

ABSTRACT
Targeted expression of genes coding for proteins specific to astrocytes, oligodendrocytes and myelin was performed in frontal cortex area 8 of Creutzfeldt-Jakob disease methionine/methionine and valine/valine (CJD MM1 and VV2, respectively) compared with controls. GFAP (glial fibrillary acidic protein) mRNA was up-regulated whereas SLC1A2 (solute carrier family 1 member 2, coding for glutamate transporter 1: GLT1), AQ4 (aquaporin 4), MPC1 (mitochondrial pyruvate carrier 1) and UCP5 (mitochondrial uncoupled protein 5) mRNAs were significantly down-regulated in CJD MM1 and CJD VV2, and GJA1 (connexin 43) in CJD VV2. OLIG1 and OLIG2 (oligodendocyte transcription factor 1 and 2, respectively), SOX10 (SRY-Box10) and oligodendroglial precursor cell (OPC) marker NG2 (neuronal/glial antigen) 2 were preserved, but GALC (coding for galactosylceramidase), SLC2A1 (solute carrier family 2 member 1: glucose transporter member 1: GLUT1) and MCT1 (monocarboxylic acid transporter 1) mRNA expression levels were significantly reduced in CJD MM1 and CJD VV2. Expression levels of most genes linked to myelin were not altered in the cerebral cortex in CJD. Immunohistochemistry to selected proteins disclosed individual variations but GFAP, Olig-2, AQ4 and GLUT1 correlated with mRNA levels, whereas GLT1 was subjected to individual variations. However, MPC1, UCP5 and MCT1 decrease was more closely related to the respective reduced neuronal immunostaining. These observations support the idea that molecular deficits linked to energy metabolism and solute transport in astrocytes and oligodendrocytes, in addition to neurons, are relevant in the pathogenesis of cortical lesions in CJD.
KEYWORDS: Creutzfeldt-Jakob disease, prion diseases, astrocytes, oligodendrocytes, myelin, energy metabolism, astrogliopathy, oligodendrogliopathy
Introduction
Prion diseases are a group of transmissible encephalopathies linked to the prion protein (PrPC) which is converted into an abnormally conformed proteinase K-resistant protein named prion (PrPSC). The human prion diseases are sporadic, iatrogenic and genetic Creutzfeld-Jakob disease (sCJD, iCJD and gCJD, respectively), Gerstmann-Straüsler-Scheinker disease (GSS) disease, and fatal familial insomnia (FFI). gCJD, GSS and FFI are due to mutations in prion protein gene (PRNP) [1–7]. The main prionopathies in animals are scrapie (typical and atypical) in sheep and goats; chronic wasting disease in deer, elk, moose and reindeer; and bovine spongiform encephalopathy (BSE) in cattle [8].Variant CJD (vCJD) is considered a form of BSE transmitted to humans [9]. CJD is not homogeneous; different subtypes are distinguished in sCJD depending on the prion type (I and 2) and the composition of codon 129 in PRNP (methionine/methionine: MM, valine/valine: VV, and methionine/valine: MV), each one with singular clinical phenotypes [10,11]. The most frequent subtypes are sCJD MM1 and sCJD VV2.
Common neuropathological lesions in CJD are neuron loss, spongiform change and deposition of PrPSC. Additional characteristics of sCJD MM1 are microvacuolar spongiform change in the frontal and occipital cortices, and molecular layer of the cerebellum, and synaptic-like PrPSC deposition. Particular features of sCJD VV2 are microvacuolar or confluent spongiform change, major involvement of the cerebellum, and synaptic, perineuronal and plaque-like PrPSC deposits [4–7]. Reactive astrocytosis and microgliosis, accompanied by a robust molecular inflammatory response, are important accompanying features [12–15].
Besides their contribution to astrocytic gliosis and inflammation, little is known about the defects of astrocytes in CJD. In addition to neuronal bodies, dendrites, synapses and axons, PrPSC can be present in astrocytes and microglia [16]. Aquaporin 1 and 4 immunoreactivity is increased [17,18], thus suggesting adaptation to water transport. Moreover, recent studies have shown that human astrocytes have the capacity to take up and degrade normal and protease-resistant prion protein [19] and that they can transfer PrPSC to neurons via nanotubules [20] thereby contributing to prion disease progression.
Regarding oligodendrocytes, less information is available in CJD. Oligodendrocytes are apparently resistant to PrPSC infectivity [21]. However, axon and myelin damage occurs in prion diseases [22], abnormal interactions between oligodendroglia and astrocytes are found in experimental CJD and scrapie [23], and engulfment of oligodendrocytes by hypertrophic astrocytes has been reported in the white matter in CJD [24]. PRPSC is also localized as arrays adjacent to myelin fibers in the cerebrum and cerebellum in CJD [25].
Based on these data, the present study was undertaken to gain understanding about transcription alteration of genes encoding proteins specifically expressed in astrocytes and oligodendrocytes in frontal cortex area 8 of CJD MM1 and CJD VV2. Selected genes include those encoding structural proteins of astrocytes and myelin, those involved in energy metabolism and axon maintenance, and genes of connexins of the gap junctions between oligodendrocytes and astrocytes. Protein expression of altered has been assessed by immunohistochemistry.
