

Abstract
Loss of Sostdc1, a growth factor paralogous to Sost, causes the formation of ectopic incisors, fused molars, abnormal hair follicles, and resistance to kidney disease. Sostdc1 is expressed in the periosteum, a source of osteoblasts, fibroblasts and mesenchymal progenitor cells, which are critically important for fracture repair. Here, we investigated the role of Sostdc1 in bone metabolism and fracture repair. Mice lacking Sostdc1 (Sostdc1−/−) had a low bone mass phenotype associated with loss of trabecular bone in both lumbar vertebrae and in the appendicular skeleton. In contrast, Sostdc1−/− cortical bone measurements revealed larger bones with higher BMD, suggesting that Sostdc1 exerts differential effects on cortical and trabecular bone. Mid-diaphyseal femoral fractures induced in Sostdc1−/− mice showed that the periosteal population normally positive for Sostdc1 rapidly expands during periosteal thickening and these cells migrate into the fracture callus at 3 days post fracture. Quantitative analysis of mesenchymal stem cell (MSC) and osteoblast populations determined that MSCs express Sostdc1, and that Sostdc1−/− 5 day calluses harbor >2-fold more MSCs than fractured wildtype controls. Histologically a fraction of Sostdc1-positive cells also expressed nestin and α-smooth muscle actin, suggesting that Sostdc1 marks a population of osteochondral progenitor cells that actively participate in callus formation and bone repair. Elevated numbers of MSCs in D5 calluses resulted in a larger, more vascularized cartilage callus at day 7, and a more rapid turnover of cartilage with significantly more remodeled bone and a thicker cortical shell at 21 days post fracture. These data support accelerated or enhanced bone formation/remodeling of the callus in Sostdc1−/− mice, suggesting that Sostdc1 may promote and maintain mesenchymal stem cell quiescence in the periosteum.
Keywords: Sostdc1, Wise, Ectodin, Sost-like, Usag-1, Sost, Wnt signaling, periosteum, bone regeneration, fracture repair
1. Introduction
Optimal fracture repair requires contribution from surrounding tissues, yet we know very little about the interactions between bone, muscle, vasculature, and the periosteum [1–3]. In particular the periosteum, a thin tissue that covers the outer cortical bone surface, contains a reservoir of progenitor cells that contribute to bone repair; further, periosteal grafts and alpha-smooth muscle actin (α-SMA)-positive cells integrate into the callus after fracture[3, 4]. Other in vivo studies have shown that periosteal and perivascular cells migrate into developing bone and bone undergoing repair, and differentiate into osteoblasts [4, 5].
Sostdc1, a paralog of Sost also known by several other names including Sost-Like, Wise, Ectodin and Usag-1 arose by segmental gene duplication and was previously studied in the context of tooth development, kidney disease, cancer progression, hair follicle formation, and embryo implantation [6–11]. Recently, we have shown that Sostdc1 also contributes to limb morphogenesis [12], and its expression in the periosteum suggests that it may also play a role in bone maintenance and repair. However, the role of Sostdc1 in fracture healing has not been investigated. Sostdc1 has been described as both a Bmp and a Wnt antagonist in a context-dependent manner [13], and it interacts with Lpr4, 5, and 6 Wnt co-receptors and with Bmp ligands [6, 13–16], in vivo. While Sostdc1 shares 55% protein sequence homology to its paralog Sost, it reportedly displays higher specificity for the Lrp4 co-receptor [14], in contrast to the preferred binding to the Lrp5 and 6 co-receptors by Sost [17, 18]. Mesenchyme-derived Sostdc1 inhibits Wnt signaling in the epithelium through the activation of Hedgehog signaling, which in turn suppresses Wnt signaling via Sostdc1 up-regulation in the mesenchyme [12, 19].
To determine the contribution of Sostdc1 to bone metabolism and repair, we characterized trabecular and cortical bone structure and fracture healing program in Sostdc1−/− mice. Herein we show data in support of Sostdc1 as both an anabolic and a catabolic agent, with distinct compartmental contributions to bone metabolism, wherein Sostdc1 deficiency results in a substantial loss of trabecular bone and a significant gain in cortical bone. We find that Sostdc1 marks a population of progenitor stem cells of mesenchymal origin that rapidly expands after injury and populates the callus up to 7 days post-fracture. In addition, we show that in the early stages of fracture repair, Sostdc1−/− cells increase β-catenin-dependent Wnt signaling and promote callus formation via enhanced progenitor cell migration and differentiation. At early stages post-fracture (3 and 7 days post fracture), Sostdc1−/− mice show enhanced intramembranous bone formation and neovascularization compared to controls, along with dramatically elevated numbers of cells expressing nestin, α-SMA, and SP7/Osterix. Although the genetic loss of Sostdc1 results in trabecular bone loss, Sostdc1 may represent a novel therapeutic target for bone formation defects that require rapid intramembranous bone formation to stabilize structural integrity.
2. Material and Methods
2.1. Animals and Femoral Fracture Model
Sostdc1−/− mice have been previously described [12]. Stabilized femoral fractures were produced in Sostdc1−/−, Sostdc1+/− and C57B/L6 wildtype control male mice at 8-weeks of age using an Einhorn closed fracture model, as previously described [20]. Fractures were confirmed radiologically (CareStream in vivo MS-FX) at the time of surgery and femora were harvested at days 3, 5, 7, 10, 14, 21 and 28 post-fracture for subsequent analysis. Animal studies were approved by the Institutional Animal Care and Use Committee of Lawrence Livermore National Laboratory (Livermore, CA, USA).
2.2. LacZ and Immunohistological Staining
The Sostdc1 knockout allele was generated by replacing both Sostdc1 exons with an in-frame LacZ reporter. LacZ stains were performed on fixed tissues (fractured and intact femora at days 3, 7, 10, 14, and 21 post-fracture, n=3 per group, per time point, male mice) of Sostdc1+/− and Sostdc1−/− mice as previously described [21] with minor modifications. Tissues were fixed and then decalcified in 0.5M EDTA until endpoint confirmation by radiotranslucency. LacZ stain was followed by fixation, dehydration, and paraffin embedding for sectioning (6μm) and histology. Sections were counterstained with alcoholic eosin. For immunostains, femora from Sostdc1−/− or Sostdc1+/− mice, with or without fractures (n=3 per group of male mice, per time point), were fixed for 72 hours in 10% neutral buffered formalin at 4°C, followed by decalcification and sectioning as above. Antigen retrieval was conducted using Uni-trieve (Innovex) for 30 minutes at 65°C unless otherwise stated. Antigen retrieval for activated β-catenin (Millipore, 05-665) included digestion with Proteinase K (15μg/ml) for 20 minutes at 37°C followed by Rodent Block M (Biocare Medical, 50-832-64). Primary antibodies for α-Smooth Muscle Actin (α-SMA) (abcam, ab5694), β-galactosidase (abcam, ab9361), nestin, (abcam, ab6142) and SP7/osterix (abcam, ab22552) were incubated on slides overnight at RT followed by secondary antibody conjugated with Alexa Fluor 488 or 596 (Molecular Probes) for green and red stains, respectively. Immunostained slides were mounted with Prolong Gold with DAPI (Molecular Probes) for imaging. Images were obtained with a color CCD QIClick camera and ImagePro Plus V7.0 imaging software. Quantification of immunostains was performed using Image J software, utilizing the Analyze Particles tool, to determine the total area of stem cell marker-positive cells. A minimum of 50 images of similar callus regions from n=3 animals per group per time point were used. This quantification was not absolute, but provided an estimate for the magnitude of the observed differences seen between genotypes.
2.3. Micro-Computed Tomography (μCT) of intact and fracture calluses
Sostdc1−/− and C57B/L6 wildtype control male mice at 5.5 months of age were analyzed by μCT at the distal femoral metaphysis, femoral mid-diaphysis, and L4 vertebral body to determine bone micro-structure parameters (n=6-10 per group) (μCT 35, SCANCO, Brüttisellen, Switzerland) according to the guidelines for μCT analysis of rodent bone structure [22]: energy 55 kVp, intensity 114 mA, integration time 900 ms, 6 μm nominal voxel size. For fracture analysis, callus measurements were compared at 28 days post fracture between Sostdc1−/− and WT, 3 month old, age-matched animals. Intact femora of Sostdc1−/− and C57B/L6 animals were also measured at 12 weeks of age. The threshold for “bone” was set at 350 (35% of maximum value), which is approximately equal to 567 mg HA/cm3. Callus volume measurements (CV) excluded the native bone volume.
2.4. Bone Histomorphometry Measurements
Dynamic bone histomorphometry measurements were obtained from femoral mid-diaphyses of 12 weeks and 5.5 month old male mice. Mice were injected with 30 mg/kg alizarin red and 10 mg/kg calcein 10- and 3- days before euthanasia. Femora were dissected and fixed in 10% neutral phosphate-buffered formaldehyde for 24 hours. Bone histomorphometry was performed using semiautomatic image analysis as described [23, 24].
