in Cathepsin D and Beclin-1 mRNA and protein expression by the excitotoxin quinolinic acid in human astrocytes and neurons. Metab Brain Dis 29: 873–883, 2014 [DOI] [PubMed] [Google Scholar]
- 45. Braidy N. and Grant R. Kynurenine pathway metabolism and neuroinflammatory disease. Neural Regen Res 12: 39–42, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Braidy N, Grant R, Adams S, Brew BJ, and Guillemin GJ. Mechanism for quinolinic acid cytotoxicity in human astrocytes and neurons. Neurotox Res 16: 77–86, 2009 [DOI] [PubMed] [Google Scholar]
- 47. Braidy N, Grant R, Adams S, and Guillemin GJ. Neuroprotective effects of naturally occurring polyphenols on quinolinic acid-induced excitotoxicity in human neurons. FEBS J 277: 368–382, 2010 [DOI] [PubMed] [Google Scholar]
- 48. Braidy N, Grant R, Brew BJ, Adams S, Jayasena T, and Guillemin GJ. Effects of Kynurenine pathway metabolites on intracellular NAD synthesis and cell death in human primary astrocytes and neurons. Int J Tryptophan Res 2: 61–69, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Braidy N, Guillemin G, and Grant R. Promotion of cellular NAD(+) anabolism: therapeutic potential for oxidative stress in ageing and Alzheimer's disease. Neurotox Res 13: 173–184, 2008 [DOI] [PubMed] [Google Scholar]
- 50. Braidy N, Guillemin GJ, and Grant R. Effects of Kynurenine pathway inhibition on NAD metabolism and cell viability in human primary astrocytes and neurons. Int J Tryptophan Res 4: 29–37, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, and Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 6: e19194, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, Guillemin GJ, Smythe G, and Sachdev P. Mapping NAD(+) metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology 15: 177–198, 2014 [DOI] [PubMed] [Google Scholar]
- 53. Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, Smythe G, Sachdev P, and Guillemin GJ. Differential expression of sirtuins in the aging rat brain. Front Cell Neurosci 9: 167, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brazill JM, Li C, Zhu Y, and Zhai RG. NMNAT: it's an NAD+ synthase … it's a chaperone … it's a neuroprotector. Curr Opin Genet Dev 44: 156–162, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brousseau KM, Filley CM, Kaye K, Kiser JJ, Adler LE, and Connick E. Dementia with features of Alzheimer's disease and HIV-associated dementia in an elderly man with AIDS. AIDS 23: 1029–1031, 2009 [DOI] [PubMed] [Google Scholar]
- 56. Brown KD, Maqsood S, Huang JY, Pan Y, Harkcom W, Li W, Sauve A, Verdin E, and Jaffrey SR. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab 20: 1059–1068, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, and Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015, 2004 [DOI] [PubMed] [Google Scholar]
- 58. Busse M, Hettler V, Fischer V, Mawrin C, Hartig R, Dobrowolny H, Bogerts B, Frodl T, and Busse S. Increased quinolinic acid in peripheral mononuclear cells in Alzheimer's dementia. Eur Arch Psychiatry Clin Neurosci 2017. DOI: 10.1007/s00406-017-0785-y [DOI] [PubMed] [Google Scholar]
- 59. Busso N, Karababa M, Nobile M, Rolaz A, Van Gool F, Galli M, Leo O, So A, and De Smedt T. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One 3: e2267, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bustamante S, Jayasena T, Richani D, Gilchrist RB, Wu LE, Sinclair DA, Sachdev P, and Braidy N. Quantifying the cellular NAD+ metabolome using a tandem liquid chromatography mass spectrometry approach. Metabolomics 14: 15, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cadenas C, Franckenstein D, Schmidt M, Gehrmann M, Hermes M, Geppert B, Schormann W, Maccoux LJ, Schug M, Schumann A, Wilhelm C, Freis E, Ickstadt K, Rahnenfuhrer J, Baumbach JI, Sickmann A, and Hengstler JG. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res 12: R44, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Calabrese EJ. Hormesis: a fundamental concept in biology. Microb Cell 1: 145–149, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Calabrese EJ. and Mattson MP. How does hormesis impact biology, toxicology, and medicine? NPJ Aging Mech Dis 3: 13, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Calabrese EJ, Mattson MP, and Calabrese V. Resveratrol commonly displays hormesis: occurrence and biomedical significance. Hum Exp Toxicol 29: 980–1015, 2010 [DOI] [PubMed] [Google Scholar]
- 65. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, and Chini EN. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab 23: 1127–1139, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, and Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, and Auwerx J. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 15: 838–847, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Canto C, Menzies KJ, and Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22: 31–53, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cao W, Hong Y, Chen H, Wu F, Wei X, and Ying W. SIRT2 mediates NADH-induced increases in Nrf2, GCL, and glutathione by modulating Akt phosphorylation in PC12 cells. FEBS Lett 590: 2241–2255, 2016 [DOI] [PubMed] [Google Scholar]
- 70. Casabona G, Sturiale L, L'Episcopo MR, Raciti G, Fazzio A, Sarpietro MG, Genazzani AA, Cambria A, and Nicoletti F. HPLC analysis of cyclic adenosine diphosphate ribose and adenosine diphosphate ribose: determination of NAD+ metabolites in hippocampal membranes. Ital J Biochem 44: 258–268, 1995 [PubMed] [Google Scholar]
- 71. Castellano-Gonzalez G, Pichaud N, Ballard JW, Bessede A, Marcal H, and Guillemin GJ. Epigallocatechin-3-gallate induces oxidative phosphorylation by activating cytochrome c oxidase in human cultured neurons and astrocytes. Oncotarget 7: 7426–7440, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Catz P, Shinn W, Kapetanovic IM, Kim H, Kim M, Jacobson EL, Jacobson MK, and Green CE. Simultaneous determination of myristyl nicotinate, nicotinic acid, and nicotinamide in rabbit plasma by liquid chromatography-tandem mass spectrometry using methyl ethyl ketone as a deproteinization solvent. J Chromatogr B Analyt Technol Biomed Life Sci 829: 123–135, 2005 [DOI] [PubMed] [Google Scholar]
- 73. Chambers DA, Martin DW, Jr., and Weinstein Y. The effect of cyclic nucleotides on purine biosynthesis and the induction of PRPP synthetase during lymphocyte activation. Cell 3: 375–380, 1974 [DOI] [PubMed] [Google Scholar]
- 74. Chapman MJ, Le Goff W, Guerin M, and Kontush A. Cholesteryl ester transfer protein: at the heart of the action of lipid-modulating therapy with statins, fibrates, niacin, and cholesteryl ester transfer protein inhibitors. Eur Heart J 31: 149–164, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chaudhuri RN. and Chakravarti H. An outbreak of pellegra syndrome in a rural area of Bengal. Ind Med Gaz 82: 657–660, 1947 [PMC free article] [PubMed] [Google Scholar]
- 76. Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des 15: 57–63, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. This reference has been deleted.