Results
Astrocytic markers
Covariance analysis (ANCOVA) of GFAP (coding for glial fibrillary acidic protein) mRNA expression levels revealed a significant group effect [F(2,19) = 4.383, P = 0.027] but did not show RIN effect [F(1,19) = 0.337, P = 0.568]. Subsequent one-way ANOVA analysis [F(2,20) = 7.918, P = 0.0029] revealed significant increase in CJD MM1 and CJD VV2 when compared with controls (P = 0.012 and P = 0.005, respectively). In contrast, ANCOVA of ALDH1L1 (coding for aldehyde dehydrogenase 1, family member L1) mRNA, used as marker of total astrocytes, had non-significant group [F(2,22) = 1.087, P = 0.355] and RIN [F(1,22) = 0.021, P = 0.887] effects; no differences of ALDH1L1 mRNA were observed between CJD and controls. ANCOVA of AQP4 (coding for aquaporin 4) mRNA showed significant group effect [F(2,21) = 4.307, P = 0.027] but not RIN effect [F(1,21) = 0.334, P = 0.569]. Subsequent one-way ANOVA analysis [F(2,22) = 8.596, P = 0.0017] showed significant decreased expression in CJD MM1 and CJD VV2 when compared with controls (P = 0.26 and P = 0.002, respectively). ANCOVA analysis of SLC1A2 (coding for glutamate transporter 1) mRNA expression showed significant group effect [F(2,21) = 6.031, P = 0.009] but not RIN effect [F(3,25) = 0.754, P = 0.395]. One-way ANOVA analysis [F(1,21) = 8.865, P = 0.0015] revealed significant down-regulation reduction of SLC1A2 in CJD VV2 when compared with controls (P = 0.001) and with CJD MM1 (P = 0.028).
ANCOVA of MCT4 (coding for solute carrier family member 16: monocarboxylic acid transporter member 4) mRNA expression revealed no significant changes with respect to group [F(2,20) = 2.110, P = 0.147] and RIN [F (1,20) = 0.195, P = 0.664] effect. Conversely, ANCOVA of MPC1 (coding for mitochondria pyruvate carrier 1) and UCP5 (coding for mitochondrial anion carrier, mitochondrial uncoupling protein 5) showed significant group effect [F(2,21) = 6.282, P = 0.007] and [F(2,21) = 9.103, P = 0.001], respectively, as well as RIN effect [F(1,21) = 4.972, P = 0.037] and [F(1,21) = 8.734, P = 0.008], respectively. Next, ANCOVA revealed significantly decreased expression of MPC1 and UCP5 in CJD MM1 (P = 0.019 and P = 0.008 respectively) and CJD VV2 (P = 0.002 and P = 0.001, respectively) when compared with controls. ANCOVA of UCP4 mRNA (coding for mitochondrial uncoupling protein 4) did not reveal significant alterations related to group [F(2,20) = 0.009, P = 0.991] but to RIN effects [F(1,20) = 11.258, P = 0.003].
Finally, the expression of GJB6 (coding for connexin 30), which was preserved (although with a trend to decrease in CJD VV2), was not dependent on group [F(2,19) = 2.418, P = 0.113] or RIN [F(1,19) = 1.158, P = 0.294] effect. ANCOVA of GJA1 (coding for connexin 43) mRNA expression indicated significant group effect [F(2,21) = 8.218, P = 0.002] but not RIN effect [F(1,21) = 4.105, P = 0.056]. Subsequent one-way ANOVA analysis [F(2,22) = 8.224, P = 0.002] revealed significantly reduced GJA1 expression in CJD VV2 when compared with controls (P = 0.007) and with CJD MM1 (P = 0.004) (Figure 1(a)).
Figure 1.
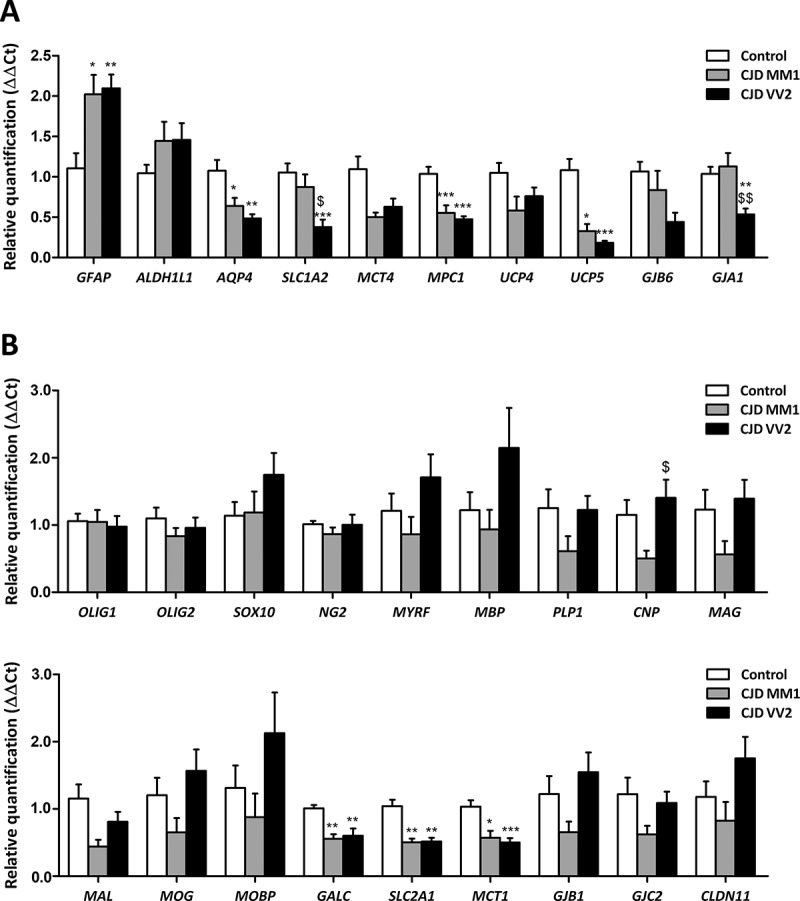