2.5. Bone Strength Measurements
Fractured and uninjured contralateral femora of male mice were mechanically tested in torsion to quantify biomechanical properties of bone (n=8-10 per group). Femora were rehydrated in isotonic saline for 5-10 minutes prior to testing, and were kept hydrated throughout testing. Both ends of each femur were embedded in Wood’s metal alloy blocks with a testing length of 6 mm, then mounted in a materials testing system (Bose ELF 3200, Eden Prairie, MN) with torsion motor (Exlar SLM Series). An axial load of 3-4 N was applied, then 10 preconditioning torsion cycles were applied to +/− 5° at 0.1 Hz, followed by a single cycle to failure in external rotation at 1°/s. Torque and rotational displacement data were collected at 50 Hz. The failure cycle was used to calculate torsional stiffness, ultimate torque, and rotation at ultimate torque.
2.6. Analysis of Stromal Bone Cells
Unfractured and fractured femora at 8-10 weeks of age male mice were dissected and placed in 1x Hank’s Balanced Salt Solution (HBSS) pH7.2 without serum. Bone digestion was performed as described [25]. Briefly, bones were crushed in 1x HBSS using a mortar and pestle and bone marrow cells were washed away from the bone chips. The bone chips were then transferred to a 50 ml conical tube with 2.0 ml of 3.0 mg/ml Type I Collagenase (Worthington, Lakewood, NJ) and digested in a shaker at 110 rpm at 37°C for 1 hour. The supernatant from the digest was transferred through a 70 micron filter into a fresh conical tube, the remaining bone chips were rinsed with additional 1x HBSS containing 2% FCS, which was added to the supernatant. Cells were then pelleted at 1200 rpm for 5 minutes at 4°C. Live cell yield was counted using a hemocytometer and Trypan blue staining. Cells were then prepared for flow cytometric analysis. Cells were transferred to 96 well V-bottom plates and stained with an antibody cocktail containing anti CD16/32 (clone 93), and PE-Cy7 conjugated anti-CD3 (2C11), CD4 (GK1.5), CD8 (53.6.7), CD11b (M1/70), CD19 (6D5), NK1.1 (PK136), Ter119 (TER119), and Gr1 (RB6-8C3) in 50 μl volume of FACS Buffer for at least 15 minutes at 4°C. Cells were then washed and pelleted for 5 minutes at 2000 rpm, and stained with a second antibody cocktail containing CD45-eFluor 450 (30F11), Sca1-FITC (D7), CD31-APC (390), and either CD51-biotin (RMV-7) or isotype-matched biotin control Ab. Cells were incubated at 4°C for 15 minutes, washed, and pelleted. Lastly, cells were stained with streptavidin-PE for 5 minutes, washed, pelleted and resuspended in FACS buffer for analysis. Propidium iodide was added to the samples before analysis as a viability stain. Live cells were analyzed on a BD LSR II flow cytometric analyzer and data analysis was performed using FlowJo software. Antibodies were purchased from BioLegend and eBioscience.
2.7. Gene Expression Analysis
Bone derived mesenchymal stem cells (MSCs) cells were stained as above, except that Sca1-Brilliant Violet 510 (BioLegend) was used. MSCs, OBs and endothelial cells were isolated using the BD FACS Aria II flow cytometry sorter, and then pelleted and resuspended in Trizol (Qiagen). Total RNA was purified using RNeasy mini Kit (Qiagen) according to manufacturer’s protocol. Samples were analyzed for purity and concentration using a NanoDrop 2000c (Thermo Scientific). Superscript III First-Strand Synthesis System (Invitrogen) was used with oligo dT primers for reverse transcription according to manufacturer’s protocol. Real-time quantitative PCR was then performed with SYBR Select Master Mix (Applied Biosystems) using a Applied Biosystems 7900HT Fast Real-Time PCR System with the following cycling conditions: 50°C for 2 min for Sybr then 95°C for 3 min (2 min for SYBR), followed by 40 cycles of 95°C for 3s (10s for SYBR) and 30s at 60°C. Reactions were run on a 2% agarose gel then DNA fragments imaged under UV light. Primers: β-galactosidase forward ACGGCCAGGACAGTCGTTTG and reverse CCGCTCATCCGCCACATATC; GAPDH forward CCAATGTGTCCGTCGTGGATCT and reverse CCTCAGTGTAGCCCAAGATGC.
2.8. Statistics
Data are expressed as the mean ± standard deviation. For statistical analysis, we used Student’s t-test with a two-tailed distribution, and two-sample equal variance (homoscedastic), for significance. p <0.05 was considered significant.
3. Results
3.1. Sostdc1 deletion decreases trabecular and increases cortical bone volume
Sostdc1−/− mice displayed 31% lower trabecular bone volume fraction (BV/TV) in the distal femoral metaphysis and 35% less in the L4 vertebral body compared to controls (p<0.05). Connectivity density (p<0.005), and trabecular number (p<0.0005) were also reduced 30% and 18%, in the distal femur, respectively. Trabecular separation (p<0.005) was increased 28% in the femur and 31% in L4, consistent with a lower trabecular bone volume phenotype. However, bone tissue BMD (mg HA/cm3) was increased in the cortical bone, relative to WT (p≤0.05) (Table 1). In contrast to the trabecular compartments, the cortical compartment indicated that the bones (total area, TA) were larger (TA 2.02 vs 2.35, p<0.05). The cortical total area was increased by 16%, and the marrow area was enlarged by 21% (p<0.01) (Table 1). These data show that Sostdc1 modulates bone metabolism differently in the cortical than in the trabecular compartments of bone.
TABLE 1.
Bone Phenotyping Based on μCT Parameters in the Cancellous Bone Compartment of the Distal Femur, L4 Vertebrae and Cortical Bone of 5.5-Month-Old Sostdc1−/− Mice Compared to WT Controls.
| Index | WT | Sostdc1−/− | |
|---|---|---|---|
| BV/TV (%) | 9.50±1.20 | 6.50±2.00* | |
| Conn. Dens. (1/mm3) | 155.827±13.975 | 108.507±38.540* | |
| SMI | 2.284±0.225 | 2.502±0.326 | |
| Femur | Tb.N (1/mm) | 4.164±0.143 | 3.387 ± 0.397* |
| Tb.Th (mm) | 0.0399 ±0.0009 | 0.039 ±0.006 | |
| Tb.Sp (mm) | 0.227±0.009 | 0.292±0.035* | |
| BMD (mg HA/cm3) | 913.452±14.108 | 935.367±15.824* | |
|
| |||
| BV/TV (%) | 19.90±0.80 | 12.80±2.40* | |
| Conn Dens. (1/mm3) | 273.007±14.759 | 211.350±46.188* | |
| SMI | 0.490±0.090 | 1.181±0.232* | |
| L4 Vertebrae | Tb.N (1/mm) | 5.270±0.154 | 4.155±0.428* |
| Tb.Th(mm) | 0.036±0.001 | 0.035±0.002 | |
| Tb.Sp (mm) | 0.179±0.006 | 0.235±0.029* | |
| BMD(mg HA/cm3) | 947.457±19.014 | 935.367±15.824* | |
|
| |||
| pMOI (mm4) | 0.47±0.063 | 0.592±0.145* | |
| TA (mm2) | 2.028±0.128 | 2.352±0.298* | |
| Cortex | BA/TA (%) | 42.80±1.80 | 40.00±2.00* |
| MA (mm2) | 1.161±0.089 | 1.414±0.208* | |
| Ct.Th (mm) | 0.184±0.009 | 0.188±0.011 | |
| BMD (mg HA/cm3) | 1113.169±18.433 | 1132.091±17.407* | |
Data represents mean ± standard deviation for parameters measured. BV=Bone Volume; TV= Total Volume; Conn. Dens.= Connectivity Density; SMI= Structural Model Index; Tb. N= Trabecular Number; Tb.Th= Trabecular Thickness; Tb.Sp= Trabecular Separation; BMD= Bone Mineral Density; pMOI= Moment of Inertia; TA= Total Area; BA= Bone Area; MA= Marrow Area. Group size n = 5-10.
p-values < 0.05
3.2. Sostdc1−/− mice exhibit enhanced cortical bone formation
Histomorphometric analysis of mid-femur regions of 5.5 month old mice on the periosteal and endocortical surfaces indicated enhanced bone formation in Sostdc1−/− mice, with a significant increase in mineralized surface (MS/BS) and 78% increase on the periosteal surface (p<0.035). An 8% increase on the endocortical surface was also observed, but this change was not statistically significant. We also observed a 30% increase in the bone formation rate (BFS/BS) on the periosteal surface of Sostdc1−/− mice compared to WT controls, indicating significantly more active bone formation on the periosteal surface compared to the endocortical surface. There was a significant 32% decrease in BFR/BS (p=0.0497) and 42% decrease in mineral apposition rate (MAR) (p<0.00013) on the endocortical surface and no change on the periosteal surface in Sostdc1−/− mice compared to WT controls (Table 2). The decreased change in both MAR and BFR/BS suggests that the endocortical region of Sostdc1−/− femora has a reduced remodeling rate, which, combined with the more active bone formation rate on the periosteal surface, contributes to the high bone mass phenotype and larger bone morphology observed in the cortices of Sostdc1−/− femora (Table 2). In addition, we compared intact mid-diaphyseal μCT data at 12 weeks of age data (Sup. Fig. 1C). Only the marrow area was significantly larger in the Sostdc1−/− femora at this time point. Cortical reconstruction images do not indicate cortical thinning or enlargement indicative of premature aging in Sostdc1−/− mice (Sup. Fig. 1A,B). These data show that lack of Sostdc1 promotes periosteal cortical bone formation, suggesting that Sostdc1, similarly to Sost, acts as a negative regulator of bone formation, although its function is regionally restricted to the diaphysis due to its periosteal expression [26].