- 78. Clementi E. and Nisoli E. Nitric oxide and mitochondrial biogenesis: a key to long-term regulation of cellular metabolism. Comp Biochem Physiol A Mol Integr Physiol 142: 102–110, 2005 [DOI] [PubMed] [Google Scholar]
- 79. Cockhill J, Jhamandas K, Boegman RJ, and Beninger RJ. Action of picolinic acid and structurally related pyridine carboxylic acids on quinolinic acid-induced cortical cholinergic damage. Brain Res 599: 57–63, 1992 [DOI] [PubMed] [Google Scholar]
- 80. Coleman MP. and Freeman MR. Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci 33: 245–267, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Collins PB. and Chaykin S. The management of nicotinamide and nicotinic acid in the mouse. J Biol Chem 247: 778–783, 1972 [PubMed] [Google Scholar]
- 82. Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, and Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325: 201–204, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, and Anderson RM. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun 5: 3557, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cooper DL, Murrell DE, Roane DS, and Harirforoosh S. Effects of formulation design on niacin therapeutics: mechanism of action, metabolism, and drug delivery. Int J Pharm 490: 55–64, 2015 [DOI] [PubMed] [Google Scholar]
- 85. Dantzer F, Ame JC, Schreiber V, Nakamura J, Menissier-de Murcia J, and de Murcia G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol 409: 493–510, 2006 [DOI] [PubMed] [Google Scholar]
- 86. Dasgupta B. and Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A 104: 7217–7222, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai S, and Seals DR. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15: 522–530, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dellinger RW, Santos SR, Morris M, Evans M, Alminana D, Guarente L, and Marcotulli E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: a randomized, double-blind, placebo-controlled study. NPJ Aging Mech Dis 3: 17, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Di Lisa F. and Ziegler M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett 492: 4–8, 2001 [DOI] [PubMed] [Google Scholar]
- 90. Di Stefano M. and Conforti L. Diversification of NAD biological role: the importance of location. FEBS J 280: 4711–4728, 2013 [DOI] [PubMed] [Google Scholar]
- 91. Diamond MP, Fletcher NM, Neubauer BR, Saed MG, H M AS, and Saed GM. Hypoxia-induced genotype switch in nicotinamide adenine dinucleotide phosphate (NADPH) oxidase through the up-regulation of cytidine deaminase regulates postoperative adhesion development. J Minim Invasive Gynecol 22: S159, 2015 [DOI] [PubMed] [Google Scholar]
- 92. Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, and Garcia JA. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 324: 1289–1293, 2009 [DOI] [PubMed] [Google Scholar]
- 93. Dragovic J, Kim SH, Brown SL, and Kim JH. Nicotinamide pharmacokinetics in patients. Radiother Oncol 36: 225–228, 1995 [DOI] [PubMed] [Google Scholar]
- 94. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, and Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112: 1049–1057, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Duarte-Pereira S, Pereira-Castro I, Silva SS, Correia MG, Neto C, da Costa LT, Amorim A, and Silva RM. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget 7: 1973–1983, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Duley JA, Christodoulou J, and de Brouwer AP. The PRPP synthetase spectrum: what does it demonstrate about nucleotide syndromes? Nucleosides Nucleotides Nucleic Acids 30: 1129–1139, 2011 [DOI] [PubMed] [Google Scholar]
- 97. Easlon E, Tsang F, Skinner C, Wang C, and Lin SJ. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev 22: 931–944, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Easton T, Richardson AJ, Hall AD, Kidane L, Das I, and Puri BK. The niacin flush test as an index of fatty acid deficiency in schizophrenia. Schizophr Res 36: 307–308, 1999 [Google Scholar]
- 99. Elvehjem CA, Madden RJ, Strong FM, and Wolley DW. The isolation and identification of the anti-black tongue factor. 1937. J Biol Chem 277: e22, 2002 [PubMed] [Google Scholar]
- 100. Elvehjem CA, Madden RJ, Strong FM, and Woolley DW. The isolation and identification of the anti-black tongue factor. Nutr Rev 32: 48–50, 1974 [DOI] [PubMed] [Google Scholar]
- 101. Fan J, Krautkramer KA, Feldman JL, and Denu JM. Metabolic regulation of histone post-translational modifications. ACS Chem Biol 10: 95–108, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, and Bohr VA. NAD(+) in aging: molecular mechanisms and translational implications. Trends Mol Med 23: 899–916, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Fatokun AA, Hunt NH, and Ball HJ. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: characteristics and potential roles in health and disease. Amino Acids 45: 1319–1329, 2013 [DOI] [PubMed] [Google Scholar]
- 104. Fazio F, Carrizzo A, Lionetto L, Damato A, Capocci L, Ambrosio M, Battaglia G, Bruno V, Madonna M, Simmaco M, Nicoletti F, and Vecchione C. Vasorelaxing action of the kynurenine metabolite, xanthurenic acid: the missing link in endotoxin-induced hypotension? Front Pharmacol 8: 214, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fedele G, Frasca L, Palazzo R, Ferrero E, Malavasi F, and Ausiello CM. CD38 is expressed on human mature monocyte-derived dendritic cells and is functionally involved in CD83 expression and IL-12 induction. Eur J Immunol 34: 1342–1350, 2004 [DOI] [PubMed] [Google Scholar]
- 106. Fernandez-Pol JA. Transition metal ions induce cell growth in NRK cells synchronized in G1 by picolinic acid. Biochem Biophys Res Commun 76: 413–419, 1976 [DOI] [PubMed] [Google Scholar]
- 107. Fu CS, Swendseid ME, Jacob RA, and McKee RW. Biochemical markers for assessment of niacin status in young men: levels of erythrocyte niacin coenzymes and plasma tryptophan. J Nutr 119: 1949–1955, 1989 [DOI] [PubMed] [Google Scholar]
- 108. Fukui M, Choi HJ, and Zhu BT. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic Biol Med 49: 800–813, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fukunaga M, Yamamoto Y, Kawasoe M, Arioka Y, Murakami Y, Hoshi M, and Saito K. Studies on tissue and cellular distribution of indoleamine 2,3-dioxygenase 2: the absence of IDO1 upregulates IDO2 expression in the epididymis. J Histochem Cytochem 60: 854–860, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, and Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell 14: 661–673, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gaertner FH. and Shetty AS. Kynureninase-type enzymes and the evolution of the aerobic tryptophan-to-nicotinamide adenine dinucleotide pathway. Biochim Biophys Acta 482: 453–460, 1977 [DOI] [PubMed] [Google Scholar]
- 112. Galassi L, Di Stefano M, Brunetti L, Orsomando G, Amici A, Ruggieri S, and Magni G. Characterization of human nicotinate phosphoribosyltransferase: kinetic studies, structure prediction and functional analysis by site-directed mutagenesis. Biochimie 94: 300–309, 2012 [DOI] [PubMed] [Google Scholar]
- 113. Gale EA, Bingley PJ, Emmett CL, and Collier T; European Nicotinamide Diabetes Intervention Trial (ENDIT) Group. European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 363: 925–931, 2004 [DOI] [PubMed] [Google Scholar]
- 114. Ganji SH, Qin S, Zhang L, Kamanna VS, and Kashyap ML. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis 202: 68–75, 2009 [DOI] [PubMed] [Google Scholar]
- 115. Gensler HL. Prevention of photoimmunosuppression and photocarcinogenesis by topical nicotinamide. Nutr Cancer 29: 157–162, 1997 [DOI] [PubMed] [Google Scholar]
- 116. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, and Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J 26: 1913–1923, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gillman J. and Gillman T. Malnutrition and pellegra in South Africa. Nutr Rev 5: 353–355, 1947 [DOI] [PubMed] [Google Scholar]
- 118. Godoy JA, Rios JA, Zolezzi JM, Braidy N, and Inestrosa NC. Signaling pathway cross talk in Alzheimer's disease. Cell Commun Signal 12: 23, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Godoy JA, Zolezzi JM, Braidy N, and Inestrosa NC. Role of Sirt1 during the ageing process: relevance to protection of synapses in the brain. Mol Neurobiol 50: 744–756, 2014 [DOI] [PubMed] [Google Scholar]
- 120. This reference has been deleted.
- 121. Goldberger J. The etiology of pellagra. 1914. Public Health Rep 121(Suppl 1): 77–79; discussion 76, 2006 [PubMed] [Google Scholar]
- 122. Gonzalez Esquivel D, Ramirez-Ortega D, Pineda B, Castro N, Rios C, and Perez de la Cruz V. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 112: 331–345, 2017 [DOI] [PubMed] [Google Scholar]
- 123. Gorrini C, Harris IS, and Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12: 931–947, 2013 [DOI] [PubMed] [Google Scholar]
- 124. Gospe SM, Jr., Olin KL, and Keen CL. Reduced GABA synthesis in pyridoxine-dependent seizures. Lancet 343: 1133–1134, 1994 [DOI] [PubMed] [Google Scholar]
- 125. Goto T, Matsuo N, and Takahashi T. CSF glutamate/GABA concentrations in pyridoxine-dependent seizures: etiology of pyridoxine-dependent seizures and the mechanisms of pyridoxine action in seizure control. Brain Dev 23: 24–29, 2001 [DOI] [PubMed] [Google Scholar]
- 126. Gougeon L, Payette H, Morais JA, Gaudreau P, Shatenstein B, and Gray-Donald K. Intakes of folate, vitamin B6 and B12 and risk of depression in community-dwelling older adults: the Quebec Longitudinal Study on Nutrition and Aging. Eur J Clin Nutr 70: 380–385, 2016 [DOI] [PubMed] [Google Scholar]
- 127. Grant RS. and Kapoor V. Murine glial cells regenerate NAD, after peroxide-induced depletion, using either nicotinic acid, nicotinamide, or quinolinic acid as substrates. J Neurochem 70: 1759–1763, 1998 [DOI] [PubMed] [Google Scholar]
- 128. Grant RS, Passey R, Matanovic G, Smythe G, and Kapoor V. Evidence for increased de novo synthesis of NAD in immune-activated RAW264.7 macrophages: a self-protective mechanism? Arch Biochem Biophys 372: 1–7, 1999 [DOI] [PubMed] [Google Scholar]
