

Abstract
Copper toxicity and copper limitation can both be effective host defense mechanisms against pathogens. Tolerance of high copper by fungi makes toxicity as a defense mechanism largely ineffective against fungal pathogens. A forward genetic screen for Histoplasma capsulatum mutant yeasts unable to replicate within macrophages showed the Ctr3 copper transporter is required for intramacrophage proliferation. Ctr3 mediates copper uptake and is required for growth in low copper. Transcription of the CTR3 gene is induced by differentiation of H. capsulatum into pathogenic yeasts and by low available copper, but not decreased iron. Low expression of a CTR3 transcriptional reporter by intracellular yeasts implies that phagosomes of non-activated macrophages have moderate copper levels. This is further supported by the replication of Ctr3-deficient yeasts within the phagosome of non-activated macrophages. However, IFN-γ activation of phagocytes causes restriction of phagosomal copper as shown by upregulation of the CTR3 transcriptional reporter and by the failure of Ctr3-deficient yeasts, but not Ctr3 expressing yeasts, to proliferate within these macrophages. Accordingly, in a respiratory model of histoplasmosis, Ctr3-deficient yeasts are fully virulent during phases of the innate immune response but are attenuated after the onset of adaptive immunity. Thus, while technical limitations prevent direct measurement of phagosomal copper concentrations and copper-independent factors can influence gene expression, both the CTR3 promoter induction and the attenuation of Ctr3-deficient yeasts indicate activation of macrophages switches the phagosome from a copper-replete to a copper-depleted environment, forcing H. capsulatum reliance on Ctr3 for copper acquisition.
Author summary
Control of primary pathogens that infect phagocytes often requires adaptive immunity, but the mechanisms that convert host cells from permissive to antimicrobial states are only partially understood. The intracellular fungal pathogen Histoplasma capsulatum resides and proliferates within the macrophage phagosome. During innate immunity, macrophages which normally control fungi prove ineffective against H. capsulatum yeasts. At this stage, the phagosome of unactivated macrophages has ample copper that facilitates intracellular growth of Histoplasma but does not cause copper toxicity. However, the onset of adaptive immunity and the subsequent activation of macrophages decreases phagosomal copper and macrophages become less permissive to Histoplasma proliferation. IFN-γ acts as a key cytokine for switching the macrophage strategy by changing phagosomes from a copper-sufficient to a copper-depleted state in order to control intracellular pathogens. In such activated macrophages, H. capsulatum yeasts upregulate expression of the Ctr3 copper transporter to enable continued acquisition of essential copper.
Introduction
To successfully infect and colonize a host, pathogens must acquire sufficient nutrients from the host to enable microbe growth and proliferation. These metabolic resources include, but are not limited to, essential metals. The nutrient-limited phagosome represents a particularly challenging environment for intracellular pathogens as mammalian hosts can sequester essential elements such as iron and zinc from pathogens. This has been termed “nutritional immunity” [1,2]. For example, host molecules such as heme, ferritin, transferrin, and lactoferrin make iron largely inaccessible to microbes [1]. However, successful pathogens have developed sophisticated strategies to combat iron limitation. For example, Mycobacterium tuberculosis and the fungal pathogen Histoplasma capsulatum secrete iron-chelating siderophores [3–5]. Accordingly, inability to synthesize siderophores severely impairs intracellular growth [5,6]. In addition, H. capsulatum maintains a slightly acidic intra-phagosomal pH which is sufficient to release iron from host transferrin [7]. Mammalian hosts also restrict available zinc by production of zinc chelating proteins such as S100 family proteins and calprotectin [8,9]. In addition, host zinc transporters (ZIPs) are employed to tightly control zinc levels in different cellular compartments [10]. Host zinc limitation mechanisms are an important aspect of activation of cellular immunity [11]. However, as with iron limitation, some pathogens have evolved efficient mechanisms to counteract zinc sequestration. High affinity transporters expressed by Salmonella species and H. capsulatum (ZnuABC and Zrt2, respectively) enable these pathogens to import zinc in environments with low zinc concentrations [12–15]. Without these zinc transporters Salmonella and H. capsulatum intracellular proliferation is significantly attenuated. Employing an alternative strategy, the fungal pathogen Candida albicans expresses zincophore (Pra1), a zinc-chelating molecule, to scavenge zinc during endothelial invasion [16].
Like iron and zinc, pathogen acquisition of copper during infection is essential, but high levels of copper are toxic. Copper killing mechanisms involve reactive oxygen-generating fenton-type reactions, nitrosative stress, or iron-sulfur cluster attack [1]. Recent evidence has shown that immune cells can utilize excessive copper as a powerful weapon to kill pathogens during innate immunity [17–19]. For pathogens, an inability to decrease cellular copper can impair pathogen virulence. For example, M. tuberculosis survival in host cells depends on copper exporter proteins [19] and Salmonella systemic infection requires detoxification of excess copper by a multi-copper-ion oxidase (CueO) [20]. The fungus Cryptococcus neoformans (grubii) utilizes copper-sequestering metallothionein (Cmt) proteins for full virulence during pulmonary infection [17]. On the other hand, there is evidence that host defenses also use copper limitation in some tissue environments. During kidney infection, C. albicans switches from copper-dependent superoxide dismutase 1 (Sod1) to expression of the copper-independent Sod3 [21]. Proliferation of C. neoformans in murine brains requires two copper transporters (Ctr1 and Ctr4) indicating that copper is limited in the mouse CNS [22,23]. Thus, maintenance of copper homeostasis in host environments with high or low copper environments is essential for pathogens to establish successful infections.
H. capsulatum is a primary fungal pathogen that is not efficiently controlled by innate immunity alone since clearance requires activation of cell-mediated immunity [24,25]. H. capsulatum resides within the phagosome of host phagocytes, an environment that is initially permissive for fungal proliferation. Through a forward genetic screen, we identified a homolog of copper transporters (Ctr3) which was required for growth of H. capsulatum in low copper and within the phagosome of host macrophages. We determined that Ctr3 enhances H. capsulatum survival in vivo specifically during the peak of the adaptive immune response to pulmonary infection. Consistent with this, expression of CTR3 increases in low copper concentrations in vitro and in activated, but not in unactivated macrophages. These findings show that copper is sufficiently available to intramacrophage H. capsulatum during innate immunity, but that activation of macrophages induces copper limitation to enact fungal control.
Results
Intramacrophage growth of H. capsulatum requires Ctr3
To identify genes required for intramacrophage growth, a genetic screen was designed to identify mutants unable to proliferate within macrophages. Insertion mutants were created using Agrobacterium tumefaciens-mediated transformation of a T-DNA element previously shown to provide relatively random and trackable mutations [26]. To facilitate efficient identification of mutants with reduced intramacrophage proliferation, two indirect assays of H. capsulatum yeast replication within macrophages were used. First, increasing fluorescence of red-fluorescence protein (RFP) expressing yeasts was used to indicate intramacrophage yeast replication [27]. Second, a lacZ-expressing macrophage cell line was used to rapidly quantify the ability of mutant yeast to lyse infected macrophages as a result of yeast replication [28]. Individual H. capsulatum mutants were added to macrophage populations to initiate infections. RFP-fluorescence was monitored daily over 7–8 days after which remaining macrophages were quantified by the remaining β-galactosidase activity. Mutants were selected that showed less than 30% increase in RFP fluorescence and/or at least 30% reduction in macrophage lysis. Of 40,000 insertion mutants, 178 had reduced intramacrophage growth and/or attenuated virulence in macrophages.
The insertion mutations were mapped to the genome by sequencing the regions flanking the T-DNA insertion, and two mutants (27H11 and 84D11) were identified which had T-DNA insertions in the promoter region of a gene encoding a putative copper transporter (193 bp and 215 bp upstream of the CDS initiation codon for 27H11 and 84D11, respectively). We designated the gene CTR3 based on phylogenetic analysis that showed the gene product was similar to copper transporters, including the high affinity Ctr3 copper transporter of Saccharomyces cerevisiae (S1 Fig). Each mutant had approximately 50% reduced RFP-fluorescence (intramacrophage fungal growth) and 60% reduced macrophage lysis compared to wild type (Fig 1A and 1B). Consistent with the RFP-based measurement of intracellular growth, CFU-based measurement of viable yeasts within macrophages in culture confirmed that the Ctr3-deficient mutant proliferated only 30% to 50% as well as wild type at 48 hours and 72 hours post-infection (Fig 1C). However, the ctr3 mutant grew as well as the wild-type CTR3 parent in liquid culture (Fig 1D). Complementation of the ctr3 mutants with the wild-type CTR3 gene restored intramacrophage proliferation (Fig 1A) and virulence in macrophages (Fig 1B) linking intramacrophage growth to Ctr3 function. We also found that Ctr3 is required for intracellular proliferation in another phylogenetically distinct H. capsulatum strain G186A, but the virulence defect observed in G186A yeasts was not as significant as that in G217B background strain (S2A Fig).
Fig 1. H. capsulatum intramacrophage growth requires Ctr3.

H. capsulatum proliferation in macrophages was measured by fluorescence of RFP-expressing yeasts (A), yeast-growth dependent lysis of the macrophage population (B), and determination of intracellular viable yeasts (C). (A) RFP-fluorescence of intracellular H. capsulatum yeasts was monitored over 72 hours following infection of P388D1 macrophages at an MOI of 1:2 with CTR3-expressing yeasts (CTR3; black), two mutants with the CTR3 gene disrupted by T-DNA insertion (ctr3 27H11 (circles) and 84D11(squares); red), or mutant strains complemented with a wild-type CTR3 gene (ctr3/CTR3; blue). (B) Survival of lacZ-expressing P388D1 macrophages was quantified after 7 days of infection by removal of culture medium and measurement of the remaining macrophage-derived β-galactosidase activity. (C) Viability of intracellular H. capsulatum yeasts over 72 hours was determined by lysing infected macrophages and plating of the lysate on solid HMM medium to enumerate colony forming units (CFU). (D) Growth of CTR3-expressing (black), ctr3 mutant (27H11; red), and the ctr3/CTR3 complemented (blue) strains in liquid culture in HMM at 37°C was measured by plating dilutions of the culture on solid medium to enumerate CFU. All data represent the average ± standard deviation of biological replicates (n = 3). Statistically significant differences between CTR3 and ctr3 strains were determined by one-tailed Student’s t-test and are indicated with asterisks (* P < 0.05, ** P < 0.01, *** P < 0.001).
Yeast growth requires Ctr3 under copper-limited conditions
Consistent with the function of Ctr3 homologs in other fungi, Ctr3 enables H. capsulatum acquisition of copper when copper is limited. Restriction of copper showed Ctr3-deficient yeasts are more sensitive to reduced copper availability; the IC50 of the copper chelator bathocuproine disulfonate (BCS) for the ctr3 mutant is over 150-fold lower than that of the CTR3 parent and ctr3/CTR3 complemented strains (Fig 2A). A similar pattern was observed in the G186A strain background; lack of Ctr3 rendered yeasts approximately 10-fold more sensitive to BCS (S2B Fig). Supplementation of BCS-chelated media with excess copper, but neither zinc nor iron, restored the growth of the ctr3 mutant demonstrating the specificity of the phenotype for copper (S3 Fig). Consistent with this, loss of Ctr3 function did not affect growth in iron restricted conditions (using the iron chelator bathophenanthroline disulfonate (BPS); Fig 2B), suggesting that Ctr3 plays no role in iron uptake (Fig 2B). In contrast to copper-chelation, loss of Ctr3 function does not affect H. capsulatum growth in high copper as both the ctr3 mutant and CTR3 parent strain grow equally well in media with millimolar concentrations of copper (Fig 2C).
Fig 2. Ctr3 enables H. capsulatum growth in limited copper.

Dose response curves for Ctr3-expressing (CTR3, black), Ctr3-deficient (ctr3, red), and the complemented (ctr3/CTR3, blue) strains grown in liquid culture with the copper-specific chelator BCS (A), the iron-specific chelator BPS (B), and with added CuSO4 (C). Yeasts were grown in HMM at 37°C in the presence of a gradient of concentrations of BPS, BCS or CuSO4. Yeast growth was monitored daily by measurement of culture turbidity (optical density at 595 nm) and growth after 5 days normalized to growth of yeasts in the absence of chelators or CuSO4. Dose-response curves were generated by non-linear regression. IC50 values for growth in BCS were 1,661 μM, 2.8 μM, and 442 μM, for CTR3, ctr3, and ctr3/CTR3, respectively. Data represent the average relative growth ± standard deviation of biological replicates (n = 3).
