

Abstract
Apolipoprotein E (APOE) is a component of lipid-transporting particles and a recognition ligand for receptors, which bind these particles. The APOE isoform ε2 is a risk factor for age-related macular degeneration; nevertheless, APOE absence in humans and mice does not significantly affect the retina. We found that retinal cholesterol biosynthesis and the levels of retinal cholesterol were increased in Apoe−/− mice, whereas cholesterol elimination by metabolism was decreased. No focal cholesterol deposits were observed in the Apoe−/− retina. Retinal proteomics identified the most abundant cholesterol-related proteins in WT mice and revealed that, of these cholesterol-related proteins, only APOA4 had increased expression in the Apoe−/− retina. In addition, there were changes in retinal abundance of proteins involved in proinflammatory and antiinflammatory responses, cellular cytoskeleton maintenance, vesicular traffic, and retinal iron homeostasis. The data obtained indicate that when APOE is absent, particles containing APOA1, APOA4, and APOJ still transport cholesterol in the intraretinal space, but these particles are not taken up by retinal cells. Therefore, cholesterol biosynthesis inside retinal cells increase, whereas metabolism to oxysterols decreases to prevent cells from cholesterol depletion. These and other compensatory changes underlie only a minor retinal phenotype in Apoe−/− mice.
Keywords: cholesterol, retina, lipoproteins, iron, cytoskeleton, vesicular traffic
APOE is a glycoprotein and constituent of lipoprotein particles (LPPs) that bind and deliver lipids from one tissue or cell type to another (supplemental Text 1) (1). In the blood, APOE is mainly associated with VLDLs, IDLs, and chylomicron remnants (2). In the brain and retina, however, the APOE-containing LPPs are different and have densities similar to those of HDLs (3, 4). APOE mediates lipid transfer by being a recognition ligand for specific cell-surface receptors, which bind and internalize the APOE-containing LPPs (5) after these particles acquire cholesterol from cells with cholesterol excess. The APOE receptors belong to the LDL receptor (LDLR) family and include LDLR, VLDLR, LDLR-related protein 1 (LRP1), LRP1B, LRP2 (megalin), LRP4 (MEGF7), LRP5, LRP6, LRP8 (APOER2), and LRP11 (SORL1) (6)
In humans, APOE exists in three isoforms (ε2, ε3, and ε4), which differ in their lipid-binding capacity, ability to integrate into LPP, and affinity for the receptors (7). APOE ε4 is a risk factor for Alzheimer’s disease, whereas APOE ε2 is protective (8–10). Conversely, APOE ε4 and ε2 decrease and increase risks, respectively, for age-related macular degeneration (AMD), a disease that affects the retina and causes irreversible vision loss in the elderly of industrialized countries (11–14). Humans can lack APOE and develop severe type III hyperlipoproteinemia, a lipid disorder disease characterized by an exceptionally high serum cholesterol and triglyceride content (15). The clinical manifestations of this disorder have been described (15), yet only recently, an APOE-deficient individual was evaluated for cognitive, neurological, and retinal functions (16). Remarkably, no defects were found in these functions, thus suggesting that either APOE is not essential for the brain and retina or that the mechanisms exist to compensate for a lack of APOE (16).
The mammalian retina is a thin layer of photosensitive tissue in the back of the eye. The retina comprises the neural retina (several layers of neurons interconnected by synapses and glial cells) and retinal pigment epithelium (RPE; a polarized monolayer of cells, whose apical side faces the neural retina). The RPE forms a part of the blood-retina barrier, which is permeable to LDL and HDL from the systemic circulation (17, 18). In addition, the RPE basal lamina is a part of Bruch’s membrane (BM), a unique penta-layered structure located between the RPE and choroid, the vascular layer of the eye, which contains connective tissues. Normally, APOE appears to be primarily expressed in retinal ganglion and Müller cells, as well as in the RPE (19, 20). In addition, in aging eyes, APOE is found in drusen (heterogeneous debris external to the RPE, a hallmark of AMD) and basal laminar deposits (a stereotypically thickened RPE basal lamina) (11, 21–23).
Mice have only one APOE protein, a form similar structurally to APOE ε4 but functionally to APOE ε3 (24), and are hypercholesterolemic when they lack APOE, even when fed a standard rodent chow (25). The retina of Apoe−/− mice is grossly normal and shows an unaltered amplitude of electroretinography (ERG) responses, an indicator of retinal function; the ERG function becomes impaired only when animals are aged and put on a high-cholesterol diet (26–28). Transgenic mice expressing human APOE isoforms have a more pronounced retinal phenotype (29, 30) with the observed abnormalities being isoform-specific and including some of the AMD-like changes (e.g., drusenoid deposits and choroidal neovascularization along with RPE atrophy, hypopigmentation, and hyperpigmetation) (30). Similarly, there is the isoform-specific effect on pathogenic subretinal inflammation in Cx3cr1−/− mice with transgenic APOE ε2 provoking and APOE ε4 inhibiting this inflammation (31, 32).
The present study was initiated by an apparent discrepancy between APOE involvement in AMD [as suggested by genetic studies (11–13) and identification in drusen (21–23)], yet generally normal retinal structure and function of the individual with a total APOE absence (16), in agreement with only a minor retinal phenotype of Apoe−/− mice (26, 28). We hypothesized that this discrepancy is due to compensatory changes in the Apoe−/− retina elicited by a lack of APOE and conducted retinal characterizations of Apoe−/− mice. We used approaches that have not been used in previous studies of this genotype and identified the compensatory mechanisms in the Apoe−/− retina. In addition, we identified proteins involved in retinal lipid transport and obtained insights into noncanonical APOE functions.
MATERIALS AND METHODS
Animals
C57BL/6J mice and Apoe−/− mice on the C57BL/6J background were from the Jackson Laboratory (catalog nos. 000664 and 002052, respectively) and were free of the Crblrd8 mutation. Mice were 6 months old, kept on a 12 h light–dark cycle, and provided food and water ad libitum. The food was either a regular rodent chow (5P76 Prolab Isopro RMH 3000, T.R. Last Co, containing no appreciable amounts of cholesterol) or a Western-type diet [WTD; catalog no. TD.120425, Teklad, containing 21% (weight %) milkfat and 0.2% (weight %) cholesterol]. Mice were maintained on WTD from 1 to 6 months of age. All animal experiments were approved by Case Western Reserve University’s IACUC and conformed to recommendations of the American Veterinary Association Panel on Euthanasia.
Retinal imaging
Ultra-high-resolution spectral-domain optical coherence tomography (SD-OCT), fluorescein angiography (FA), and transmission electron microscopy (TEM) were performed as described (33, 34) and utilized a 840HHP SD-OCT system (Bioptigen), a scanning laser ophthalmoscope (Spectralis HRA+OCT, Heidelberg Engineering), and a 1200EX transmission electron microscope (JEOL Ltd.), respectively. Tissue fixation for TEM was performed as described (35) and included sequential incubations in 0.1 M Na cacodylate buffer, pH 7.4, containing first 3% glutaraldehyde and then 1% OsO4, followed by incubations with 1% tannic acid in 0.05 M Na cacodylate, pH 7.4, and 1% para-phenylenediamine in 70% ethanol.
Retinal isolation and sterol quantifications
Retinal isolation and processing were performed as described (36), as were subsequent sterol quantifications by isotope dilution GC/MS (37). Both unesterified and total sterols (a sum of unesterified and esterified forms) were measured using deuterated sterol analogs as internal standards.
Stains for unesterified cholesterol and iron
Unesterified cholesterol (UC) was visualized as described (38) using filipin (Cayman Chemical). Iron was detected using the Perls’ Prussian blue stain enhanced with the very intense purple reagent (VIP, Vector Laboratories) (39, 40). Briefly, paraffin sections were incubated at room temperature for 30 min in 5% potassium ferrocyanide and 5% hydrochloric acid followed by a 30 min incubation at room temperature with the very intense purple reagent.
Quantitative RT-PCR
Total RNA (1 μg) was isolated using the TRIzol Reagent (Life Technologies) and was then converted to cDNA by SuperScript III reverse transcriptase (Invitrogen) according to manufacturer’s instructions. PCRs were performed in triplicate using cDNA, a pair of gene-specific primers (supplemental Table S1), and a FastStart Universal SYBR Green Master (Rox) (Roche Life Science) as specified by the manufacturer. Gene expression was normalized to β-actin. Changes in relative mRNA level were calculated by the 2−ΔΔCt method (41).
Immunohistochemistry
Stains for glial fibrillary acidic protein (GFAP) and ionized calcium binding adaptor molecule (Iba1) were performed as described (33, 36) with the dilution and source of Abs being as follows: 1:1,000 for chicken anti-GFAP (catalog no. PA1-10004, Thermo Fisher Scientific), 1:1,000 for rabbit anti-Iba1 (catalog no. 019-1974, Wako), 1:200 for donkey anti-chicken Alexa Fluor 488 (catalog no. 703-545-155, Jackson Immune Res Inc.), and 1:200 for goat anti-rabbit Alexa Fluor 647 (catalog no. 111-605-144, Jackson Immune Res Inc.). Stain for H-ferritin (FTH1) was also performed as described (42) and used the following dilutions and sources of Abs: 1:500 for rabbit anti-mouse FTH1 (Z17; gift of Paolo Arosio, University of Brescia, Italy) and 1:200 for donkey anti-rabbit Cy3 (catalog no. 711-165-152, Jackson Immune Res Inc.).
Label-free analysis
Mouse retinas (three biological replicates per genotype, each containing two retinas, one retina from one mouse) were frozen, cryo-pulverized, and lysed with 3% SDS as described (43). Samples were then cleaned of detergent using a 10 kDa molecular mass cutoff filter according to a filter-aided sample preparation protocol (Millipore) (44). The buffer was exchanged with 8 M urea in 50 mM Tris-HCl buffer, pH 8.0, to a final volume of 50 µl, and proteins were reduced on a filter with 10 mM dithiothreitol for 1 h at 37°C, followed by alkylation with 25 mM iodoacetaminde for 30 min in the dark. The 8 M urea was then diluted to 4 M with 50 mM Tris-HCl buffer, pH 8.0, and samples were concentrated to a final volume of 50 μl. Protein concentration was measured using a Bradford assay according to the manufacturer’s instructions (Bio-Rad). Next, 20 μg of total protein was digested with Lysyl Endopeptidase (Wako Chemicals) for 2 h at 37°C at an enzyme:substrate ratio of 1:20. The urea concentration was next adjusted to 2 M with 50 mM Tris-HCl buffer, pH 8, followed by an overnight trypsin digestion at 37°C using sequencing-grade trypsin (Promega) at an enzyme:substrate ratio of 1:20. Tryptic peptides were stored at −80°C until further analysis by LC/MS/MS.