- 129. Guan X, Lin P, Knoll E, and Chakrabarti R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: computational and experimental studies. PLoS One 9: e107729, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Guan XH, Hong X, Zhao N, Liu XH, Xiao YF, Chen TT, Deng LB, Wang XL, Wang JB, Ji GJ, Fu M, Deng KY, and Xin HB. CD38 promotes angiotensin II-induced cardiac hypertrophy. J Cell Mol Med 21: 1492–1502, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Guan XH, Liu XH, Hong X, Zhao N, Xiao YF, Wang LF, Tang L, Jiang K, Qian YS, Deng KY, Ji GJ, Fu MG, and Xin HB. CD38 deficiency protects the heart from ischemia/reperfusion injury through activating SIRT1/FOXOs-mediated antioxidative stress pathway. Oxid Med Cell Longev 2016: 7410257, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Guillemin GJ. and Brew BJ. Implications of the kynurenine pathway and quinolinic acid in Alzheimer's disease. Redox Rep 7: 199–206, 2002 [DOI] [PubMed] [Google Scholar]
- 133. Guyton JR. Niacin in cardiovascular prevention: mechanisms, efficacy, and safety. Curr Opin Lipidol 18: 415–420, 2007 [DOI] [PubMed] [Google Scholar]
- 134. Ha C, Miller LT, and Kerkvliet NI. The effect of vitamin B6 deficiency on cytotoxic immune responses of T cells, antibodies, and natural killer cells, and phagocytosis by macrophages. Cell Immunol 85: 318–329, 1984 [DOI] [PubMed] [Google Scholar]
- 135. Haag F, Adriouch S, Brass A, Jung C, Moller S, Scheuplein F, Bannas P, Seman M, and Koch-Nolte F. Extracellular NAD and ATP: partners in immune cell modulation. Purinergic Signal 3: 71–81, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blander G, Wolberger C, Prolla TA, Weindruch R, Alt FW, and Guarente L. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126: 941–954, 2006 [DOI] [PubMed] [Google Scholar]
- 137. Hallows WC, Yu W, Smith BC, Devries MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, Zhao S, Guan KL, and Denu JM. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol Cell 41: 139–149, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hammen PK, Allali-Hassani A, Hallenga K, Hurley TD, and Weiner H. Multiple conformations of NAD and NADH when bound to human cytosolic and mitochondrial aldehyde dehydrogenase. Biochemistry 41: 7156–7168, 2002 [DOI] [PubMed] [Google Scholar]
- 139. Han MK, Song EK, Guo Y, Ou X, Mantel C, and Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell 2: 241–251, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Han Q, Robinson H, and Li J. Biochemical identification and crystal structure of kynurenine formamidase from Drosophila melanogaster. Biochem J 446: 253–260, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Hansson O. Tryptophan load test and pyridoxine treatment in epileptic children. Acta Neurol Scand 43(Suppl 31):65, 1967 [DOI] [PubMed] [Google Scholar]
- 142. Hara N, Yamada K, Shibata T, Osago H, Hashimoto T, and Tsuchiya M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J Biol Chem 282: 24574–24582, 2007 [DOI] [PubMed] [Google Scholar]
- 143. Harkcom WT, Ghosh AK, Sung MS, Matov A, Brown KD, Giannakakou P, and Jaffrey SR. NAD+ and SIRT3 control microtubule dynamics and reduce susceptibility to antimicrotubule agents. Proc Natl Acad Sci U S A 111: E2443–E2452, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, Washida N, Tokuyama H, Hayashi K, and Itoh H. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun 372: 51–56, 2008 [DOI] [PubMed] [Google Scholar]
- 145. Hassa PO, Haenni SS, Elser M, and Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev 70: 789–829, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. He W, Wang Y, Zhang MZ, You L, Davis LS, Fan H, Yang HC, Fogo AB, Zent R, Harris RC, Breyer MD, and Hao CM. Sirt1 activation protects the mouse renal medulla from oxidative injury. J Clin Invest 120: 1056–1068, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hegab Z, Mohamed TMA, Stafford N, Mamas M, Cartwright EJ, and Oceandy D. Advanced glycation end products reduce the calcium transient in cardiomyocytes by increasing production of reactive oxygen species and nitric oxide. FEBS Open Bio 7: 1672–1685, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hegyi J, Schwartz RA, and Hegyi V. Pellagra: dermatitis, dementia, and diarrhea. Int J Dermatol 43: 1–5, 2004 [DOI] [PubMed] [Google Scholar]
- 149. Herrero-Yraola A, Bakhit SM, Franke P, Weise C, Schweiger M, Jorcke D, and Ziegler M. Regulation of glutamate dehydrogenase by reversible ADP-ribosylation in mitochondria. EMBO J 20: 2404–2412, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hershberger KA, Martin AS, and Hirschey MD. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol 13: 213–225, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV, Jr., Alt FW, Kahn CR, and Verdin E. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464: 121–125, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hong S, Moreno-Navarrete JM, Wei X, Kikukawa Y, Tzameli I, Prasad D, Lee Y, Asara JM, Fernandez-Real JM, Maratos-Flier E, and Pissios P. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat Med 21: 887–894, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hong SJ, Dawson TM, and Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci 25: 259–264, 2004 [DOI] [PubMed] [Google Scholar]
- 154. Hopkins FG. and Cole SW. A contribution to the chemistry of proteids: Part I. A preliminary study of a hitherto undescribed product of tryptic digestion. J Physiol 27: 418–428, 1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Horenstein AL, Sizzano F, Lusso R, Besso FG, Ferrero E, Deaglio S, Corno F, and Malavasi F. CD38 and CD157 ectoenzymes mark cell subsets in the human corneal limbus. Mol Med 15: 76–84, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, Leone TC, Pagliarini DJ, Muoio DM, Bedi KC, Jr., Margulies KB, Coon JJ, and Kelly DP. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2: e84897, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Houtkooper RH, Canto C, Wanders RJ, and Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31: 194–223, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Hunt GE, Van Nieuwenhuijzen PS, Chan-Ling T, and McGregor IS. “When an old rat smells a cat”: a decline in defense-related, but not accessory olfactory, Fos expression in aged rats. Neurobiol Aging 32: 737–749, 2011 [DOI] [PubMed] [Google Scholar]
- 159. Hwang ES. and Song SB. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell Mol Life Sci 74: 3347–3362, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Imai S. and Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab 15(Suppl 3): 26–33, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ishidoh K, Kamemura N, Imagawa T, Oda M, Sakurai J, and Katunuma N. Quinolinate phosphoribosyl transferase, a key enzyme in de novo NAD(+) synthesis, suppresses spontaneous cell death by inhibiting overproduction of active-caspase-3. Biochim Biophys Acta 1803: 527–533, 2010 [DOI] [PubMed] [Google Scholar]
- 162. Iwai K. and Taguchi H. Distribution of quinolinate phosphoribosyl-transferase in animals, plants and microorganisms. J Nutr Sci Vitaminol (Tokyo) 19: 491–499, 1973 [DOI] [PubMed] [Google Scholar]
- 163. Jackson TM, Rawling JM, Roebuck BD, and Kirkland JB. Large supplements of nicotinic acid and nicotinamide increase tissue NAD+ and poly(ADP-ribose) levels but do not affect diethylnitrosamine-induced altered hepatic foci in Fischer-344 rats. J Nutr 125: 1455–1461, 1995 [DOI] [PubMed] [Google Scholar]
- 164. Jacob RA, Swendseid ME, McKee RW, Fu CS, and Clemens RA. Biochemical markers for assessment of niacin status in young men: urinary and blood levels of niacin metabolites. J Nutr 119: 591–598, 1989 [DOI] [PubMed] [Google Scholar]
- 165. Jayaram HN, Kusumanchi P, and Yalowitz JA. NMNAT expression and its relation to NAD metabolism. Curr Med Chem 18: 1962–1972, 2011 [DOI] [PubMed] [Google Scholar]
- 166. Jin D, Liu HX, Hirai H, Torashima T, Nagai T, Lopatina O, Shnayder NA, Yamada K, Noda M, Seike T, Fujita K, Takasawa S, Yokoyama S, Koizumi K, Shiraishi Y, Tanaka S, Hashii M, Yoshihara T, Higashida K, Islam MS, Yamada N, Hayashi K, Noguchi N, Kato I, Okamoto H, Matsushima A, Salmina A, Munesue T, Shimizu N, Mochida S, Asano M, and Higashida H. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446: 41–45, 2007 [DOI] [PubMed] [Google Scholar]
- 167. Kaanders JH, Stratford MR, Liefers J, Dennis MF, van der Kogel AJ, van Daal WA, and Rojas A. Administration of nicotinamide during a five- to seven-week course of radiotherapy: pharmacokinetics, tolerance, and compliance. Radiother Oncol 43: 67–73, 1997 [DOI] [PubMed] [Google Scholar]
- 168. Kalikin L. and Calvo KC. Inhibition of quinolinate phosphoribosyl transferase by pyridine analogs of quinolinic acid. Biochem Biophys Res Commun 152: 559–564, 1988 [DOI] [PubMed] [Google Scholar]
- 169. Kamanna VS. and Kashyap ML. Mechanism of action of niacin on lipoprotein metabolism. Curr Atheroscler Rep 2: 36–46, 2000 [DOI] [PubMed] [Google Scholar]
- 170. Kamanna VS. and Kashyap ML. Nicotinic acid (niacin) receptor agonists: will they be useful therapeutic agents? Am J Cardiol 100: S53–S61, 2007 [DOI] [PubMed] [Google Scholar]
- 171. Kamanna VS. and Kashyap ML. Mechanism of action of niacin. Am J Cardiol 101: 20B–26B, 2008 [DOI] [PubMed] [Google Scholar]
- 172. Kang L. and Da Vanzo JP. Is the tryptophan load test a valid index for a chemically-induced vitamin B6 deficiency? Proc Soc Exp Biol Med 123: 340–343, 1966 [DOI] [PubMed] [Google Scholar]
- 173. Kang-Lee YA, McKee RW, Wright SM, Swendseid ME, Jenden DJ, and Jope RS. Metabolic effects of nicotinamide administration in rats. J Nutr 113: 215–221, 1983 [DOI] [PubMed] [Google Scholar]
- 174. Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Jr., Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, and Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Khan NA, Khan I, Abid SMA, Zaib S, Ibrar A, Andleeb H, Hameed S, and Iqbal J. Quinolinic carboxylic acid derivatives as potential multi-target compounds for neurodegeneration: monoamine oxidase and cholinesterase inhibition. Med Chem 14: 74–85, 2018 [DOI] [PubMed] [Google Scholar]
- 176. Khan S, Pandey SS, Jyotshna , Shanker K, Khan F, and Rahman LU. Cloning and functional characterization of quinolinic acid phosphoribosyl transferase (QPT) gene of Nicotiana tabacum. Physiol Plant 160: 253–265, 2017 [DOI] [PubMed] [Google Scholar]
- 177. Kimura N, Fukuwatari T, Sasaki R, and Shibata K. Comparison of metabolic fates of nicotinamide, NAD+ and NADH administered orally and intraperitoneally; characterization of oral NADH. J Nutr Sci Vitaminol (Tokyo) 52: 142–148, 2006 [DOI] [PubMed] [Google Scholar]