To directly show Ctr3-dependent copper acquisition, we measured intracellular copper levels in yeasts by inductively-coupled plasma mass spectrometry (ICP-MS). Ctr3-deficient yeasts have lower overall copper levels compared to Ctr3-expressing yeasts during exponential growth in medium containing 10 nM copper (Fig 3A), and starvation of yeasts for copper reduces intracellular copper to baseline levels. Upon replenishment of copper, Ctr3-expressing but not Ctr3-deficient yeasts accumulate intracellular copper (Fig 3A). Both Ctr3-expressing and Ctr3-deficient yeasts have equivalent iron levels in exponential growth and after iron starvation and accumulate iron upon supplementation (Fig 3B), showing the specificity of Ctr3 for copper, but not iron acquisition. Together these data indicate that Ctr3 functions as a copper-specific importer to facilitate growth of yeasts when available copper is low.
Fig 3. Histoplasma Ctr3 enables copper import.
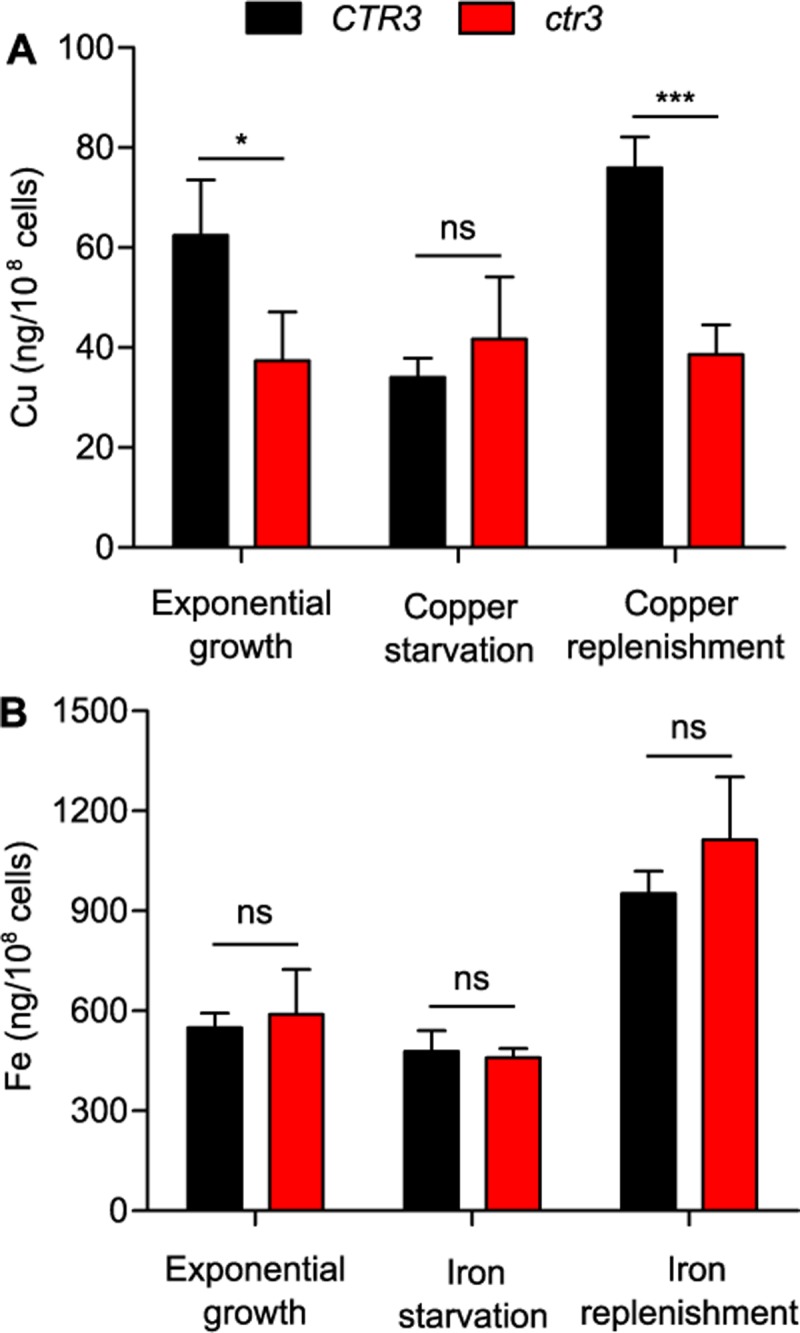
Copper (A) and iron (B) acquisition of Ctr3-expressing (CTR3, black) and Ctr3-deficient (ctr3, red) yeasts. Total cellular metal levels were measured by ICP-MS of yeasts in exponential growth in HMM (which contains 10 nM CuSO4 and 3 μM FeSO4) before and after metal depletion (treatment of yeasts for 24 hours with 2 mM BCS (A) or 16 μM BPS (B)) and after 3 hours with replenishment of copper and iron in the growth medium. Data represent the average level of copper or iron per 108 yeasts ± standard deviations among biological replicates (n = 3). Asterisks represent significant differences in metal levels between Ctr3-expressing and Ctr3-deficient yeasts (* P < 0.05, *** P < 0.001, ns P > 0.05) as determined by two-tailed Student’s t-test.
Copper limitation and differentiation into pathogenic yeasts both induce CTR3 expression
The Ctr3 requirement for yeast growth in low copper suggests CTR3 expression may be regulated by copper concentrations. Bioinformatic analysis of the H. capsulatum genome identified two additional putative copper transporters which were designated Ctr1 and Ctr2 (S1 Fig). Examination of CTR1, CTR2, and CTR3 gene expression by qRT-PCR showed that low copper (10 nM) significantly increased mRNA levels of all three CTR genes compared to high copper (10 μM) conditions; CTR1, CTR2, and CTR3 were all induced in low copper media compared to high copper media, regardless of whether cells were grown as yeasts or mycelia (Fig 4A). Interestingly, CTR3 had the highest overall expression of the CTR genes. In mycelia, CTR3 expression was induced by low copper and was expressed at similar levels to that of CTR1 and CTR2. However, in yeast cells, the expression of CTR3 in high copper was 2.5-fold higher than that of CTR1 and CTR2 (Fig 4A) and CTR3 expression was further induced nearly 10-fold when yeasts were grown in low copper (Fig 4A). These data indicate that while expression of CTR1, CTR2, and CTR3 are all induced by low available copper, differentiation of H. capsulatum cells into pathogenic yeasts establishes an overall higher baseline of expression.
Fig 4. Copper limitation and differentiation into pathogenic yeast phase induce CTR3 expression.
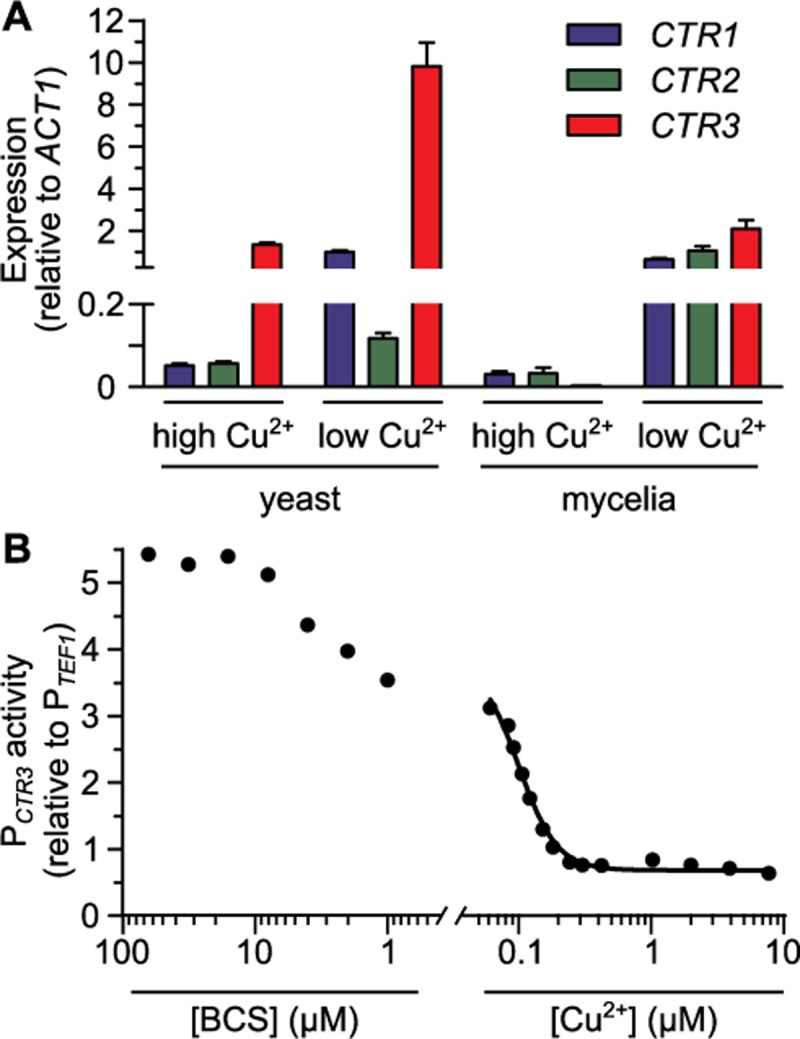
Expression of CTR genes based on qRT-PCR (A) or a CTR3 promoter fusion to gfp (B). Expression was calculated relative to the constitutive expression of the TEF1 gene. (A) H. capsulatum cells in the host-infecting form (yeasts, 37°C) and environmental form (mycelia, 25°C) were grown in 3M media with low (10 nM) or high (10 μM) CuSO4 added and the CTR1 (blue), CTR2 (green), and CTR3 (red) transcript levels were quantified by qRT-PCR and calculated relative to ACT1 transcript levels. Data represent the average expression ± standard deviation of results from biological replicates (n = 3). (B) CTR3 promoter activity (PCTR3) of yeasts grown in 3M medium with different copper concentrations was measured by fluorescence and normalized to the fluorescence of a strain with a TEF1 promoter-gfp fusion (PTEF1). Fluorescence intensity was measured under different CuSO4 concentrations with concentrations lower than 60 nM (the trace amount present in 3M medium) achieved by addition of the copper chelator BCS. A non-linear regression standard curve (black line) was fit to the data with known copper concentrations. Data represent average CTR3 promoter activity of replicates (n = 3).
Since copper regulates CTR3 expression, we created a green-fluorescent protein (gfp) transcriptional fusion to the H. capsulatum CTR3 promoter as a fluorescent indicator of copper availability. CTR3 promoter activity, as indicated by GFP fluorescence of yeast cells, was measured by microscopy after growth in liquid culture and normalized to the fluorescence of an isogenic strain in which gfp expression was controlled by the constitutive H. capsulatum TEF1 promoter. Consistent with the transcriptional analysis, decreasing copper concentrations increased the CTR3 promoter activity, and addition of BCS further increased the reporter GFP-fluorescence to levels at least 5-fold greater than expression in high copper (Fig 4B). Conversely, addition of copper greatly decreases but does not eliminate CTR3 promoter activity. The CTR3 promoter responds to changes in copper concentrations but is not affected by changes in iron or zinc (S4 Fig). In addition, the CTR3 promoter activity is not affected by reactive oxygen stress (S5A Fig) or changes in pH (S5B Fig), two physiologically-relevant environmental changes encountered by Histoplasma in the phagosome. These data indicate the specificity of the CTR3 promoter regulation for available copper.
To provide quantitative estimates of phagosomal copper levels, the fluorescence of the gfp reporter strains was measured in a gradient of copper concentrations. Analysis of media by inductively coupled plasma mass spectrometry (ICP-MS) showed that media without any metal addition had 60 nM trace copper. To reduce copper concentrations below 60 nM necessitated culture of cells in increasing concentrations of BCS. The dose response-data for increasing copper was used to generate a curve of the CTR3-promoter (PCTR3)-controlled GFP fluorescence in 60 nM to 10 μM copper, which showed that CTR3 promoter activity decreased to baseline levels at concentrations above 240 nM copper (Fig 4B). Maximal CTR3 promoter activity reached a plateau of approximately 5-fold higher relative expression in media containing at least 8 μM of BCS (Fig 4B). These data show that the CTR3 promoter is regulated by copper and the CTR3 promoter-dependent fluorescence of gfp reporter yeasts provides an estimate of the available copper in H. capsulatum’s environment.