TABLE 2.
Histomorphometric analysis of Periosteal and Endocortical regions of 5.5 Month-Old Sostdc1−/− Femurs Compared to WT Controls.
| Region | Index | WT | Sostdc1−/− | % change |
|---|---|---|---|---|
| MS/BS (%) | 19.576±5.042 | 34.89±14.517* | +78 | |
| Periosteal | MAR (μm/d) | 0.025±0.005 | 0.019±0.005 | −23 |
| BFR/BS (μm3/μm2/d) | 1.794±0.667 | 2.333±1.115 | +30 | |
|
| ||||
| MS/BS (%) | 47.023±9.881 | 51.185±13.328 | +8 | |
| Endocortical | MAR (μm/d) | 0.027±0.004 | 0.016±0.003* | −42 |
| BFR/BS (μm3/μm2/d) | 4.605±0.905 | 3.090±1.394* | −32 | |
Data represents mean ± standard deviation for parameters measured. MS = mineralizing surface; BS = bone surface; MAR = mineral apposition rate; BFR = bone formation rate. Group size n = 6.
p-values < 0.05
We examined osteoclast differentiation in these mice as well, and scored hematopoietic progenitors by flow cytometry (Sup. Fig. 2A, B). We found no significant differences (p<0.05) in numbers of osteoclast progenitors of the types indicated. In addition, TRAP stains on 12-week old mice (Sup. Fig. 2C) indicate that there are no gross differences in mature osteoclast number. These data corroborate evidence by histomorphometry that do not suggest increased resorption in Sostdc1−/− compared to mice WT.
3.3. Sostdc1-positive cells participate in early fracture repair
Sostdc1 expression in vivo was examined by tracking the LacZ reporter from the Sostdc1 knock-in allele [8]. In neonatal Sostdc1+/− mice, LacZ expression was observed in tissues adjacent or near the bone, including the adipose and periarticular cartilage (Sup. Fig. 3A-E). We also noted LacZ expression in the proliferating chondrocytes of the neonatal epiphysis [12] (Sup. Fig. 3A, E); this expression did not persist into the adult growth plate or articular cartilage (Sup. Fig. 3F, J). In intact adult Sostdc1+/− femora, LacZ was observed in the periosteum, muscle, and vasculature (Sup. Fig. 3F-J).
To examine the functional consequence of losing Sostdc1 expression in adult mice, we performed transverse femoral fractures in Sostdc1−/− mice, and tracked the Sostdc1-deficient cells using LacZ. In unfractured Sostdc1−/− femora, LacZ was expressed in the same pattern as the LacZ determined for Sostdc1+/− femora, primarily in the periosteum (Fig. 1A), muscle and vasculature (Fig. 1E). At 3 days post fracture (D3), the rapidly expanded periosteum contained primarily LacZ-positive cells (Fig. 1B). Furthermore, LacZ-positive cells appeared to migrate from the interstitial space of the adjacent injured skeletal muscle and the vasculature (Fig. 1B-C, F-G, I-J). The elevated density of LacZ-positive cells was confined to the fractured limb (B, F, I), in regions adjacent to the injury site, and was not observed in the contralateral limb (Fig. 1A, E). We found no expression LacZ-positive cells in the chondrocytes of uninjured animals, including in the growth plate (Sup. Fig. 4A). However, patellar chondrocytes distal to the injury site on the broken limb activated LacZ expression shortly after injury (Sup. Fig. 4B). This suggests that Sostdc1 transcriptional activation is mediated locally by the traumatic injury.
Fig. 1.
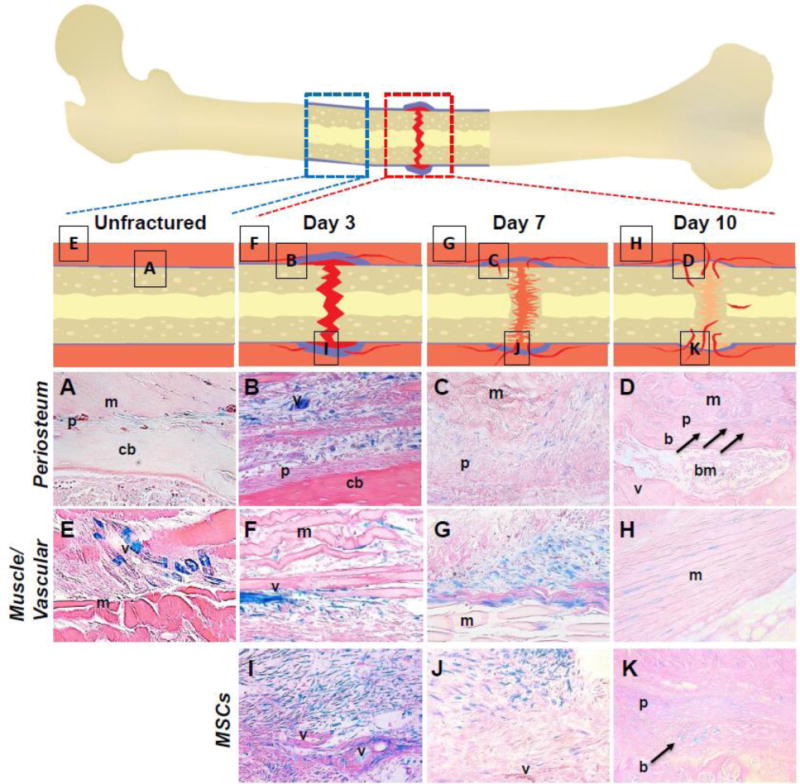
Histological characterization of early time points in fracture repair of Sostdc1−/− mice shows LacZ expression [as a surrogate for Sostdc1 expression] in and around the early fracture callus. Schematics are shown for unfractured, and the fractured femora at 3, 7, and 10 days post-fracture; boxes within this schematic indicate regions visualized for each time point. Deep orange- soft tissue outside bone; blue- periosteum and undifferentiated cells; red- clot or vasculature; light orange- cartilage callus; yellow- bone. Control unfractured Sost−/− limb (A, E) shows LacZ expression in the periosteum (A), and in intermittent cells in muscle tissue and peripheral vascular tissue (E). Images taken at D3 post-fracture show LacZ expression in the expanded periosteum (B) and evidence of cell migration into the callus (I). Cells within muscle and periosteum distal to the fracture site are also strongly LacZ-positive at this early time point (F). In nearby muscle, LacZ-positive cells appear to occupy the interstitial space, and appear to migrate out from the vasculature (B, F, I). At D7 LacZ expression begins to fade (C, G, J). The outside of the callus is marked by LacZ-positive cells (C). Undifferentiated cells continue to be found near the periphery of the callus in undefined tissue (G). At the edges of the muscle, positive cells are located in distinct cluster near connective tissue surrounding muscle bundles (G, J). By D10 post-fracture, LacZ expression is reduced both in terms of numbers and intensity (D, H, K). Newly formed periosteum, embedding osteoblasts, and nearby developing muscle show cells around the boundaries that express LacZ (arrows) (D, H). Undifferentiated cells near the periphery of the callus retain LacZ expression (K). LacZ expression is also found in the healing muscle tissue outside the callus proper (D). Images are shown at 40× magnification. m muscle; v blood vessel; p periosteum; pa patella; cb cortical bone; hc hypertrophic chondrocytes; bm bone marrow; b newly formed bone; ch chondrocytes.