Expression levels, as revealed by RT-qPCR, of genes coding for specific proteins of astrocytes (a) and oligodendrocytes (b) in frontal cortex area 8 of CJD MM1 and CJD VV2 compared with expression levels in controls. Statistical analysis of the expression data between groups uses one-way analysis of variance (ANOVA) followed by Tukey post-test for GFAP, AQP4, SLC1A2, MPC1, UCP4, GJB6, GJA1, OLIG1, OLIG2, NG2, CNP, MAL, GALC, MCT1, GJB1 and GJC2. Kruskal-Wallis test followed by Dunns post-hoc test was used for ALDH1L1, MCT4, UCP5, SOX10, MYRF, MBP, PLP1, MAG, MOG, MOBP, SLC2A1 and CLDN11 (SPSS software. IBM SPSS Statistics for Windows, Version 21.0). All data are expressed as mean ± SEM. Differences between groups are considered statistically significant at * P < 0.05, ** P < 0.01, *** P < 0.001 when comparing CJD cases with controls; and set at $ P < 0.05 and $$ P < 0.01 when comparing CJD MM1 with CJD VV2.
Oligodendrocytic markers
ANCOVA showed no group or RIN effect on mRNA expression levels of the oligodendroglial markers OLIG1 [F(2,23) = 0.221, P = 0.804] and [F(1,23) = 0.336, P = 0.568], OLIG2 [F(2,23) = 0.801, P = 0.461] and [F(1,23) = 0.276, P = 0.604], and SOX10 [F(2,23) = 1.369, P = 0.274] and [F(1,23) = 3.930, P = 0.059]; and the OPC marker NG2 [F(2,23) = 0.412, P = 0.667] and [F(1,23) = 0.001, P = 0.978]. Expression levels of OLIG1, OLIG2 and NG2 were similar in CJD and controls.
Likewise, ANCOVA showed no alterations in the expression levels of genes coding for different myelin proteins, such as MYRF (coding for myelin regulatory factor), MBP (coding for myelin basic protein), PLP1 (coding for proteolipid protein 1), MAL (coding for Mal), MOG (myelin oligodendrocyte glycoprotein) and MOBP (myelin-associated oligodendrocyte basic protein) due to group effect [F(2,23) = 2.692, P = 0.0.89], [F(2,22) = 2.214, P = 0.133], [F(2,23) = 2.356, P = 0.117], [F(2,23) = 3.373, P = 0.052], [F(2,23) = 3.146, P = 0.062] and [F(2,23) = 2.409, P = 0.112], respectively, or due to RIN effect [F(1,23) = 3.664, P = 0.068], [F(1,22) = 0.918, P = 0.349], [F(1,23) = 0.991, P = 0.330], [F(1,23) = 0.311, P = 0.582], [F(1,23) = 1.968, P = 0.174] and [F(1,23) = 3.916, P = 0.060], respectively. However, ANCOVA analysis revealed significant group effect on CNP (coding for 2ʹ,3ʹ-cyclic nucleotide 3ʹ phosphodiesterase) and MAG (myelin associated glycoprotein) mRNA expression [F(2,21) = 3.970, P = 0.034] and [F(2,22) = 3.786, P = 0.039], respectively; but not RIN effect [F(1,21) = 1.697, P = 0.207], [F(1,22) = 3.307, P = 0.083], respectively. Subsequent analysis revealed no differences in CNP [F(2,22) = 2.328, P = 0.12] and MAG [KG(2,23) = 5.391, P = 0.0675] mRNA expression levels between CJD and controls. Regarding GALC (coding for galactosylceramidase), covariance analysis showed a significant effect of group [F(2,21) = 12.680, P = 0.000] and RIN [F(1,21) = 10.171, P = 0.004]. ANCOVA’s contrast analysis revealed significant GALC mRNA down-regulation in CJD MM1 and CJD VV2 (P = 0.001 and P = 0.000, respectively).
ANCOVA applied to expression levels of SLC2A1 (coding for solute carrier family 2: glucose transporter, member 1) and MCT1 (coding for solute carrier family 16: monocarboxylic acid transporter, member 1) revealed significant group effect [F(2,22) = 9.304, P = 0.001] and [F(2,21) = 3.468, P = 0.050], respectively; but no RIN effect [F(1,22) = 0.034, P = 0.855] and [F(1,21) = 3.924, P = 0.061], respectively. Post- variance analysis of SLC2A1 and MCT1 ([KG(2,23) = 15.26, P = 0.0005] and [F(2,22) = 11.333, P = 0.000], respectively) showed significant SLC2A1 and MCT1 decrease in CJD MM1 (P = 0.01 and P = 0.04, respectively) and CJD VV2 (P = 0.01 and P = 0.001, respectively) when compared with control group.