LC/MS/MS was carried out using 400 ng of each sample injected into a LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific) equipped with a nanoAcquityTM ultra-high-pressure liquid chromatography system (Waters). Blank injections were run after each sample to minimize carryover between samples. Mobile phases were aqueous phase A (0.1% formic acid in water) and organic phase B (0.1% formic acid in acetonitrile). Peptides were loaded onto a nanoACQUITY UPLC® 2G-V/M C18 desalting trap column (180 μm × 20 mm nano column, 5 μm, 100 Å) at a flow rate of 0.3 µl/min. Subsequently, peptides were resolved in a nanoACQUITY UPLC® BEH300 C18 reversed-phase column (75 μm × 250 mm nano column, 1.7 μm, 100 Å (Waters), followed by a gradient elution of 1–90% of phase B over 240 min (isocratic at 1% B, 0–1 min; 2–42% B, 2–212 min; 42–90% B, 212–223 min; and 90–1% B, 223–240 min). A nano electrospray ion source was utilized to ionize peptides at a flow rate of 300 nl/min, 1.5 kV spray voltage, and 270°C capillary temperature. Full-scan MS spectra (m/z 380–1,800) were acquired at a resolution of 60,000 at m/z 400 followed by 20 data-dependent MS/MS scans. MS/MS spectra were generated in the ion-trap detector by collision-induced dissociation of the peptide ions (normalized collision energy = 35%; activation Q = 0.250; activation time = 20 ms) to generate a series of b and y ions as major fragments. The dynamic exclusion list was confined to a maximum of 500 entries with exclusion duration of 60 s and mass accuracy of 10 ppm for the precursor monoisotopic mass. LC/MS/MS raw data were acquired using the Xcalibur software (Thermo Fisher Scientific, version 2.2 SP1). To facilitate the peak volume-dependent label-free quantitative analysis and to ensure that the peak retention times (RTs) and the MS instrument performance were maintained within restricted tolerances, an equal amount of internal standard (Pierce Retention Time Calibration Mixture 88321, Thermo Scientific) was spiked into each sample. The RT, peak intensity, and mass accuracy of five peptides from the spiked internal standard were monitored in all samples. These peptides showed less than a 5 min RT drift, less than 4 ppm mass drift, and consistent peak widths and area under the curve as previously described (45). LC/MS/MS raw data were then acquired using the Xcalibur software (Thermo Fisher Scientific, version 2.2 SP1).
The LC/MS/MS raw files (one for each sample) were imported into Rosetta Elucidator™ (Rosetta Bio-software, version 3.3.0.1.SP.25) and processed as previously described (45). The peak list (.dta) files were searched by Mascot (version 2. 1, Matrix Science) against the mouse Uniprot database (538,585 sequences). Mascot search settings were as follows: trypsin enzyme specificity; mass accuracy window for precursor ion, 10 ppm; mass accuracy window for fragment ions, 0.8 Da; carbamidomethylation of cysteines as fixed modifications; oxidation of methionine as variable modification; and one missed cleavage. Peptide identification criteria were a mass accuracy of ≤10 ppm, an expectation value of P < 0.05, and an estimated false discovery rate of less than 2%. The search results were imported back into Elucidator, and automated differential quantification of peptides was then accomplished with Rosetta Elucidator as previously described (45). RT alignment, feature identification (RT × m/z surface), extraction of the peptide ion peak, background subtraction, and smoothing across both RT and m/z dimensions were performed using the PeakTellerTM algorithm. Normalization of signal intensities across samples was based on the average signal intensities in each sample. For each peptide, the average intensity for all replicates within a genotype was calculated, and the fold change between the genotypes was then determined using these average intensities. Statistical analysis of peptide intensity differences between the Apoe−/− and WT retinas was by one-way ANOVA.
Quantification and statistical analyses
All images are representative of studies in three to five animals per genotype. The quantitative data represent the mean ± SD; the number of animals (n) is indicated in each figure or figure legend. Data were analyzed either by a two-tailed, unpaired Student’s t-test or by one-way ANOVA. Statistical significance was defined as * P ≤ 0.05; ** P ≤ 0.01; and *** P ≤ 0.001.
RESULTS
In vivo characterizations of Apoe−/− mice
The Apoe−/− retina did not seem to be previously characterized by high-resolution SD-OCT and FA; therefore, we used these imaging modalities to investigate retinal gross structure and the status of blood vessels. Apoe−/− mice fed regular chow had normal retinal appearance on both SD-OCT and FA (Fig. 1A, B). Similarly, no major abnormalities were detected in the Apoe−/− retina of mice fed a WTD (Fig. 1C, D). Thus, a lack of APOE does not seem to cause changes of clinical relevance in retinal structure and vasculature.
Fig. 1.

In vivo imaging of C57BL/6J (WT) and Apoe−/− mice on normal rodent chow and WTD. A, C: Ultra-high-resolution SD-OCT of mouse fundi (left) and retinal cross-sections (right). B, D: Fundus images of early (left), intermediate (center), and late (right) stages of fluorescein angiography. All images are representative (n = 3–5 mice per genotype).
Serum and retinal sterol levels in Apoe−/− mice
High levels of serum cholesterol are well documented in Apoe−/− mice (25, 28); yet, their retinal cholesterol levels have never been measured. We found that in Apoe−/− animals on regular chow, total retinal cholesterol was increased from 2.8-fold (female mice) to 2.9-fold (male mice) as compared with the corresponding WT mice (Fig. 2A). However, in the serum, cholesterol increases showed gender differences and were more pronounced in male mice (3.9-fold increase) than female mice (2.6-fold increase) (Fig. 2D). Notably, in WT or Apoe−/− male mice on a WTD, a further increase in serum total cholesterol did not lead to an increase in retinal total cholesterol (Fig. 2A, D).
Fig. 2.

Sterol profile in the retina and serum of C57BL/6J (WT) and Apoe−/− mice. Retinal levels of cholesterol (A), lathosterol and desmosterol (B), as well as 24-hydroxycholesterol (24HC) and 5-cholestenoic acid (5CA) (C) in female and male mice on regular chow and WTD fed for 5 months (from 1 to 6 months of age). Serum levels of cholesterol (D), lathosterol and desmosterol (E), as well as 24HC (F) in female and male mice on regular chow and Western diet. Results are means ± SD; n, number of mice. For retinal cholesterol, lathosterol, desmosterol, and 24HC measurements, number of mice equals the number of retinas; for 5CA measurements, three and four samples for C57BL/6J and Apoe−/− genotypes, respectively, were individually quantified, each containing two retinas from three or four mice. Significance was calculated using a two-tailed, unpaired Student’s t-test. ** P ≤ 0.01; *** P ≤ 0.001. EC, esterified cholesterol.
The levels of other serum and retinal sterols were measured to gain insights into homeostatic responses in the retina elicited by a lack of APOE (Fig. 2B, C, E, F). Lathosterol (cholest-7-en-3β-ol, Lipid Maps ID: LMST01010089) and desmosterol (cholest-5,24-dien-3β-ol, Lipid Maps ID: LMST01010016) are cholesterol precursors, whereas 24S-hydroxycholesterol (cholest-5-en-3β,24S-diol, Lipid Maps ID: LMST01010019) and 5-cholestenoic acid (3β-hydroxycholest-5-en-25R-26-oic acid, Lipid Maps ID: LMST04030072) are cholesterol metabolites generated by cytochrome P450 enzymes CYP46A1 and CYP27A1, respectively. In the serum, total lathosterol is an indicator of cholesterol biosynthesis in whole body (46), and total 24-hydroxycholesterol is a marker of cholesterol elimination from the brain (47). In the retina, lathosterol reflects cholesterol biosynthesis in neurons (48, 49), and desmosterol could be a marker of cholesterol biosynthesis in astrocytes (3). The 24-hydroxycholesterol and 5-cholestenoic acid indicate retinal cholesterol elimination by metabolism, which along with cholesterol removal by LPP is an important contributor to retinal cholesterol maintenance (4, 37, 50, 51). Neither total lathosterol nor desmosterol levels were changed in the serum of Apoe−/− mice fed regular chow or a WTD (Fig. 2E). Yet, both total lathosterol and desmosterol amounts were increased in the Apoe−/− retina (Fig. 2B), whereas retinal total 24-hydroxycholesterol levels were unchanged, and those of free 5-cholestenoic acid were decreased >4-fold (Fig. 2C). These changes suggested that there was cholesterol deprivation in retinal cells in Apoe−/− mice; hence, retinal cells increased their cholesterol biosynthesis and decreased cholesterol elimination by metabolism.
Retinal distribution of UC in Apoe−/− mice
Only UC, which constitutes the majority of cholesterol in the retina (Fig. 2A), was studied using a fluorescent antibiotic filipin. The pattern of filipin staining was similar in WT and Apoe−/− mice (Fig. 3A, B), except in the latter, the staining seemed to be more pronounced in the photoreceptor outer segments (OSs). No focal cholesterol deposits were noted in the Apoe−/− retina, an indication that retinal cholesterol excess in this genotype was present on the LPPs circulating in the intraretinal space. Furthermore, these circulating LPPs did not seem to be taken up by retinal cells due to APOE absence; therefore, cholesterol biosynthesis in retinal cells was increased, and cholesterol catabolism was decreased (Fig. 2B, C).
Fig. 3.

Retinal distribution of UC. A fluorescent antibiotic filipin was used for staining (cyan). A: Cross-section of the C57BL/6J (WT) retina. B: Cross-section of the Apoe−/− retina. OPL, outer plexiform layer. All images are representative (n = 3 mice per genotype). Scale bars: 50 μm.
Retinal proteomics of WT mice
The label-free analysis, a proteomics approach, was used. This approach quantifies relative peptide intensity and thus enables estimates of relative protein abundance. Peptides from 18 cholesterol-related proteins were detected in the retina of C57BL/6J mice (Table 1). Four of these proteins were apolipoproteins (APOA1, APOA4, APOE, and APOJ). Two proteins were the enzymes from the pathways of cholesterol biosynthesis (DHCR7) and metabolism (CYP27A1). Four proteins [LRP1, α2-macroglobulin receptor-associated protein 1 (LRPAP1), mesoderm development candidate 2 (MESDC2), and SORT1] were related to receptors from the LDLR family. Two proteins (PON1 and PON2) pertained to HDL. One protein (SCARB2) was from the scavenger receptor family, which binds different molecules including oxidized LPPs. Finally, five proteins (OSBPs 1 and 2 along with OSBPLs 1A, L2, and L8) were from the oxysterol binding protein families. In addition, the label-free analysis provided insight into the groups of the most abundant retinal proteins, whose relative expression was comparable or even higher than that of the housekeeping proteins. These were the nuclear histone proteins (HIST1H1C, HIST1H1E, HIST1H2AF, HIST1H2BB, HIST1H3A, and HIST1H4K), the chaperone proteins (CRYAA2 and CRYBB2), the enzymes involved in glycolysis/gluconeogenesis as well as ATP production/utilization (ALDOA, ATP5B, CKB, ENO1, LDHA, PGAM1, PGK1, and PKM), and of course vision-related proteins (GNAT1, GNB1, GNT1, RHO, RLBP1, and SAG). Thus, APOE is not the only major apolipoprotein in the retina, and three other retinal apolipoproteins (APOA1, APOA4, and APOJ) could carry cholesterol excess in the Apoe−/− retina to compensate for a lack APOE.
TABLE 1.