- 178. Kirkland JB. Niacin status and treatment-related leukemogenesis. Mol Cancer Ther 8: 725–732, 2009 [DOI] [PubMed] [Google Scholar]
- 179. Kirkland JB. Niacin status impacts chromatin structure. J Nutr 139: 2397–2401, 2009 [DOI] [PubMed] [Google Scholar]
- 180. Kirkland JB. Niacin requirements for genomic stability. Mutat Res 733: 14–20, 2012 [DOI] [PubMed] [Google Scholar]
- 181. Kirkland JB. Niacin status and genomic instability in bone marrow cells; mechanisms favoring the progression of leukemogenesis. Subcell Biochem 56: 21–36, 2012 [DOI] [PubMed] [Google Scholar]
- 182. Kitaoka Y, Munemasa Y, Kojima K, Hirano A, Ueno S, and Takagi H. Axonal protection by Nmnat3 overexpression with involvement of autophagy in optic nerve degeneration. Cell Death Dis 4: e860, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Kitay BM, McCormack R, Wang Y, Tsoulfas P, and Zhai RG. Mislocalization of neuronal mitochondria reveals regulation of Wallerian degeneration and NMNAT/WLD(S)-mediated axon protection independent of axonal mitochondria. Hum Mol Genet 22: 1601–1614, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Komrower GM, Wilson V, Clamp JR, and Westall RG. Hydroxykynureninuria: a case of abnormal tryptophan metabolism probably due to a deficiency of kynureninase. Arch Dis Child 39: 250–256, 1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, and Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321, 2002 [DOI] [PubMed] [Google Scholar]
- 186. Kourtzidis IA, Stoupas AT, Gioris IS, Veskoukis AS, Margaritelis NV, Tsantarliotou M, Taitzoglou I, Vrabas IS, Paschalis V, Kyparos A, and Nikolaidis MG. The NAD(+) precursor nicotinamide riboside decreases exercise performance in rats. J Int Soc Sports Nutr 13: 32, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Koyama T, Kume S, Koya D, Araki S, Isshiki K, Chin-Kanasaki M, Sugimoto T, Haneda M, Sugaya T, Kashiwagi A, Maegawa H, and Uzu T. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic Biol Med 51: 1258–1267, 2011 [DOI] [PubMed] [Google Scholar]
- 188. Krieger I. and Statter M. Tryptophan deficiency and picolinic acid: effect on zinc metabolism and clinical manifestations of pellagra. Am J Clin Nutr 46: 511–517, 1987 [DOI] [PubMed] [Google Scholar]
- 189. Krishnaswamy K. and Bapurao S. Effect of leucine at different levels of pyridoxine on hepatic quinolinate phosphoribosyl transferase (EC 2.4.2.19) and leucine aminotransferase (EC 2.6.1.6) in rats. Br J Nutr 39: 61–64, 1978 [DOI] [PubMed] [Google Scholar]
- 190. Kulikova V, Shabalin K, Nerinovski K, Dolle C, Niere M, Yakimov A, Redpath P, Khodorkovskiy M, Migaud ME, Ziegler M, and Nikiforov A. Generation, release, and uptake of the NAD precursor nicotinic acid riboside by human cells. J Biol Chem 290: 27124–27137, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kume S, Haneda M, Kanasaki K, Sugimoto T, Araki S, Isono M, Isshiki K, Uzu T, Kashiwagi A, and Koya D. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic Biol Med 40: 2175–2182, 2006 [DOI] [PubMed] [Google Scholar]
- 192. Lan F, Cacicedo JM, Ruderman N, and Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem 283: 27628–27635, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Lau C, Niere M, and Ziegler M. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front Biosci (Landmark Ed) 14: 410–431, 2009 [DOI] [PubMed] [Google Scholar]
- 194. Lee HJ, Hong YS, Jun W, and Yang SJ. Nicotinamide riboside ameliorates hepatic metaflammation by modulating NLRP3 inflammasome in a rodent model of type 2 diabetes. J Med Food 18: 1207–1213, 2015 [DOI] [PubMed] [Google Scholar]
- 195. Lee MC, Ting KK, Adams S, Brew BJ, Chung R, and Guillemin GJ. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One 5: e14123, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lehmann S, Costa AC, Celardo I, Loh SH, and Martins LM. PARP mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson's disease. Cell Death Dis 7: e2166, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Li D, Tian YJ, Guo J, Sun WP, Lun YZ, Guo M, Luo N, Cao Y, Cao JM, Gong XJ, and Zhou SS. Nicotinamide supplementation induces detrimental metabolic and epigenetic changes in developing rats. Br J Nutr 110: 2156–2164, 2013 [DOI] [PubMed] [Google Scholar]
- 198. Li F, He X, Ye D, Lin Y, Yu H, Yao C, Huang L, Zhang J, Wang F, Xu S, Wu X, Liu L, Yang C, Shi J, He X, Liu J, Qu Y, Guo F, Zhao J, Xu W, and Zhao S. NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Mol Cell 60: 661–675, 2015 [DOI] [PubMed] [Google Scholar]
- 199. Li X, Zhang S, Blander G, Tse JG, Krieger M, and Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28: 91–106, 2007 [DOI] [PubMed] [Google Scholar]
- 200. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, and Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38: 864–878, 2010 [DOI] [PubMed] [Google Scholar]
- 201. Lischke T, Heesch K, Schumacher V, Schneider M, Haag F, Koch-Nolte F, and Mittrucker HW. CD38 controls the innate immune response against Listeria monocytogenes. Infect Immun 81: 4091–4099, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Liu B, Che W, Xue J, Zheng C, Tang K, Zhang J, Wen J, and Xu Y. SIRT4 prevents hypoxia-induced apoptosis in H9c2 cardiomyoblast cells. Cell Physiol Biochem 32: 655–662, 2013 [DOI] [PubMed] [Google Scholar]
- 203. Liu D, Gharavi R, Pitta M, Gleichmann M, and Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med 11: 28–42, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Liu ZX, Azhipa O, Okamoto S, Govindarajan S, and Dennert G. Extracellular nicotinamide adenine dinucleotide induces T cell apoptosis in vivo and in vitro. J Immunol 167: 4942–4947, 2001 [DOI] [PubMed] [Google Scholar]
- 205. Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, Yang Y, Chen Y, Hirschey MD, Bronson RT, Haigis M, Guarente LP, Farese RV, Jr., Weissman S, Verdin E, and Schwer B. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 27: 8807–8814, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, and Kroemer G. The hallmarks of aging. Cell 153: 1194–1217, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B, and Chen Y. PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal 13: 1011–1022, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, and Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107: 137–148, 2001 [DOI] [PubMed] [Google Scholar]
- 209. Luo YX, Tang X, An XZ, Xie XM, Chen XF, Zhao X, Hao DL, Chen HZ, and Liu DP. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J 38: 1389–1398, 2017 [DOI] [PubMed] [Google Scholar]
- 210. Lynn EG, McLeod CJ, Gordon JP, Bao JJ, and Sack MN. SIRT2 is a negative regulator of anoxia-reoxygenation tolerance via regulation of 14-3-3 zeta and BAD in H9c2 cells. FEBS Lett 582: 2857–2862, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, and Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4: 1199–1206, 2001 [DOI] [PubMed] [Google Scholar]
- 212. Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, and Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci 61: 19–34, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Magni G, Amici A, Emanuelli M, Raffaelli N, and Ruggieri S. Enzymology of NAD+ synthesis. Adv Enzymol Relat Areas Mol Biol 73: 135–182, xi, 1999 [DOI] [PubMed] [Google Scholar]
- 214. Magni G, Amici A, and Orsomando G. The enzymology of cytosolic pyrimidine 5′-nucleotidases: functional analysis and physiopathological implications. Curr Med Chem 20: 4304–4316, 2013 [DOI] [PubMed] [Google Scholar]
- 215. Maier KG. Nicotinamide adenine dinucleotide phosphate oxidase and diabetes: vascular implications. Vasc Endovascular Surg 42: 305–313, 2008 [DOI] [PubMed] [Google Scholar]
- 216. Majmundar AJ, Wong WJ, and Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 40: 294–309, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Maltos AL, Portari GV, Moraes GV, Monteiro MC, Vannucchi H, and da Cunha DF. Niacin metabolism and indoleamine 2,3-dioxygenase activation in malnourished patients with flaky paint dermatosis. Nutrition 31: 890–892, 2015 [DOI] [PubMed] [Google Scholar]
- 218. Mansour H, McColm JR, Cole L, Weible M, 2nd, Korlimbinis A, and Chan-Ling T. Connexin 30 expression and frequency of connexin heterogeneity in astrocyte gap junction plaques increase with age in the rat retina. PLoS One 8: e57038, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Marohnic CC, Bewley MC, and Barber MJ. Engineering and characterization of a NADPH-utilizing cytochrome b5 reductase. Biochemistry 42: 11170–11182, 2003 [DOI] [PubMed] [Google Scholar]
- 220. Martire S, Fuso A, Mosca L, Forte E, Correani V, Fontana M, Scarpa S, Maras B, and d'Erme M. Bioenergetic impairment in animal and cellular models of Alzheimer's disease: PARP-1 inhibition rescues metabolic dysfunctions. J Alzheimers Dis 54: 307–324, 2016 [DOI] [PubMed] [Google Scholar]
- 221. Martire S, Mosca L, and d'Erme M. PARP-1 involvement in neurodegeneration: a focus on Alzheimer's and Parkinson's diseases. Mech Ageing Dev 146–148: 53–64, 2015 [DOI] [PubMed] [Google Scholar]
- 222. Marzetti E, Wohlgemuth SE, Aulisa AG, Bernabei R, Pahor M, and Leeuwenburgh C. Calorie restriction for optimal cardiovascular aging: the weight of evidence. Curr Cardiovasc Risk Rep 4: 340–346, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, and Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7: e42357, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Massudi H, Grant R, Guillemin GJ, and Braidy N. NAD+ metabolism and oxidative stress: the golden nucleotide on a crown of thorns. Redox Rep 17: 28–46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, and Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 104: 14855–14860, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, Roth GS, Ingram DK, Weindruch R, de Cabo R, and Anderson RM. Caloric restriction improves health and survival of rhesus monkeys. Nat Commun 8: 14063, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, Barnard D, Ward WF, Qi W, Ingram DK, and de Cabo R. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 489: 318–321, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. McClure JM, Wierman MB, Maqani N, and Smith JS. Isonicotinamide enhances Sir2 protein-mediated silencing and longevity in yeast by raising intracellular NAD+ concentration. J Biol Chem 287: 20957–20966, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. McLin JP, Thompson LM, and Steward O. Differential susceptibility to striatal neurodegeneration induced by quinolinic acid and kainate in inbred, outbred and hybrid mouse strains. Eur J Neurosci 24: 3134–3140, 2006 [DOI] [PubMed] [Google Scholar]