Finally, ANCOVA of expression of genes coding for connexins 32 and 47 (GJB1 and GJC2), and claudin 11 (CLDN11) did not reveal significant group [F(2,22) = 3.271, P = 0.057], [F(2,23) = 2.461, P = 0.107] and [F(2,23) = 2.608, P = 0.095], and RIN [F(1,22) = 1.326, P = 0.262], [F(1,23) = 0.730, P = 0.402] and [F(1,23) = 0.719, P = 0.405] effects, respectively. GJB1, GJC2 and CLDN11 expression was not modified in CJD, although there was a trend to decrease in CJD MM1, when compared with controls (Figure 1(b)).
Immunohistochemistry
Paraffin sections processed for immunohistochemistry showed marked increase in GFAP immunoreactivity in CJD when compared with controls, as expected. GFAP-immunoreacytive astrocytes were increased in number, and the amount of GFAP was increased per astrocyte with robust immunostaining of astrocyte processes (Figure 2). In contrast, OLig-2 immunostaining in the cerebral cortex did not reveal major differences between CJD and controls (Figure 2). Aquaporin 4 (AQP4) immunoreactive localized in astrocytes delineating fine and varicose network processes in control cerebral cortex. AQP4 immunoreactivity in CJD was variable from one case to another yet preserving the same morphological profile (Figure 2). In advanced cases, AQP4 immunoreactivity was increased and blurred as reported elsewhere. GLT1 immunoreactivity in control cases was seen as delicate meshwork of fine and varicose processes around the nuclei of astrocytes. This pattern was preserved in CJD, although with marked differences from one case to another; in some cases GLT1 immunoreactivity practically disappeared in areas with severe spongiform change while it was practically preserved in others; Moreover variations were also observed in different areas in the same tissue section (Figure 2). MPC1 was mainly localized in the cytoplasm of neurons in the cerebral cortex of control cases as a punctuate immunostaining consistent with a mitochondrial localization. MPC1 immunoreactivity was dramatically reduced in neurons in CJD but increased in a few cortical glial cells (Figure 2). Similarly, UPC5 immunostaining mainly decorated neurons in controls. However, UPC5 immunoreactivity decreased in neurons but increased in glial cells, mainly in cells with astrocyte morphology, in CJD (Figure 2).
Figure 2.
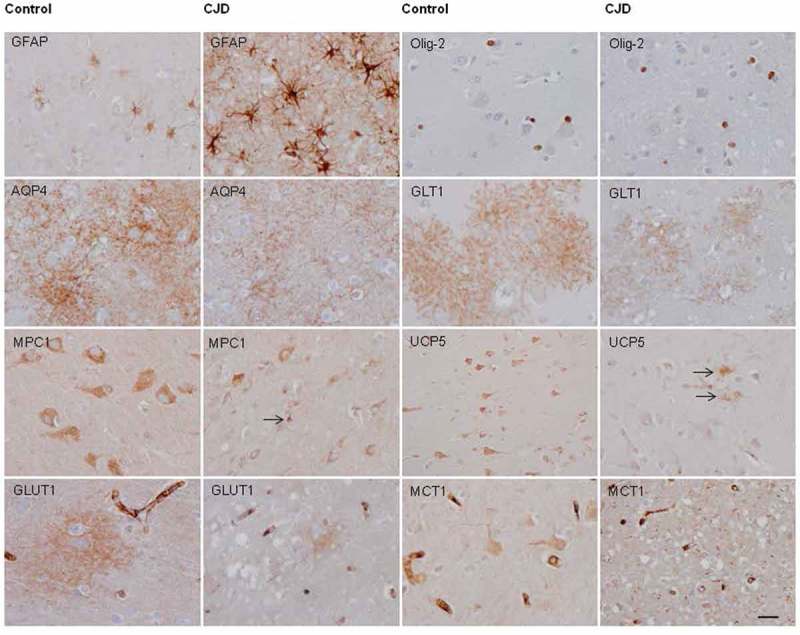
Representative images of glial fibrillary acidic protein (GFAP), Olig-2, aquaporin 4 (AQP4), solute carrier family 1, member 1 (SLC1A2: glial affinity glutamate transporter: GLT1), mitochondrial pyruvate carrier 1 (MPC1), mitochondrial uncoupling protein 5 (UCP5), solute carrier family 2 member 1 (glucose transporter 1: GLUT1) and solute carrier family 16 member 1 (monocarboxylic acid transporter 1: MCT1) immunoreactivity in the frontal cortex area 8 of controls and cases with CJD MM1. Paraffin sections processed for immunohistochemistry and slightly stained with hematoxylin, bar = 25 μm.
GLUT1 was mainly found in the walls of small blood vessels consistent with endothelial localization as expected. In addition, GLUT1 immunoractivity was found as blurred patches in the cortical neuropil in control cases; these patches were markedly reduced in size and intensity in CJD (Figure 2). Finally, MCT1 was mainly localized in endothelial cells and to a lesser degree in neurons and glial cells in controls. Marked reduction in MCT1 immunoractivity was found in neurons and glial cells, but not in endothelial cells, in CJD (Figure 2).
Discussion
The present study is focused on astrocytes and oligodendrocytes of the frontal cortex area 8 in CJD MM1 and VV2 compared to controls. In spite of the optically-monitored separation of the cerebral cortex from the underlying white matter, contamination by subcortical U fibers cannot be ruled out in our study. This is convenient remark as astrocytes and oligodendrocytes in grey matter differ from astrocytes and oligodendrocytes in white matter, and from one region to another.