H. capsulatum virulence in vivo requires Ctr3 during the adaptive immune response
To determine the role of Ctr3 in H. capsulatum virulence, Ctr3-producing and Ctr3-deficient yeasts were tested in a murine model of pulmonary histoplasmosis. Respiratory infections of mice were established using a mixed inoculum of the wild-type and ctr3 mutant strains and the fungal burden in lungs determined over time. To enable measurement of the relative fitness of the Ctr3-deficient ctr3 strain, the ctr3 mutant strain was marked with constitutive GFP-fluorescence, and the viable colony forming units (cfu) of wild-type versus mutant strains were differentiated by colony fluorescence. At day 6 post-infection, a time point before significant adaptive immune responses, the ctr3 mutant showed equivalent lung infection as wild type (Fig 5A). However, after the peak of the adaptive immune response (day 9–21 post-infection), the ctr3 mutant was less fit compared to the co-infecting wild-type strain. Complementation of the ctr3 mutant with the CTR3 gene restored the virulence of the mutant (S6 Fig) indicating the loss of fitness was due to loss of Ctr3 function. These data show that Ctr3 is required for full virulence, specifically at time points following activation of cell-mediated immunity. The requirement for Ctr3 function in H. capsulatum growth both in limited copper (Fig 2A) and for pathogenesis during adaptive immune response stages suggests that copper becomes limiting in the phagosome of phagocytes during adaptive immunity.
Fig 5. H. capsulatum virulence in vivo requires Ctr3 during the adaptive immune response.
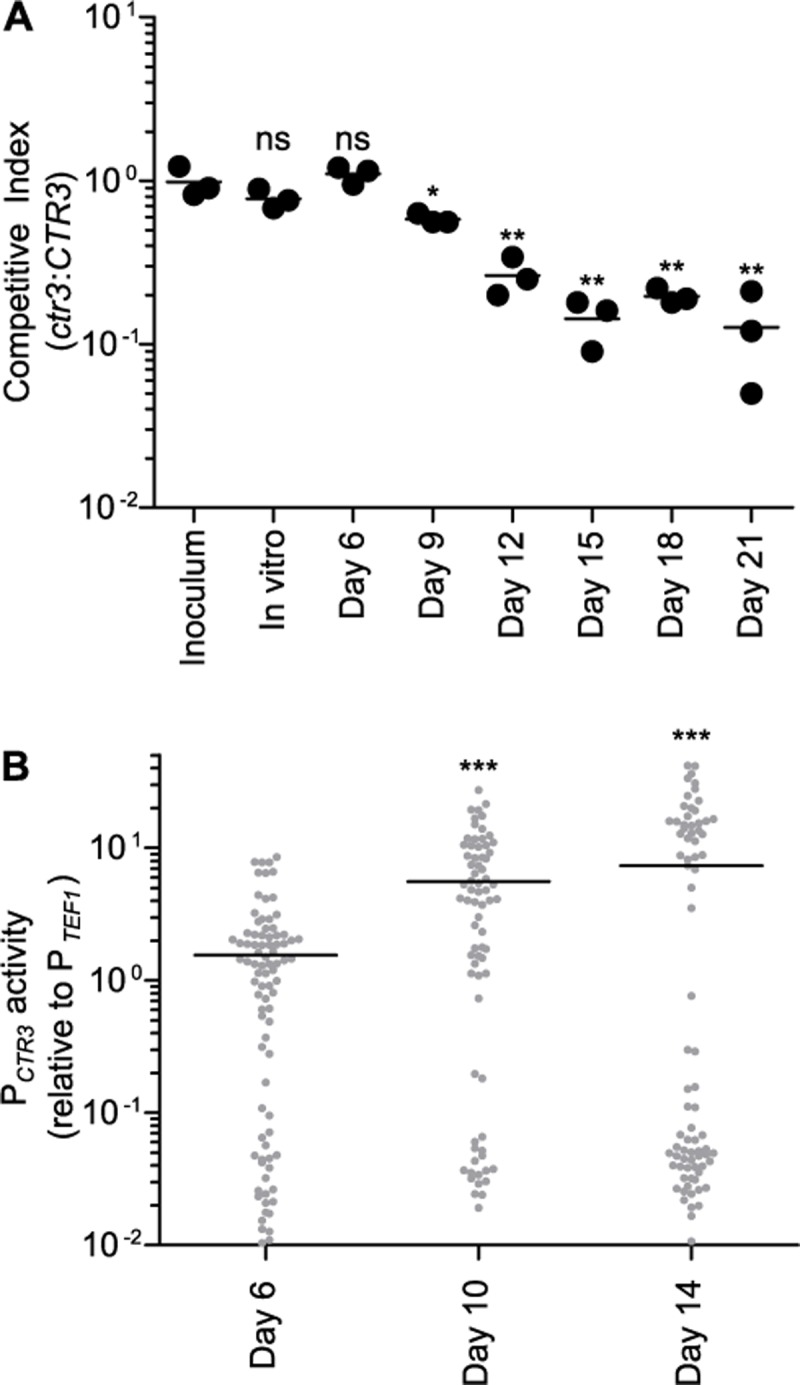
Proliferation of H. capsulatum yeasts (A) and CTR3 promoter activity (B) in vivo following respiratory infection of mice. (A) The relative fitness (competitive index) of Ctr3-deficient (ctr3) compared to Ctr3-producing (CTR3) yeasts was determined by co-infecting mice intranasally with equal amounts of CTR3 (non fluorescent) and ctr3 (GFP-fluorescent) yeasts (1x104 each). Fungal burdens were determined at 6, 9, 12, 15, 18, and 21 days post-infection by harvesting lungs, plating lung homogenates on solid medium, and enumerating fluorescent and non-fluorescent CFU. Data points represent the ratio of GFP-fluorescent to GFP-negative CFU at each time point (n = 3 mice) with the average ratio indicated (horizontal bar). Asterisks indicate significant (* P < 0.05, ** P < 0.01) differences compared to the ratio of the inoculum as determined by one-tailed Student’s t-test. (B) CTR3 promoter activity in vivo was determined by infecting mice intranasally with wild-type yeasts expressing either the CTR3 promoter-gfp fusion (PCTR3) or a TEF1 promoter-gfp fusion (PTEF1) and collecting lung tissue at 6, 10, and 14 days post infection. Lung cells were lysed to release intracellular yeasts and the fluorescence of yeasts quantified by microscopy. Fluorescence of individual yeasts with the CTR3 promoter-gfp fusion was normalized to the average fluorescence of yeast with the TEF1 promoter-gfp fusion. Data points represent the CTR3 promoter activity of individual yeasts and bars represent the average results from replicate infections (n = 3). For each infection, at least 60 yeasts were analyzed. Asterisks indicate significant (*** P < 0.001) differences compared to the CTR3 promoter activity at day 6 post-infection as determined by one-tailed Student’s t-test.
To probe the intraphagosomal copper concentration during H. capsulatum infection, we used the copper-regulated CTR3 promoter-gfp fusion to measure the CTR3 promoter activity in vivo. Following respiratory infections in mice, H. capsulatum yeasts were collected from lung tissue and the fluorescence of the CTR3 promoter-gfp yeasts measured. Consistent with the equivalent fitness of the wild type and the Ctr3-deficient strain during innate immunity (Fig 5A), the CTR3 promoter activity remained low at 6 days post-infection (Fig 5B). Comparing the yeast GFP fluorescence to the copper concentration dose-response curve for the CTR3 promoter (Fig 4B) estimates the copper concentration H. capsulatum encounters at day 6 post-infection is approximately 100 nM, a concentration sufficient to allow yeast growth without Ctr3 function (Fig 2A). However, the CTR3 promoter activity at day 10 and day 14 post-infection was 3- to 5-fold higher than that at day 6 post-infection indicating less available copper in the H. capsulatum-containing phagosome at these time points (Fig 5B). At day 14 post-infection, the fluorescence distribution appears bimodal. The low fluorescent yeasts may reflect a sub-population that is inhibited for growth due to macrophage activation or that not all phagocytes have equivalent changes in phagosomal copper. The average CTR3 promoter activity measured in yeast in vivo at day 10 and 14 (including the low-fluorescent population) was similar to growth in liquid medium containing at least 64 μM BCS, a concentration which induces the CTR3 promoter (Fig 4B) and at which the ctr3 mutant cannot grow (Fig 2A). Together these data suggest that phagosomal copper becomes significantly limited in phagocytes during the adaptive immune response.
Cytokine activation of macrophages decreases phagosomal copper levels
As one of the central features of the adaptive immune response involves cytokine activation of phagocytes, we tested which cytokines induce phagosomal copper restriction in H. capsulatum-infected macrophages. For these experiments, the H. capsulatum CTR3 promoter-regulated GFP fluorescence was used to indicate the levels of available copper within the macrophage phagosome. H. capsulatum yeasts within the phagosome of non-activated bone marrow-derived macrophages (BMDMs) expressed GFP at moderate levels (Fig 6A). Quantification of the CTR3-driven GFP fluorescence and correlation with the in vitro-derived dose-response data (Fig 4B) estimates the phagosomes of unactivated macrophages is around 80 nM. Treatment of H. capsulatum-infected BMDMs with IFN-γ increased CTR3 promoter activity in a dose-dependent manner indicating phagocyte activation with IFN-γ stimulates restriction of phagosomal copper availability (Fig 6A and 6B). However, treatment with TNF-α or GM-CSF did not significantly increase CTR3 promoter activity, suggesting these cytokines do not significantly influence phagocyte phagosomal copper concentrations (Fig 6B).
Fig 6. Activation of macrophages decreases phagosomal copper availability and impairs proliferation of Ctr3-deficient yeasts.

Phagosome copper dynamics following cytokine activation of macrophages (A, B, and D) or in vivo administration of IFN-γ (C). (A) Representative differential interference contrast (DIC) and fluorescence (GFP) images of intracellular H. capsulatum yeasts with the CTR3 promoter-gfp fusion in non-activated BMDMs (top panel) or BMDMs activated with 1000 U/mL IFN-γ (bottom panel). Scale bar represents 10 μm. (B) BMDMs were treated with IFN-γ, TNF-α, or GM-CSF and infected with H. capsulatum yeast (MOI 1:2) with TEF1 (PTEF1) or CTR3 (PCTR3) promoter-gfp fusions. After 48 hours CTR3 promoter activity of intramacrophage yeasts was determined by lysis of macrophages, recovery of yeasts, and measurement of GFP-fluorescence by microscopy (n > 100 yeasts for each sample) and normalized to the average TEF1 promoter activity. (C) Activation of the CTR3 promoter in vivo following IFN-γ treatment was determined by infecting mice intranasally with wild-type H. capsulatum yeasts with the PTEF1 or PCTR3 promoter gfp fusions. 2000 U of IFN-γ was delivered to the lungs at day 4 and 5 post-infection, and at 6 days post-infection, lungs were harvested, lung cells lysed to release intracellular yeasts, and the GFP fluorescence quantified by microscopy (n > 100 yeasts). (B-C) Data represent the CTR3 promoter activity among biological replicates (n = 3) after normalization to TEF1 promoter activity. Box plot shows upper and lower quartiles with the median value (horizontal bar). Vertical bars indicate 10% to 90% of the data distribution. Asterisks indicate significant differences compared to non-activated macrophages (* P < 0.05, ** P < 0.01, *** P < 0.001). (D) BMDMs were activated with IFN-γ, TNF-α, or both cytokines and infected with Ctr3-expressing (CTR3, black), Ctr3-deficient (ctr3, red) and complemented (ctr3/CTR3, blue) H. capsulatum yeasts (MOI 1:2). After 48 hours, intracellular yeasts were recovered and enumerated by plating for CFU. Data represent the average fold change ± standard deviation compared to yeasts at the start of the assay among biological replicate infections (n = 3). Asterisks indicate significant differences compared to the intracellular proliferation of CTR3 yeasts (* P < 0.05).
IFN-γ-induced phagosomal copper restriction also occurs in vivo. At day 6 post-infection, the CTR3 promoter activity is normally low (Fig 5B). However, administration of IFN-γ to mice increased the CTR3 promoter activity of H. capsulatum yeasts (Fig 6C); the average fluorescence was at least two-fold higher with just two IFN-γ treatments indicating that IFN-γ is sufficient to induce copper restriction in vivo. Copper concentration estimation using the dose-response curve (Fig 4B) indicates that this IFN-γ treatment causes copper concentration to decrease from 100 nM to well below 60 nM.