- 230. Mehler AH. Formation of picolinic and quinolinic acids following enzymatic oxidation of 3-hydroxyanthranilic acid. J Biol Chem 218: 241–254, 1956 [PubMed] [Google Scholar]
- 231. Menon RM, Adams MH, Gonzalez MA, Tolbert DS, Leu JH, and Cefali EA. Plasma and urine pharmacokinetics of niacin and its metabolites from an extended-release niacin formulation. Int J Clin Pharmacol Ther 45: 448–454, 2007 [DOI] [PubMed] [Google Scholar]
- 232. Menon RM, Gonzalez MA, Adams MH, Tolbert DS, Leu JH, and Cefali EA. Effect of the rate of niacin administration on the plasma and urine pharmacokinetics of niacin and its metabolites. J Clin Pharmacol 47: 681–688, 2007 [DOI] [PubMed] [Google Scholar]
- 233. Mesripour A, Hajhashemi V, and Kuchak A. Effect of concomitant administration of three different antidepressants with vitamin B6 on depression and obsessive compulsive disorder in mice models. Res Pharm Sci 12: 46–52, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Monfrecola G, Gaudiello F, Cirillo T, Fabbrocini G, Balato A, and Lembo S. Nicotinamide downregulates gene expression of interleukin-6, interleukin-10, monocyte chemoattractant protein-1, and tumour necrosis factor-alpha gene expression in HaCaT keratinocytes after ultraviolet B irradiation. Clin Exp Dermatol 38: 185–188, 2013 [DOI] [PubMed] [Google Scholar]
- 235. Moresco RM, Lavazza T, Belloli S, Lecchi M, Pezzola A, Todde S, Matarrese M, Carpinelli A, Turolla E, Zimarino V, Popoli P, Malgaroli A, and Fazio F. Quinolinic acid induced neurodegeneration in the striatum: a combined in vivo and in vitro analysis of receptor changes and microglia activation. Eur J Nucl Med Mol Imaging 35: 704–715, 2008 [DOI] [PubMed] [Google Scholar]
- 236. Mori V, Amici A, Mazzola F, Di Stefano M, Conforti L, Magni G, Ruggieri S, Raffaelli N, and Orsomando G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS One 9: e113939, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Morigi M, Perico L, Rota C, Longaretti L, Conti S, Rottoli D, Novelli R, Remuzzi G, and Benigni A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J Clin Invest 125: 715–726, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Moroni F, Carpenedo R, and Chiarugi A. Kynurenine hydroxylase and kynureninase inhibitors as tools to study the role of kynurenine metabolites in the central nervous system. Adv Exp Med Biol 398: 203–210, 1996 [DOI] [PubMed] [Google Scholar]
- 239. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills KD, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, and Alt FW. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124: 315–329, 2006 [DOI] [PubMed] [Google Scholar]
- 240. Myint AM. and Kim YK. Network beyond IDO in psychiatric disorders: revisiting neurodegeneration hypothesis. Prog Neuropsychopharmacol Biol Psychiatry 48: 304–313, 2014 [DOI] [PubMed] [Google Scholar]
- 241. Nahalkova J. Novel protein-protein interactions of TPPII, p53, and SIRT7. Mol Cell Biochem 409: 13–22, 2015 [DOI] [PubMed] [Google Scholar]
- 242. Nakagawa T, Lomb DJ, Haigis MC, and Guarente L. SIRT5 deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137: 560–570, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Nakahata Y. and Bessho Y. The circadian NAD+ metabolism: impact on chromatin remodeling and aging. Biomed Res Int 2016: 3208429, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Nakamichi R, Miranda EP, Lobo SM, Tristao VR, Dalboni MA, Quinto BM, and Batista MC. Action of nicotinic acid on the reversion of hypoxic-inflammatory link on 3T3-L1 adipocytes. Lipids Health Dis 15: 91, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Nematollahi A, Sun G, Jayawickrama GS, and Church WB. Kynurenine aminotransferase isozyme inhibitors: a review. Int J Mol Sci 17: 946, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Nemoto S, Fergusson MM, and Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem 280: 16456–16460, 2005 [DOI] [PubMed] [Google Scholar]
- 247. Niere M, Kernstock S, Koch-Nolte F, and Ziegler M. Functional localization of two poly(ADP-ribose)-degrading enzymes to the mitochondrial matrix. Mol Cell Biol 28: 814–824, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW, and Verdin E. SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell 59: 321–332, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, Horvath TL, Sinclair DA, Pfluger PT, and Tschop MH. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev 92: 1479–1514, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. O'Neill AB, Morgan SJ, and Brioni JD. Histological and behavioral protection by (-)-nicotine against quinolinic acid-induced neurodegeneration in the hippocampus. Neurobiol Learn Mem 69: 46–64, 1998 [DOI] [PubMed] [Google Scholar]
- 251. Oblong JE. The evolving role of the NAD+/nicotinamide metabolome in skin homeostasis, cellular bioenergetics, and aging. DNA Repair (Amst) 23: 59–63, 2014 [DOI] [PubMed] [Google Scholar]
- 252. Obrenovitch TP. Quinolinic acid accumulation during neuroinflammation. Does it imply excitotoxicity? Ann N Y Acad Sci 939: 1–10, 2001 [DOI] [PubMed] [Google Scholar]
- 253. Obrenovitch TP. and Urenjak J. Accumulation of quinolinic acid with neuroinflammation: does it mean excitotoxicity? Adv Exp Med Biol 527: 147–154, 2003 [DOI] [PubMed] [Google Scholar]
- 254. Ohashi K, Kawai S, and Murata K. Secretion of quinolinic acid, an intermediate in the kynurenine pathway, for utilization in NAD+ biosynthesis in the yeast Saccharomyces cerevisiae. Eukaryot Cell 12: 648–653, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Oliver E. Diets of cereals & other vegetable proteins & cutaneous pellegra in our country [in Spanish]. Rev Esp Enferm Apar Dig Nutr 17: 68–73, 1958 [PubMed] [Google Scholar]
- 256. Olvera-Garcia G, Espinosa E, Sieg SF, and Lederman MM. Cytomegalovirus-specific responses of CD38(+) memory T cells are skewed towards IFN-gamma and dissociated from CD154 in HIV-1 infection. AIDS 28: 311–316, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Palcic MM, Antoun M, Tanizawa K, Soda K, and Floss HG. Stereochemistry of the kynureninase reaction. J Biol Chem 260: 5248–5251, 1985 [PubMed] [Google Scholar]
- 258. Penberthy WT. Nicotinic acid-mediated activation of both membrane and nuclear receptors towards therapeutic glucocorticoid mimetics for treating multiple sclerosis. PPAR Res 2009: 853707, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Penberthy WT. and Tsunoda I. The importance of NAD in multiple sclerosis. Curr Pharm Des 15: 64–99, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Peng Q, Jia SH, Parodo J, Ai Y, and Marshall JC. Pre-B cell colony enhancing factor induces Nampt-dependent translocation of the insulin receptor out of lipid microdomains in A549 lung epithelial cells. Am J Physiol Endocrinol Metab 308: E324–E333, 2015 [DOI] [PubMed] [Google Scholar]
- 261. Peters JC. Tryptophan nutrition and metabolism: an overview. Adv Exp Med Biol 294: 345–358, 1991 [DOI] [PubMed] [Google Scholar]
- 262. Phillips RS. Structure and mechanism of kynureninase. Arch Biochem Biophys 544: 69–74, 2014 [DOI] [PubMed] [Google Scholar]
- 263. Pissios P. Nicotinamide N-methyltransferase: more than a vitamin B3 clearance enzyme. Trends Endocrinol Metab 28: 340–353, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Puri BK, Hirsch SR, Easton T, and Richardson AJ. A volumetric biochemical niacin flush-based index that noninvasively detects fatty acid deficiency in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 26: 49–52, 2002 [DOI] [PubMed] [Google Scholar]
- 265. Qin W, Yang T, Ho L, Zhao Z, Wang J, Chen L, Zhao W, Thiyagarajan M, MacGrogan D, Rodgers JT, Puigserver P, Sadoshima J, Deng H, Pedrini S, Gandy S, Sauve AA, and Pasinetti GM. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J Biol Chem 281: 21745–21754, 2006 [DOI] [PubMed] [Google Scholar]
- 266. Qiu X, Brown K, Hirschey MD, Verdin E, and Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 12: 662–667, 2010 [DOI] [PubMed] [Google Scholar]
- 267. Rahman A, Ting K, Cullen KM, Braidy N, Brew BJ, and Guillemin GJ. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS One 4: e6344, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Rajakumar SV, Lu B, Crikis S, Robson SC, d'Apice AJ, Cowan PJ, and Dwyer KM. Deficiency or inhibition of CD73 protects in mild kidney ischemia-reperfusion injury. Transplantation 90: 1260–1264, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, Hottiger MO, and Gupta MP. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol 29: 4116–4129, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Ramautar A, Mabandla M, Blackburn J, and Daniels WM. Inhibition of HIV-1 tat-induced transactivation and apoptosis by the divalent metal chelators, fusaric acid and picolinic acid-implications for HIV-1 dementia. Neurosci Res 74: 59–63, 2012 [DOI] [PubMed] [Google Scholar]
- 271. Rankin PW, Jacobson EL, Benjamin RC, Moss J, and Jacobson MK. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J Biol Chem 264: 4312–4317, 1989 [PubMed] [Google Scholar]
- 272. Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, Auwerx J, Yanes O, Brenner C, and Canto C. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun 7: 13103, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Rawling JM, ApSimon MM, and Kirkland JB. Lung poly(ADP-ribose) and NAD+ concentrations during hyperoxia and niacin deficiency in the Fischer-344 rat. Free Radic Biol Med 20: 865–871, 1996 [DOI] [PubMed] [Google Scholar]
- 274. Rawling JM, Jackson TM, Driscoll ER, and Kirkland JB. Dietary niacin deficiency lowers tissue poly(ADP-ribose) and NAD+ concentrations in Fischer-344 rats. J Nutr 124: 1597–1603, 1994 [DOI] [PubMed] [Google Scholar]
- 275. Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, and Imai S. Nampt/PBEF/visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 6: 363–375, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, and Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1 alpha and SIRT1. Nature 434: 113–118, 2005 [DOI] [PubMed] [Google Scholar]
- 277. This reference has been deleted.