Relative protein abundance (in alphabetical order) in the C57BL/6J retina as assessed by label free quantifications
| Protein | Number of Peptides | Peptide Intensity (a.u.) × 106 |
| Cholesterol-related proteins | ||
| APOA1 | 5 | 9.4 ± 8.1 |
| APOA4 | 3 | 1.4 ± 1.2 |
| APOE | 6 | 4.2 ± 1.2 |
| APOJ | 8 | 6.6 ± 3.4 |
| CYP27A1 | 1 | 2.3 ± 1.8 |
| DHCR7 | 2 | 2.4 ± 1.1 |
| LRP1 | 4 | 3.3 ± 0.9 |
| LRPAP1 | 5 | 5.7 ± 4.0 |
| MESDC2 | 5 | 3.2 ± 1.8 |
| OSBP1 | 1 | 2.9 ± 1.4 |
| OSBP2 | 1 | 3.7 ± 1.8 |
| OSBPL1A | 3 | 4.7 ± 4.5 |
| OSBPL2 | 1 | 3.8 ± 2.3 |
| OSBPL8 | 1 | 2.5 ± 1.8 |
| PON1 | 5 | 8.5 ± 2.3 |
| PON2 | 2 | 6.3 ± 3.3 |
| SCARB2 | 2 | 5.2 ± 2.0 |
| SORT1 | 1 | 4.5 ± 2.5 |
| Housekeeping Proteins | ||
| ACTB | 31 | 578.1 ± 773.1 |
| GAPDH | 35 | 1,445.4 ± 760.4 |
| TUBA1A | 47 | 500.5 ± 766.8 |
| Most Abundant Retinal Proteins | ||
| HIST1H1C | 3 | 4,832.0 ± 6,081.8 |
| HIST1H1E | 3 | 5,149 ± 4,426.7 |
| HIST1H2AF | 5 | 5,881.7 ± 8,425.4 |
| HIST1H2BB | 10 | 3,743.2 ± 4,806.4 |
| HIST1H3A | 8 | 2,463,4 ± 3,603.4 |
| HIST1H4K | 15 | 2,509.5 ± 3,787.6 |
| CRYAA2 | 5 | 733.6 ± 707.7 |
| CRYBB2 | 16 | 1,486.7 ± 1626.5 |
| ALDOA | 29 | 482.4 ± 490.3 |
| ATP5B | 49 | 309.4 ± 331.0 |
| CKB | 30 | 615.2 ± 599.2 |
| ENO1 | 43 | 1,014 ± 1,353.5 |
| LDHA | 21 | 599,8 ± 532,8 |
| PGAM1 | 16 | 537.5 ± 588.6 |
| PGK1 | 31 | 476.2 ± 480.7 |
| PKM | 45 | 730.4 ± 1,062,5 |
| Vision-related proteins | ||
| GNB1 | 16 | 1,093.4 ± 841.1 |
| GNGT1 | 6 | 814,4 ± 1,195.8 |
| RHO | 8 | 959.9 ± 1,139.2 |
| SAG | 27 | 788.5 ± 1,037.8 |
a.u., arbitrary units.
Retinal proteomics of Apoe−/− versus WT mice
The label-free analysis of the Apoe−/− retina enabled the identification of proteins that were expressed differentially as compared with the WT retina. Proteins were considered as differentially expressed, if: i) two or more peptides per protein showed a significant change (P ≤ 0.05) in abundance in Apoe−/− versus WT retina; and ii) some of the remaining peptides in the Apoe−/− retina had nonsignificant changes in abundance of the same direction as that of the significantly changed peptides. Based on these criteria, 14 differentially expressed proteins were identified in the Apoe−/− retina: 8 with a decreased expression and 6 with an increased expression as compared with the WT retina (Table 2). As an exception, we have also added ankyrin-2 (ANK2) to the group of proteins with decreased abundance in the Apoe−/− retina; of the 57 peptides generated from ANK2, 13 had a decreased retinal abundance (3 peptides with significant and 10 peptides with nonsignificant changes) and 5 peptides had an increased retinal abundance (1 peptide with significant and 4 peptides with nonsignificant changes) as compared with WT mice.
TABLE 2.
Alphabetical list of proteins with differential abundance in the Apoe−/− (KO) versus WT retina
| Protein (total number of detected peptides) | Peptides with Significant Changes in Abundance | Peptide Intensity (a.u.) × 106 | Peptide Intensity Ratio, KO/WT | Number of Peptides with Nonsignificant Changes in Abundance, KO/WT | |
| Apoe-/-, Mean ± SD | WT, Mean ± SD | ||||
| Proteins with Decreased Abundance in the Apoe−/− versus WT Retina | |||||
| ANK2 (57) | NGLSPLHMAAQGDHVECVK | 0.1 ± 0.1 | 4.7 ± 4.1 | 0.02 | 10 (↓) and 4 (↑) |
| SAALLLQNDHNADVQSK | 0.2 ± 0.4 | 3.8 ± 2.4 | 0.06 | ||
| CFCMTDDK | 2.5 ± 1.2 | 3.8 ± 2.7 | 0.63 | ||
| MNEEIQEEPATSEDK | 5.7 ± 1.7 | 3.7 ± 0.3 | 1.54 | ||
| ApoE (6) | LGADMEDLR | 0.0 ± 0.0 | 4.4 ± 4.2 | WT only | None |
| LGPLVEQGR | 0.4 ± 0.8 | 5.7 ± 3.8 | 0.07 | ||
| MEEQTQQIR | 0.0 ± 0.0 | 3.1 ± 2.4 | WT only | ||
| NEVHTMLGQSTEEIR | 0.0 ± 0.0 | 2.5 ± 1.4 | WT only | ||
| LQAEIFQAR | 0.0 ± 0.0 | 4.7 ± 2.7 | WT only | ||
| TANLGAGAAQPLR | 0.0 ± 0.0 | 4.7 ± 2.4 | WT only | ||
| ATP1A2 (11) | SPEFTHENPLETR | 4.5 ± 2.5 | 8.5 ± 5.0 | 0.53 | 7 (↓) |
| LCFVGLMSMIDPPR | 1.5 ± 0.9 | 3.2 ± 1.5 | 0.46 | ||
| EMQDAFQNAYMELGGLGER | 1.7 ± 0.9 | 4.3 ± 3.1 | 0.40 | ||
| FDTDELNFPTEK | 5.9 ± 2.0 | 9.7 ± 5.0 | 0.61 | ||
| CRYBA1 (5) | RMEFTSSCPNVSER | 29.1 ± 39.4 | 223.2 ± 148.2 | 0.13 | |
| ENFIGRQWEICDDYPSLQAM | 1.2 ± 1.0 | 12.7 ± 3.8 | 0.09 | 3 (↓) | |
| GWFNNEVGSMK | |||||
| DPYSL4 (21) | GVNSFLVFMAYK | 0.4 ± 0.3 | 0.9 ± 1.3 | 0.44 | 4 (↓) |
| NLHQSGFSLSGSQADDHIAR | 26.1 ± 8.8 | 40.1 ± 13.3 | 0.65 | ||
| EZR (9) | GTDLWLGVDALGLNIYEK | 3.8 ± 1.8 | 6.00 ± 1.1 | 0.63 | 2 (↓) |
| FYPEDVAEELIQDITQK | 4.3 ± 0.9 | 5.9 ± 0.4 | 0.73 | ||
| FGG (6) | FEGNCAEQDGSGWWMNK | 1.2 ± 0.5 | 2.0 ± 1.0 | 0.60 | 1 (↓) |
| SSTTNGFDDGIIWATWK | 1.3 ± 0.5 | 2.3 ± 2.3 | 0.57 | ||
| FN3KRP (5) | EALELWSALQLK | 5.0 ± 3.3 | 10.0 ± 5.9 | 0.50 | 3 (↓) |
| MFEGEMASLTAILK | 4.5 ± 2.8 | 8.3 ± 2.6 | 0.54 | ||
| KRT15 (3) | AGLENSLAEVECR | 0.6 ± 0.7 | 2.3 ± 0.4 | 0.26 | None |
| EVASNTEMIQTSK | 0.7 ± 0.6 | 2.8 ± 0.6 | 0.25 | ||
| Proteins with Increased Abundance in the Apoe−/− versus WT Retina | |||||
| APOA4 (3) | LGDASTYADGVHNK | 1.2 ± 0.3 | 0.3 ± 0.2 | 4.0 | 1 (↑) |
| SLAPLTVGVQEK | 3.6 ± 1.1 | 1.1 ± 0.7 | 3.3 | ||
| FTH1 (3) | IFLQDIK | 6.7 ± 3.3 | 3.4 ± 2.0 | 1.97 | None |
| MGAPEAGMAEYLFDK | 16.8 ± 3.7 | 10.5 ± 0.9 | 1.60 | ||
| QNYHQDAEAAINR | 4.2 ± 1.3 | 0.4 ± 0.2 | 10.5 | ||
| HMGN5 (10) | EGQPEEDGKEDQPEEDGK | 1.3 ± 1.9 | 0.3 ± 0.5 | 4.33 | 5 (↑) |
| SEDAEVSKDEEEK | 1.6 ± 0.7 | 0.8 ± 0.6 | 2.00 | ||
| LZIC (3) | VSTELGSGDK | 4.9 ± 1.2 | 2.5 ± 0.4 | 1.96 | None |
| LMQQLQDLEECREELDADEYEETK | 16.2 ± 3.1 | 9.1 ± 1.1 | 1.78 | ||
| PTMS (3) | TAEEEDEADPK | 31.7 ± 17.7 | 8.3 ± 3.5 | 3.82 | 1 (↑) |
| TAEEEDEADPKR | 18.5 ± 13.3 | 8.1 ± 4.2 | 2.28 | ||
| WDFY1 (4) | CEQPFFWNIK | 231.0 ± 326.7 | 32.0 ± 45.3 | 7.22 | None |
| LEQNTCSVITTLK | 229.0 ± 310.0 | 44.0 ± 31.2 | 5.20 | ||
Two or more peptides in each of these proteins had a significantly different abundance (P ≤ 0.05) between the genotypes, and a number of the remaining peptides showed the same direction of change in abundance (nonsignificant); in the latter, the KO/WT peptide intensity ratio was ≤0.7 (↓) or ≥1.3 (↑). a.u., arbitrary units.
Of nine proteins with decreased expression in the Apoe−/− retina, one was APOE, consistent with the Apoe−/− genotype. The remaining eight proteins were ANK2, Na+/K+-transporting ATPase subunit α2 (ATP1A2), β-crystallin A1 (CRYBA1), dihydropyrimidinase-related protein 4 (DPYSL4), ezrin (EZR), fibrinogen γ chain (FGG), ketosamine-3-kinase (FN3KRP), and keratin, type I cytoskeletal 15 (KRT15). These eight proteins have different functions, yet six of them (ANK2, ATP1A2, CRYBA1, DPYSL4, EZR, and FGG) could be linked to ATP1A2 (supplemental Fig. S1).
Five proteins with increased retinal expression in Apoe−/− mice were APOA4 , FTH1, high mobility group nucleosome-binding domain-containing protein 5 (HMGN5), leucine zipper and ICAT homologous domain-containing protein (LZIC), parathymosin (PTMS), and WD repeat and FYVE domain-containing protein 1 (WDFY1). The APOA4 upregulation in the Apoe−/− retina could be a compensatory response to a lack of APOE, whereas an increased expression of the remaining four proteins could be a consequence of an increased ferritin expression (supplemental Fig. S2).
Next, we investigated whether additional differentially expressed proteins could be identified, if the inclusion criteria are relaxed and include proteins with a significant change in abundance in only one peptide with two or more of the remaining peptides showing a change in abundance of the same direction. Only eight such proteins were identified: six with a decreased expression and two with an increased expression in the Apoe−/− retina (Table 3). The downregulated proteins were β-crystallin B1 (CRYBB1), β-crystallin B2 (CRYBB2), fibrinogen β chain (FGB), gelsolin (GSN), histone H4 (HIST1H4K), and receptor expression-enhancing protein 5 (REEP5). The upregulated proteins were secretogranin-2 (SCG2) and zinc finger CCCH domain-containing protein 4 (ZC3H4). The downregulation of CRYBB1 and CRYBB2 as well as FGB is consistent with those of CRYBA1 and FGG, respectively, identified based on the more stringent inclusion criteria (Table 2). Similarly, the upregulation of SCG2 and ZC3H4 involved in macrophage activation (52–54) may reflect a low-grade retinal inflammation, which was already indicated by increased levels of PTMS and WDFV1 (Table 2). Finally, the downregulation of GSN, an actin-binding protein whose fragment forms a complex with APOE (55), could reflect a lack of APOE.