Astrogliosis is one of the characteristic neuropathological lesions in cerebral cortex CJD which is here manifested by increased significant GFAP mRNA expression, as previously reported [15]. However, no significant differences are seen in the expression levels of the gene coding for aldehyde dehydrogenase 1 family member L1 (ALDH1L1) used as a marker of the total astrocyte population. Aquaporin 4 protein levels, as revealed by gel electrophoresis and western blotting, are markedly increased in CJD [17]. This is in contrast with the down-regulation of AQP4 mRNA in CJD MM1 and CJD VV2, here observed. Yet, AQP4 immunoreactivity is variable from one case to another:
AQP4 immunorecativity is apparently decreased in some cases, increased in others, and even increased and blurred in cases with severe spongiform change. We do not have an explanation for this point; enhanced translation and/or reduced degradation can account for discrepancies between mRNA and protein expression, and for case-to-case and area differences in protein immunoreactivity. Expression of solute carrier family 1A2 (SLC1A2: GLT1) is reduced in CJD VV2, pointing to the possibility of unbalanced glutamate concentrations between neurons and astrocytes in certain forms of CJD. However, immunohistochemistry reveals individual variations and areal variations in GLT1 expression in the same tissue section. Therefore, further studies in animal models are needed to evaluate GLT1 mRNA and protein expression in prion diseases.
In addition to these aspects, new information has been obtained in this study. Present quantitative data show altered expression of several genes coding for key specific proteins of astrocytes and oligodendrocytes in frontal cortex area 8 in CJD which may have functional implications in the pathogenesis of the disease. Reduced expression of mitochondrial pyruvate carrier 1 (MPC1) and mitochondrial uncoupling protein 5 (UCP5, SLC25A14, BMCP1: brain mitochondrial uncoupling protein 1) suggest altered mitochondrial function in CJD MM1 and CJD VV2. Reduced expression of solute carrier family 2A1 (SLC2A1 or GLUT1) points to altered glucose transport, whereas reduced expression of solute carrier family 16 (MCT1) suggests impaired monocarboxylic acid transport. These are important aspects as blood-derived glucose through GLUT1 is metabolized via glycolysis to produce piruvate and lactate which are delivered to the axons through specific solute carriers, the monocarboxylase transporters (MCTs) located in cell membranes [26,27]. In glia, MCT1 is mainly expressed in developing oligodendrocytes and MCT4 in astrocytes [28,29]. Inhibition of MCT1 in organotypic cultures of the spinal cord in glucose-deprived media is toxic to neurons; the effects on neurons are reversed with the addition of lactate into the medium [29]; moreover, reduction of MCT1 activity in vivo results in axonal damage [29].
These changes are not accompanied by altered expression of oligodendroglial markers OLIG1, OLIG2 and SOX10 [30]; and adult oligodendroglial precursors (NG2) [31,32]. The expression of genes encoding main myelin proteins is also preserved in frontal cortex in CJD, including myelin basic protein (MBP), proteolipid protein (PLP), 2ʹ,3ʹ-cyclic nucleotide-3ʹphosphodiesterase (CNP), myelin-associated glycoprotein (MAG), myelin oligodendrocyte glycoprotein (MOG) and myelin/oligodendrocyte basic protein (MOBP) [33–35].The expression of CLDN11, the gene encoding for claudin 11, principal component of tight junctions of myelin connections at their edges [36,37], is not modified in CJD. The expression of MYRF, encoding myelin regulatory factor which triggers myelination following binding to several promoters of genes coding for myelin proteins [38], is maintained in CJD. In contrast, the expression of the gene coding for galactocerebrosidase (GALC) is reduced in CJD MM1 and VV2, thus indicating possible impairment in glycosphingolipid metabolism. To our knowledge, most studies of brain lipids in CJD were carried out many years ago and the main focus was on ganglioside alterations [39–42]. Since the present studies are focused on the cerebral cortex, further investigation is needed to get information about brain lipid alterations in the white matter in prion diseases.
Oligodendrocytes are connected with each other and with astrocytes through gap junctions thus forming a glial syncytium in the white matter tracts [43,44]. Gap junctions are composed of connexins (Cx); Cx32 and Cx47 are synthesized by oligodendrocytes, and Cx30 and Cx43 by astrocytes [45]. The expression of genes coding for Cx32 (GJB1) and Cx47 (GJC2), and Cx30 (GJB6) is similar in CJD and in control cases, but GJA1 (encoding Cx43) mRNA expression is reduced in CJD VV2 when compared with controls and CJD MM1. Whether these changes have implications in the normal function of the glial syncitium deserves further attention.
Interpretation of some of the present data must be taken with caution. MPC1 is expressed in brain [46] principally in neurons in controls. MPC1 immunoreactivity is reduced in neurons but increased in glial cells in CJD. Therefore, the origin of decreased MPC1 mRNA expression in CJD here observed may be due to damaged neurons, whereas MPC1 protein is increased in glial cells. Similarly, UC5P in the brain is mainly expressed in neurons [47]; reduced UCP5 mRNA expression here observed in CJD is accompanied by reduced UPC5 immunoreactivity in neurons, but this accompanied by increased immunoreactivity in glial cells with the morphology of astrocytes.