We used the growth of Ctr3-deficient yeasts as an alternate indicator of cytokine-induced changes to phagosomal copper availability. Intramacrophage growth of Ctr3-deficient yeasts, which are sensitive to low copper, was compared to that of wild-type H. capsulatum yeasts. Without cytokine treatment, Ctr3-deficient yeasts proliferate equally as well in BMDMs as Ctr3-expressing yeasts (Fig 6D). This indicates copper is not limited in the phagosomes of these macrophages and is consistent with the CTR3 promoter activity measurements (Fig 6A and 6B). Treatment of BMDMs with IFN-γ, but not TNF-α, restricted the growth of Ctr3-deficient H. capsulatum yeasts (Fig 6D), demonstrating that IFN-γ triggers restriction of available copper in the phagosome to levels which impair the growth of Ctr3-deficient yeasts. These results are consistent with the CTR3 promoter activity data in IFN-γ-treated macrophages (Fig 6A and 6B) indicating that IFN-γ activation of macrophages changes the available phagosomal copper from high to low concentrations (significantly less than 60 nM).
For quantifying CTR3 promoter activity, GFP fluorescence driven by the CTR3 promoter was normalized to fluorescence of intracellular yeasts with a GFP-promoter fusion to the TEF1 promoter to control for any changes in global gene expression due to the state of intracellular yeast cells. The activity of the TEF1 promoter is not affected by copper levels (S7A Fig) or by residence within unactivated or activated macrophages in culture (S7A Fig). Furthermore, normalization of the GFP fluorescence driven by the CTR3 promoter to a different housekeeping gene (H2B) promoter fusion showed a similar increase of the CTR3 promoter in intramacrophage yeasts before and after activation (S7B Fig). Finally, GFP-fluorescence driven by the CTR3 promoter was normalized to RFP-fluorescence driven by the TEF1 promoter within the same yeast cells. This also showed the same induction of the CTR3 promoter in activated BMDMs (S7C Fig). These data indicate that TEF1 promoter activity serves as an accurate normalization factor to account for global transcription variation due to intracellular residence of yeasts.
Available intraphagosomal copper concentration differs among primary macrophages
Surveying primary murine phagocytes as well as common macrophage cell lines showed that CTR3 promoter activity of intracellular H. capsulatum yeasts was high in cultured macrophage cell lines, indicating significantly restricted phagosomal copper even without cytokine treatment in these cells (Fig 7). A similar pattern among cell lines was also observed when the H2B promoter was used for normalization (S8A Fig) or when the CTR3-driven GFP fluorescence was normalized to the TEF1 promoter activity within the same cells (S8B Fig). Among primary cells, resident peritoneal macrophages and alveolar macrophages had high phagosomal copper concentrations (Fig 7) estimated at 280 nM and 320 nM, based on the copper dose-response curve for the CTR3 promoter (Fig 4B). This is consistent with the equivalent in vivo proliferation of the Ctr3-deficient and Ctr3-expressing yeasts when H. capsulatum yeasts are primarily present in alveolar macrophages before phagocytes are activated.
Fig 7. Phagosomal copper concentration differs among primary macrophages and macrophage cell lines.

Common macrophage cell lines (P388D1, RAW264.7, J774.1, and PMA-differentiated THP-1 cells) and unactivated primary cells (bone marrow derived macrophages (BMDMs), peritoneal macrophages (PMs), and alveolar macrophages (AMs)) were infected with H. capsulatum yeasts (MOI 1:2) with TEF1 or CTR3 promoter-gfp fusions for 48 hours. Macrophages were lysed, intracellular yeasts recovered, and the GFP fluorescence of individual yeast was quantified by microscopy (n > 100 yeasts for each sample). CTR3 promoter activity as indicated by the CTR3 promoter-gfp fusion was normalized to the average fluorescence of the TEF1 promoter-gfp fusion. Horizontal bars represent the average CTR3 promoter activity.
Discussion
In order to establish infections and proliferate in macrophages, H. capsulatum yeasts must acquire essential metals within the phagosomal environment. H. capsulatum secretes siderophores and expresses zinc transporters to combat host limitation of iron and zinc, respectively. In this study, we demonstrate that growth in macrophages also imposes challenges on yeasts to maintain copper homeostasis. Specifically, H. capsulatum yeasts rely on the Ctr3 copper transporter to acquire sufficient copper when copper becomes limiting, both in liquid culture and within macrophages. Besides Ctr3, the H. capsulatum genome encodes two additional putative copper transporters (Ctr1 and Ctr2). However, Ctr1 and Ctr2 are not simply redundant with Ctr3; Ctr1 and Ctr2 are not as highly expressed as Ctr3, and they are not sufficient for copper acquisition when phagosomal copper levels become severely limited. Thus, Ctr3 is the primary transporter involved in copper acquisition as part of H. capsulatum’s pathogenesis program.
Two aspects related to H. capsulatum pathogenesis contribute to Ctr3 expression. First, differentiation of H. capsulatum into pathogenic yeasts induces Ctr3 expression independent of copper levels, consistent with a virulence role facilitating H. capsulatum infection of macrophages and persisting within a copper-limited environment. Second, restriction of available copper further increases Ctr3 expression above the level set by yeast-phase differentiation. In support of this dual regulation of Ctr3 transcript levels, the Ctr3 promoter contains putative binding sites for two transcription factors, Ryp1 and Mac1, which have been implicated in yeast-phase gene regulation and fungal transcriptional responses to copper concentration, respectively [17,29,30]. Furthermore, ChIP-chip analysis showed that two yeast-phase transcriptional regulators, Ryp1 and Ryp4, preferentially interact with the CTR3 locus in the yeast phase compared to mycelia [29]. While we can not rule out the possibility that other copper-independent features of macrophage infection do not impact the CTR3 promoter activity, the data suggest that yeast-phase expression of CTR3 is regulated primarily by available copper. Other physiologically-relevant conditions encountered by yeasts within the phagosome (i.e., iron and zinc concentrations, reactive oxygen, and pH changes) do not influence the CTR3 promoter. Consistent with our results, CTR3 is part of a copper-responsive regulon in a microarray-based study of copper-regulated genes [31], although in this study only yeast responses were examined. H. capsulatum strain differences in CTR3 expression are due to trans-acting factors [32], likely from variations in either Ryp or Mac1 production or activity among strains. Together, these data are consistent with the model that yeast phase differentiation primes H. capsulatum cells for pathogenesis by inducing basal Ctr3 expression and the level of Ctr3 production is further tuned to the precise level of copper availability in the phagosome.
Technical challenges in direct measurement of phagosomal copper required the use of surrogate indicators of copper availability for intracellular H. capsulatum. Using a transcriptional gfp fusion as a semi-quantitative reporter of available copper, we determined that during innate immune stages, H. capsulatum resides within a phagosomal environment with copper concentrations above 100 nM. Consistent with this, CTR3 transcription is lowest for yeast within alveolar macrophages (Fig 7). At 10 days post-infection, which coincides with the onset of adaptive immune responses, copper becomes restricted. Both in vitro and in vivo, IFN-γ is sufficient to trigger copper restriction. Comparison of in vivo CTR3 transcription to a standard curve generated in vitro estimates phagosomal copper concentrations become significantly lower than 60 nM. As H. capsulatum yeasts are found almost exclusively within phagocytes during mammalian infection [33], these levels reflect copper concentration within the phagosomal environment at these two points of infection. We note that these copper concentrations are inferred using regulation of the CTR3 promoter, which assumes differential expression is not influenced by copper-independent changes during infection. However, our conclusions about CTR3 transcription reflecting copper dynamics are completely supported by the differential growth of Ctr3-deficient and Ctr3-expressing H. capsulatum strains as a second indicator of copper availability within the phagosome; Ctr3-mediated copper transport is required for H. capsulatum yeast proliferation in vivo at 9 days post-infection but not before. While higher intraphagosomal copper concentration can be microbicidal to some intracellular pathogens, H. capsulatum can tolerate high copper (up to mM levels) and thus copper toxicity mechanisms of immune defense are ineffective.
As a nearly exclusive intracellular pathogen, H. capsulatum responses to copper levels provide unique insights into the dynamics of the phagosomal environment. Like H. capsulatum, C. neoformans (grubii) yeasts up-regulate transcription of copper transporters (Ctr4 and Ctr1 which are homologs of H. capsulatum Ctr3 and Ctr1, respectively) in response to copper restriction [30,34,35]. For C. neoformans, these transcriptional responses vary by tissue with CTR4 promoter activity increasing in the CNS environment but not in the lung environment, suggesting limited copper in the CNS, but not the lung [17,22,35]. These results are corroborated by the reciprocal expression profile of C. neoformans copper-binding metallothioneins (Cmt1 and Cmt2) which are induced by high copper concentrations; C. neoformans yeasts in the lung have elevated Cmt1 and Cmt2 expression but not in the brain [17,22]. Multiple functional studies with mutants of C. neoformans support the transcriptional profiles since Ctr4- and Ctr1-deficient C. neoformans yeasts, which are unable to grow in limited copper [23,30], are impaired in CNS but not lung infection [22,23,30,36]. In contrast, C. neoformans mutants lacking the Cmt1 and Cmt2 metallothioneins are attenuated in lung infection. Together these studies indicate copper concentrations in the lungs during C. neoformans infection are sufficiently high to not require the Ctr1 and Ctr4 transporters. Initially, these findings appear to contrast with those we observe with H. capsulatum yeasts. However, while H. capsulatum yeasts are nearly exclusively intracellular during infection [33], C. neoformans has multiple mechanisms to avoid long-term residence within macrophages (e.g., formation of phagocytosis-resistant titan cells [37], production of the anti-phagocytic capsule [38], secretion of anti-phagocytic protein App1 [39], and vomocytosis [40]). Thus, C. neoformans infection studies indicating the lung is not copper limiting likely include the general extracellular environment, whereas H. capsulatum yeasts indicate phagosome-specific copper concentrations. Indeed, C. neoformans infection of RAW264.7 or J774.1 macrophages in vitro show up-regulation of CTR4 but not CMT1 expression in intracellular yeasts consistent with our data showing the phagosome in these macrophage cell lines have low available copper [23,35]. In addition, Ding et al. found that expression of the mammalian phagosomal copper transporter ATP7A decreased in bronchoalveolar lavage cells at day 14 following Cryptococcus infection consistent with reduced transport of copper into the phagosome during adaptive immunity [17]. These findings and our results with Histoplasma establish a general model that while the extracellular lung environment has ample copper, the phagosome of lung phagocytes becomes copper limiting, particularly following IFN-γ activation.
Copper restriction as a mechanism to control fungal pathogens contrasts with copper toxicity as a means to control bacterial pathogens. Phagosomes of macrophages infected with the intracellular bacterial pathogen M. tuberculosis have approximately 400 μM Cu after 1 hour which decreases to 20 μM after 24 hours [41]. Despite this decrease in copper, 20 μM is still a considerably high amount of copper. Consistent with elevated copper levels in the M. tuberculosis-containing phagosome, M. tuberculosis bacteria which have lost the outer membrane copper export protein have reduced tolerance to copper and reduced virulence compared to wild type [19]. Similarly, M. tuberculosis mutants in the RicR regulon are inhibited by high copper (> 60 μM) in vitro and are attenuated in vivo [42]. Loss of the CsoR regulon improves copper resistance of M. tuberculosis enabling full virulence during early stages of infection. These data indicate that the M. tuberculosis-containing phagosome contains relatively high concentrations of copper. Supporting this, treatment of macrophages with LPS or IFN-γ increases the host copper transporting protein ATP7A on the phagosome membrane [43]. Even though these studies lack direct measurement of intraphagosomal copper concentrations, they are consistent with findings of elevated copper within latex bead-containing phagosomes and a requirement for ATP7A for phagocyte killing of Escherichia coli [43]. In contrast, 14 days following C. neoformans pulmonary infection (a time point consistent with IFN-γ production) alveolar macrophages have decreased ATP7A levels [17]. These differences and our data with intramacrophage H. capsulatum yeasts suggest that macrophages may differentiate between bacterial and fungal pathogens and employ copper toxicity or copper limitation, respectively, in their attempts to limit replication of these two classes of pathogens.
With the involvement of adaptive immunity, host utilization of copper for control of fungi switches from copper toxicity to copper restriction. Aspergillus fumigatus conidia infection of alveolar macrophages increases ATP7A expression consistent with elevation of phagosomal copper [18] as an initial phagocyte response. A. fumigatus conidia lacking the AceA transcription factor are less tolerant of high copper and accordingly are less virulent in vivo [18]. Aspergillus cells lacking two Ctr transporters homologous to the fungal Ctr3/Ctr4 and Ctr1 proteins are unable to grow in low copper but lack any virulence defects in vivo [44]. These data indicate that copper toxicity is the primary host defense initially employed against fungal cells (i.e., during the innate immune response). In contrast to H. capsulatum yeasts, innate immune mechanisms are sufficient for control of A. fumigatus infections and cells that escape initial clearance by phagocytes grow as extracellular hyphae. H. capsulatum yeasts, on the other hand, are not controlled by innate immunity and are primarily intracellular. Instead restriction of H. capsulatum, as well as C. neoformans, requires activation of phagocytes by the adaptive immune system. Our data shows that IFN-γ is key to the switch of macrophage phagosomes from a high copper environment to a copper-limited environment, and it explains, in part, how adaptive immunity contributes to the control of intracellular primary pathogens.