TABLE 3.
Alphabetical list of proteins, which may have differential abundance in the Apoe−/− (KO) versus WT retina
| Protein (total number. of detected peptides) | Peptides with Significant Changes in Abundance | Peptide Intensity (a.u.) × 106 | Peptide Intensity Ratio, KO/WT | Number of Peptides with Nonsignificant Changes in Abundance, KO/WT | |
| Apoe-/-, Mean ± SD | WT, Mean ± SD | ||||
| Proteins with decreased abundance in the Apoe−/− versus WT retina | |||||
| CRYBB1 (8) | VGSITVSGGTWVGYQYPGYR | 0.8 ± 0.5 | 6.7 ± 3.3 | 0.12 | 7 (↓) |
| CRYBB2 (9) | GEYPRWDSWTSSR | 0.7 ± 0.7 | 22.4 ± 18.2 | 0.03 | 8 (↓) |
| FGB (4) | YCGLPGEYWLGNDK | 3.3 ± 1.5 | 5.0 ± 4.2 | 0.66 | 2 (↓) |
| GSN (11) | SEDCFILDHGR | 2.2 ± 3.8 | 9.5 ± 3.3 | 0.24 | 10 (↓) |
| HIST1H4K (14) | VFLENVIRDAVTYTEHAK | 67.5 ± 114.8 | 1,378.0 ± 399.4 | 0.05 | 10 (↓) |
| REEP5 (4) | HESQVDSVVK | 3.0 ± 2.3 | 5.1 ± 4.4 | 0.59 | 3 (↓) |
| Proteins with Increased Abundance in the Apoe−/− versus WT Retina | |||||
| SCG2 (5) | PNGLVEPEQDLELAVDLDDIPEADLDRPDMFQSK | 3.0 ± 0.7 | 2.0 ± 0.8 | 1.5 | 3 (↑) |
| ZC3H4 (7) | LDSFSQVGPGSETVTQK | 3.2 ± 1.2 | 1.6 ± 0.3 | 1.90 | 6 (↑) |
One peptide in each of these proteins had a significantly different abundance (P ≤ 0.05) between the genotypes, and two or more of the remaining peptides showed the same direction of change in abundance (nonsignificant); in the latter, the KO/WT peptide intensity ratio was ≤0.7 (↓) or ≥1.3 (↑). a.u., arbitrary units.
Retinal gene expression in Apoe−/− versus WT mice
Two groups of genes, cholesterol- and inflammation-related, were evaluated, both encoding proteins, whose peptides were not detected by the label-free analysis. The first group included Apoa2, Apob, Apoc3, Apod, and Apof (all apolipoproteins); Lxr (liver X receptors) α and β, transcription factors that control the expression of Apoe, Lpl, and other genes of pertinence to lipoprotein-mediated cholesterol transport; Idol (inducible degrader of LDLR); Lpl (LPL that hydrolyzes triglycerides on chylomicrons and VLDL), and Ldlr (Fig. 4A). Abrogation of Apoe upregulated the expression of Apob and downregulated the expression of Apoc3, Apod, and Idol; the expression of the remaining genes was not changed. These data indicate that the Apoe−/− retina may have compensatory changes in the expression of other apolipoproteins (in addition to changes in the APOA4 abundance; Table 2) as well an increased expression of LDLR, whose levels are controlled by IDOL (56).
Fig. 4.

Retinal gene expression in C57BL/6J (WT) and Apoe−/− mice. Relative expression of cholesterol-related (A) and proinflammatory (B) genes after normalization to the expression of β-actin. Results are means ± SD of triplicate measurements on pooled retinal samples from six (C57BL/6J) or five (Apoe−/−) mice, a total of 12 and 10 retinas, respectively. Significance was calculated using a two-tailed, unpaired Student’s t-test. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001.
The second group of the evaluated genes included Il-1β and Il-6 (both interleukins), Tnfα, Ccl2 (C-C motif chemokine 2), Cox-2 (prostaglandin G/H synthase 2), and iNos (inducible NO synthase). The expression of Il-1β, Il-6, Tnfα, and Ccl2 was significantly downregulated, whereas that of Cox-2 was upregulated in the Apoe−/− retina; the expression of iNos was unchanged (Fig. 4B). Such mixed pattern of changes suggests that both proinflammatory and antiinflammatory processes likely take place in the Apoe−/− retina, consistent with the proteomics data, i.e., a simultaneous increase in the expression of proinflammatory PTMS, WDFV1, SCG2, and ZC3H4 and antiinflammatory LZIC and HMGN5 (Tables 2, 3 and supplemental Figs. S1, S2).
Immunochemistry for GFAP and Iba1
Retinal proteomics of the Apoe−/− versus WT retina did not allow a conclusion about changes in the levels of GFAP and Iba1, markers for activated Müller cells and microglia/macrophages, respectively (57, 58). There was significant data variability for the former and a lack of peptides from the latter. Hence, retinal immunochemical localizations of GFAP and Iba1 were carried out. The immunoreactivity for both GFAP and Iba1 was essentially absent in the WT retina but present in the Apoe−/− retina (Fig. 5B, C, E, F). For GFAP, the immunoreactivity was mainly in the ganglion cell layer (GCL), consistent with predominant GFAP expression in the end feet of retinal Müller cells (57). For Iba1, the immunoreactivity was around a blood vessel in the GCL. Thus, it is possible that APOE absence activates retinal Müller cells where this apolipoprotein is expressed (19) and leads to microglia/macrophage accumulation in or around some blood vessels due to high serum cholesterol content (25). Overall, the immunohistochemistry data suggested that there is only a minor retinal inflammation in Apoe−/− mice.
Fig. 5.

Retinal Müller cells and microglia/macrophage status in C57BL/6J (WT) and Apoe−/− mice. A, D: Control (Ctrl) stains with preimmune serum only. B, E: Retinal GFAP immunolocalizations (green). C, F: Retinal Iba1 immunolocalizations (red). Nuclei were stained with DAPI (blue). All images are representative (n = 3 mice per genotype). Scale bars: 50 μm.
Ultrastructural changes in the Apoe−/− retina
No more than mild alterations in the Apoe−/− retina were detected previously by TEM. These were: i) increases in the incidence and amount of basal linear deposit-like debris in BM in aged mice (28); ii) some condensation of nuclear chromatin and focal perinuclear vacuolization in the inner nuclear layer (INL) (26); and iii) a decrease by 17% in cell number in the outer nuclear layer (ONL) (26). Retinal proteomics indicated that the RPE cytoskeleton and vesicular traffic could be disturbed in Apoe−/− mice (supplemental Figs. S1, S2). Accordingly, we used TEM with osmium and tannic acid-p-phenylenediamine (OTAP), an approach that did not seem to be used previously in studies of the Apoe−/− retina and whose advantage is that it preserves and enhances the visualization of membranous structures (tannic acid) and osmium-treated neutral lipids (p-phenylenediamine) (35). We found that in the Apoe−/− retina, some of the RPE microvilli appeared shorter than others, and there were numerous membrane-bounded vesicles inside the RPE and at the RPE-OS interface (Fig. 6B). Remarkably, the observed changes in the Apoe−/− retina were reminiscent of those in the brain of Apoe−/− mice, which had a decreased integrity of the dendritic cytoskeleton with dendritic processes showing abundant vacuolization and distortion of their morphology (59). Consequently, inside the RPE, the detected vesicles could represent endosomes and multivesicular bodies, whereas outside the RPE, the membrane-bounded vesicles could be the vesiculated microvilli. The altered size of these vesicles in Apoe−/− mice may also be an indicator of disturbed vesicular traffic.
Fig. 6.

Retinal ultrastructure of C57BL/6J (WT) and Apoe−/− mice. A, B, D: TEM of RPE and interface with the OSs. BM is also shown. Magenta circles denote some of the membrane-bounded vesicles; arrowheads denote some of the microvilli with altered morphology. C: Toluidine blue stain of an Apoe−/− retinal cross-section showing the areas with a more intense stain (white rectangles). The ultrastructure of the left rectangle is shown in D. A, B: Images are representative of two mice per genotype. C, D: Changes are found in one of the two mice analyzed. Scale bars: 2 μm (A, B); 50 μm (C); and 5 μm (D).
In addition, we found a region on TEM with a more intense RPE staining (Fig. 6D), which looked condensed as compared with the adjacent region with a normal intensity of staining. Hence, we next analyzed the toluidine-blue plastic section that encompassed this TEM region with increased intensity (Fig. 6C). Similar to the TEM section, the staining intensity of this region with toluidine blue was also stronger as compared with the remaining RPE, and there were two additional regions in the section that had a more intense toluidine-blue staining. Toluidine blue is an acidophilic metachromatic dye whose staining intensity depends on pH (60), and there may be intracellular acidification due to inhibition of Na+/K+-ATPase (61). Accordingly, changes in staining intensity with toluidine blue could reflect focal decreases in the ATP1A2 abundance in the Apoe−/− retina (Table 2) and subsequent cell acidification. Changes on TEM are more difficult to interpret because they could reflect the effect of pH on tissue fixation or alteration in the RPE caused by cellular acidification.
Immunochemistry for FTH1 and Perls’ stain
An increase in retinal abundance of FTH1 in Apoe−/− mice prompted retinal FTH1 immunolocalizations. In the WT retina, the strongest immunoreactive signal was observed in the photoreceptor inner segment (IS) and the innermost inner plexiform layer (IPL) (Fig. 7A, B). In the Apoe−/− retina, the signal intensity in the IS was significantly diminished, whereas that in the GCL became more prominent (Fig. 7C, D). Thus, there are changes in the FTH1 expression pattern in the Apoe−/− retina, and these changes could reflect iron accumulation in the GCL.
Fig. 7.
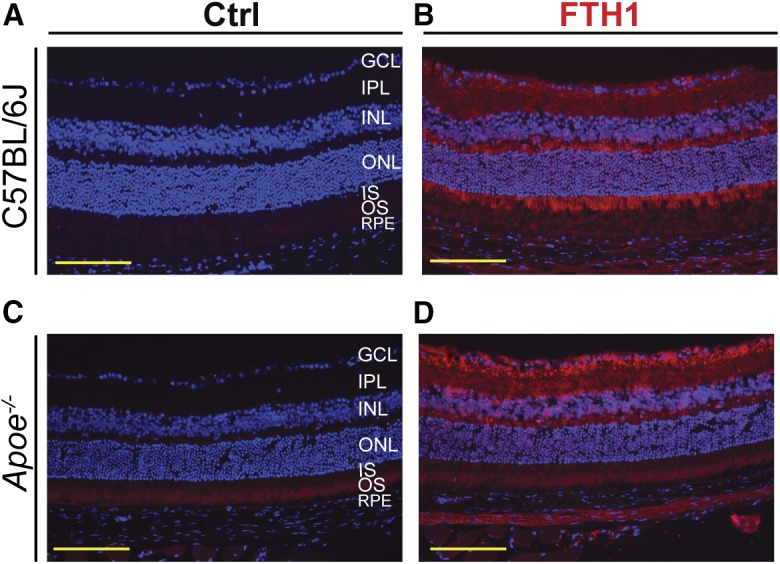
Retinal stains for FTH1 in C57BL/6J (WT) and Apoe−/− mice. A, C: Control (Ctrl) stains without primary antibody. B, D: Retinal FTH1 immunolocalizations (red). Nuclei were stained with DAPI (blue). All images are representative (n = 3 mice per genotype). Scale bars: 100 μm.