GLUT1 is largely expressed in red blood cells and endothelial cells, including brain microvessels [48,49]. No alterations in GLUT1 immunoreactivity is found in CJD microvessels; reduction of GLUT1 mRNA correlates with decreased GLUT1 immunoreactivity in the neuropil in CJD. Finally, MCT1 is widely expressed in endothelial cells in brain [50–52], but also in glial cells especially during development [53,54]. MCT1 immunoreactivity in the adult brain is also observed in neurons. MCT1 immunoractivity is preserved in endothelial cells in CJD when compared with controls; reduced MCT1 mRNA and protein in CJD can be ascribed to reduced expression in neurons.
The concepts of astrocytopathy and oligodendrocytopathy, referring to molecular alterations in astrocytes and oligodendrocytes, are appropriate in neurodegenerative diseases with abnormal protein aggregates [55–60]. This study supports the idea that molecular flaws in astrocytes and oligodendrocytes, in addition to neurons, are relevant in the pathogenesis of cortical alterations in CJD.
Material and methods
Human cases
CJD cases were clinically diagnosed on the basis of rapid dementia together with variable accompanying symptoms, neuroimaging alterations and characteristic CSF biomarkers; all of them were tested for homozygosity of codon 129 in PRNP. Verification of clinical diagnosis was obtained after post-mortem neuropathological study of the brain. Controls did not show neurological symptoms; moreover, cases with metabolic syndrome, autoimmune diseases, fever and prolonged agonal state were not included in this series. Brain samples were obtained from the Brain Banks of the Institute of Neuropathology HUB-ICO-IDIBELL Biobank and the Hospital Clinic-IDIBAPS Biobank following the guidelines of the Spanish legislation on this matter and the approval of the local ethics committees. The post-mortem interval between death and tissue processing was between 2h 45min and 22h 40min. One hemisphere was fixed by immersion in 4% buffered formalin for 3 weeks. The neuropathological study in control and CJD cases was carried out on, at a minimum, twenty selected 4μm-thick de-waxed paraffin sections of representative regions of the frontal, temporal, parietal, motor, primary visual, anterior cingulate and entorhinal cortices, hippocampus, amygdala, basal forebrain, caudate, putamen, globus pallidus, thalamus, midbrain, pons, medulla oblongata, cerebellar vermis, hilus and cerebral white matter. Sections were stained with haematoxylin and eosin, Klüver-Barrera, or processed for immunohistochemistry for microglia (Iba-1, Wako, Richmond, VA, USA), glial acidic protein (GFAP, Dako, Gostrup, Denmark), β-amyloid (Dako, clone 6F/3D), phospho-tau (Thermo Scientific, Rockford, USA, clone AT8), α-synuclein (Novocastra, Newcastle, UK, clone KM51), TDP-43 (Abnova, Taipei, Taiwan, clone 2E2-D3), PrP (clon 3F4, Millipore, Darmstadt, GE) following proteinase K incubation, ubiquitin (Dako, Polyclonal Rabbit) and p62 (BD Biosciences, San Jose, USA, Purified Mouse Anti-p62 LCK ligand) using EnVision+ System peroxidase (Dako), and diaminobenzidine and H2O2.
Samples of the frontal cortex and cerebellum of the other hemisphere were rapidly dissected, frozen on metal plates over dry ice and stored at −80ºC until use. Part of this material was used for gel electrophoresis and western blotting for identification of protease-resistant PrP type. CJD was diagnosed following well-established neuropathological criteria, codon 129 genotype and characteristics of PrP pattern on western blots [61]. None of the cases had suffered from panencephalopathic CJD [62]. Additional brain pathology consisting of neurofibrillary tangles at stages I-II of Braak and Braak, argyrophilic grain disease stage 2, mild small blood vessel disease, and Lewy bodies in the brain stem in some individuals were expected with the age of the patients. Similar lesions were found in the control group and, therefore, they were not contemplated as discriminating variables.
The whole series included 7 CJD MM1 (4 men and 3 women; age: 70.5 ± 5.5), 10 CJD VV2 (4 men and 6 women; age 66.5 ± 6.85) and 10 controls (6 men and 4 women, age 67.1 ± 7.6). A summary of cases is shown in Table 1.
Table 1.
Summary of cases M: man; W: woman; Age in years; CJD: Creutzfeldt-Jakob disease, subtypes MM1 and VV2; PMD: post-mortem delay; RIN: RNA integrity number.