Materials and methods
H. capsulatum strains and growth
H. capsulatum strains used in this study are listed in the S1 Table and were derived from the G217B and G186A clinical isolates. H. capsulatum yeasts were grown in H. capsulatum-macrophage medium (HMM, which contains 10 nM CuSO4) or in 3M media [45] without added copper for metal supplementation tests with FeSO4, ZnSO4, or CuSO4 as appropriate. For growth of uracil auxotrophs, HMM was supplemented with 100 μg/ml uracil. Yeasts were grown with continuous shaking (200 rpm) at 37°C and mycelia cultures at 25°C. Cultures were grown to exponential phase for use in infection studies. For dose-response tests with chelators and metals, yeasts were grown at 37°C in microtiter plates with twice-daily agitation [46]. For growth on solid medium, HMM was solidified with 0.6% agarose and supplemented with 25 μM FeSO4.
Growth of G217B derived strains in liquid culture was quantified by measurement of culture turbidity (optical density at 595 nm) or enumerating viable CFU by plating dilutions on solid HMM. Growth of G186A derived strains was determined by resazurin-based yeast metabolic assay [46]. Briefly, 100 μM resazurin was added to the yeast culture at 37°C and resorufin fluorescence (530 nm excitation, 590 nm emission) was measured over 90 minutes.
Macrophage cell culture
LacZ-expressing P388D1 cell line was created from mouse cell line P388D1 (ATCC CCL-46, [29]). LacZ-expressing P388D1 RAW264.7 (ATCC TIB-71) and J774.1 (ATCC TIB-67) macrophage cell lines were maintained in Ham’s F-12 medium supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals). L929 cells (ATCC CCL-1) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% FBS. THP-1 cells (ATCC TIB-202) were maintained in RPMI-1640 medium supplemented with 10% (FBS) and were differentiated in 10 ng/ml phorbol 12-myristate 13-acetate (PMA) for 48 h before use. All cell lines were cultured at 37°C in 5% CO2/95% air. For infection experiments, macrophage cell lines were co-cultured with yeasts in Ham’s F-12 medium supplemented with 10% FBS.
Peritoneal macrophages were obtained from wild-type C57BL/6 mice by peritoneal lavage with phosphate-buffered saline (PBS). For elicitation of macrophages, peritoneal injection of 3% protease peptone was performed 4 days prior to lavage. Bone marrow cells were isolated from femurs of C57BL/6 mice (Charles River) and differentiated by culturing in Dulbecco’s modified Eagle medium (DMEM) supplemented with 30% L929 cell culture supernatant for 7 days to obtain bone marrow derived macrophages (BMDMs). Non-adherent cells were removed from plastic dishes by washing with PBS. Alveolar macrophages were obtained from C57BL/6 mice by bronchoalveolar lavage (BAL) with PBS. All primary cells were cultured in DMEM at 37°C in 5% CO2/95% air.
Mutagenesis and isolation of H. capsulatum mutants with attenuated intramacrophage growth
H. capsulatum strain OSU233 was used as the genetic background for insertional mutagenesis. OSU233 was constructed by A. tumefaciens-mediated transformation of H. capsulatum yeasts [47] with plasmid pQS01 which contains the apt3 gene for selection (G418-resistance) and the tdTomato red-fluorescent protein transgene under control of the H. capsulatum TEF1 constitutive promoter. OSU233 yeasts were mutagenized by A. tumefaciens-mediated transformation [48] using strain LBA1100 harboring plasmid pBHt2 [26]. Bacteria and yeasts were co-cultured for 40 hours on solid Agrobacterium induction medium containing 0.1 mM acetosyringone at 25°C. Cells were then transferred to HMM medium containing 100 μg/ml uracil, 100 μg/ml hygromycin to select for H. capsulatum transformants, and 10 μg/ml tetracycline to counter select A. tumefaciens. Plates were incubated at 37°C for 10–12 days until transformants appeared. Individual transformants were picked into liquid HMM with 100 μg/ml uracil in wells of a 96-well microtiter plate and incubated at 37°C for 5 days.
Monolayers of P388D1 lacZ-expressing macrophage cells in 96-well microtiter plates [28] were then inoculated with mutant yeasts at a multiplicity of infection (MOI) of 1:1 (yeasts:macrophages). Intramacrophage growth of yeasts was monitored daily by measuring RFP fluorescence (530 nm excitation, 590 nm emission) with a Synergy 2 microplate reader (Biotek). After 7 days, surviving macrophages were quantified by removal of culture media from the infected macrophages, lysis of the remaining macrophages with 0.1% Triton X-100, addition of 1 mg/mL o-nitrophenyl-β-D-galactopyranoside (ONPG), and determination of the β-galactosidase activity (absorbance at 420 nm with correction at 600 nm). Mutants with at least 30% reduction in intramacrophage growth or in lysis of the macrophages were retained as candidate attenuated strains.
Mapping of H. capsulatum T-DNA insertional mutants
The location of the T-DNA insertion in individual mutants was determined by thermal asymmetric interlaced PCR (TAIL-PCR; [49]). 100 ng of genomic DNA was used as the template for primary PCR, with a T-DNA left or right border-specific primer (LB11 or RB9) and one of four semi-random primers (LAD1-4). The primary PCR reaction was diluted 500-fold and used as the template for the secondary PCR with nested left- or right-border primers (LB12 or RB10) and the AC1 primer. PCR products were sequenced and aligned to the H. capsulatum genome sequence. T-DNA insertion at the CTR3 locus was confirmed by PCR and sequencing using CTR3-specific primers in conjunction with LB11 and RB9. Primer sequences are listed in the Key Resource Table.
Complementation of the ctr3 mutation
A 1.7 kb fragment consisting of the wild-type CTR3 gene and 825 bp of upstream sequence was amplified by PCR from H. capsulatum G217B genomic DNA using CTR3-specific primers (ORF9-3 and CTR-13) and cloned into a URA5-based T-DNA plasmid fusing the CTR3 gene with sequence encoding a C-terminal FLAG epitope. Either the CTR3 complementation vector (pDT06) or a control gfp-expression vector (pCR628) were transformed by A. tumefaciens-mediated transformation into the ctr3 mutants and Ura+ transformants were selected by plating on solid HMM.
Phylogenetic analysis of fungal copper transporters
Reciprocal BLAST searches of fungal genomes using the H. capsulatum Ctr3 protein sequence and other known fungal copper transporters (e.g., Saccharomyces Ctr proteins) were used to identify copper transporter homologs in Saccharomyces cerevisiae, Schizosaccharomyces pombe, Candida albicans, Neurospora crassa, Aspergillus nidulans, Aspergillus fumigatus, Blastomyces dermatitidis, Paracoccidioides braziliensis, Magnaporthe oryzae, Trichophyton rubrum, Ustilago maydis, and Cryptococcus neoformans var grubii. Proteins with E-values less than 10−4 with and at least 50% coverage were aligned and used for construction of a phylogenetic tree (Clustal Omega).
Determination of yeast sensitivity to metal ion chelation and toxicity
H. capsulatum sensitivity to bathocuproine disulfonate (BCS), bathophenanthroline disulfonate (BPS), or excess CuSO4 was assayed by addition of two-fold dilutions of chelators or CuSO4 to HMM in 48-well plates containing 4 × 104 H. capsulatum yeasts/ml. Plates were incubated at 37°C with continuous shaking (200 rpm) for 5 days. Yeast proliferation was quantified by measurement of culture turbidity (optical density at 595nm) with a Synergy 2 microplate reader (Biotek). Relative growth in the presence of chelators or CuSO4 was determined by normalization of growth to wells lacking BCS, BPS or CuSO4. Dose-response curves were determined by non-linear regression of the data and the 50% inhibitory concentrations calculated from the regression curve.
Intracellular copper or iron measurement by inductively coupled plasma mass spectrometry (ICP-MS)
Histoplasma yeasts were pre-grown in HMM to late exponential phase and subsequently treated with 2 mM BCS or 16 μM BPS for 24 h to deplete residual copper or iron carried over from HMM. Thereafter, yeast cells were washed 3 times in PBS to remove BCS or BPS and resuspended in HMM for 3 h or 24 h. Yeast cell concentrations were determined by plating serial dilutions on solid HMM. To measure the intracellular total iron or copper, the yeasts were heated at 95°C to remove all the water. To each yeast sample, 0.1 ml of concentrated ultrapure nitric acid (Fisher Scientific Tracemetal grade distilled at TERL using a Savillex DST-1000) was added. Samples were digested by floating in a 100°C water bath using a floating tube rack for 15–30 min. Samples were visually inspected to ensure complete digestion until no solids were seen. Samples were cooled and diluted by 10-fold in deionized water spiked with 11.11ppb Indium. Final Indium concentration was 10 ppb which was used as an internal standard.
All samples were analyzed on a Thermo Finnigan Element 2 Inductively Coupled Plasma Sector Field Mass Spectrometer. Samples were introduced to the ICP at 100 μl/min using a PFA-100 Microflow self aspirating nebulizer (Elemental Scientific) pumped at 100 μl/min using 0.25 mm I.D. pvc pump tubing and a Gilson 3 peristaltic pump. Iron was measured at m/z 54, 56 and 57 in medium resolution (R = 4000) and copper was measured at m/z 63 and 65 in medium resolution (R = 4000). Calibration standards were prepared by dilution from commercially available single element 1000 μg/ml Fe and 1000 μg/ml Cu (Inorganic Ventures) into 10% v/v nitric acid to match the sample solvent. Calibration standards also included 10 ppb Indium (Inorganic Ventures) as an internal standard. Intracellular total copper or iron were calculated based on 108 yeast cells.
CTR gene expression determination
CTR gene transcriptional analyses were determined by quantitative RT-PCR (qRT-PCR) or by CTR3 promoter-gfp reporter fusions. For qRT-PCR, wild-type yeasts or mycelia were grown in HMM containing low (10 nM) or high (10 μM) concentrations of CuSO4. RNA was isolated from fungal cells by mechanical disruption with 0.5 mm glass beads, extraction with RiboZol (Amresco) and alcohol precipitation of nucleic acids. Following DNA removal with DNase, RNA was reversed transcribed with Maxima reverse transcriptase (Thermo Scientific) primed with random pentadecamers. Quantitative PCR was carried out using CTR gene specific primer pairs with SYBR green-based visualization of product amplification (Bioline). Changes in CTR transcript levels relative to actin (ACT1) were determined using the ΔΔCt method [50] after normalization of cycle thresholds to that of the TEF1 gene.
For construction of the CTR3 promoter-gfp fusion, 1453 bp of sequence upstream of the CTR3 coding sequence was used to drive gfp transcription (PCTR3-gfp; pMK32, [32]). For normalization purposes, a similar gfp reporter fusion was created using 661 bp of sequence upstream of the TEF1 gene (PTEF1-gfp). Constructs were cloned into a Ura5+ T-DNA vector and transformed by A. tumefaciens-mediated transformation into the WU15 ura5 auxotroph. For relative quantification of CTR3 expression, GFP-fluorescence of PCTR3-gfp transformed yeasts was normalized to GFP-fluorescence of PTEF1-gfp transformed yeasts grown in identical conditions (in vitro and in vivo). For population-based measurements in multi-well plates, GFP fluorescence (485/20 nm excitation, 528/20 nm emission) was measured and the TEF1 or CTR3 promoter activity was calculated by normalization to the number of yeasts present (OD595). CTR3 promoter activity was then normalized to that of the constitutively TEF1 promoter. For determination of the GFP-fluorescence of individual yeasts, 0.1% Uvitex 3BSA was added to yeast suspensions to label the cell wall and yeasts were examined by microscopy (Nikon E400). GFP-fluorescent (480/30 nm excitation, 535/40 nm emission) and Uvitex-fluorescent images (350/50 nm excitation, 460/50 nm emission) were captured with identical exposure settings and the average GFP fluorescence contained within the Uvitex cell outline was measured (Micro-manager Studio v1.4.5) and ImageJ [51]. Relative CTR3 promoter activity was determined as the ratio of PCTR3 GFP fluorescence of individual yeasts to the average PTEF1 GFP fluorescence.