Because FTH1 protein translation can be upregulated by increased intracellular iron (62), we performed the VIP-enhanced Perls’ histochemical stain for iron in retinas from WT and Apoe−/− mice. There was no detectable signal in retinas from either genotype (data not shown). However, this does not rule out a moderate increase in retinal iron in the KO mice, because the Perls’ stain is a less-sensitive measure of retinal intracellular iron accumulation than immunohistochemistry for ferritin (63).
DISCUSSION
The present work led to several novel findings. First, retinal cholesterol was documented to be increased almost 3-fold in Apoe−/− mice, yet did not form visible focal deposits in the retina (Figs. 2, 3). Second, the most abundant retinal cholesterol-related proteins were identified (Table 1), including major apolipoproteins (APOA1, APOA4, APOE, and APOJ) and receptors for retinal LPP (SCARB2 and LRP1), as well as proteins that control the expression of the LPP receptors (LRPAP1, MESDC2, and SORT1). Third, comparative studies of the retinal sterol, protein, and mRNA levels in Apoe−/− versus WT mice (Figs. 2, 4, 5 and Tables 2, 3) revealed several compensatory mechanisms that are likely triggered by a lack of APOE and underlie, at least in part, only a minor retinal phenotype of Apoe−/− mice. Finally, retinal proteomics pointed to noncanonical functions of APOE (supplemental Figs. S1, S2), which were tested in part in the present work and suggested that APOE may play a role in the maintenance of the RPE cytoskeleton and vesicular traffic as well as retinal iron homeostasis (Figs. 6, 7).
The retina synthesizes cholesterol locally (Fig. 8A) (64, 65) as well as takes up cholesterol from the systemic circulation (Fig. 8B), mainly via the RPE and receptors present on the RPE basolateral side: LDLR, SRB1 (now SCARB1), and CD36 (now SCARB2) (17, 18, 66–68). Once in the RPE, systemic cholesterol is likely reassembled and secreted basolaterally and apically in the form of LPPs (Fig. 8C, D). The basolaterally secreted LPPs (called BM-LP and composed of APOA1, APOB, APOC1, APOC2, and APOE) traffic to the choroid (69, 70). The apically secreted LPPs (called HDL-like particles and composed of APOA1, APOE, and probably other apolipoproteins) traffic inside the retina and deliver lipids to different cell types (Fig. 8E) (4). An almost 3-fold increase in retinal cholesterol in Apoe−/− mice (Fig. 2A) confirmed retinal importance of APOE as a recognition ligand for the LPP uptake (Fig. 8E′) and suggested that this increase is likely due to an increase in the amount of cholesterol on LPPs circulating in the intraretinal space (Fig. 8F′).
Fig. 8.

Putative links between the proteins, genes, and sterols discussed in the present work. The biological processes in WT and Apoe−/− mice are labeled with bold small letters in maroon and small letters with primes in maroon, respectively. Proteins and sterols are in capital and small letters in black, respectively; all genes are italicized. The vertical upwards and downwards arrow indicate increases and decreases, respectively. 5CA, 5-cholestenoic acid. All other abbreviations are expanded in supplemental Text 1.
To compensate for retinal cholesterol increase, several homeostatic responses probably became operative in the Apoe−/− retina. The first was the upregulation of retinal APOA4 (Table 2, Fig. 8F′), the apolipoprotein that was also upregulated in the serum of an APOE-deficient individual and found on his VLDL and IDL, the particles that normally contain APOE but not APOA4 (16). This upregulation suggests that APOA4 and APOE could have redundant functions in the retina as carriers of intracellular cholesterol, which prevent increased intraretinal cholesterol content in Apoe−/− mice from focal deposition (Fig. 3). Yet, APOA4 does not seem to replace APOE as a recognition ligand on retinal LPPs, which stay in Apoe−/− mice in the intraretinal space and increase retinal cholesterol levels (Fig. 8F′). In addition, APOA4, an antiinflammatory agent and a potent inhibitor of lipid oxidation (71–73), may protect the retina from cholesterol oxidation and subsequent inflammation. These functions would be particularly important for the Apoe−/− retina, which has a cholesterol overload, and be consistent with the proinflammatory gene downregulation (Il-1β, Il-6, Tnfα, and Ccl2; Figs. 4, 8G′), as well as moderate immunoreactivity for GFAP and Iba1 (Figs. 5, 8H′). Furthermore, the downregulation of Apoc3 [also downregulated on serum VLDL and IDL of the APOE-deficient individual (16)] and Apod (Figs. 4, 8F′) suggests that not only APOA4 but other apolipoproteins could be a part of this compensatory response.
Additional compensatory mechanisms that were triggered in the Apoe−/− retina likely pertain to those inside retinal cells and the RPE. In the former, these mechanisms probably reflect cell deprivation from intraretinal cholesterol because retinal cells can no longer acquire LPPs lacking APOE (Fig. 8E′). Therefore, the rates of cholesterol biosynthesis in neurons (lathosterol) and astrocytes (desmosterol) were increased in the Apoe−/− retina (Figs. 2B, 8A′), whereas cholesterol elimination by metabolism to 5-cholestenoic acid was decreased (Figs. 2C, 8I′). In the RPE, a decrease in the Idol expression (Figs. 4, 8B′), if translated into a decrease in the protein expression, should increase the LDLR levels and hence the uptake of serum LDL to compensate for a decreased uptake of APOA1/APOE-containing HDL. Because LDLR could induce APOB degradation (74), increased LDLR levels in the Apoe−/− retina would lead to a compensatory Apob upregulation (Fig. 8B′), which may also serve as a mechanism to increase the uptake by choroidal blood vessels of BM-LP lacking APOE (Fig. 8C′). Collectively, these and other homeostatic responses in Apoe−/− mice probably diminished the impact of APOE absence on the retina and led only to a minor retinal phenotype. Conversely, if functional APOE was expressed and presented by an isoform (such as in humans or transgenic mice), the homeostatic responses would be weaker (if any at all) and lead to the isoform-specific phenotype. Thus, we agree with the conclusion of the study of the APOE-lacking individual that having no APOE is better than having the deleterious APOE isoform and that the targeted knockdown of APOE in the CNS might indeed be a therapeutic modality in neurodegenerative disorders (16).
Besides homeostatic responses specific to the Apoe−/− retina, we have potentially identified a general mechanism for protecting the retina from the increased content of serum cholesterol. Serum LDL is known to be avidly taken up by the RPE (17, 18) and is increased 2.3- and 1.4-fold in WT and Apoe−/− mice, respectively, fed a WTD (25). We put both genotypes on a WTD and found that retinal levels of their lathosterol and desmosterol were decreased as compared with animals fed regular chow (Fig. 2B). This result suggests that when serum cholesterol is increased, there is a compensatory downregulation of cholesterol biosynthesis, likely in the RPE, to balance an increased uptake of serum LDL. This cholesterol biosynthesis downregulation provides an explanation for a lack of consistent association between serum lipids and the incidence or progression of AMD (75).
Retinal detection by the label-free approach of the 18 cholesterol-related proteins (Table 1) was in agreement with previous studies in the field, which already documented the expression in the retina of some of the detected proteins (APOA1/4, APOE, CYP27A1, DHCR7, and SCARB2) (4, 20, 33, 68, 76–78). In addition, these proteomics data pointed to cholesterol-related proteins that have not yet been considered for their role in retinal cholesterol maintenance. So far, the main focus in retinal research was on APOE as the major apolipoprotein in the retina (26–28). Retinal studies of APOA1 and APOJ were conducted in the noncholesterol context (76, 77, 79, 80), and APOA4 was not investigated at all. Our work showed that not only APOE but also APOJ, APOA1, and APOA4 are present in the retina and should further be studied for their roles in retinal lipid transport (Fig. 8F). All four apolipoproteins are abundant in the brain (3) and integrate into APOAs/APOE- or APOJ-containing LPPs (81). If the same integration occurs in the retina, which like the brain is a neural tissue, there is probably still an uptake of the APOJ-containing LPPs in the Apoe−/− retina (Fig. 8E′), and this uptake involves LRP8 (ApoER2), VLDLR, and possibly LRP2 (megalin) (Fig. 8E) (82). The three receptors are known to be expressed in the retina (83–85) and recognize both APOE and APOJ. Measurements of retinal cholesterol in Apoj−/− mice would be required to gain insight into relative contributions of APOE and APOJ to the uptake of retinal LPP.
Research on retinal/RPE receptors for LPPs has been limited so far to those known to bind serum LPPs (LDLR, SCARB1, and SCARB2; Fig. 8B) (17, 18, 66–68). Our work highlighted LRP1, another LPP receptor, as well as LRPAP1 and MESDC2, the LRP chaperones (Table 1, Fig. 8E). LRP1, a scavenger receptor for diverse ligands including α2-macroglobulin and APOE (86), is a major LPP receptor in the brain (3). In the retina, LRP1 was minimally detectable by immunohistochemistry but was found to be significantly upregulated in diabetic retinopathy, sickle cell retinopathy, and proliferative vitreoretinopathy (87–89). In addition, LRP1 was shown to be protective against normal tension glaucoma by preventing retinal ganglion cells from death (90). Thus, similar to the brain, LRP1 seems to be of importance for the retina, where it probably interacts with LRPAP1, a chaperone, which is required for normal functional expression of LRP1 [LRPAP1 deficiency leads to LRP1 deficiency and severe myopia (91)]. The other function of LRPAP1 is to prevent all known ligands from binding to LRP1 (92–94). Yet, the role of LRPAP1 in cellular uptake of retinal LPP has not yet been evaluated. Similarly, not much is known about cholesterol-related significance of MESDC2, a chaperone for LRPs 2, 4, 5, and 6 (Fig. 8E) (95, 96), whose functions are similar to those of LRPAP1, i.e., facilitation of receptor folding and regulation of ligand-receptor interactions. MESDC2 was only shown to be released from the shed OSs and bind to these shed segments to facilitate their phagocytic clearance by the RPE (97). The identification of LRP1, LRPAP1, and MESDC2 as abundant cholesterol-related retinal proteins enhances our knowledge about retinal cholesterol maintenance and links retinal cholesterol to other retinal processes, some of which have not yet been considered for association with this lipid.
Of importance is perhaps retinal detection of SORT1 and the OSBPs (OSBPs 1 and 2 as well as OSBPLs 1A, L2, and L80) (Table 1, Fig. 8J). SORT1 is a member of the Vps10p (vacuolar protein sorting 10 protein) receptor family, which can bind to a variety of protein ligands and transport them from trans-Golgi network to lysosome or plasma membranes by endosomal traffic (98). These functions posttranslationally regulate plasma VLDL and LDL levels and tissue levels of LDLR (98, 99). In the retina, SORT1 was shown to be involved in the regulation of the cell-surface levels of LRP1 (100) and thereby to be a posttranslational regulator of retinal cholesterol input via LPP uptake. Previously, we found that a posttranslational mechanism (protein ubiquitination) plays a role in retinal control of cellular cholesterol biosynthesis (48). In the present work, we detected SORT1 and thus obtained a novel insight that both pathways of retinal cholesterol input (cellular cholesterol biosynthesis and cellular uptake of cholesterol-containing LPPs) could be controlled posttranslationally. Like SORT1, OSBPs and OSBPLs (Fig. 8J) transport molecules inside cells, but these molecules are lipids, rather than proteins (e.g., oxysterols and cholesterol). OSBPs facilitate cholesterol exchange between membrane junctions inside cells (101, 102) and received so far only little attention in the retinal field (103). Yet, five members from this family were found to be abundant in the retina (Table 1), thus suggesting that retinal significance of intracellular retinal sterol transport is currently underestimated and requires additional investigations.