At 7 days post injury (D7) many LacZ-positive cells remained in the periphery of the developing callus (Fig. 1C, G, J). Migratory cells remained present in the soft tissue surrounding the bone injury (Fig. 1G, J). Fewer LacZ-positive cells were observed in the vasculature (Fig. 1J). By 10 days post fracture (D10), weak LacZ expression emerged in the developing cartilage callus (Fig. 1K), and LacZ-positive cells had embedded into the newly formed bone surface (Fig. 1D, K; arrows). Histologically, we observed bone formation as cells with mesenchymal morphology, yet surrounded by abundant matrix, increasing their cell-to-cell spacing (Fig. 1D, K), in contrast to the tightly packed mesenchymal cells observed in the D3 and D7 callus (Fig. 1B-C; F-G, I-J). At this stage of repair, LacZ expression decreased in the fracture callus and in the surrounding tissues. LacZ expression primarily marked the boundary between muscle and bone at the edge of the callus and the soft tissue near the skin (Fig. 1D, H, K). Immature chondrocytes near the callus edge continued to show weak LacZ expression, yet no LacZ expression was observed in the hypertrophic chondrocytes of the callus, consistent with the lack of expression in uninjured adult bones (Sup. Fig. 4C, D).
LacZ-positive cells were confined to the periosteal and soft tissue compartments of the Sostdc1−/− limb, at all time points examined. No LacZ-positive cells were found on the endosteal surfaces or in the marrow cavity of contralateral, uninjured limbs, nor was LacZ activated in these regions of the injured limb, in response to injury. No LacZ-positive cells were seen in the metaphyseal trabecular niche of the uninjured bones, or in the injured bones in response to injury at any time point examined (Supporting Fig. 3A, D, F). During fracture repair, we conclude that LacZ-positive cells in Sostdc1−/− injured femora, and hence Sostdc1-positive cells in injured WT femora, appear to migrate from the soft tissues surrounding the injury, including periosteum, vasculature, and/or muscle, and participate in early fracture repair events. Participation of these LacZ-positive cells diminished at later milestones of healing, and was not present in the mature cartilage or in bone cells, suggesting that Sostdc1 may mark a population of periosteal osteochondral progenitor cells needed for fracture repair.
3.4. Fracture healing program is altered in Sostdc1−/− mice
Histological analysis of Sostdc1−/− calluses revealed several differences between Sostdc1−/− mice and WT or Sostdc1−/+ controls throughout the 28-day fracture healing interval examined (Fig. 2). Starting at D3, Sostdc1−/− mice had an enhanced periosteal reaction, as characterized by a thicker layer of undifferentiated cells in the periosteal areas adjacent to the injury (Fig. 2A, F). By D7, the cartilage callus was larger in Sostdc1−/− mice and intramembranous bone formation was more robust (Fig. 2B), with pronounced vascular invasion, as indicated by the presence of red blood cells in vessels (Fig. 2B; arrow). In contrast, WT calluses displayed insignificant intramembranous bone formation or neovascularization (Fig. 2G). At D14, while vascular invasion was occurring only at the periphery of the cartilage callus of WT animals (Fig. 2H), Sostdc1−/− mice had evident neovasculature deeper within the interior of the callus (Fig. 2C). By D21, the calluses of each genotype looked distinctly different, with Sostdc1−/− calluses showing a thick cortical shell surrounding the callus, very little woven bone in the interior of the callus, and original cortical bone that was remodeling, compared to WT (Fig. 2D, I; arrows). These mature calluses in Sostdc1−/− mice showed no evidence of persistent cartilage in the callus relative to matched controls. These data suggest that the absence of Sostdc1 accelerates and/or enhances intramembranous bone formation and neovascularization in the callus.
Fig. 2.
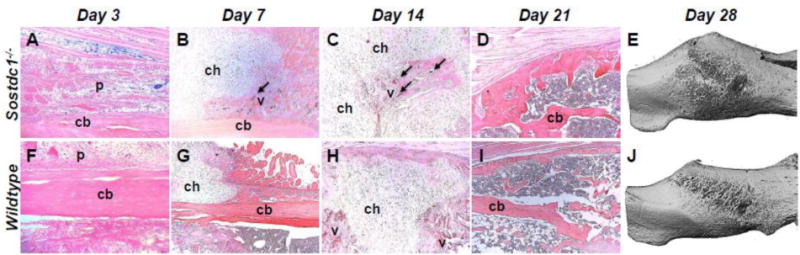
Fracture callus is larger at 28 days post-fracture in Sostdc1−/− mice and H&E stains indicate differential progress during healing milestones. Periosteal reaction is more extensive in Sostdc1−/− compared to WT controls at D3 post-fracture (A, F). Chondrogenesis during early callus formation (D7) shows increased vascular invasion (arrow) in Sostdc1−/− mice compared to controls (B, G). Vascular invasion at D14 progresses to the middle of the cartilaginous callus in Sostdc1−/− mice, while neovascularization is occurring in the peripheral callus in WTs (arrows) (C, H). At D21, a thick cortical shell around the callus is evident in Sostdc1−/− mice (arrow), a reduced amount of trabecular bone formation in the interior of the callus is also present, and the original cortical bone is remodeling (arrow) while in WTs there is more trabecular bone, thinner cortical shell (arrow), and the original cortex remains unremodeled (D, I). Micro-CT analysis of 28-day calluses shows a greater callus volume in Sostdc1−/− (p<0.005) (E, J) (See also Table 2). p periosteum; cb cortical bone; chondrocytes; v blood vessel.
3.5. Sostdc1−/− mice have larger, more mineralized fracture calluses with normal mechanical strength
Sostdc1−/− fractured limbs had a significantly increased callus size (60%, p<0.05) and increased apparent bone mineral density (3%; p<0.0005) at D28, compared to WT calluses (Fig. 2E, J; Table 3). While bone volume was increased (p<0.01), BV/TV was increased by 36%, but this value was not significantly different between Sostdc1−/− and WT controls, due to large animal to animal variation. Mechanical testing of intact bones and calluses revealed that Sostdc1−/− femora and calluses were not mechanically different from WT controls (Table 3), as determined by torsional stiffness, ultimate torque at failure and rotation at ultimate failure quantification. Larger calluses (Fig. 2E, J), with an increased mineral content indicated they were structurally mature. These data suggests that Sostdc1−/− fractured femora have an expedited healing program compared to WT controls, and that the healed calluses may yield stronger repaired bones.
TABLE 3.
Biomechanical properties determined by μCT and Torsional Testing of the Mature Callus Compartment of Sostdc−/− Mice Compared to WT Controls, at 28 Days Post-Fracture.
| Index | WT | Sostdc1−/− | |
|---|---|---|---|
| Callus Volume (mm3) | 17.495±5.328 | 28.760±9.413* | |
| Callus | BV (mm3) | 3.804±0.966 | 5.197±1.042* |
| BV/TV (%) | 23.30±0.88 | 19.10±4.40 | |
| BMD (mg HA/cm3) | 992.936±11.032 | 1022.581±11.220* | |
|
| |||
| Stiffness (Nm/deg) | 0.00171±0.0005 | 0.00192±0.0007 | |
| Control | Ult. Torque (Nm) | 0.032±0.006 | 0.032±0.009 |
| Rot. Ult. Torque (degrees) | 24.983±6.020 | 20.888±5.371 | |
|
| |||
| Stiffness (Nm/deg) | 0.00073±0.0005 | 0.00064±0.0003 | |
| Fractured | Ult. Torque (Nm) | 0.021±0.009 | 0.018±0.005 |
| Rot. Ult. Torque (Degrees) | 47.200±17.339 | 35.500±14.343 | |
Data represents mean ± standard deviation for parameters measured. BV=Bone Volume; TV= Total Volume; BMD= Bone Mineral Density; Ult. Torque= Ultimate Torque at failure; Rot. Ult. Torque= Rotation at Ultimate Torque.
p-values < 0.05
3.6. Sostdc1 marks a population of osteochondral progenitor cells
Since Sostdc1-positive cells display migratory, proliferative, and differentiation capabilities consistent with those of mesenchymal stem cells (MSCs), we next examined co-localization of stem cell markers nestin and alpha smooth muscle actin (α-SMA) with LacZ as a surrogate of Sostdc1 expression, in D3 and D7 calluses (Fig. 3). We compared LacZ expression in Sostdc1−/− and Sostdc1−/+ calluses using an antibody specific for its gene product, β-galactosidase. Nestin is a mesenchymal cell marker used in flow cytometry to identify mesenchymal stem cell populations [27] and α-SMA marks progenitor cells that can differentiate into chondrocytes and osteoblasts [2]. β-galactosidase antibody stain (Fig. 3G-R; red) closely resembled the β-galactosidase enzymatic activity (Fig. 3A-C). While nestin and β-galactosidase co-localized in only a few periosteal cells in Sostdc1−/− intact femora (Fig. 3G, J), at D3 there was a discernable increase in the population of nestin - LacZ double positive cells, in the expanded periosteal region of Sostdc1−/− fractures (Fig. 3H, K). By D7, a large fraction of the Sostdc1−/− callus harbored nestin - LacZ double positive cells (Fig. 3I), and now a smaller such population emerged in the Sostdc1−/+ callus (Fig. 3L), suggesting that Sostdc1 may mark a unique population of periosteal derived stem cells. In contrast, at D3, α-SMA - LacZ marked mostly cells resembling well-developed blood vessels at the muscle-bone interface of Sostdc1−/− calluses (Fig. 3N, Q), with insignificant evidence of neovascularization at this time-point in Sostdc1−/+ controls. By D7, the population of α-SMA – LacZ-positive increased in Sostdc1−/− calluses without a correspondent increase in the Sostdc1−/+ control calluses (Fig. 3O, R). The overlapping expression of β-galactosidase with either nestin or α-SMA initiated at ~D3 and showed similar elevated levels near the site of injury at D7, suggesting that they mark a small population of rapidly dividing cells that are migrating from the periosteum and/or vasculature into the injured site.