Estimation of phagosome copper concentrations
A standard curve of CTR3 promoter activity at different copper concentrations was generated by incubation of H. capsulatum yeasts with TEF1 or CTR3 promoter-gfp fusions in 3M medium with a gradient of copper concentrations. After 48 hours, GFP fluorescence and culture turbidity (OD595) were measured. TEF1 and CTR3 promoter activities were determined per OD595 unit, and the CTR3 promoter activity was normalized to that of the constitutively expressed TEF1 gene. The actual available copper in the 3M-based media was determined using inductively coupled plasma mass spectrometry (ICP-MS). Media samples were introduced into a Perkin Elmer Nexion 350D ICP-UCT mass spectrometer at the speed of 400 μl/min after being spiked with 10 ppb indium as an internal standard. Copper was measured in DRC (dynamic reaction cell) mode using ammonia gas (0.35 ml/min) to reduce polyatomic and molecular overlaps. Copper concentration analysis showed that 3M medium without any copper addition contained 60 nM copper. Copper concentrations lower than 60 nM were achieved by adding increasing amounts of BCS. For copper concentrations above 60 nm, a curve was fit to the data by four-variable non-linear regression.
For determination of CTR3 promoter activity in macrophages, macrophages were infected with PTEF1-gfp or PCTR3-gfp yeasts at an MOI of 1:2 in 6-well microtiter plates. Phagocytes were previously seeded into 6-well plates at 5 × 105 (P388D1, RAW 264.7, J774.1, and THP-1) or 3 × 106 (peritoneal macrophages, and BMDMs). Due to the limited yield of alveolar macrophages, 3 × 104 alveolar macrophages were seeded into 96-well plates. For activated macrophages, BMDMs were treated with IFN-γ (200 U/ml and 1000 U/ml), TNF-α (20 U/ml and 100 U/ml) or GM-CSF (2 ng/ml or 10 ng/ml) for 24 hours before infection. 48 hours following macrophage infection, extracellular yeasts were removed by washing macrophages with PBS and intracellular yeasts released by lysis of macrophages with 1% Triton-X100. Yeasts in the macrophage lysates were stained by addition of 0.1% Uvitex and the GFP fluorescence of individual yeasts determined by microscopy and fluorescence quantification. Phagosomal copper concentrations were estimated by comparison of the CTR3 promoter activity of yeasts recovered from macrophages to the in vitro fluorescence versus copper concentration standard curve.
For determination of the phagosome copper concentration in vivo, C57BL/6 mice were intranasally infected with 2 × 104 PTEF1-gfp or PCTR3-gfp yeasts. At day 6, 10 and 14 post-infection, intracellular yeasts were recovered by euthanizing mice, collection and homogenization of lungs in water, and filtration of the homogenate through a 70 μM cell strainer to remove large debris. The filtrate was treated with collagenase and DNAse for 60 minutes. 0.1% Uvitex was added to the filtrate to label yeasts and the GFP-fluorescence of individual yeast was imaged and measured by microscopy. For IFN-γ treatment of mice, infected mice received 2000 U of IFN-γ intranasally at days 4 and 5 post-infection, with control mice receiving PBS in parallel. At day 6 post-infection, intracellular yeasts were recovered from lung homogenates and the GFP-fluorescence measured by microscopy as above.
Intramacrophage proliferation of H. capsulatum yeasts
Macrophage monolayers were established in 96-well plates by seeding with 3 × 104 (P388D1), 2 × 105 (peritoneal macrophages) or 1 × 105 (BMDMs) cells. For experiments with activated macrophages, BMDMs were treated with IFN-γ (1000 U/ml), TNF-α (100 U/ml) or both for 24 hours before infection. Macrophages were then infected with Ctr3-expressing or Ctr3-deficient H. capsulatum yeasts at an MOI of 1:2. Yeast-infected macrophages were incubated at 37°C for up to 72 hours. At different time points, intracellular yeasts were quantified by removal of any extracellular yeasts with the culture supernatant followed by lysis of the macrophages with sterile H2O and plating of the macrophage lysate on solid HMM to enumerate H. capsulatum CFU.
Murine model of pulmonary histoplasmosis
To measure in vivo fitness of Ctr3-deficient yeasts compared to Ctr3-expressing yeasts, wild type C57BL/6 mice were infected with H. capsulatum by intranasal delivery of approximately 2 × 104 yeast cells consisting of equal numbers of Ctr3-expressing (GFP-negative) and Ctr3-deficient yeasts (GFP-fluorescent). Actual numbers and ratios of yeasts delivered were determined by plating serial dilutions of the inocula on solid media for enumeration of CFU. For fungal burden determination at 6, 9, 12, 15, 18 and 21 days post-infection, mice were euthanized, lungs were collected and homogenized in HMM, and serial dilutions of the homogenates were plated on solid HMM to determine the fungal burden (CFU) and fluorescence of recovered colonies. Colony fluorescence was determined with a modified transilluminator and image capture system [52]. The competitive index was calculated as the number of fluorescent colonies (ctr3) divided by the number of non-fluorescent colonies (CTR3). For testing complementation of the ctr3 mutant in vivo, the competition assay was repeated using the ctr3/CTR3 complemented strain and wild type at day 6, 10 and 14 post-infection.
Quantification and statistical analyses
Data were tabulated and analyzed by Student’s t-test (Prism v5, GraphPad Software) for determination of statistically significant differences which are indicated in graphs with asterisk symbols (*, P < 0.05; **, P < 0.01; *** P < 0.001). Dose-response curves were generated by four-variable non-linear regression. The number of mice and the number of biological replicates used in experiments is specified in the relevant figure legends.
Ethical statements
H. capsulatum G217B (ATCC 26032) and H. capsulatum G186A (ATCC 26029) were obtained from American Type Culture Collection (ATCC). All animals were housed in Ohio State University’s AAALAC- and OLAW-accredited animal research facilities (OLAW assurance # A3261-01). All experiments involving mice followed standards in the Public Health Service (PHS) “Guide for the Care and Use of Laboratory Animals” and were approved by The Institutional Animal Care and Use Committee (IACUC) at Ohio State University (protocol # 2007A0241). Mice were anesthetized by inhalation of isoflurane and were euthanized by CO2 inhalation as described in the AVMA "Guidelines for the Euthanasia of Animals”
Supporting information
Putative and proven copper transporter proteins of Saccharomyces cerevisiae (Sce), Schizosaccharomyces pombe (Spo), Candida albicans (Cal), Neurospora crassa (Ncr), Aspergillus nidulans (Ani), Aspergillus fumigatus (Afu), H. capsulatum (Hca), Blastomyces dermatitidis (Bde), Paracoccidioides braziliensis (Pbr), Magnaporthe oryzae (Mor), Trichophyton rubrum (Tru), Ustilago maydis (Uma), and Cryptococcus neoformans (grubii) (Cne) were aligned with Clustal Omega and a Neighbor-joining phylogenetic tree constructed to infer relatedness. Clades were identified representing the Ctr1 (blue), Ctr2 (green), and Ctr3 (red) homologs. Predictions of hypothetical (Hyp), or metal transporters (transporter) from genomic sequencing efforts are listed with gene designation numbers. Protein accession numbers are listed in brackets.
(PDF)
Growth of the Ctr3-expressing parent strain (CTR3, black) and a strain in which the ctr3 gene was deleted (ctr3Δ, red) of the G186A genetic background in macrophages (A) or in liquid media with limited copper (B). (A) A strain in which the CTR3 locus was deleted was generated by allelic replacement [53] with a hygromycin expression cassette flanked by 2 kb upstream and downstream of the CTR3 gene. Intracellular CTR3 and ctr3 mutant H. capsulatum yeasts were quantified over 72 hours following infection of P388D1 macrophages (MOI 1:1). Macrophages were lysed, the intracellular yeasts recovered, and the lysate plated on solid HMM medium to enumerate colony forming units (CFU). Data represent the average intramacrophage CFU ± standard deviation among infections with biological replicates (n = 3). Statistically significant differences between CTR3 and ctr3 proliferation at each day were determined by one-tailed Student’s t-test and are indicated with asterisks (* P < 0.05). (B) Growth of Ctr3-expressing (CTR3, black) and the ctr3 mutant (ctr3, red) strains grown in liquid HMM with the copper chelator BCS were determined by measurement of yeast metabolic conversion of resazurin to fluorescent resorufin (quantified by fluorescence: 530 nm excitation and 590 nm emission 90 minutes following addition of 1 mM resazurin) after 5 days of growth at 37°C. Relative growth was determined by normalization of yeast-dependent resazurin metabolism for each BCS concentration to that of yeasts grown in the absence of BCS. Dose-response curves were generated by non-linear regression and the IC50 for BCS treatment of CTR3 and ctr3 strains determined as 2,156 μM and 263 μM, respectively. Data represent average growth ± standard deviation among biological replicates (n = 3).
(PDF)
Growth of Ctr3-deficient yeasts (ctr3) in BCS-containing media with and without supplementation with copper (A), zinc (B) or iron (C). Ctr3-deficient yeasts were incubated at 37°C in liquid HMM (black symbols) or HMM containing 100 μM BCS (red symbols) with (squares) and without (circles) 100 μM CuSO4 (+Cu), 100 μM ZnSO4 (+Zn) or 100 μM FeSO4 (+Fe). Yeast growth was monitored by culture turbidity (optical density at 595 nm) over 6 days. Data represent the average growth ± standard deviation among biological replicates (n = 3).
(PDF)
CTR3 promoter activity in 1 μM CuSO4 (low CTR3 promoter activity, black bars) or 10 nM CuSO4 (high CTR3 promoter activity, red bars) with high and low concentrations of zinc (A), iron (B), or the iron-specific chelator BPS (C). (A) H. capsulatum yeasts were incubated in 3M medium (containing 4 μM FeSO4 and 1 μM CuSO4 or 10 nM CuSO4) with different ZnSO4 concentrations (0.3 μM to 64 μM). (B) H. capsulatum yeasts were incubated in 3M medium (containing 4 μM ZnSO4 and 1 μM CuSO4 or 10 nM CuSO4) with different FeSO4 concentrations (0.3 μM to 64 μM). (C) H. capsulatum yeasts were incubated in 3M medium (containing 4 μM ZnSO4 and 1 μM CuSO4 or 10 nM CuSO4) with different BPS concentrations (0 μM to 16 μM). The CTR3 promoter activity was assessed by fluorescence of wild-type yeasts with the CTR3 promoter-gfp fusion (PCTR3) after normalization to yeasts with the TEF1 promoter-gfp fusion (PTEF1) grown in identical conditions. After 72 hours incubation at 37°C, culture turbidity (optical density at 595nm) and GFP fluorescence (485 nm excitation, 528 emission) were measured. TEF1 or CTR3 promoter activity (GFP fluorescence) was normalized to the yeast density (OD595) and the CTR3 promoter activity then compared to that of the constitutively expressed TEF1 promoter. Data represent the average relative CTR3 promoter activity ± standard deviation among biological replicates (n = 3).
(PDF)
CTR3 promoter activity in 1 μM CuSO4 (low CTR3 promoter activity, black bars) or 10 nM CuSO4 (high CTR3 promoter activity, red bars) at a range of H2O2 concentrations (A) and pH (B). (A) H. capsulatum yeasts were incubated in 3M medium (containing 1 μM CuSO4 or 10 nM CuSO4) with H2O2 (0 μM to 250 μM). (B) H. capsulatum yeasts were incubated in 3M medium (containing 1 μM CuSO4 or 10 nM CuSO4) buffered to different pH with MES (4.5 to 6.0) or HEPES (6.5 to 7.0). The CTR3 promoter activity was assessed by fluorescence of wild-type yeasts with the CTR3 promoter-gfp fusion (PCTR3) after normalization to yeasts with the TEF1 promoter-gfp fusion (PTEF1) grown in identical conditions. After 72 hours incubation at 37°C, culture turbidity (optical density at 595nm) and GFP fluorescence (485 nm excitation, 528 emission) were measured. TEF1 or CTR3 promoter activity (GFP fluorescence) was normalized to the yeast density (OD595) and the CTR3 promoter activity then compared to that of the constitutively expressed TEF1 promoter. Data represent the average relative CTR3 promoter activity ± standard deviation among biological replicates (n = 3).
(PDF)
Wild-type C57BL/6 mice were infected intranasally with 2×104 yeasts consisting of an equal amount of wild-type CTR3 (RFP-negative) and ctr3/CTR3 complemented (RFP-expressing) yeasts. At days 6 and 14 post-infection, the pulmonary fungal burden was measured by collecting lungs and plating lung homogenates on solid media for enumeration of RFP-fluorescent and non-fluorescent colony forming units (CFU). Data points represent the individual ratio of RFP-negative (ctr3/CTR3) yeasts and RFP-fluorescent (CTR3) yeasts at each time point (n = 3 mice) with horizontal bars representing the average ratio. No significant differences in the ratio of complemented and wild-type yeasts compared to the ratio of the number of yeasts in the inoculum were found by one-tailed Student’s t-tests.