Finally, APOE was shown to be important for microtubular stability in the brain due to its interaction with tau and MAP2C (59, 104, 105), the microtubule-associated proteins. We detected a decreased abundance of ATP1A2, ANK2, and EZR in the Apoe−/− versus WT retina (Table 2) and linked these changes to the RPE cytoskeleton maintenance and vesicular traffic (Fig. 8K′). We hypothesized that these processes affect phagocytosis and endocytosis (Fig. 8L) and ultimately lead to iron accumulation in the Apoe−/− retina (supplemental Figs. S1, S2). We used TEM with OTAP and found that Apoe−/− mice had numerous membrane-bounded vesicles inside the RPE and at the RPE-OS interface (Figs. 6, 7). We also carried out FTH1 immunohistochemistry and showed a change in FTH1 localization, with retinas from Apoe−/− mice showing less signal in photoreceptors and more signal in retinal ganglion cells (Fig. 7). These results suggest that Apoe−/− mice may have affected intercellular retinal iron trafficking. Ferritin can also be upregulated by inflammation, but the Apoe−/− retinas showed only limited, if any, inflammation. Additional studies are required to further test our proteomics interpretations; nevertheless, our work points to the noncanonical and novel APOE roles.
In summary, we confirmed retinal importance of APOE and demonstrated that the retinal impact of APOE deficiency is minimized by homeostatic responses. We identified some of these responses and found them to be similar to those in the APOE-deficient individual. The data obtained support the notion that targeted silencing of APOE in the central nervous system might be a therapeutic approach for Alzheimer’s disease and AMD. In addition, we found key apolipoproteins and receptors involved in the intraretinal lipid transfer. Abundant cholesterol-related proteins from other pathways of retinal cholesterol maintenance were discovered in the retina as well and pointed to the gaps in our knowledge about retinal cholesterol homeostasis. Finally, evidence was obtained that APOE might have roles beyond those of lipid transfer.
Supplementary Material
Acknowledgments
The authors thank the Visual Sciences Research Center Core Facilities (supported by National Institutes of Health Grant P30 EY11373) for assistance with mouse breeding (Heather Butler and Kathryn Franke), animal genotyping (John Denker), tissue sectioning (Catherine Doller), and microscopy (Anthony Gardella). The authors also thank Dr. Hisashi Fujioka (Electron Microscopy Core Facility) for help with studies of retinal ultrastructure and Danie Schlatzer (Proteomics and Small Molecule Mass Spectrometry Core) for conducting retinal label-free analysis.
Footnotes
Abbreviations:
- AMD
- age-related macular degeneration
- ANK2
- ankyrin-2
- ATP1A2
- Na+/K+-transporting ATPase subunit α2
- BM
- Bruch’s membrane
- CCL2
- C-C motif chemokine 2
- CRYBA1
- β-crystallin A1
- CRYBB1
- β-crystallin B1
- CRYBB2
- β-crystallin B2
- DPYSL4
- dihydropyrimidinase-related protein 4
- EZR
- ezrin
- FA
- fluorescein angiography
- FGB
- fibrinogen β chain
- FGG
- fibrinogen γ chain
- FN3KRP
- ketosamine-3-kinase
- FTH1
- H-ferritin
- GCL
- ganglion cell layer
- GFAP
- glial fibrillary acidic protein
- GSN
- gelsolin
- HIST1H4K
- histone H4
- HMGN5
- high mobility group nucleosome-binding domain-containing protein 5
- Iba1
- ionized calcium binding adaptor molecule
- IDOL
- inducible degrader of LDL receptor
- IL
- interleukin
- INL
- inner nuclear layer
- IPL
- inner plexiform layer
- IS
- photoreceptor inner segment
- KRT15
- keratin, type I cytoskeletal 15
- LDLR
- LDL receptor
- LPP
- lipoprotein particle
- LRP1
- LDL receptor-related protein 1
- LRPAP1
- α2-macroglobulin receptor-associated protein 1
- LXR
- liver X receptor
- LZIC
- leucine zipper and ICAT homologous domain-containing protein
- MESDC2
- mesoderm development candidate 2
- ONL
- outer nuclear layer
- OS
- photoreceptor outer segment
- OSBP
- oxysterol-binding protein
- PTMS
- parathymosin
- RPE
- retinal pigment epithelium
- RT
- retention time
- SCG2
- secretogranin-2
- SD-OCT
- resolution spectral-domain optical coherence tomography
- TEM
- transmission electron microscopy
- UC
- unesterified cholesterol
- WTD
- Western-type diet
- ZC3H4
- zinc finger CCCH domain-containing protein 4
This work was supported in part by National Institutes of Health Grants EY018383, EY11373, (both to I.A.P.) and EY015240; Research to Prevent Blindness; and the F. M. Kirby Foundation (J.L.D). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Mahley R. W., and Rall S. C. Jr.. 2000. Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1: 507–537. [DOI] [PubMed] [Google Scholar]
- 2.Mahley R. W. 1988. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 240: 622–630. [DOI] [PubMed] [Google Scholar]
- 3.Pfrieger F. W., and Ungerer N.. 2011. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50: 357–371. [DOI] [PubMed] [Google Scholar]
- 4.Tserentsoodol N., Gordiyenko N. V., Pascual I., Lee J. W., Fliesler S. J., and Rodriguez I. R.. 2006. Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol. Vis. 12: 1319–1333. [PubMed] [Google Scholar]
- 5.Windler E., Chao Y., and Havel R. J.. 1980. Regulation of the hepatic uptake of triglyceride-rich lipoproteins in the rat. Opposing effects of homologous apolipoprotein E and individual C apoproteins. J. Biol. Chem. 255: 8303–8307. [PubMed] [Google Scholar]
- 6.Bu G. 2009. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10: 333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toops K. A., Tan L. X., and Lakkaraju A.. 2016. Apolipoprotein E isoforms and AMD. Adv. Exp. Med. Biol. 854: 3–9. [DOI] [PubMed] [Google Scholar]
- 8.Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., and Pericak-Vance M. A.. 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 261: 921–923. [DOI] [PubMed] [Google Scholar]
- 9.Mayeux R., Stern Y., Ottman R., Tatemichi T. K., Tang M. X., Maestre G., Ngai C., Tycko B., and Ginsberg H.. 1993. The apolipoprotein epsilon 4 allele in patients with Alzheimer’s disease. Ann. Neurol. 34: 752–754. [DOI] [PubMed] [Google Scholar]
- 10.Corder E. H., Saunders A. M., Risch N. J., Strittmatter W. J., Schmechel D. E., Gaskell P. C. Jr., Rimmler J. B., Locke P. A., Conneally P. M., Schmader K. E., et al. . 1994. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7: 180–184. [DOI] [PubMed] [Google Scholar]
- 11.Klaver C. C., Kliffen M., van Duijn C. M., Hofman A., Cruts M., Grobbee D. E., van Broeckhoven C., and de Jong P. T.. 1998. Genetic association of apolipoprotein E with age-related macular degeneration. Am. J. Hum. Genet. 63: 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Souied E. H., Benlian P., Amouyel P., Feingold J., Lagarde J. P., Munnich A., Kaplan J., Coscas G., and Soubrane G.. 1998. The epsilon4 allele of the apolipoprotein E gene as a potential protective factor for exudative age-related macular degeneration. Am. J. Ophthalmol. 125: 353–359. [DOI] [PubMed] [Google Scholar]
- 13.McKay G. J., Patterson C. C., Chakravarthy U., Dasari S., Klaver C. C., Vingerling J. R., Ho L., de Jong P. T., Fletcher A. E., Young I. S., et al. . 2011. Evidence of association of APOE with age-related macular degeneration: a pooled analysis of 15 studies. Hum. Mutat. 32: 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pascolini D., Mariotti S. P., Pokharel G. P., Pararajasegaram R., Etya’ale D., Negrel A. D., and Resnikoff S.. 2004. 2002 global update of available data on visual impairment: a compilation of population-based prevalence studies. Ophthalmic Epidemiol. 11: 67–115. [DOI] [PubMed] [Google Scholar]
- 15.Blum C. B. 2016. Type III hyperlipoproteinemia: still worth considering? Prog. Cardiovasc. Dis. 59: 119–124. [DOI] [PubMed] [Google Scholar]
- 16.Mak A. C., Pullinger C. R., Tang L. F., Wong J. S., Deo R. C., Schwarz J. M., Gugliucci A., Movsesyan I., Ishida B. Y., Chu C., et al. . 2014. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol. 71: 1228–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elner V. M. 2002. Retinal pigment epithelial acid lipase activity and lipoprotein receptors: effects of dietary omega-3 fatty acids. Trans. Am. Ophthalmol. Soc. 100: 301–338. [PMC free article] [PubMed] [Google Scholar]
- 18.Tserentsoodol N., Sztein J., Campos M., Gordiyenko N. V., Fariss R. N., Lee J. W., Fliesler S. J., and Rodriguez I. R.. 2006. Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol. Vis. 12: 1306–1318. [PubMed] [Google Scholar]
- 19.Amaratunga A., Abraham C. R., Edwards R. B., Sandell J. H., Schreiber B. M., and Fine R. E.. 1996. Apolipoprotein E is synthesized in the retina by Muller glial cells, secreted into the vitreous, and rapidly transported into the optic nerve by retinal ganglion cells. J. Biol. Chem. 271: 5628–5632. [DOI] [PubMed] [Google Scholar]
- 20.Anderson D. H., Ozaki S., Nealon M., Neitz J., Mullins R. F., Hageman G. S., and Johnson L. V.. 2001. Local cellular sources of apolipoprotein E in the human retina and retinal pigmented epithelium: implications for the process of drusen formation. Am. J. Ophthalmol. 131: 767–781. [DOI] [PubMed] [Google Scholar]
- 21.Li C. M., Clark M. E., Chimento M. F., and Curcio C. A.. 2006. Apolipoprotein localization in isolated drusen and retinal apolipoprotein gene expression. Invest. Ophthalmol. Vis. Sci. 47: 3119–3128. [DOI] [PubMed] [Google Scholar]
- 22.Crabb J. W., Miyagi M., Gu X., Shadrach K., West K. A., Sakaguchi H., Kamei M., Hasan A., Yan L., Rayborn M. E., et al. . 2002. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA. 99: 14682–14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L., Clark M. E., Crossman D. K., Kojima K., Messinger J. D., Mobley J. A., and Curcio C. A.. 2010. Abundant lipid and protein components of drusen. PLoS One. 5: e10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raffai R. L., Dong L. M., Farese R. V. Jr., and Weisgraber K. H.. 2001. Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc. Natl. Acad. Sci. USA. 98: 11587–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plump A. S., Smith J. D., Hayek T., Aalto-Setala K., Walsh A., Verstuyft J. G., Rubin E. M., and Breslow J. L.. 1992. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 71: 343–353. [DOI] [PubMed] [Google Scholar]
- 26.Ong J. M., Zorapapel N. C., Rich K. A., Wagstaff R. E., Lambert R. W., Rosenberg S. E., Moghaddas F., Pirouzmanesh A., Aoki A. M., and Kenney M. C.. 2001. Effects of cholesterol and apolipoprotein E on retinal abnormalities in ApoE-deficient mice. Invest. Ophthalmol. Vis. Sci. 42: 1891–1900. [PubMed] [Google Scholar]