- 278. Roessler C, Tuting C, Meleshin M, Steegborn C, and Schutkowski M. A Novel continuous assay for the deacylase sirtuin 5 and other deacetylases. J Med Chem 58: 7217–7223, 2015 [DOI] [PubMed] [Google Scholar]
- 279. Rojas A, Stratford MR, Bentzen SM, and Denekamp J. Is sensitization with nicotinamide and carbogen dependent on nicotinamide concentration at the time of irradiation? Int J Radiat Biol 80: 499–506, 2004 [DOI] [PubMed] [Google Scholar]
- 280. Romashkan SV, Das SK, Villareal DT, Ravussin E, Redman LM, Rochon J, Bhapkar M, and Kraus WE; CALERIE Study Group. Safety of two-year caloric restriction in non-obese healthy individuals. Oncotarget 7: 19124–19133, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Rongvaux A, Shea RJ, Mulks MH, Gigot D, Urbain J, Leo O, and Andris F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur J Immunol 32: 3225–3234, 2002 [DOI] [PubMed] [Google Scholar]
- 282. Rothgiesser KM, Erener S, Waibel S, Luscher B, and Hottiger MO. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci 123: 4251–4258, 2010 [DOI] [PubMed] [Google Scholar]
- 283. Ruddock MW. and Hirst DG. Nicotinamide relaxes vascular smooth muscle by inhibiting myosin light chain kinase-dependent signaling pathways: implications for anticancer efficacy. Oncol Res 14: 483–489, 2004 [DOI] [PubMed] [Google Scholar]
- 284. Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, and Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med 11: 1306–1313, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Sakiyama Y. Purine nucleoside phosphorylase deficiency [in Japanese]. Ryoikibetsu Shokogun Shirizu 32: 43–45, 2000 [PubMed] [Google Scholar]
- 286. Sanchez WY, Obispo C, Ryan E, Grice JE, and Roberts MS. Changes in the redox state and endogenous fluorescence of in vivo human skin due to intrinsic and photo-aging, measured by multiphoton tomography with fluorescence lifetime imaging. J Biomed Opt 18: 061217, 2013 [DOI] [PubMed] [Google Scholar]
- 287. Sartini D, Santarelli A, Rossi V, Goteri G, Rubini C, Ciavarella D, Lo Muzio L, and Emanuelli M. Nicotinamide N-methyltransferase upregulation inversely correlates with lymph node metastasis in oral squamous cell carcinoma. Mol Med 13: 415–421, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Sasaki Y, Araki T, and Milbrandt J. Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J Neurosci 26: 8484–8491, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Sasaki Y, Vohra BP, Lund FE, and Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci 29: 5525–5535, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, and Imai S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 18: 416–430, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Sauer AV, Brigida I, Carriglio N, and Aiuti A. Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency. Front Immunol 3: 265, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Sauve AA, Moir RD, Schramm VL, and Willis IM. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol Cell 17: 595–601, 2005 [DOI] [PubMed] [Google Scholar]
- 293. Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, Mangerich A, Wilson MA, Mattson MP, Bergersen LH, Cogger VC, Warren A, Le Couteur DG, Moaddel R, Wilson DM, 3rd, Croteau DL, de Cabo R, and Bohr VA. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 20: 840–855, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Schlicker C, Gertz M, Papatheodorou P, Kachholz B, Becker CF, and Steegborn C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J Mol Biol 382: 790–801, 2008 [DOI] [PubMed] [Google Scholar]
- 295. Schreiber V, Dantzer F, Ame JC, and de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 7: 517–528, 2006 [DOI] [PubMed] [Google Scholar]
- 296. Schweiger M, Hennig K, Lerner F, Niere M, Hirsch-Kauffmann M, Specht T, Weise C, Oei SL, and Ziegler M. Characterization of recombinant human nicotinamide mononucleotide adenylyl transferase (NMNAT), a nuclear enzyme essential for NAD synthesis. FEBS Lett 492: 95–100, 2001 [DOI] [PubMed] [Google Scholar]
- 297. Seman M, Adriouch S, Scheuplein F, Krebs C, Freese D, Glowacki G, Deterre P, Haag F, and Koch-Nolte F. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity 19: 571–582, 2003 [DOI] [PubMed] [Google Scholar]
- 298. Semba RD. The discovery of the vitamins. Int J Vitam Nutr Res 82: 310–315, 2012 [DOI] [PubMed] [Google Scholar]
- 299. Semba RD. The historical evolution of thought regarding multiple micronutrient nutrition. J Nutr 142: 143S–156S, 2012 [DOI] [PubMed] [Google Scholar]
- 300. Sharma A, Diecke S, Zhang WY, Lan F, He C, Mordwinkin NM, Chua KF, and Wu JC. The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. J Biol Chem 288: 18439–18447, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Shetty PK, Galeffi F, and Turner DA. Nicotinamide pre-treatment ameliorates NAD(H) hyperoxidation and improves neuronal function after severe hypoxia. Neurobiol Dis 62: 469–478, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Shi H, Sun N, Mayevsky A, Zhang Z, and Luo Q. Preclinical evidence of mitochondrial nicotinamide adenine dinucleotide as an effective alarm parameter under hypoxia. J Biomed Opt 19: 17005, 2014 [DOI] [PubMed] [Google Scholar]
- 303. Shi W, Hegeman MA, van Dartel DA, Tang J, Suarez M, Swarts H, van der Hee B, Arola L, and Keijer J. Effects of a wide range of dietary nicotinamide riboside (NR) concentrations on metabolic flexibility and white adipose tissue (WAT) of mice fed a mildly obesogenic diet. Mol Nutr Food Res 61: 1600878, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Shibata K. True niacin deficiency in quinolinic acid phosphoribosyltransferase (QPRT) knockout mice. J Nutr Sci Vitaminol (Tokyo) 61(Suppl): S145–S147, 2015 [DOI] [PubMed] [Google Scholar]
- 305. Shibata K. and Fukuwatari T. Large amounts of picolinic acid are lethal but small amounts increase the conversion of tryptophan-nicotinamide in rats. J Nutr Sci Vitaminol (Tokyo) 60: 334–339, 2014 [DOI] [PubMed] [Google Scholar]
- 306. Shibata K, Mushiage M, Kondo T, Hayakawa T, and Tsuge H. Effects of vitamin B6 deficiency on the conversion ratio of tryptophan to niacin. Biosci Biotechnol Biochem 59: 2060–2063, 1995 [DOI] [PubMed] [Google Scholar]
- 307. Shibata K, Ohno T, Sano M, and Fukuwatari T. The urinary ratio of 3-hydroxykynurenine/3-hydroxyanthranilic acid is an index to predicting the adverse effects of d-tryptophan in rats. J Nutr Sci Vitaminol (Tokyo) 60: 261–268, 2014 [DOI] [PubMed] [Google Scholar]
- 308. Siegel MP, Kruse SE, Knowels G, Salmon A, Beyer R, Xie H, Van Remmen H, Smith SR, and Marcinek DJ. Reduced coupling of oxidative phosphorylation in vivo precedes electron transport chain defects due to mild oxidative stress in mice. PLoS One 6: e26963, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Sinclair DA. and Guarente L. Extrachromosomal rDNA circles—a cause of aging in yeast. Cell 91: 1033–1042, 1997 [DOI] [PubMed] [Google Scholar]
- 310. Sociali G, Raffaghello L, Magnone M, Zamporlini F, Emionite L, Sturla L, Bianchi G, Vigliarolo T, Nahimana A, Nencioni A, Raffaelli N, and Bruzzone S. Antitumor effect of combined NAMPT and CD73 inhibition in an ovarian cancer model. Oncotarget 7: 2968–2984, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, Tanokura M, Denu JM, and Prolla TA. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143: 802–812, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Song J, Keppler BD, Wise RR, and Bent AF. PARP2 is the predominant poly(ADP-ribose) polymerase in Arabidopsis DNA damage and immune responses. PLoS Genet 11: e1005200, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Sono M. Enzyme kinetic and spectroscopic studies of inhibitor and effector interactions with indoleamine 2,3-dioxygenase. 2. Evidence for the existence of another binding site in the enzyme for indole derivative effectors. Biochemistry 28: 5400–5407, 1989 [DOI] [PubMed] [Google Scholar]
- 314. Soudijn W, van Wijngaarden I, and Ijzerman AP. Nicotinic acid receptor subtypes and their ligands. Med Res Rev 27: 417–433, 2007 [DOI] [PubMed] [Google Scholar]
- 314a. Southam CM. and Ehrlich J. Effects of extracts of western red-cedar heartwood on certain wood-decaying fungi in culture. Phytopathology 33: 517–524, 1943 [Google Scholar]