Fig. 3.
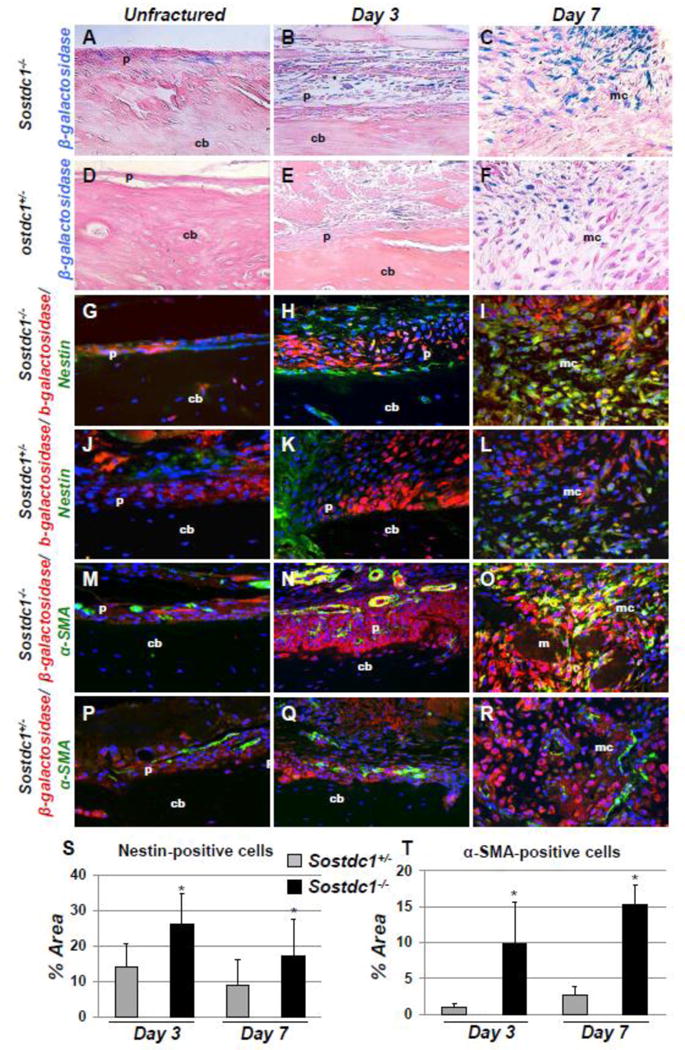
Nestin and α-smooth muscle actin co-localize with Sostdc1 [using LacZ from the knocked in allele]. LacZ-stained bones show positive cells on the periosteal surface of the unbroken bone (A). LacZ–positive cells participate in the periosteal expansion at (B). This population of LacZ -positive cells expands into the undifferentiated callus tissue at D7 (C). LacZ expression in Sostdc1+/− mice is qualitatively reduced in intact periosteum, D3 and D7 fracture calluses, suggesting the pool of LacZ-positive cells is expanded in Sostdc1−/− mice (D-F). Dual marker immunofluorescent staining indicates the presence of both LacZ and Nestin at the periosteal surface in unfractured femora, while there are fewer Nestin(+) cells in Sostdc1+/− (G, J). During the periosteal reaction, some cells expressing Nestin also show LacZ expression (H, K), while the two groups of cells are mutually exclusive in Sostdc1+/−. By D7, many cells in the undifferentiated callus region of Sostdc1−/− are positive for both markers, while substantially fewer double-labeled cells are present in Sostdc1+/− samples (I, L). [red β-galactosidase (LacZ); green Nestin (G-L); green α-Smooth muscle actin (α-SMA) (M-R)]. α-SMA is also shown in periosteum of unfractured femora of Sostdc1+/− and Sostdc1−/− mice (M, P). Cells and vessels positive for α-SMA are much more abundant, have more dual label, and vessel are of a larger diameter in Sostdc1−/− mice at D3 post-fracture (N, Q). There is significant dual-labeling of undifferentiated cells in the D7 fracture callus of Sostdc1−/− mice, while there is very little overlap and very few α-SMA-positive cells in Sostdc1+/− calluses (O, R). Quantitation of Nestin signal in D3 and D7 calluses of Sostdc1−/− mice revealed a significantly higher percentage of image area covered by Nestin(+) cells compared to Sostdc1−/− calluses at both time points (S). Quantitation of α-SMA signal in D3 and D7 calluses of Sostdc1−/− mice revealed a significantly higher percentage of image area covered by α-SMA -positive cells compared to Sostdc1−/+ calluses at both time points (T). [p periosteum; cb cortical Bone; mc mesenchyme.]
Quantitative analysis of nestin and α-SMA-positive cells, revealed a significant increase in the number of stem cell marker positive cells in Sostdc1−/− mice, compared to controls (Fig. 3S, T) which suggests that lack of Sostdc1 promotes or accelerates progenitor or stem cell response to injury and supports a role for Sostdc1 in stem cell maintenance. Protein expression of Osterix (Sp7/Osx), a transcription factor essential for the differentiation of pre-osteoblasts into mature osteoblasts [28], was similarly analyzed. Significant differences were observed in intact femora, where Sostdc1−/− periosteal cells expressed higher levels of Osx than controls (Fig. 4A, D). At D3, the periosteal region closest to the cortical bone showed increased expression of Osx in Sostdc1−/− mice (Fig. 4B, E). Significantly more Osx-positive cells remained near the periosteal surface of Sostdc1−/− D7 calluses, compared to Sostdc1+/− controls (Fig. 4C, F, G) suggesting that lack of Sostdc1 promotes differentiation towards the osteoblast lineage.
Fig. 4.
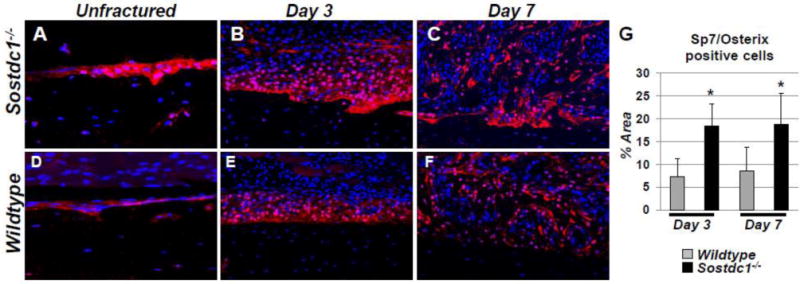
SP7/Osterix (Osx) osteoblast precursor levels are dramatically increased in Sostdc1−/− mice during fracture repair. Immunostains for Osx in Sostdc1−/− mice show an increased number of positive cells at the resting periosteal bone surface of unbroken bones compared to WT controls (A, D). The layer of cells that participate in the periosteal reaction at D3 post fracture is thicker and has more Osx-positive cells, especially at the periosteal surface, compared to controls (B, E). At D7 post-fracture, Osx levels are activated in developing intramembranous callus, which is larger and contains more positive cells in Sostdc1−/− mice, especially at the periosteal surface (C, F). Quantitation of signal area at D3 and D7 revealed an increased area of Osx-positive cells in Sostdc1−/− mice at both time points examined (G).