| Case | Sex | Age | Diagnosis | PMD | RIN |
|---|---|---|---|---|---|
| 1 | M | 66 | Control | 18 h 00 min | 6.4 |
| 2 | M | 61 | Control | 03 h 40 min | 7.0 |
| 3 | M | 74 | Control | 06 h 40 min | 7.2 |
| 4 | M | 65 | Control | 05 h 15 min | 6.8 |
| 5 | M | 63 | Control | 08 h 05 min | 7,1 |
| 6 | W | 79 | Control | 03 h 35 min | 6.8 |
| 7 | W | 67 | Control | 05 h 20 min | 6,2 |
| 8 | M | 70 | Control | 03 h 45 min | 7.2 |
| 9 | M | 52 | Control | 04 h 40 min | 7.2 |
| 10 | W | 74 | Control | 02 h 45 min | 5.7 |
| 11 | M | 69 | CJD MM1 | 15 h 00 min | 6.4 |
| 12 | M | 70 | CJD MM1 | N/A | 4.7 |
| 13 | W | 64 | CJD MM1 | 14 h 00 min | 5.6 |
| 14 | M | 64 | CJD MM1 | 14 h 00 min | 4.7 |
| 15 | M | 77 | CJD MM1 | 07 h 10 min | 6,0 |
| 16 | W | 72 | CJD MM1 | 08 h 00 min | 7.0 |
| 17 | M | 78 | CJD MM1 | 22 h 40 min | 5.1 |
| 18 | W | 76 | CJD VV2 | 05 h 00 min | 5.5 |
| 19 | M | 65 | CJD VV2 | 06 h 00 min | 5.3 |
| 20 | W | 65 | CJD VV2 | 07 h 15 min | 5.4 |
| 21 | W | 72 | CJD VV2 | 06 h 00 min | 6.2 |
| 22 | W | 62 | CJD VV2 | 08 h 45 min | 4.7 |
| 23 | M | 71 | CJD VV2 | 09 h 00 min | 5.8 |
| 24 | M | 66 | CJD VV2 | 05 h 00 min | 6.8 |
| 25 | M | 52 | CJD VV2 | 06 h 30 min | 6.5 |
| 26 | W | 59 | CJD VV2 | 09 h 00 min | 6,7 |
| 27 | W | 67 | CJD VV2 | 12 h 30 min | 5.5 |
Biochemical studies were carried out in the frontal cortex area 8 after our best optically-monitored dissection of the cerebral cortex and white matter. However, inclusion of white matter subcortical U fibers cannot be ruled out in the present study.
RNA purification
RNA from frozen frontal cortex area 8 was extracted following the instructions of the supplier (RNeasy Mini Kit, Qiagen® GmbH, Hilden, Germany). RNA integrity and 28S/18S ratios were determined with the Agilent Bioanalyzer (Agilent Technologies Inc, Santa Clara, CA, USA). RIN values are shown in Table 1: control 6.76 ± 0.5; CJD MM1 5.68 ± 0.8 and CJD VV2 5.84 ± 0.6). Samples were treated with DNase digestion, and RNA concentration was evaluated using a NanoDrop™ Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Rt-qPCR
TaqMan RT-qPCR assays were performed in duplicate for each gene on cDNA samples in 384-well optical plates using an ABI Prism 7900 Sequence Detection system (Applied Biosystems, Life Technologies, Waltham, MA, USA). Probes are listed in Table 2. For each 10μL TaqMan reaction, 4.5μL cDNA was mixed with 0.5μL 20x TaqMan Gene Expression Assays and 5μL of 2x TaqMan Universal PCR Master Mix (Applied Biosystems). Values of GUS-β were used as internal controls for normalization [63]. The parameters of the reactions were 50°C for 2min, 95°C for 10min, and 40 cycles of 95°C for 15sec and 60°C for 1min. Finally, capture of all TaqMan PCR data used the Sequence Detection Software (SDS version 2.2.2, Applied Biosystems). For the data analysis, threshold cycle (CT) values for each sample were processed to obtain the double delta CT (ΔΔCT) values. First, delta CT (ΔCT) values were calculated as the normalized CT values of each target gene in relation to the CT of endogenous controls GUS-β. Then, ΔΔCT values were obtained from the ΔCT of each sample minus the mean ΔCT of the population of control samples.
Table 2.
TaqMan probes: gene identification, full name and reference.
| Gene | Full name | Reference |
|---|---|---|
| ALDH1L1 | Aldehyde Dehydrogenase 1 Family Member L1 | Hs01003842_m1 |
| AQP4 | Aquaporin-4 | Hs00242342_m1 |
| CLDN11 | Claudin 11 | Hs00194440_m1 |
| CNP | 2ʹ,3ʹ-Cyclic Nucleotide 3ʹ Phosphodiesterase | Hs00263981_m1 |
| GALC | Galactosylceramidase | Hs00164660_m1 |
| GFAP | Glial fibrillary acidic protein | Hs00909233_m1 |
| GJA1 | Gap junction alpha-1 protein/connexin-43 | Hs00748445_s1 |
| GJB1 | Gap junction beta-1 protein/connexin-32 | Hs00939759_s1 |
| GJC2 | Gap junction beta-2 protein/connexin-47 | Hs00252713_s1 |
| GJB6 | Gap junction beta-6 protein/connexin-30 | Hs00922742_s1 |
| GUS-β | β-glucuronidase | Hs00939627_m1 |
| MAG | Myelin Associated Glycoprotein | Hs01114387_m1 |
| MAL | Mal, T-Cell Differentiation Protein | Hs00360838_m1 |
| MBP | Myelin Basic Protein | Hs00921945_m1 |
| MCT1 | Solute Carrier Family 16 (Monocarboxylic Acid Transporters), Member 1 | Hs01560299_m1 |
| MCT4 | Solute Carrier Family 16 (Monocarboxylic Acid Transporters), Member 4 | Hs01006127_m1 |
| MOBP | Myelin-Associated Oligodendrocyte Basic Protein | Hs01094434_m1 |
| MOG | Myelin Oligodendrocyte Glycoprotein | Hs01555268_m1 |
| MPC1 | Mitochondrial Pyruvate Carrier 1 | Hs00211484_m1 |
| MYRF | Myelin Regulatory Factor | Hs00973739_m1 |
| NG2 | Neural/glial antigen 2 | Hs00426981_m1 |
| OLIG1 | Oligodendrocyte Transcription Factor 1 | Hs00744293_s1 |
| OLIG2 | Oligodendrocyte Lineage Transcription Factor 2 | Hs00377820_m1 |
| PLP1 | Proteolipid Protein 1 | Hs00166914_m1 |
| SLC1A2 | Solute Carrier Family 1 (Glial High Affinity Glutamate Transporter), Member 2 | Hs01102423_m1 |
| SLC2A1 | Solute Carrier Family 2 (Facilitated Glucose Transporter), Member 1 | Hs01102423_m1 |
| SOX-10 | SRY-Box 10 | Hs00366918_m1 |
| UCP4 | Mitochondrial Uncoupling Protein 4 | Hs00188687_m1 |
| UCP5 | Mitochondrial Uncoupling Protein 5 | Hs00605850_m1 |
Statistical analysis
The normality of distribution of fold change values was analyzed with the Kolmogorov–Smirnov test. Pearson’s correlation coefficient was used to assess a possible linear association between two continuous quantitative variables. To determine the relationship between gene expression and RIN values according to pathologic variables, we used the analysis of covariance (ANCOVA). Statistical analysis of the expression data between groups was performed using one-way analysis of variance (ANOVA) followed by Tukey post-test or Kruskal-Wallis test followed by Dunns post-hoc test when required using the SPSS software (IBM Corp. Released 2013. IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp.). Outliers were detected using the GraphPad software QuickCalcs (P < 0.05). All data were expressed as mean values ± SEM. Differences between controls and CJD MM1 or CJD VV2 were considered statistically significant at * P < 0.05, ** P < 0.01, *** P < 0.001; and set at $ P < 0.05 and $$ P < 0.01when comparing CJD MM1 and CJD VV2.