(PDF)
(A) TEF1 promoter activity in liquid culture or in BMDMs with and without IFN-γ activation. (B and C) CTR3 promoter activity of intracellular H. capsulatum yeasts in BMDMs with and without IFN-γ activation. (A) H. capsulatum TEF1 promoter activity was measured by fluorescence of the PTEF1-gfp fusion in yeasts cultured in high (10 μM) or low (10 nM) copper media or in BMDMs with and without activation by IFN-γ (1000U/mL). (B) CTR3 promoter activity of intracellular yeasts was measured by the fluorescence produced by the PCTR3-gfp reporter after normalization to H2B promoter activity (PH2B-gfp) of a parallel population of intracellular yeasts (C) CTR3 promoter activity of intracellular yeasts was measured by the GFP fluorescence produced by the PCTR3-gfp reporter fusion after normalization to the RFP fluorescence produced by the PTEF1-rfp reporter fusion within the same yeast cells. In all experiments, BMDMs were infected with H. capsulatum yeasts (MOI 1:2) and the fluorescence of intracellular yeasts measured after 48 hours by lysis of macrophages, recovery of yeasts, and measurement of GFP or RFP fluorescence in individual yeasts by microscopy (n > 100 yeasts for each sample). Box plots represent quartiles and median fluorescence of the population with lines showing the 10–90% range of the data. Asterisks indicate significant differences in promoter activity compared to non-activated macrophages (*** P < 0.001) using Student’s t-test and “ns” indicates no significant difference among the experimental groups (P > 0.05) using one-way ANOVA with Tukey's Honest Significant Difference test.
(PDF)
CTR3 promoter activity in P388D1 or peritoneal macrophages following normalization to H2B (A) or TEF1 (B) promoter activity in separate or the same yeast cells, respectively. Cell line (P388D1) and primary (peritoneal macrophages (PM)) macrophages were infected with H. capsulatum yeast (MOI 1:2) and the CTR3 promoter activity of intracellular yeasts measured by fluorescence of the PCTR3-gfp reporter fusion. (A) Intracellular yeast GFP fluorescence produced by the PCTR3-gfp reporter fusion was normalized to the GFP fluorescence of a population of PH2B-gfp reporter fusion yeasts from parallel infections. (B) GFP fluorescence produced by the PCTR3-gfp reporter fusion was normalized to the RFP fluorescence produced by the PTEF1-rfp reporter fusion within the same yeast cell. Data points represent the CTR3 promoter activity of individual yeasts (n > 100 for each sample) measured by microscopy of intracellular yeasts recovered after lysis of macrophages. Horizontal bars indicate the population mean. Asterisks (*** P < 0.001) indicate significant differences in the CTR3 promoter activity between yeasts recovered from P388D1 cells and from peritoneal macrophages as determined by two-tailed Student’s t-test.
(PDF)
(PDF)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This study was supported in part by funds from grant AI111015 to CAR from the National Institutes of Health (www.niaid.nih.gov) and by a predoctoral fellowship to QS from the American Heart Association (www.heart.org). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen–host interface. Nat Rev Microbiol. 2012;10: 525–537. 10.1038/nrmicro2836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinberg ED. Nutritional Immunity: Host’s Attempt to Withhold Iron From Microbial Invaders. JAMA. 1975;231: 39–41. 10.1001/jama.1975.03240130021018 [DOI] [PubMed] [Google Scholar]
- 3.Gobin J, Horwitz M. Exochelins of Mycobacterium tuberculosis remove iron from human iron- binding proteins and donate iron to mycobactins in the M. tuberculosis cell wall. J Exp Med. 1996;183: 1527–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heymann P, Gerads M, Schaller M, Dromer F, Winkelmann G, Ernst JF. The siderophore iron transporter of Candida albicans (Sit1p/Arn1p) mediates uptake of ferrichrome-type siderophores and is required for epithelial invasion. Infect Immun. 2002;70: 5246–5255. 10.1128/IAI.70.9.5246-5255.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang LH, Mayfield JA, Rine J, Sil A. Histoplasma Requires SID1, a Member of an Iron-Regulated Siderophore Gene Cluster, for Host Colonization. PLOS Pathog. 2008;4: e1000044 10.1371/journal.ppat.1000044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hilty J, George Smulian A, Newman SL. Histoplasma capsulatum utilizes siderophores for intracellular iron acquisition in macrophages. Med Mycol. 2011;49: 633–642. 10.3109/13693786.2011.558930 [DOI] [PubMed] [Google Scholar]
- 7.Newman SL, Gootee L, Brunner G, Deepe GS. Chloroquine induces human macrophage killing of Histoplasma capsulatum by limiting the availability of intracellular iron and is therapeutic in a murine model of histoplasmosis. J Clin Invest. 1994;93: 1422–1429. 10.1172/JCI117119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319: 962–965. 10.1126/science.1152449 [DOI] [PubMed] [Google Scholar]
- 9.Gläser R, Harder J, Lange H, Bartels J, Christophers E, Schröder J-M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol. 2005;6: 57–64. 10.1038/ni1142 [DOI] [PubMed] [Google Scholar]
- 10.Wilson D, Citiulo F, Hube B. Zinc Exploitation by Pathogenic Fungi. PLOS Pathog. 2012;8: e1003034 10.1371/journal.ppat.1003034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS. Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity. 2013;39: 697–710. 10.1016/j.immuni.2013.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ammendola S, Pasquali P, Pistoia C, Petrucci P, Petrarca P, Rotilio G, et al. High-affinity Zn2+ uptake system ZnuABC is required for bacterial zinc homeostasis in intracellular environments and contributes to the virulence of Salmonella enterica. Infect Immun. 2007;75: 5867–5876. 10.1128/IAI.00559-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campoy S, Jara M, Busquets N, Pérez De Rozas AM, Badiola I, Barbé J. Role of the high-affinity zinc uptake znuABC system in Salmonella enterica serovar typhimurium virulence. Infect Immun. 2002;70: 4721–4725. 10.1128/IAI.70.8.4721-4725.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dade J, DuBois JC, Pasula R, Donnell AM, Caruso JA, Smulian AG, et al. HcZrt2, a zinc responsive gene, is indispensable for the survival of Histoplasma capsulatum in vivo. Med Mycol. 2016;54: 865–875. 10.1093/mmy/myw045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE, et al. Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe. 2012;11: 227–239. 10.1016/j.chom.2012.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Citiulo F, Jacobsen ID, Miramón P, Schild L, Brunke S, Zipfel P, et al. Candida albicans Scavenges Host Zinc via Pra1 during Endothelial Invasion. PLOS Pathog. 2012;8: e1002777 10.1371/journal.ppat.1002777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding C, Festa RA, Chen Y-L, Espart A, Palacios Ò, Espín J, et al. Cryptococcus neoformans copper detoxification machinery is critical for fungal virulence. Cell Host Microbe. 2013;13: 265–276. 10.1016/j.chom.2013.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiemann P, Perevitsky A, Lim FY, Shadkchan Y, Knox BP, Figueora JAL, et al. Aspergillus fumigatus Copper Export Machinery and Reactive Oxygen Intermediate Defense Counter Host Copper-Mediated Oxidative Antimicrobial Offense. Cell Rep. 2017;19: 1008–1021. 10.1016/j.celrep.2017.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, et al. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2011;108: 1621–1626. 10.1073/pnas.1009261108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Achard MES, Tree JJ, Holden JA, Simpfendorfer KR, Wijburg OLC, Strugnell RA, et al. The multi-copper-ion oxidase CueO of Salmonella enterica serovar Typhimurium is required for systemic virulence. Infect Immun. 2010;78: 2312–2319. 10.1128/IAI.01208-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li CX, Gleason JE, Zhang SX, Bruno VM, Cormack BP, Culotta VC. Candida albicans adapts to host copper during infection by swapping metal cofactors for superoxide dismutase. Proc Natl Acad Sci U S A. 2015;112: E5336–5342. 10.1073/pnas.1513447112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun T-S, Ju X, Gao H-L, Wang T, Thiele DJ, Li J-Y, et al. Reciprocal functions of Cryptococcus neoformans copper homeostasis machinery during pulmonary infection and meningoencephalitis. Nat Commun. 2014;5: 5550 10.1038/ncomms6550 [DOI] [PubMed] [Google Scholar]
- 23.Waterman SR, Park Y-D, Raja M, Qiu J, Hammoud DA, O’Halloran TV, et al. Role of CTR4 in the Virulence of Cryptococcus neoformans. mBio. 2012;3: e00285–12. 10.1128/mBio.00285-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garfoot AL, Rappleye CA. Histoplasma capsulatum surmounts obstacles to intracellular pathogenesis. FEBS J. 2016;283: 619–633. 10.1111/febs.13389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou P, Sieve MC, Bennett J, Kwon-Chung KJ, Tewari RP, Gazzinelli RT, et al. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-gamma. J Immunol Baltim Md 1950. 1995;155: 785–795. [PubMed] [Google Scholar]
- 26.Kemski MM, Stevens B, Rappleye CA. Spectrum of T-DNA integrations for insertional mutagenesis of Histoplasma capsulatum. Fungal Biol. 2013;117: 41–51. 10.1016/j.funbio.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards JA, Kemski MM, Rappleye CA. Identification of an Aminothiazole with Antifungal Activity against Intracellular Histoplasma capsulatum. Antimicrob Agents Chemother. 2013;57: 4349–4359. 10.1128/AAC.00459-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edwards JA, Zemska O, Rappleye CA. Discovery of a Role for Hsp82 in Histoplasma Virulence through a Quantitative Screen for Macrophage Lethality ▿. Infect Immun. 2011;79: 3348–3357. 10.1128/IAI.05124-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beyhan S, Gutierrez M, Voorhies M, Sil A. A temperature-responsive network links cell shape and virulence traits in a primary fungal pathogen. PLoS Biol. 2013;11: e1001614 10.1371/journal.pbio.1001614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding C, Yin J, Tovar EMM, Fitzpatrick DA, Higgins DG, Thiele DJ. The copper regulon of the human fungal pathogen Cryptococcus neoformans H99. Mol Microbiol. 2011;81: 1560–1576. 10.1111/j.1365-2958.2011.07794.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gebhart D, Bahrami AK, Sil A. Identification of a copper-inducible promoter for use in ectopic expression in the fungal pathogen Histoplasma capsulatum. Eukaryot Cell. 2006;5: 935–944. 10.1128/EC.00028-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards JA, Chen C, Kemski MM, Hu J, Mitchell TK, Rappleye CA. Histoplasma yeast and mycelial transcriptomes reveal pathogenic-phase and lineage-specific gene expression profiles. BMC Genomics. 2013;14: 695 10.1186/1471-2164-14-695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deepe GS, Gibbons RS, Smulian AG. Histoplasma capsulatum manifests preferential invasion of phagocytic subpopulations in murine lungs. J Leukoc Biol. 2008;84: 669–678. 10.1189/jlb.0308154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ory JJ, Griffith CL, Doering TL. An efficiently regulated promoter system for Cryptococcus neoformans utilizing the CTR4 promoter. Yeast Chichester Engl. 2004;21: 919–926. 10.1002/yea.1139 [DOI] [PubMed] [Google Scholar]
- 35.Waterman SR, Hacham M, Hu G, Zhu X, Park Y-D, Shin S, et al. Role of a CUF1/CTR4 copper regulatory axis in the virulence of Cryptococcus neoformans. J Clin Invest. 2007;117: 794–802. 10.1172/JCI30006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang P, Zhang D, Zhao X, Wei D, Wang Y, Zhu X. Effects of CTR4 deletion on virulence and stress response in Cryptococcus neoformans. Antonie Van Leeuwenhoek. 2016;109: 1081–1090. 10.1007/s10482-016-0709-2 [DOI] [PubMed] [Google Scholar]
- 37.Okagaki LH, Strain AK, Nielsen JN, Charlier C, Baltes NJ, Chrétien F, et al. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 2010;6: e1000953 10.1371/journal.ppat.1000953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell TG, Friedman L. In Vitro Phagocytosis and Intracellular Fate of Variously Encapsulated Strains of Cryptococcus neoformans. Infect Immun. 1972;5: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luberto C, Martinez-Mariño B, Taraskiewicz D, Bolaños B, Chitano P, Toffaletti DL, et al. Identification of App1 as a regulator of phagocytosis and virulence of Cryptococcus neoformans. J Clin Invest. 2003;112: 1080–1094. 10.1172/JCI18309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez M, Casadevall A. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr Biol CB. 2006;16: 2161–2165. 10.1016/j.cub.2006.09.061 [DOI] [PubMed] [Google Scholar]