3.7. Sostdc1−/− hastens the expansion and differentiation of mesenchymal cells during fracture repair
Using cell surface signatures established for mesenchymal stem cells (MSC) [25] and mature osteoblasts (OB) [25] we quantified MSC and OB populations in Sostdc1−/− and WT femora in intact contralateral limbs and 5, 6, 7 days post fracture. No significant differences were observed in the frequency of MSCs or OBs in Sostdc1−/− and WT intact femora or at D7 (Table 4). At D5, while the MSC and OB populations remained at baseline levels in WT femora, the Sostdc1−/− injured bones had significantly more MSCs (>2-fold; p<0.0105) and OBs (>3-fold; p<0.00246). At D6, the WT MSC population reached 8-fold above baseline; whereas the MSC population in the Sostdc1−/− was significantly lower than WT (Table 4, Fig. 5B). The Sostdc1−/− OB population tended to outpace the magnitude observed in the WT, at D6, but was stabilized between the genotypes by D7, with significant differences only observed at D5 (Table 4). We also quantified the populations of endothelial cells and found no significant differences between WT and Sostdc1−/−, at all time points examined. Furthermore, we found LacZ expression only within MSCs, consistent with Sostdc1 marking a subpopulation of osteochondral progenitor cells (Fig. 5A).
TABLE 4.
Cell populations in the Femur during Fracture Repair in Sostdc1−/− Mice Compared to WT Controls.
| Days Post Fracture | (N) | Mesenchymal Stem Cells | Osteoblasts | Endothelial | |||
|---|---|---|---|---|---|---|---|
| WT | Sostdc1−/− | WT | Sostdc1−/− | WT | Sostdc1−/− | ||
| Unfractured | 11 | 2.14±1.82 | 2.40±2.51 | 8.74±4.95 | 7.80±4.67 | 9.77±5.98 | 14.33±12.48 |
| Day 5 | 4 | 1.98±0.68 | 5.50±1.67* | 8.68±4.89 | 26.63±2.78* | 5.77±1.99 | 11.32±5.7 |
| Day 6 | 3 | 16.35±1.43 | 11.06±1.02* | 36.31±5.89 | 44.49±10.83 | 17.03±2.92 | 15.98±6.59 |
| Day 7 | 4 | 3.41±3.04 | 1.37±1.08 | 45.38±11.98 | 48.58±25.06 | 9.25±3.29 | 8.25±5.11 |
| Area Under the Curve# | 48.83 | 48.13 | 864.67 | 957.74* | 129.95 | 168.96 | |
Data represents mean percentages of cells in whole digested femurs ± standard deviation for parameters measured. All contralateral unfractured femurs for all time-points [days 5, 6, and 7] were averaged to obtain the unfractured values for WT and Sostdc1−/− mice.
p-values < 0.05;
to calculate are under the curve unfractured values were used for day 0, day 14 [MSCs; ENDO] and day 35 [OBs] under the assumption that MSC and OBs would revert to unfractured levels at these time points (Figure 5B).
Fig. 5.
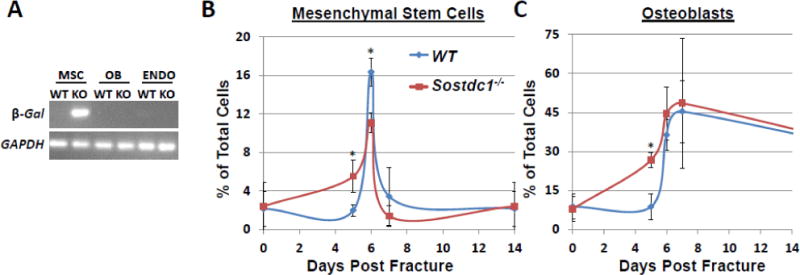
Quantification of mesenchymal stem cells (MSC) and osteoblasts (OB) populations during fracture repair. Purified MSCs from Sostdc1−/− femurs were found to express β-galactosidase, but no expression was detected in OB or endothelial (ENDO) cells, suggesting that Sostdc1 is expressed in MSCs only (A). A time course quantification of MSCs (B) and OBs (C) determined that both MSC and OB expansion curves are shifted to the left, suggestive of earlier expansion of MSCs during fracture repair.
We next questioned whether loss of Sostdc1 enhances fracture repair by enhancing the differentiation of MSCs in a cell autonomous fashion, or whether the enhanced fracture repair resulted from increased number of both MSCs and OBs in Sostdc1−/− mice. To discriminate between the two potential scenarios, we plotted frequency of each cell type as a function of days post fracture. The resulting curve for MSCs exhibited a left shift in Sostdc1−/− compared to WT mice (Fig. 5B), as did the curve of OBs (Fig. 5C), however, computing the area under the curve generated highly similar values for Sostdc1−/− and WT MSC profiles (Table 4). This temporal shift in MSC expansion can also be visually observed in the nestin and α-SMA immunostains where the double positive population is always higher in the Sostdc1−/− (Fig. 3H, I, K, L, N, O, R) than in the Sostdc1−/+ calluses. However, the immunostain quantification indicates both significantly more nestin+ cells and α-SMA+ cells (indicators of stem cells) in Sostdc1−/− at D7 compared to Sostdc1−/+ (Fig. 3). This difference may be due to the use of whole broken bones for the flow cytometry analysis, compared to fracture-callus-only for immunostain quantification. In addition, the immunostain quantification used single markers, and both nestin and α-SMA may also indicate the presence of endothelial cells [29] while the cell surface markers define endothelial cells as a separate population, and could contribute to the differences seen at D7 between the two analyses. The temporal shift in MSCs populating the callus is consistent with a rapid shift in cellular identity which contributes to the accelerated differentiation into OB in the Sostdc1−/− calluses (see also Fig. 4) and is reconciled as a greater area under the curve for Sostdc1−/− OBs (Fig. 5, Table 4). As later time points did not have osteoblast quantification by flow cytometry, it is not possible to determine whether there is a greater absolute number of osteoblasts throughout fracture healing in Sostdc1−/− compared to WT, or if there is simply an early shift toward differentiation in the Sostdc1−/− calluses. However, at late time points during repair, Sostdc1−/− mice show enhanced callus volume, BV, and mineral density (Table 3) suggesting that increased osteoblast differentiation, or relative number early in repair, translates to more bone formation (Table 3).
3.8. Loss of Sostdc1 activates Wnt signaling during fracture repair
Sostdc1 had been described as both a BMP and a Wnt antagonist [16], and we have previously shown that Sostdc1 does not behave as a canonical Wnt antagonist in the context of limb development [12]. We examined the expression pattern of activated β-catenin in the fracture callus at D3 and D7 to determine whether Sostdc1 behaves similarly in the context of fracture repair. In contrast to our limb analyses, we found dramatically increased levels of activated β-catenin in Sostdc1−/− mice, compared to controls (Fig. 6). In unbroken bones, we found increased signal at the periosteal surface and in the metaphyseal niche of Sostdc1−/− mice, compared to controls (Fig. 6A, B, F, G). At D3 post-fracture, higher levels of activated β-catenin were observed in the periosteum as well as in the cortical bone of Sostdc1−/− femora (Fig. 6C, H). By D7, increased levels of activated β-catenin were observed in the periphery of the developing cartilaginous callus (excluding chondrocytes), and the expression intensified in regions resembling blood vessels in the callus (Fig 6D, I; brackets/arrows). Furthermore, the developing neovascular network in the callus consisted of larger diameter vessels compared to controls. Woven bone present in the marrow space of D7 injured femurs also contained elevated levels of activated β-catenin in Sostdc1−/− mice (Fig. 6E, J). The increase in activated β-catenin levels in the metaphysis and cortical bone suggests that Sostdc1 affects canonical Wnt signaling in a non-cell-autonomous manner, and it implies that the increase in cortical bone is due to elevated Wnt signaling.
Fig. 6.
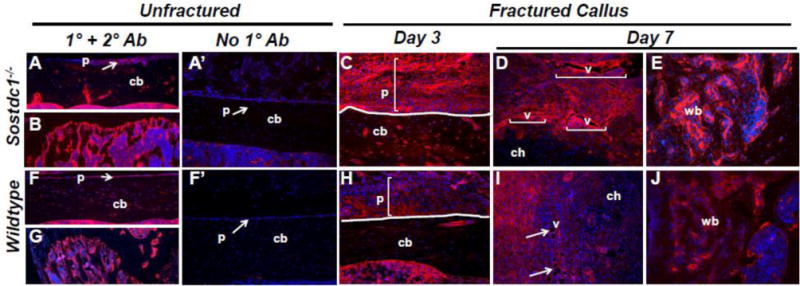
Higher Levels of Activated β-catenin are detected in Sostdc1−/− mice during fracture repair. Immunostains for activate β-catenin in Sostdc1−/− and WT mice show increased β-catenin levels at the resting periosteal and metaphyseal bone surfaces in unbroken bones compared to WT controls (A, B, F, G). The layer of cells that participate in the periosteal reaction at D3 post-fracture is thicker and has more robust activated β-catenin compared to controls (brackets) (C, H). At D7 post-fracture, β-catenin is activated in the developing neovasculature, which is larger and more plentiful in Sostdc1−/− calluses compared to WT (brackets, arrows). Chondrocytes are negative for activated β –catenin (D, I). Sostdc1−/− show more activated β-catenin in woven bone at D7 post-fracture compared to controls (E, J). Negative controls stained with the secondary antibody only show insignificant background stain in the periosteum of both Sostdc1−/− (A’) and WT (F’) unfractured femurs [p periosteum; cb cortical bone; ch chondrocytes; v blood vessel; vb woven bone.]