- 315. Spaans SK, Weusthuis RA, van der Oost J, and Kengen SW. NADPH-generating systems in bacteria and archaea. Front Microbiol 6: 742, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Spinnler R, Gorski T, Stolz K, Schuster S, Garten A, Beck-Sickinger AG, Engelse MA, de Koning EJ, Korner A, Kiess W, and Maedler K. The adipocytokine Nampt and its product NMN have no effect on beta-cell survival but potentiate glucose stimulated insulin secretion. PLoS One 8: e54106, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Stratford MR, Dennis MF, Hoskin P, Phillips H, Hodgkiss RJ, and Rojas A. Nicotinamide pharmacokinetics in humans: effect of gastric acid inhibition, comparison of rectal vs oral administration and the use of saliva for drug monitoring. Br J Cancer 74: 16–21, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Sun S, Cheng S, Zhu Y, Zhang P, Liu N, Xu T, Sun C, and Lv Y. Identification of PRKDC (protein kinase, DNA-activated, catalytic polypeptide) as an essential gene for colorectal cancer (CRCs) cells. Gene 584: 90–96, 2016 [DOI] [PubMed] [Google Scholar]
- 319. Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, and Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest 119: 2758–2771, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Surjana D, Halliday GM, and Damian DL. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J Nucleic Acids 2010: 157591, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Surjana D, Halliday GM, Martin AJ, Moloney FJ, and Damian DL. Oral nicotinamide reduces actinic keratoses in phase II double-blinded randomized controlled trials. J Invest Dermatol 132: 1497–1500, 2012 [DOI] [PubMed] [Google Scholar]
- 322. Suzuki K. and Koike T. Mammalian Sir2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: a crucial role of tubulin deacetylation. Neuroscience 147: 599–612, 2007 [DOI] [PubMed] [Google Scholar]
- 323. Szkudelski T, Zywert A, and Szkudelska K. Metabolic disturbances and defects in insulin secretion in rats with streptozotocin-nicotinamide-induced diabetes. Physiol Res 62: 663–670, 2013 [DOI] [PubMed] [Google Scholar]
- 324. Takahashi R. Anti-aging studies on the senescence accelerated mouse (SAM) strains [in Japanese]. Yakugaku Zasshi 130: 11–18, 2010 [DOI] [PubMed] [Google Scholar]
- 325. Tang K, Sham H, Hui E, and Kirkland JB. Niacin deficiency causes oxidative stress in rat bone marrow cells but not through decreased NADPH or glutathione status. J Nutr Biochem 19: 746–753, 2008 [DOI] [PubMed] [Google Scholar]
- 326. Tang KS, Suh SW, Alano CC, Shao Z, Hunt WT, Swanson RA, and Anderson CM. Astrocytic poly(ADP-ribose) polymerase-1 activation leads to bioenergetic depletion and inhibition of glutamate uptake capacity. Glia 58: 446–457, 2010 [DOI] [PubMed] [Google Scholar]
- 327. Taniguchi A. PNP (purine nucleoside phosphorylase) deficiency [in Japanese]. Nihon Rinsho 61(Suppl 1): 372–376, 2003 [PubMed] [Google Scholar]
- 328. Tanno O, Ota Y, Kitamura N, Katsube T, and Inoue S. Nicotinamide increases biosynthesis of ceramides as well as other stratum corneum lipids to improve the epidermal permeability barrier. Br J Dermatol 143: 524–531, 2000 [DOI] [PubMed] [Google Scholar]
- 329. Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, and Gius D. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40: 893–904, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Tao R, Kim SH, Honbo N, Karliner JS, and Alano CC. Minocycline protects cardiac myocytes against simulated ischemia-reperfusion injury by inhibiting poly(ADP-ribose) polymerase-1. J Cardiovasc Pharmacol 56: 659–668, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Tempel W, Rabeh WM, Bogan KL, Belenky P, Wojcik M, Seidle HF, Nedyalkova L, Yang T, Sauve AA, Park HW, and Brenner C. Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLoS Biol 5: e263, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Terada N, Kinoshita K, Taguchi S, and Tokuda Y. Wernicke encephalopathy and pellagra in an alcoholic and malnourished patient. BMJ Case Rep 2015: bcr2015209412, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Ting KK, Brew BJ, and Guillemin GJ. Effect of quinolinic acid on human astrocytes morphology and functions: implications in Alzheimer's disease. J Neuroinflammation 6: 36, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Tong DL, Zhang DX, Xiang F, Teng M, Jiang XP, Hou JM, Zhang Q, and Huang YS. Nicotinamide pretreatment protects cardiomyocytes against hypoxia-induced cell death by improving mitochondrial stress. Pharmacology 90: 11–18, 2012 [DOI] [PubMed] [Google Scholar]
- 335. Trammell SA. and Brenner C. Targeted, LCMS-based metabolomics for quantitative measurement of NAD(+) metabolites. Comput Struct Biotechnol J 4: e201301012, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Trammell SA. and Brenner C. NNMT: a bad actor in fat makes good in liver. Cell Metab 22: 200–201, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, and Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun 7: 12948, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Trammell SA, Yu L, Redpath P, Migaud ME, and Brenner C. Nicotinamide riboside is a major NAD+ precursor vitamin in cow milk. J Nutr 146: 957–963, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, and Parikh SM. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531: 528–532, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Tummala KS, Gomes AL, Yilmaz M, Grana O, Bakiri L, Ruppen I, Ximenez-Embun P, Sheshappanavar V, Rodriguez-Justo M, Pisano DG, Wagner EF, and Djouder N. Inhibition of de novo NAD(+) synthesis by oncogenic URI causes liver tumorigenesis through DNA damage. Cancer Cell 26: 826–839, 2014 [DOI] [PubMed] [Google Scholar]
- 341. Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, and Yalcin A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Abeta(1–42)-induced rat model of Alzheimer's disease. Free Radic Res 48: 146–158, 2014 [DOI] [PubMed] [Google Scholar]
- 342. Ugur S, Ulu R, Dogukan A, Gurel A, Yigit IP, Gozel N, Aygen B, and Ilhan N. The renoprotective effect of curcumin in cisplatin-induced nephrotoxicity. Ren Fail 37: 332–336, 2015 [DOI] [PubMed] [Google Scholar]
- 343. Vaccari CS, Nagamia S, Thoenes M, Oguchi A, Hammoud R, and Khan BV. Efficacy of controlled-release niacin in treatment of metabolic syndrome: correlation to surrogate markers of atherosclerosis, vascular reactivity, and inflammation. J Clin Lipidol 1: 605–613, 2007 [DOI] [PubMed] [Google Scholar]
- 344. Vakhrusheva O, Smolka C, Gajawada P, Kostin S, Boettger T, Kubin T, Braun T, and Bober E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ Res 102: 703–710, 2008 [DOI] [PubMed] [Google Scholar]
- 345. van der Hoorn JW, de Haan W, Berbee JF, Havekes LM, Jukema JW, Rensen PC, and Princen HM. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol 28: 2016–2022, 2008 [DOI] [PubMed] [Google Scholar]
- 346. Vaquero A, Sternglanz R, and Reinberg D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 26: 5505–5520, 2007 [DOI] [PubMed] [Google Scholar]
- 347. Vazquez BN, Thackray JK, and Serrano L. Sirtuins and DNA damage repair: SIRT7 comes to play. Nucleus 8: 107–115, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science 350: 1208–1213, 2015 [DOI] [PubMed] [Google Scholar]
- 349. Vohra BP, Sasaki Y, Miller BR, Chang J, DiAntonio A, and Milbrandt J. Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J Neurosci 30: 13729–13738, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Wang F, Nguyen M, Qin FX, and Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 6: 505–514, 2007 [DOI] [PubMed] [Google Scholar]
- 351. Wang F. and Zhang WP. Research progress on nicotinamide phosphoribosyl transferase involved in aging and age-related diseases [in Chinese]. Zhejiang Da Xue Xue Bao Yi Xue Ban 40: 680–684, 2011 [DOI] [PubMed] [Google Scholar]
- 352. Wang H, Liang X, Luo G, Ding M, and Liang Q. Protection effect of nicotinamide on cardiomyoblast hypoxia/re-oxygenation injury: study of cellular mitochondrial metabolism. Mol Biosyst 12: 2257–2264, 2016 [DOI] [PubMed] [Google Scholar]