- 27.Ong J. M., Zorapapel N. C., Aoki A. M., Brown D. J., Nesburn A. B., Rich K. A., and Kenney C. M.. 2003. Impaired electroretinogram (ERG) response in apolipoprotein E-deficient mice. Curr. Eye Res. 27: 15–24. [DOI] [PubMed] [Google Scholar]
- 28.Dithmar S., Curcio C. A., Le N. A., Brown S., and Grossniklaus H. E.. 2000. Ultrastructural changes in Bruch’s membrane of apolipoprotein E-deficient mice. Invest. Ophthalmol. Vis. Sci. 41: 2035–2042. [PubMed] [Google Scholar]
- 29.Kliffen M., Lutgens E., Daemen M. J., de Muinck E. D., Mooy C. M., and de Jong P. T.. 2000. The APO(*)E3-Leiden mouse as an animal model for basal laminar deposit. Br. J. Ophthalmol. 84: 1415–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malek G., Johnson L. V., Mace B. E., Saloupis P., Schmechel D. E., Rickman D. W., Toth C. A., Sullivan P. M., and Bowes Rickman C.. 2005. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc. Natl. Acad. Sci. USA. 102: 11900–11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy O., Calippe B., Lavalette S., Hu S. J., Raoul W., Dominguez E., Housset M., Paques M., Sahel J. A., Bemelmans A. P., et al. . 2015. Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol. Med. 7: 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levy O., Lavalette S., Hu S. J., Housset M., Raoul W., Eandi C., Sahel J. A., Sullivan P. M., Guillonneau X., and Sennlaub F.. 2015. APOE isoforms control pathogenic subretinal inflammation in age-related macular degeneration. J. Neurosci. 35: 13568–13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Omarova S., Charvet C. D., Reem R. E., Mast N., Zheng W., Huang S., Peachey N. S., and Pikuleva I. A.. 2012. Abnormal vascularization in mouse retina with dysregulated retinal cholesterol homeostasis. J. Clin. Invest. 122: 3012–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charvet C. D., Saadane A., Wang M., Salomon R. G., Brunengraber H., Turko I. V., and Pikuleva I. A.. 2013. Pretreatment with pyridoxamine mitigates isolevuglandin-associated retinal effects in mice exposed to bright light. J. Biol. Chem. 288: 29267–29280. [Erratum. 2013. J. Biol. Chem. 288: 34054.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guyton J. R., and Klemp K. F.. 1988. Ultrastructural discrimination of lipid droplets and vesicles in atherosclerosis: value of osmium-thiocarbohydrazide-osmium and tannic acid-paraphenylenediamine techniques. J. Histochem. Cytochem. 36: 1319–1328. [DOI] [PubMed] [Google Scholar]
- 36.Saadane A., Mast N., Charvet C., Omarova S., Zheng W., Huang S. S., Kern T. S., Peachey N. S., and Pikuleva I. A.. 2014. Retinal and non-ocular abnormalities in Cyp27a1−/− Cyp64a1−/− mice with dysfunctional metabolism of cholesterol. Am. J. Pathol. 184: 2403–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mast N., Reem R., Bederman I., Huang S., DiPatre P. L., Bjorkhem I., and Pikuleva I. A.. 2011. Cholestenoic acid is an important elimination product of cholesterol in the retina: comparison of retinal cholesterol metabolism with that in the brain. Invest. Ophthalmol. Vis. Sci. 52: 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng W., Reem R. E., Omarova S., Huang S., DiPatre P. L., Charvet C. D., Curcio C. A., and Pikuleva I. A.. 2012. Spatial distribution of the pathways of cholesterol homeostasis in human retina. PLoS One. 7: e37926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dentchev T., Hahn P., and Dunaief J. L.. 2005. Strong labeling for iron and the iron-handling proteins ferritin and ferroportin in the photoreceptor layer in age-related macular degeneration. Arch. Ophthalmol. 123: 1745–1746. [DOI] [PubMed] [Google Scholar]
- 40.Bhisitkul R. B., Winn B. J., Lee O. T., Wong J., Pereira Dde S., Porco T. C., He X., Hahn P., and Dunaief J. L.. 2008. Neuroprotective effect of intravitreal triamcinolone acetonide against photoreceptor apoptosis in a rabbit model of subretinal hemorrhage. Invest. Ophthalmol. Vis. Sci. 49: 4071–4077. [DOI] [PubMed] [Google Scholar]
- 41.Pfaffl M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hahn P., Dentchev T., Qian Y., Rouault T., Harris Z. L., and Dunaief J. L.. 2004. Immunolocalization and regulation of iron handling proteins ferritin and ferroportin in the retina. Mol. Vis. 10: 598–607. [PubMed] [Google Scholar]
- 43.Tomechko S. E., Lundberg K. C., Jarvela J., Bebek G., Chesnokov N. G., Schlatzer D., Ewing R. M., Boom W. H., Chance M. R., and Silver R. F.. 2015. Proteomic and bioinformatics profile of paired human alveolar macrophages and peripheral blood monocytes. Proteomics. 15: 3797–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiśniewski J. R., Zougman A., Nagaraj N., and Mann M.. 2009. Universal sample preparation method for proteome analysis. Nat. Methods. 6: 359–362. [DOI] [PubMed] [Google Scholar]
- 45.Azzam S., Schlatzer D., Maxwell S., Li X., Bazdar D., Chen Y., Asaad R., Barnholtz-Sloan J., Chance R. M., and Sieg F. S.. 2016. Proteome and protein network analyses of memory T cells find altered translation and cell stress signaling in treated human immunodeficiency virus patients exhibiting poor CD4 recovery. Open Forum Infect. Dis. 3: ofw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kempen H. J., Glatz J. F., Gevers Leuven J. A., van der Voort H. A., and Katan M. B.. 1988. Serum lathosterol concentration is an indicator of whole-body cholesterol synthesis in humans. J. Lipid Res. 29: 1149–1155. [PubMed] [Google Scholar]
- 47.Bretillon L., Lutjohann D., Stahle L., Widhe T., Bindl L., Eggertsen G., Diczfalusy U., and Bjorkhem I.. 2000. Plasma levels of 24S-hydroxycholesterol reflect the balance between cerebral production and hepatic metabolism and are inversely related to body surface. J. Lipid Res. 41: 840–845. [PubMed] [Google Scholar]
- 48.Zheng W., Mast N., Saadane A., and Pikuleva I. A.. 2015. Pathways of cholesterol homeostasis in mouse retina responsive to dietary and pharmacologic treatments. J. Lipid Res. 56: 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mast N., Bederman I. R., and Pikuleva I. A.. 2018. Retinal cholesterol content is reduced in Simvastatin-treated mice due to inhibited local biosynthesis albeit increased uptake of serum cholesterol. Drug Metab. Dispos. 46: 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li C. M., Chung B. H., Presley J. B., Malek G., Zhang X., Dashti N., Li L., Chen J., Bradley K., Kruth H. S., et al. . 2005. Lipoprotein-like particles and cholesteryl esters in human Bruch’s membrane: initial characterization. Invest. Ophthalmol. Vis. Sci. 46: 2576–2586. [DOI] [PubMed] [Google Scholar]
- 51.Li C. M., Presley J. B., Zhang X., Dashti N., Chung B. H., Medeiros N. E., Guidry C., and Curcio C. A.. 2005. Retina expresses microsomal triglyceride transfer protein: implications for age-related maculopathy. J. Lipid Res. 46: 628–640. [DOI] [PubMed] [Google Scholar]
- 52.Yang X., Wang J., Zhou Z., Jiang R., Huang J., Chen L., Cao Z., Chu H., Han B., Cheng Y., et al. . 2018. Silica-induced initiation of circular ZC3H4 RNA/ZC3H4 pathway promotes the pulmonary macrophage activation. FASEB J. 32: 3264–3277. [DOI] [PubMed] [Google Scholar]
- 53.Storch M. K., Fischer-Colbrie R., Smith T., Rinner W. A., Hickey W. F., Cuzner M. L., Winkler H., and Lassmann H.. 1996. Co-localization of secretoneurin immunoreactivity and macrophage infiltration in the lesions of experimental autoimmune encephalomyelitis. Neuroscience. 71: 885–893. [DOI] [PubMed] [Google Scholar]
- 54.Schratzberger P., Woll E., Reinisch N., Kahler C. M., and Wiedermann C. J.. 1996. Secretoneurin-induced in vitro chemotaxis of human monocytes is inhibited by pertussis toxin and an inhibitor of protein kinase C. Neurosci. Lett. 214: 208–210. [DOI] [PubMed] [Google Scholar]
- 55.Soto C., Castano E. M., Prelli F., Kumar R. A., and Baumann M.. 1995. Apolipoprotein E increases the fibrillogenic potential of synthetic peptides derived from Alzheimer’s, gelsolin and AA amyloids. FEBS Lett. 371: 110–114. [DOI] [PubMed] [Google Scholar]
- 56.Zelcer N., Hong C., Boyadjian R., and Tontonoz P.. 2009. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 325: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis G. P., and Fisher S. K.. 2003. Up-regulation of glial fibrillary acidic protein in response to retinal injury: its potential role in glial remodeling and a comparison to vimentin expression. Int. Rev. Cytol. 230: 263–290. [DOI] [PubMed] [Google Scholar]
- 58.Santos A. M., Calvente R., Tassi M., Carrasco M. C., Martin-Oliva D., Marin-Teva J. L., Navascues J., and Cuadros M. A.. 2008. Embryonic and postnatal development of microglial cells in the mouse retina. J. Comp. Neurol. 506: 224–239. [DOI] [PubMed] [Google Scholar]
- 59.Roses A. D., Einstein G., Gilbert J., Goedert M., Han S. H., Huang D., Hulette C., Masliah E., Pericak-Vance M. A., Saunders A. M., et al. . 1996. Morphological, biochemical, and genetic support for an apolipoprotein E effect on microtubular metabolism. Ann. N. Y. Acad. Sci. 777: 146–157. [DOI] [PubMed] [Google Scholar]
- 60.Sridharan G., and Shankar A. A.. 2012. Toluidine blue: a review of its chemistry and clinical utility. J. Oral Maxillofac. Pathol. 16: 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D., Cragoe E. J. Jr., and Smith T. W.. 1987. Relations among sodium pump inhibition, Na-Ca and Na-H exchange activities, and Ca-H interaction in cultured chick heart cells. Circ. Res. 60: 185–193. [DOI] [PubMed] [Google Scholar]
- 62.Hentze M. W., Rouault T. A., Caughman S. W., Dancis A., Harford J. B., and Klausner R. D.. 1987. A cis-acting element is necessary and sufficient for translational regulation of human ferritin expression in response to iron. Proc. Natl. Acad. Sci. USA. 84: 6730–6734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hahn P., Qian Y., Dentchev T., Chen L., Beard J., Harris Z. L., and Dunaief J. L.. 2004. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. USA. 101: 13850–13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fliesler S. J., Florman R., Rapp L. M., Pittler S. J., and Keller R. K.. 1993. In vivo biosynthesis of cholesterol in the rat retina. FEBS Lett. 335: 234–238. [DOI] [PubMed] [Google Scholar]