4. Discussion
Sost is a potent negative regulator of bone formation and Sost deficiency contributes to high bone mass and enhanced fracture healing phenotypes; yet its paralog Sostdc1, which is expressed in the periosteum, has not yet been examined for its potential contribution to bone repair despite the established role of periosteal cells in fracture healing [4, 30]. Here we report that global loss of Sostdc1 results in a complex skeletal phenotype characterized by an increase in femoral cortical bone structure and BMD, but with reduced trabecular bone mass. Since periosteal osteoblasts are responsible for outward expansion of long bones, our results suggest that lack of Sostdc1 increases the osteoblast activity locally in the periosteum, resulting in larger, thicker bone cortices. The increased moment of inertia (pMOI) in Sostdc1−/− femurs suggested that the increase in periosteal activity among Sostdc1−/− mice might translate into improved bending properties for the diaphysis. These findings are consistent with results published by He et al., who correlated a polymorphism in Sostdc1 with low lumbar BMD but not with femoral neck or total hip BMD, in Chinese women [31]. Since lumbar vertebrae are primarily composed of trabecular bone, this human association study suggests that mutations that interfere with Sostdc1 function may negatively influence trabecular BMD to a greater extent than cortical BMD.
Previous studies have shown that both Sostdc1 and Sost inhibit Wnt signaling by binding to multiple Lrp co-receptors (Lrp1, Lrp4, Lrp5 and Lrp6) [32–34], and that Sost primarily functions in a cell non-autonomous manner wherein it is secreted by osteocytes and binds to receptors on the osteoblast surface. The loss of Sost results in a robust response in trabecular bone, contributing to strengthening and replenishment of trabeculae in the case of osteoporosis. Loss of Sost, as with Sclerostin-neutralizing antibody treatment, also directs fracture calluses and stem cells toward enhanced bone formation, although it is not yet clear by what mechanism [35, 36]. Thus, we speculated that due to its expression in periosteum, Sostdc1 may exert its effects on periosteal osteoblasts in a similar fashion, where it is secreted by periosteal cells and binds to Lrp co-receptors on the neighboring osteoblasts residing on the periosteal bone surface. Since osteoblasts residing on the periosteal surface of cortical bones are likely to be exposed to both Sostdc1 from the periosteum and to Sost from the underlying osteocytes, the periosteal osteoblasts may be more sensitive to levels of Wnt antagonists, and therefore may be more likely to upregulate β-catenin-dependent Wnt signaling in the absence of Sostdc1 than osteoblasts residing on the trabecular surfaces. Immunohistological analysis of activated β-catenin expression supports this hypothesis, where we observe greater levels of activated β-catenin on the periosteal surface of Sostdc1−/− than in Sostdc1+/− controls (Fig. 6A, F).
Since the cambium layer of the periosteum is also a major source of osteoblast and chondrocyte progenitors during fracture healing, we also examined whether Sostdc1-deficient periosteal cells interfere with normal fracture healing. After injury, a typical periosteal thickening was observed accompanied by a rapid expansion of Sostdc1-LacZ positive cells with mesenchymal morphology that populated most of the callus forming region at D3. Beyond D7, these LacZ positive cells became restricted to regions closer to the periosteum, suggesting that the Sostdc1-expressing cells rapidly expand during periosteal reaction and are subsequently recruited into the fracture site where they participate in the soft callus formation. FACS analysis of MSCs in combination with histological staining with stem cell markers nestin and α-SMA indicate that Sostdc1-positive cells in the early fracture callus mark a subpopulation of multipotent mesenchymal stem cells that migrate into the callus from the periosteum. The behavior of Sostdc1-positive cells is similar to osteochondroprogenitor cells in the periosteum that, using lineage tracing, make a major contribution to the soft callus [30]. The role of Sostdc1 as a WNT antagonist, and the known role of WNT/β-catenin signaling to promote osteogenic[37] and chondrogenic [38, 39] differentiation and maturation, suggests that Sostdc1 may act as the rate-limiting inhibitor of the differentiation of pluripotent periosteal cells during fracture repair. Indeed, we observe a temporal shift in the frequency of MSC and OB subpopulations during fracture repair in Sostdc1−/− mice, as well as greater area-under-curve of OBs, but not of MSCs, indicating more rapid activation of migration, proliferation and differentiation of MSCs and OBs, and further supporting the role of Sostdc1 as an osteochondral progenitor gatekeeper. Further exploration of Sostdc1 may define it as a morphogen that functions to maintain stem cells in a progenitor state [40].
5. Conclusion
This work describes Sostdc1 activity in a new context, highlighting its potential role in the metabolism and repair of the skeleton. In addition, for the first time we have linked Sostdc1 to the behavior of mesenchymal stem cells, which is consistent with, and mechanistically may explain Sostdc1-related phenotypes noted by other published studies, such as in cancer prognosis, tooth development, kidney injury resistance, and diet-induced obesity resistance. We have shown that Sostdc1LacZ is expressed in Sostdc1−/− MSCs, therefore, Sostdc1 expression marks a subpopulation of osteochondral progenitor cells, and have shown that stem cell response is enhanced in the absence of Sostdc1 after injury, accelerating bone repair. We have demonstrated that Sostdc1 is important for trabecular bone maintenance, bone formation, early fracture repair events, and the lack of Sostdc1 influences mesenchymal stem cell behavior in response to injury, in vivo. Future work may show a synergy between the loss of Sost and Sostdc1 to combine enhanced trabecular bone and enhanced cortical bone in the treatment of fractures and osteoporosis.
Supplementary Material
Highlights.
Sostdc1 is expressed in the periosteum
Sostdc1−/− mice exhibit increased cortical bone volume caused by an increase in bone formation
Sostdc1-positive periosteal cells participate in early fracture repair
Sostdc1−/− mice have larger, more mineralized fracture calluses with normal mechanical strength
Sostdc1 marks a population of osteochondral progenitor cells
Lack of Sostdc1 elevates Wnt signaling in the fracture callus
Acknowledgments
We would like to thank the National Institutes of Health (NIH) Knock-Out Mouse Program (KOMP) and Regeneron for providing the Sost and Sostdc1 knockout mice. We are also grateful to David Gravano for his assistance with FACS analysis. NMC, CSY, DKM and GGL were supported in part by NIH grant DK075730. NMC and GGL were also supported in part by LLNL LDRD ER (11-ERD-060). This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
Author Contributions
NMC and GGL conceptualized the hypothesis, designed the experiments, analyzed data and wrote the article. NMC, CSY, DKM, NRH, LX, ANE, JOM, AGR, BAC conducted experiments and analyzed data. NMC, CSY and DKM conducted animal study and animal care.