- 353. Wang P, Du H, Zhang RY, Guan YF, Xu TY, Xu QY, Su DF, and Miao CY. Circulating and local visfatin/Nampt/PBEF levels in spontaneously hypertensive rats, stroke-prone spontaneously hypertensive rats and Wistar-Kyoto rats. J Physiol Sci 60: 317–324, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Wang P. and Miao CY. NAMPT as a therapeutic target against stroke. Trends Pharmacol Sci 36: 891–905, 2015 [DOI] [PubMed] [Google Scholar]
- 355. Wang P, Xu TY, Guan YF, Su DF, Fan GR, and Miao CY. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res 81: 370–380, 2009 [DOI] [PubMed] [Google Scholar]
- 356. Wang X, Hu X, Yang Y, Takata T, and Sakurai T. Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res 1643: 1–9, 2016 [DOI] [PubMed] [Google Scholar]
- 357. Warburg O. and Christian W. Pyridine, the hydrogen transferring element of fermentation enzymes (Pyridine-nucleotide.). Biochemische Zeitschrift 287: 291–328, 1936 [Google Scholar]
- 358. Wardman P. Hypoxic cell radiosensitizers—chemical aspects and in vitro models. Strahlentherapie Sonderb 75: 20–26, 1978 [PubMed] [Google Scholar]
- 359. Wardman P. Chemical radiosensitizers for use in radiotherapy. Clin Oncol (R Coll Radiol) 19: 397–417, 2007 [DOI] [PubMed] [Google Scholar]
- 360. Wei CC, Kong YY, Li GQ, Guan YF, Wang P, and Miao CY. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci Rep 7: 717, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, and Longo VD. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet 4: e13, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Weidele K, Beneke S, and Burkle A. The NAD+ precursor nicotinic acid improves genomic integrity in human peripheral blood mononuclear cells after X-irradiation. DNA Repair (Amst) 52: 12–23, 2017 [DOI] [PubMed] [Google Scholar]
- 363. Whitacre CM, Hashimoto H, Tsai ML, Chatterjee S, Berger SJ, and Berger NA. Involvement of NAD-poly(ADP-ribose) metabolism in p53 regulation and its consequences. Cancer Res 55: 3697–3701, 1995 [PubMed] [Google Scholar]
- 364. Wiley CA. Quinolinic acid and neurodegeneration in AIDS. J Neurovirol 1: 328–329, 1995 [DOI] [PubMed] [Google Scholar]
- 365. Williams AC, Hill LJ, and Ramsden DB. Nicotinamide, NAD(P)(H), and methyl-group homeostasis evolved and became a determinant of ageing diseases: hypotheses and lessons from pellagra. Curr Gerontol Geriatr Res 2012: 302875, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Williams AC. and Ramsden DB. Autotoxicity, methylation and a road to the prevention of Parkinson's disease. J Clin Neurosci 12: 6–11, 2005 [DOI] [PubMed] [Google Scholar]
- 367. Wojcik M, Seidle HF, Bieganowski P, and Brenner C. Glutamine-dependent NAD+ synthetase. How a two-domain, three-substrate enzyme avoids waste. J Biol Chem 281: 33395–33402, 2006 [DOI] [PubMed] [Google Scholar]
- 368. Wood JD. and Peesker SJ. The effect on GABA metabolism in brain of isonicotinic acid hydrazide and pyridoxine as a fuction of time after administration. J Neurochem 19: 1527–1537, 1972 [DOI] [PubMed] [Google Scholar]
- 369. Wu W, Nicolazzo JA, Wen L, Chung R, Stankovic R, Bao SS, Lim CK, Brew BJ, Cullen KM, and Guillemin GJ. Expression of tryptophan 2,3-dioxygenase and production of kynurenine pathway metabolites in triple transgenic mice and human Alzheimer's disease brain. PLoS One 8: e59749, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Yang H, Zuo XZ, Tian C, He DL, Yi WJ, Chen Z, Zhang PW, Ding SB, and Ying CJ. Green tea polyphenols attenuate high-fat diet-induced renal oxidative stress through SIRT3-dependent deacetylation. Biomed Environ Sci 28: 455–459, 2015 [DOI] [PubMed] [Google Scholar]
- 371. Yang SJ, Choi JM, Kim L, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, and Park CY. Nicotinamide improves glucose metabolism and affects the hepatic NAD-sirtuin pathway in a rodent model of obesity and type 2 diabetes. J Nutr Biochem 25: 66–72, 2014 [DOI] [PubMed] [Google Scholar]
- 372. Yao Z, Yang W, Gao Z, and Jia P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci Lett 647: 133–140, 2017 [DOI] [PubMed] [Google Scholar]
- 373. Yin F, Boveris A, and Cadenas E. Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal 20: 353–371, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Yoon J, Lee KJ, Oh GS, Kim GH, and Kim SW. Regulation of Nampt expression by transcriptional coactivator NCOA6 in pancreatic beta-cells. Biochem Biophys Res Commun 487: 600–606, 2017 [DOI] [PubMed] [Google Scholar]
- 375. Yoshino J, Baur JA, and Imai SI. NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab 27: 513–528, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Yoshino J, Mills KF, Yoon MJ, and Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab 14: 528–536, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Young GS, Choleris E, Lund FE, and Kirkland JB. Decreased cADPR and increased NAD+ in the Cd38−/− mouse. Biochem Biophys Res Commun 346: 188–192, 2006 [DOI] [PubMed] [Google Scholar]
- 378. Young GS, Jacobson EL, and Kirkland JB. Water maze performance in young male Long-Evans rats is inversely affected by dietary intakes of niacin and may be linked to levels of the NAD+ metabolite cADPR. J Nutr 137: 1050–1057, 2007 [DOI] [PubMed] [Google Scholar]
- 379. Yu J, Sadhukhan S, Noriega LG, Moullan N, He B, Weiss RS, Lin H, Schoonjans K, and Auwerx J. Metabolic characterization of a Sirt5 deficient mouse model. Sci Rep 3: 2806, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Zeng J, Libien J, Shaik F, Wolk J, and Hernandez AI. Nucleolar PARP-1 expression is decreased in Alzheimer's disease: consequences for epigenetic regulation of rDNA and cognition. Neural Plast 2016: 8987928, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, Schulze KL, Verstreken P, and Bellen HJ. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol 4: e416, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, and Bellen HJ. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452: 887–891, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Zhang B, Muller-Steffner H, Schuber F, and Potter BVL. Nicotinamide 2-fluoroadenine dinucleotide unmasks the NAD(+) glycohydrolase activity of Aplysia californica adenosine 5′-diphosphate ribosyl cyclase. Biochemistry 46: 4100–4109, 2007 [DOI] [PubMed] [Google Scholar]
- 384. Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, and Mostoslavsky R. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140: 280–293, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Zhong L, Yeh TY, Hao J, Pourtabatabaei N, Mahata SK, Shao J, Chessler SD, and Chi NW. Nutritional energy stimulates NAD+ production to promote tankyrase-mediated PARsylation in insulinoma cells. PLoS One 10: e0122948, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Zhou SS, Li D, Sun WP, Guo M, Lun YZ, Zhou YM, Xiao FC, Jing LX, Sun SX, Zhang LB, Luo N, Bian FN, Zou W, Dong LB, Zhao ZG, Li SF, Gong XJ, Yu ZG, Sun CB, Zheng CL, Jiang DJ, and Li ZN. Nicotinamide overload may play a role in the development of type 2 diabetes. World J Gastroenterol 15: 5674–5684, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Zhou SS, Li D, and Zhou Y. Letter to the editor: high serum N(1)-methylnicotinamide in obesity and diabetes: a consequence of excess nicotinamide? J Clin Endocrinol Metab 100: L78–L79, 2015 [DOI] [PubMed] [Google Scholar]
- 388. Zhou Y, Wang L, Yang F, Lin X, Zhang S, and Zhao ZK. Determining the extremes of the cellular NAD(H) level by using an Escherichia coli NAD(+)-auxotrophic mutant. Appl Environ Microbiol 77: 6133–6140, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Zhu XH, Lu M, Lee BY, Ugurbil K, and Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A 112: 2876–2881, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Zhuo L, Fu B, Bai X, Zhang B, Wu L, Cui J, Cui S, Wei R, Chen X, and Cai G. NAD blocks high glucose induced mesangial hypertrophy via activation of the sirtuins-AMPK-mTOR pathway. Cell Physiol Biochem 27: 681–690, 2011 [DOI] [PubMed] [Google Scholar]
- 391. Zykova TA, Zhu F, Zhai X, Ma WY, Ermakova SP, Lee KW, Bode AM, and Dong Z. Resveratrol directly targets COX-2 to inhibit carcinogenesis. Mol Carcinog 47: 797–805, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]