- 65.Lin J. B., Mast N., Bederman I. R., Li Y., Brunengraber H., Bjorkhem I., and Pikuleva I. A.. 2016. Cholesterol in mouse retina originates primarily from in situ de novo biosynthesis. J. Lipid Res. 57: 258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duncan K. G., Hosseini K., Bailey K. R., Yang H., Lowe R. J., Matthes M. T., Kane J. P., LaVail M. M., Schwartz D. M., and Duncan J. L.. 2009. Expression of reverse cholesterol transport proteins ATP-binding cassette A1 (ABCA1) and scavenger receptor BI (SR-BI) in the retina and retinal pigment epithelium. Br. J. Ophthalmol. 93: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rudolf M., Winkler B., Aherrahou Z., Doehring L. C., Kaczmarek P., and Schmidt-Erfurth U.. 2005. Increased expression of vascular endothelial growth factor associated with accumulation of lipids in Bruch’s membrane of LDL receptor knockout mice. Br. J. Ophthalmol. 89: 1627–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Houssier M., Raoul W., Lavalette S., Keller N., Guillonneau X., Baragatti B., Jonet L., Jeanny J. C., Behar-Cohen F., Coceani F., et al. . 2008. CD36 deficiency leads to choroidal involution via COX2 down-regulation in rodents. PLoS Med. 5: e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Curcio C. A., Messinger J. D., Sloan K. R., McGwin G., Medeiros N. E., and Spaide R. F.. 2013. Subretinal drusenoid deposits in non-neovascular age-related macular degeneration: morphology, prevalence, topography, and biogenesis model. Retina. 33: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Curcio C. A., Johnson M., Huang J. D., and Rudolf M.. 2010. Apolipoprotein B-containing lipoproteins in retinal aging and age-related macular degeneration. J. Lipid Res. 51: 451–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vowinkel T., Mori M., Krieglstein C. F., Russell J., Saijo F., Bharwani S., Turnage R. H., Davidson W. S., Tso P., Granger D. N., et al. . 2004. Apolipoprotein A-IV inhibits experimental colitis. J. Clin. Invest. 114: 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Qin X., Swertfeger D. K., Zheng S., Hui D. Y., and Tso P.. 1998. Apolipoprotein AIV: a potent endogenous inhibitor of lipid oxidation. Am. J. Physiol. 274: H1836–H1840. [DOI] [PubMed] [Google Scholar]
- 73.Ostos M. A., Conconi M., Vergnes L., Baroukh N., Ribalta J., Girona J., Caillaud J. M., Ochoa A., and Zakin M. M.. 2001. Antioxidative and antiatherosclerotic effects of human apolipoprotein A-IV in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 21: 1023–1028. [DOI] [PubMed] [Google Scholar]
- 74.Blasiole D. A., Oler A. T., and Attie A. D.. 2008. Regulation of ApoB secretion by the low density lipoprotein receptor requires exit from the endoplasmic reticulum and interaction with ApoE or ApoB. J. Biol. Chem. 283: 11374–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klein R., Myers C. E., Buitendijk G. H., Rochtchina E., Gao X., de Jong P. T., Sivakumaran T. A., Burlutsky G., McKean-Cowdin R., Hofman A., et al. . 2014. Lipids, lipid genes, and incident age-related macular degeneration: the three continent age-related macular degeneration consortium. Am. J. Ophthalmol. 158: 513–524.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simó R., García-Ramírez M., Higuera M., and Hernández C.. 2009. Apolipoprotein A1 is overexpressed in the retina of diabetic patients. Am. J. Ophthalmol. 147: 319–325.e1. [DOI] [PubMed] [Google Scholar]
- 77.Miyara N., Shinzato M., Yamashiro Y., Iwamatsu A., Kariya K. I., and Sawaguchi S.. 2008. Proteomic analysis of rat retina in a steroid-induced ocular hypertension model: potential vulnerability to oxidative stress. Jpn. J. Ophthalmol. 52: 84–90. [DOI] [PubMed] [Google Scholar]
- 78.Fliesler S. J. 2010. Retinal degeneration in a rat model of Smith-Lemli-Opitz Syndrome: thinking beyond cholesterol deficiency. Adv. Exp. Med. Biol. 664: 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jomary C., Murphy B. F., Neal M. J., and Jones S. E.. 1993. Abnormal distribution of retinal clusterin in retinitis pigmentosa. Brain Res. Mol. Brain Res. 20: 274–278. [DOI] [PubMed] [Google Scholar]
- 80.Shanmugaratnam J., Berg E., Kimerer L., Johnson R. J., Amaratunga A., Schreiber B. M., and Fine R. E.. 1997. Retinal Muller glia secrete apolipoproteins E and J which are efficiently assembled into lipoprotein particles. Brain Res. Mol. Brain Res. 50: 113–120. [DOI] [PubMed] [Google Scholar]
- 81.Elliott D. A., Weickert C. S., and Garner B.. 2010. Apolipoproteins in the brain: implications for neurological and psychiatric disorders. Clin. Lipidol. 51: 555–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leeb C., Eresheim C., and Nimpf J.. 2014. Clusterin is a ligand for apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) and signals via the Reelin-signaling pathway. J. Biol. Chem. 289: 4161–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gil S. Y., Youn B. S., Byun K., Huang H., Namkoong C., Jang P. G., Lee J. Y., Jo Y. H., Kang G. M., Kim H. K., et al. . 2013. Clusterin and LRP2 are critical components of the hypothalamic feeding regulatory pathway. Nat. Commun. 4: 1862. [DOI] [PubMed] [Google Scholar]
- 84.Trotter J. H., Klein M., Jinwal U. K., Abisambra J. F., Dickey C. A., Tharkur J., Masiulis I., Ding J., Locke K. G., Rickman C. B., et al. . 2011. ApoER2 function in the establishment and maintenance of retinal synaptic connectivity. J. Neurosci. 31: 14413–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heckenlively J. R., Hawes N. L., Friedlander M., Nusinowitz S., Hurd R., Davisson M., and Chang B.. 2003. Mouse model of subretinal neovascularization with choroidal anastomosis. Retina. 23: 518–522. [DOI] [PubMed] [Google Scholar]
- 86.Emonard H., Theret L., Bennasroune A. H., and Dedieu S.. 2014. Regulation of LRP-1 expression: make the point. Pathol. Biol. (Paris). 62: 84–90. [DOI] [PubMed] [Google Scholar]
- 87.Barcelona P. F., Jaldin-Fincati J. R., Sanchez M. C., and Chiabrando G. A.. 2013. Activated alpha2-macroglobulin induces Muller glial cell migration by regulating MT1-MMP activity through LRP1. FASEB J. 27: 3181–3197. [DOI] [PubMed] [Google Scholar]
- 88.Barcelona P. F., Luna J. D., Chiabrando G. A., Juarez C. P., Bhutto I. A., Baba T., McLeod D. S., Sanchez M. C., and Lutty G. A.. 2010. Immunohistochemical localization of low density lipoprotein receptor-related protein 1 and alpha(2)-Macroglobulin in retinal and choroidal tissue of proliferative retinopathies. Exp. Eye Res. 91: 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sánchez M. C., Barcelona P. F., Luna J. D., Ortiz S. G., Juarez P. C., Riera C. M., and Chiabrando G. A.. 2006. Low-density lipoprotein receptor-related protein-1 (LRP-1) expression in a rat model of oxygen-induced retinal neovascularization. Exp. Eye Res. 83: 1378–1385. [DOI] [PubMed] [Google Scholar]
- 90.Hayashi H., Eguchi Y., Fukuchi-Nakaishi Y., Takeya M., Nakagata N., Tanaka K., Vance J. E., and Tanihara H.. 2012. A potential neuroprotective role of apolipoprotein E-containing lipoproteins through low density lipoprotein receptor-related protein 1 in normal tension glaucoma. J. Biol. Chem. 287: 25395–25406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aldahmesh M. A., Khan A. O., Alkuraya H., Adly N., Anazi S., Al-Saleh A. A., Mohamed J. Y., Hijazi H., Prabakaran S., Tacke M., et al. . 2013. Mutations in LRPAP1 are associated with severe myopia in humans. Am. J. Hum. Genet. 93: 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bu G., Geuze H. J., Strous G. J., and Schwartz A. L.. 1995. 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 14: 2269–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Willnow T. E., Armstrong S. A., Hammer R. E., and Herz J.. 1995. Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc. Natl. Acad. Sci. USA. 92: 4537–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Willnow T. E., Rohlmann A., Horton J., Otani H., Braun J. R., Hammer R. E., and Herz J.. 1996. RAP, a specialized chaperone, prevents ligand-induced ER retention and degradation of LDL receptor-related endocytic receptors. EMBO J. 15: 2632–2639. [PMC free article] [PubMed] [Google Scholar]
- 95.Lu W., Liu C. C., Thottassery J. V., Bu G., and Li Y.. 2010. Mesd is a universal inhibitor of Wnt coreceptors LRP5 and LRP6 and blocks Wnt/beta-catenin signaling in cancer cells. Biochemistry. 49: 4635–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoshi T., Tezuka T., Yokoyama K., Iemura S., Natsume T., and Yamanashi Y.. 2013. Mesdc2 plays a key role in cell-surface expression of Lrp4 and postsynaptic specialization in myotubes. FEBS Lett. 587: 3749–3754. [DOI] [PubMed] [Google Scholar]
- 97.Chen X., Guo F., LeBlanc M. E., Ding Y., Zhang C., Shakya A., and Li W.. 2016. Mesd extrinsically promotes phagocytosis by retinal pigment epithelial cells. Cell Biol. Toxicol. 32: 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gao A., Cayabyab F. S., Chen X., Yang J., Wang L., Peng T., and Lv Y.. 2017. Implications of Sortilin in Lipid Metabolism and Lipid Disorder Diseases. DNA Cell Biol. 36: 1050–1061. [DOI] [PubMed] [Google Scholar]
- 99.Gustafsen C., Kjolby M., Nyegaard M., Mattheisen M., Lundhede J., Buttenschon H., Mors O., Bentzon J. F., Madsen P., Nykjaer A., et al. . 2014. The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab. 19: 310–318. [DOI] [PubMed] [Google Scholar]
- 100.Actis Dato V., Grosso R. A., Sanchez M. C., Fader C. M., and Chiabrando G. A.. 2018. Insulin-induced exocytosis regulates the cell surface level of low-density lipoprotein-related protein-1 in Muller glial cells. Biochem. J. 475: 1669–1685. [DOI] [PubMed] [Google Scholar]
- 101.Antonny B., Bigay J., and Mesmin B.. 2018. The oxysterol-binding protein cycle: burning off PI(4)P to transport cholesterol. Annu. Rev. Biochem. 87: 809–837. [DOI] [PubMed] [Google Scholar]
- 102.Kentala H., Weber-Boyvat M., and Olkkonen V. M.. 2016. OSBP-related protein family: mediators of lipid transport and signaling at membrane contact sites. Int. Rev. Cell Mol. Biol. 321: 299–340. [DOI] [PubMed] [Google Scholar]
- 103.Moreira E. F., Jaworski C., Li A., and Rodriguez I. R.. 2001. Molecular and biochemical characterization of a novel oxysterol-binding protein (OSBP2) highly expressed in retina. J. Biol. Chem. 276: 18570–18578. [DOI] [PubMed] [Google Scholar]
- 104.Scott B. L., Welch K., deSerrano V., Moss N. C., Roses A. D., and Strittmatter W. J.. 1998. Human apolipoprotein E accelerates microtubule polymerization in vitro. Neurosci. Lett. 245: 105–108. [DOI] [PubMed] [Google Scholar]
- 105.Fleming L. M., Weisgraber K. H., Strittmatter W. J., Troncoso J. C., and Johnson G. V.. 1996. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp. Neurol. 138: 252–260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.