References
- 1.Colnot C. Cell sources for bone tissue engineering: insights from basic science. Tissue Eng Part B Rev. 2011;17:449–57. doi: 10.1089/ten.teb.2011.0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grcevic D, Pejda S, Matthews BG, Repic D, Wang L, Li H, Kronenberg MS, Jiang X, Maye P, Adams DJ, Rowe DW, Aguila HL, Kalajzic I. In vivo fate mapping identifies mesenchymal progenitor cells. Stem Cells. 2012;30:187–96. doi: 10.1002/stem.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthews BG, Grcevic D, Wang L, Hagiwara Y, Roguljic H, Joshi P, Shin DG, Adams DJ, Kalajzic I. Analysis of alphaSMA-labeled progenitor cell commitment identifies notch signaling as an important pathway in fracture healing. J Bone Miner Res. 2014;29:1283–94. doi: 10.1002/jbmr.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colnot C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res. 2009;24:274–82. doi: 10.1359/jbmr.081003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19:329–44. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurikkala J, Kassai Y, Pakkasjarvi L, Thesleff I, Itoh N. Identification of a secreted BMP antagonist, ectodin, integrating BMP, FGF, and SHH signals from the tooth enamel knot. Dev Biol. 2003;264:91–105. doi: 10.1016/j.ydbio.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 7.Yanagita M, Okuda T, Endo S, Tanaka M, Takahashi K, Sugiyama F, Kunita S, Takahashi S, Fukatsu A, Yanagisawa M, Kita T, Sakurai T. Uterine sensitization-associated gene-1 (USAG-1), a novel BMP antagonist expressed in the kidney, accelerates tubular injury. J Clin Invest. 2006;116:70–9. doi: 10.1172/JCI25445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka M, Endo S, Okuda T, Economides AN, Valenzuela DM, Murphy AJ, Robertson E, Sakurai T, Fukatsu A, Yancopoulos GD, Kita T, Yanagita M. Expression of BMP-7 and USAG-1 (a BMP antagonist) in kidney development and injury. Kidney Int. 2008;73:181–91. doi: 10.1038/sj.ki.5002626. [DOI] [PubMed] [Google Scholar]
- 9.Clausen KA, Blish KR, Birse CE, Triplette MA, Kute TE, Russell GB, D’Agostino RB, Jr, Miller LD, Torti FM, Torti SV. SOSTDC1 differentially modulates Smad and beta-catenin activation and is down-regulated in breast cancer. Breast Cancer Res Treat. 2011;129:737–46. doi: 10.1007/s10549-010-1261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahn Y, Sims C, Logue JM, Weatherbee SD, Krumlauf R. Lrp4 and Wise interplay controls the formation and patterning of mammary and other skin appendage placodes by modulating Wnt signaling. Development. 2013;140:583–93. doi: 10.1242/dev.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmons DG, Kennedy TG. Uterine sensitization-associated gene-1: a novel gene induced within the rat endometrium at the time of uterine receptivity/sensitization for the decidual cell reaction. Biol Reprod. 2002;67:1638–45. doi: 10.1095/biolreprod.102.006858. [DOI] [PubMed] [Google Scholar]
- 12.Collette NM, Yee C, Murugesh D, Sebastian A, Taher L, Gale NW, Economides AN, Harland RM, Loots GG. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev Biol. 2013 doi: 10.1016/j.ydbio.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itasaki N, Jones CM, Mercurio S, Rowe A, Domingos PM, Smith JC, Krumlauf R. Wise, a context-dependent activator and inhibitor of Wnt signalling. Development. 2003;130:4295–305. doi: 10.1242/dev.00674. [DOI] [PubMed] [Google Scholar]
- 14.Ohazama A, Johnson EB, Ota MS, Choi HY, Porntaveetus T, Oommen S, Itoh N, Eto K, Gritli-Linde A, Herz J, Sharpe PT. Lrp4 modulates extracellular integration of cell signaling pathways in development. PLoS One. 2008;3:e4092. doi: 10.1371/journal.pone.0004092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahn Y, Sanderson BW, Klein OD, Krumlauf R. Inhibition of Wnt signaling by Wise (Sostdc1) and negative feedback from Shh controls tooth number and patterning. Development. 2010;137:3221–31. doi: 10.1242/dev.054668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lintern KB, Guidato S, Rowe A, Saldanha JW, Itasaki N. Characterization of wise protein and its molecular mechanism to interact with both Wnt and BMP signals. J Biol Chem. 2009;284:23159–68. doi: 10.1074/jbc.M109.025478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Dinther M, Zhang J, Weidauer SE, Boschert V, Muth EM, Knappik A, de Gorter DJ, van Kasteren PB, Frisch C, Mueller TD, ten Dijke P. Anti-Sclerostin antibody inhibits internalization of Sclerostin and Sclerostin-mediated antagonism of Wnt/LRP6 signaling. PLoS One. 2013;8:e62295. doi: 10.1371/journal.pone.0062295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collette NM, Genetos DC, Murugesh D, Harland RM, Loots GG. Genetic evidence that SOST inhibits WNT signaling in the limb. Dev Biol. 2010;342:169–79. doi: 10.1016/j.ydbio.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho SW, Kwak S, Woolley TE, Lee MJ, Kim EJ, Baker RE, Kim HJ, Shin JS, Tickle C, Maini PK, Jung HS. Interactions between Shh, Sostdc1 and Wnt signaling and a new feedback loop for spatial patterning of the teeth. Development. 2011;138:1807–16. doi: 10.1242/dev.056051. [DOI] [PubMed] [Google Scholar]
- 20.Bonnarens F, Einhorn TA. Production of a standard closed fracture in laboratory animal bone. J Orthop Res. 1984;2:97–101. doi: 10.1002/jor.1100020115. [DOI] [PubMed] [Google Scholar]
- 21.Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A. Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 1999;208:281–92. doi: 10.1006/dbio.1999.9209. [DOI] [PubMed] [Google Scholar]
- 22.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468–86. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 23.Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28:2–17. doi: 10.1002/jbmr.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kedlaya R, Veera S, Horan DJ, Moss RE, Ayturk UM, Jacobsen CM, Bowen ME, Paszty C, Warman ML, Robling AG. Sclerostin inhibition reverses skeletal fragility in an Lrp5-deficient mouse model of OPPG syndrome. Sci Transl Med. 2013;5:211ra158. doi: 10.1126/scitranslmed.3006627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schepers K, Hsiao EC, Garg T, Scott MJ, Passegue E. Activated Gs signaling in osteoblastic cells alters the hematopoietic stem cell niche in mice. Blood. 2012;120:3425–35. doi: 10.1182/blood-2011-11-395418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collette NM, Yee CS, Murugesh D, Sebastian A, Taher L, Gale NW, Economides AN, Harland RM, Loots GG. Sost and its paralog Sostdc1 coordinate digit number in a Gli3-dependent manner. Dev Biol. 2013;383:90–105. doi: 10.1016/j.ydbio.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birbrair A, Wang ZM, Messi ML, Enikolopov GN, Delbono O. Nestin-GFP transgene reveals neural precursor cells in adult skeletal muscle. PLoS One. 2011;6:e16816. doi: 10.1371/journal.pone.0016816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baek WY, Lee MA, Jung JW, Kim SY, Akiyama H, de Crombrugghe B, Kim JE. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. J Bone Miner Res. 2009;24:1055–65. doi: 10.1359/jbmr.081248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lama G, Mangiola A, Proietti G, Colabianchi A, Angelucci C, D’Alessio A, De Bonis P, Geloso MC, Lauriola L, Binda E, Biamonte F, Giuffrida MG, Vescovi A, Sica G. Progenitor/Stem Cell Markers in Brain Adjacent to Glioblastoma: GD3 Ganglioside and NG2 Proteoglycan Expression. J Neuropathol Exp Neurol. 2016 doi: 10.1093/jnen/nlv012. [DOI] [PubMed] [Google Scholar]
- 30.Murao H, Yamamoto K, Matsuda S, Akiyama H. Periosteal cells are a major source of soft callus in bone fracture. J Bone Miner Metab. 2013;31:390–8. doi: 10.1007/s00774-013-0429-x. [DOI] [PubMed] [Google Scholar]
- 31.He JW, Yue H, Hu WW, Hu YQ, Zhang ZL. Contribution of the sclerostin domain-containing protein 1 (SOSTDC1) gene to normal variation of peak bone mineral density in Chinese women and men. J Bone Miner Metab. 2011;29:571–81. doi: 10.1007/s00774-010-0253-5. [DOI] [PubMed] [Google Scholar]
- 32.Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S, Krumlauf R. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006;21:1738–49. doi: 10.1359/jbmr.060810. [DOI] [PubMed] [Google Scholar]
- 33.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005;280:26770–5. doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 35.Yee CS, Xie L, Hatsell S, Hum N, Murugesh D, Economides AN, Loots GG, Collette NM. Sclerostin Antibody Treatment Improves Fracture Outcomes in a Type I Diabetic Mouse Model. Bone. 2015 doi: 10.1016/j.bone.2015.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Rui Y, Cheng TY, Huang S, Xu L, Meng F, Lee WY, Zhang T, Li N, Li C, Ke H, Li G. Effects of Sclerostin Antibody on the Healing of Femoral Fractures in Ovariectomised Rats. Calcif Tissue Int. 2016;98:263–74. doi: 10.1007/s00223-015-0085-3. [DOI] [PubMed] [Google Scholar]
- 37.Friedman MS, Oyserman SM, Hankenson KD. Wnt11 promotes osteoblast maturation and mineralization through R-spondin 2. J Biol Chem. 2009;284:14117–25. doi: 10.1074/jbc.M808337200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S, Zhang E, Yang M, Lu L. Overexpression of Wnt11 promotes chondrogenic differentiation of bone marrow-derived mesenchymal stem cells in synergism with TGF-beta. Mol Cell Biochem. 2014;390:123–31. doi: 10.1007/s11010-014-1963-0. [DOI] [PubMed] [Google Scholar]
- 39.Yano F, Kugimiya F, Ohba S, Ikeda T, Chikuda H, Ogasawara T, Ogata N, Takato T, Nakamura K, Kawaguchi H, Chung UI. The canonical Wnt signaling pathway promotes chondrocyte differentiation in a Sox9-dependent manner. Biochem Biophys Res Commun. 2005;333:1300–8. doi: 10.1016/j.bbrc.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 40.Boland GM, Perkins G, Hall DJ, Tuan RS. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J Cell Biochem. 2004;93:1210–30. doi: 10.1002/jcb.20284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.