- 41.Wagner D, Maser J, Lai B, Cai Z, Barry CE, Höner Zu Bentrup K, et al. Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J Immunol Baltim Md 1950. 2005;174: 1491–1500. [DOI] [PubMed] [Google Scholar]
- 42.Shi X, Festa RA, Ioerger TR, Butler-Wu S, Sacchettini JC, Darwin KH, et al. The copper-responsive RicR regulon contributes to Mycobacterium tuberculosis virulence. mBio. 2014;5 10.1128/mBio.00876-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White C, Lee J, Kambe T, Fritsche K, Petris MJ. A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem. 2009;284: 33949–33956. 10.1074/jbc.M109.070201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park Y-S, Lian H, Chang M, Kang C-M, Yun C-W. Identification of high-affinity copper transporters in Aspergillus fumigatus. Fungal Genet Biol FG B. 2014;73: 29–38. 10.1016/j.fgb.2014.09.008 [DOI] [PubMed] [Google Scholar]
- 45.Worsham PL, Goldman WE. Quantitative plating of Histoplasma capsulatum without addition of conditioned medium or siderophores. J Med Vet Mycol Bi-Mon Publ Int Soc Hum Anim Mycol. 1988;26: 137–143. [PubMed] [Google Scholar]
- 46.Goughenour KD, Balada-Llasat J-M, Rappleye CA. Quantitative Microplate-Based Growth Assay for Determination of Antifungal Susceptibility of Histoplasma capsulatum Yeasts. J Clin Microbiol. 2015;53: 3286–3295. 10.1128/JCM.00795-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zemska O, Rappleye CA. Agrobacterium-mediated insertional mutagenesis in Histoplasma capsulatum. Methods Mol Biol Clifton NJ. 2012;845: 51–66. 10.1007/978-1-61779-539-8_4 [DOI] [PubMed] [Google Scholar]
- 48.Garfoot AL, Zemska O, Rappleye CA. Histoplasma capsulatum depends on de novo vitamin biosynthesis for intraphagosomal proliferation. Infect Immun. 2014;82: 393–404. 10.1128/IAI.00824-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y-G, Chen Y. High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. BioTechniques. 2007;43: 649–650, 652, 654 passim. 10.2144/000112601 [DOI] [PubMed] [Google Scholar]
- 50.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- 51.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Youseff BH, Rappleye CA. RNAi-based gene silencing using a GFP sentinel system in Histoplasma capsulatum. Methods Mol Biol Clifton NJ. 2012;845: 151–164. 10.1007/978-1-61779-539-8_10 [DOI] [PubMed] [Google Scholar]
- 53.Sebghati TS, Engle JT, Goldman WE. Intracellular parasitism by Histoplasma capsulatum: fungal virulence and calcium dependence. Science. 2000;290: 1368–1372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Putative and proven copper transporter proteins of Saccharomyces cerevisiae (Sce), Schizosaccharomyces pombe (Spo), Candida albicans (Cal), Neurospora crassa (Ncr), Aspergillus nidulans (Ani), Aspergillus fumigatus (Afu), H. capsulatum (Hca), Blastomyces dermatitidis (Bde), Paracoccidioides braziliensis (Pbr), Magnaporthe oryzae (Mor), Trichophyton rubrum (Tru), Ustilago maydis (Uma), and Cryptococcus neoformans (grubii) (Cne) were aligned with Clustal Omega and a Neighbor-joining phylogenetic tree constructed to infer relatedness. Clades were identified representing the Ctr1 (blue), Ctr2 (green), and Ctr3 (red) homologs. Predictions of hypothetical (Hyp), or metal transporters (transporter) from genomic sequencing efforts are listed with gene designation numbers. Protein accession numbers are listed in brackets.
(PDF)
Growth of the Ctr3-expressing parent strain (CTR3, black) and a strain in which the ctr3 gene was deleted (ctr3Δ, red) of the G186A genetic background in macrophages (A) or in liquid media with limited copper (B). (A) A strain in which the CTR3 locus was deleted was generated by allelic replacement [53] with a hygromycin expression cassette flanked by 2 kb upstream and downstream of the CTR3 gene. Intracellular CTR3 and ctr3 mutant H. capsulatum yeasts were quantified over 72 hours following infection of P388D1 macrophages (MOI 1:1). Macrophages were lysed, the intracellular yeasts recovered, and the lysate plated on solid HMM medium to enumerate colony forming units (CFU). Data represent the average intramacrophage CFU ± standard deviation among infections with biological replicates (n = 3). Statistically significant differences between CTR3 and ctr3 proliferation at each day were determined by one-tailed Student’s t-test and are indicated with asterisks (* P < 0.05). (B) Growth of Ctr3-expressing (CTR3, black) and the ctr3 mutant (ctr3, red) strains grown in liquid HMM with the copper chelator BCS were determined by measurement of yeast metabolic conversion of resazurin to fluorescent resorufin (quantified by fluorescence: 530 nm excitation and 590 nm emission 90 minutes following addition of 1 mM resazurin) after 5 days of growth at 37°C. Relative growth was determined by normalization of yeast-dependent resazurin metabolism for each BCS concentration to that of yeasts grown in the absence of BCS. Dose-response curves were generated by non-linear regression and the IC50 for BCS treatment of CTR3 and ctr3 strains determined as 2,156 μM and 263 μM, respectively. Data represent average growth ± standard deviation among biological replicates (n = 3).
(PDF)
Growth of Ctr3-deficient yeasts (ctr3) in BCS-containing media with and without supplementation with copper (A), zinc (B) or iron (C). Ctr3-deficient yeasts were incubated at 37°C in liquid HMM (black symbols) or HMM containing 100 μM BCS (red symbols) with (squares) and without (circles) 100 μM CuSO4 (+Cu), 100 μM ZnSO4 (+Zn) or 100 μM FeSO4 (+Fe). Yeast growth was monitored by culture turbidity (optical density at 595 nm) over 6 days. Data represent the average growth ± standard deviation among biological replicates (n = 3).
(PDF)
CTR3 promoter activity in 1 μM CuSO4 (low CTR3 promoter activity, black bars) or 10 nM CuSO4 (high CTR3 promoter activity, red bars) with high and low concentrations of zinc (A), iron (B), or the iron-specific chelator BPS (C). (A) H. capsulatum yeasts were incubated in 3M medium (containing 4 μM FeSO4 and 1 μM CuSO4 or 10 nM CuSO4) with different ZnSO4 concentrations (0.3 μM to 64 μM). (B) H. capsulatum yeasts were incubated in 3M medium (containing 4 μM ZnSO4 and 1 μM CuSO4 or 10 nM CuSO4) with different FeSO4 concentrations (0.3 μM to 64 μM). (C) H. capsulatum yeasts were incubated in 3M medium (containing 4 μM ZnSO4 and 1 μM CuSO4 or 10 nM CuSO4) with different BPS concentrations (0 μM to 16 μM). The CTR3 promoter activity was assessed by fluorescence of wild-type yeasts with the CTR3 promoter-gfp fusion (PCTR3) after normalization to yeasts with the TEF1 promoter-gfp fusion (PTEF1) grown in identical conditions. After 72 hours incubation at 37°C, culture turbidity (optical density at 595nm) and GFP fluorescence (485 nm excitation, 528 emission) were measured. TEF1 or CTR3 promoter activity (GFP fluorescence) was normalized to the yeast density (OD595) and the CTR3 promoter activity then compared to that of the constitutively expressed TEF1 promoter. Data represent the average relative CTR3 promoter activity ± standard deviation among biological replicates (n = 3).
(PDF)
CTR3 promoter activity in 1 μM CuSO4 (low CTR3 promoter activity, black bars) or 10 nM CuSO4 (high CTR3 promoter activity, red bars) at a range of H2O2 concentrations (A) and pH (B). (A) H. capsulatum yeasts were incubated in 3M medium (containing 1 μM CuSO4 or 10 nM CuSO4) with H2O2 (0 μM to 250 μM). (B) H. capsulatum yeasts were incubated in 3M medium (containing 1 μM CuSO4 or 10 nM CuSO4) buffered to different pH with MES (4.5 to 6.0) or HEPES (6.5 to 7.0). The CTR3 promoter activity was assessed by fluorescence of wild-type yeasts with the CTR3 promoter-gfp fusion (PCTR3) after normalization to yeasts with the TEF1 promoter-gfp fusion (PTEF1) grown in identical conditions. After 72 hours incubation at 37°C, culture turbidity (optical density at 595nm) and GFP fluorescence (485 nm excitation, 528 emission) were measured. TEF1 or CTR3 promoter activity (GFP fluorescence) was normalized to the yeast density (OD595) and the CTR3 promoter activity then compared to that of the constitutively expressed TEF1 promoter. Data represent the average relative CTR3 promoter activity ± standard deviation among biological replicates (n = 3).
(PDF)
Wild-type C57BL/6 mice were infected intranasally with 2×104 yeasts consisting of an equal amount of wild-type CTR3 (RFP-negative) and ctr3/CTR3 complemented (RFP-expressing) yeasts. At days 6 and 14 post-infection, the pulmonary fungal burden was measured by collecting lungs and plating lung homogenates on solid media for enumeration of RFP-fluorescent and non-fluorescent colony forming units (CFU). Data points represent the individual ratio of RFP-negative (ctr3/CTR3) yeasts and RFP-fluorescent (CTR3) yeasts at each time point (n = 3 mice) with horizontal bars representing the average ratio. No significant differences in the ratio of complemented and wild-type yeasts compared to the ratio of the number of yeasts in the inoculum were found by one-tailed Student’s t-tests.
(PDF)
(A) TEF1 promoter activity in liquid culture or in BMDMs with and without IFN-γ activation. (B and C) CTR3 promoter activity of intracellular H. capsulatum yeasts in BMDMs with and without IFN-γ activation. (A) H. capsulatum TEF1 promoter activity was measured by fluorescence of the PTEF1-gfp fusion in yeasts cultured in high (10 μM) or low (10 nM) copper media or in BMDMs with and without activation by IFN-γ (1000U/mL). (B) CTR3 promoter activity of intracellular yeasts was measured by the fluorescence produced by the PCTR3-gfp reporter after normalization to H2B promoter activity (PH2B-gfp) of a parallel population of intracellular yeasts (C) CTR3 promoter activity of intracellular yeasts was measured by the GFP fluorescence produced by the PCTR3-gfp reporter fusion after normalization to the RFP fluorescence produced by the PTEF1-rfp reporter fusion within the same yeast cells. In all experiments, BMDMs were infected with H. capsulatum yeasts (MOI 1:2) and the fluorescence of intracellular yeasts measured after 48 hours by lysis of macrophages, recovery of yeasts, and measurement of GFP or RFP fluorescence in individual yeasts by microscopy (n > 100 yeasts for each sample). Box plots represent quartiles and median fluorescence of the population with lines showing the 10–90% range of the data. Asterisks indicate significant differences in promoter activity compared to non-activated macrophages (*** P < 0.001) using Student’s t-test and “ns” indicates no significant difference among the experimental groups (P > 0.05) using one-way ANOVA with Tukey's Honest Significant Difference test.
(PDF)
CTR3 promoter activity in P388D1 or peritoneal macrophages following normalization to H2B (A) or TEF1 (B) promoter activity in separate or the same yeast cells, respectively. Cell line (P388D1) and primary (peritoneal macrophages (PM)) macrophages were infected with H. capsulatum yeast (MOI 1:2) and the CTR3 promoter activity of intracellular yeasts measured by fluorescence of the PCTR3-gfp reporter fusion. (A) Intracellular yeast GFP fluorescence produced by the PCTR3-gfp reporter fusion was normalized to the GFP fluorescence of a population of PH2B-gfp reporter fusion yeasts from parallel infections. (B) GFP fluorescence produced by the PCTR3-gfp reporter fusion was normalized to the RFP fluorescence produced by the PTEF1-rfp reporter fusion within the same yeast cell. Data points represent the CTR3 promoter activity of individual yeasts (n > 100 for each sample) measured by microscopy of intracellular yeasts recovered after lysis of macrophages. Horizontal bars indicate the population mean. Asterisks (*** P < 0.001) indicate significant differences in the CTR3 promoter activity between yeasts recovered from P388D1 cells and from peritoneal macrophages as determined by two-tailed Student’s t-test.
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.