Immunohistochemistry
De-waxed sections, 4 microns thick, were processed for immunohistochemistry (control n = 8; CJD MM1 n = 6; CJD VV2 n = 2). The sections were boiled in citrate buffer (20min) to retrieve tau antigenicity. Endogenous peroxidases were blocked by incubation in 10% methanol-1% H2O2 solution (15min) followed by 3% normal horse serum solution. Then the sections were incubated at 4ºC overnight with one of the primary antibodies against glial fibrillary acidic protein (GFAP) (rabbit polyclonal, used at 1:500, Dako, Glostrup, DK), Olig-2 (rabbit polyclonal, used at 1:500, Abcam, Cambridge, UK), aquaporine 4 (AQP4) (monoclonal, used at 1:400, Sigma, St Louis, Missouri, USA), solute carrier family 1 member 2 (GLT1) (guinea pig used at 1:100, Merck-Millipore, Billerica, MA, USA), mitochondrial pyruvate carrier 1 (MPC1) MPC1) (polyclonal, used at 1:100, Cell Signaling Technology, Danvers, MA, USA), mitochondrial uncoupled protein 5 (UCP5) (polyclonal, used at 1:10, Novus Biological, Littleton, CO, USA), solute carrier family 2 member 1 (GLUT1) (polyclonal, used at 1:100, Abcam, Cambridge, CB, UK) and solute carrier family 16 member 1 (MCT1) (monoclonal, used at 1:50, Sigma, St Louis, Missouri, USA). Following incubation with Following incubation with the primary antibody, the sections were incubated with EnVision + system peroxidase (Dako, DK) for 30min at room temperature. The peroxidase reaction was visualized with diaminobenzidine and H2O2. Control of the immunostaining included omission of the primary antibody; no signal was obtained following incubation with only the secondary antibody. Peptides for pre-absorption studies were not available.
Funding Statement
Part of this work was supported by the Ministry of Economy and Competitiveness, Institute of Health Carlos III (co-funded by European Regional Development Fund, ERDF, a way to build Europe): FIS PI17/00809, IFI15/00035 fellowship to PA-B and co-financed by ERDF under the program Interreg Poctefa: RedPrion 148/16; Institute of Health Carlos III [FIS PI17/00809,].
Acknowledgments
We wish to thank T. Yohannan for editorial assistance
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Prusiner SB. An introduction to prion biology and diseases In: Prusiner SB, editor. Prion biology and diseases, 2nd ed. New York (NY): Cold Spring Harbor Laboratory; 2004. p. 1–87. [Google Scholar]
- [2].Aguzzi A, Sigurdson C, Heikenwaelder M.. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol. 2008;3:11–40. [DOI] [PubMed] [Google Scholar]
- [3].Gambetti P, Cali I, Notari S, et al. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011;121:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Budka H, Head MW, Ironside JW. Sporadic Creutzfeldt-Jakob disease Dickson DW, Weller RO, et al, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester, West Sussex:Willey-Blackwell; 2011. p.322–335. [Google Scholar]
- [5].Parchi P, Gambetti P, Capellari S. Genetic Creutzfeldt-Jakob disease In: Dickson DW, Weller RO, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester, West Sussex: Willey-Blackwell; 2011. p. 336–345. [Google Scholar]
- [6].Ghetti B, Tagliavini F, Kovacs GG. Gerstmann-Straüssler-Scheinker Dickson DW, Weller RO, et al, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester, West Sussex:Willey-Blackwell; 2011. p.364–377. [Google Scholar]
- [7].Head MW, Ironside JW, Ghetti B. Prion diseases Love S, Budka H, Ironside J, et al, editors. Greenfield’s neuropathology. 9th ed. Boca Raton, Florida:CRC Press; 2015. p.1016–1086. [Google Scholar]
- [8].Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ironside JW, Head MW, Will RG. Variant Creutzfeldt-Jakob disease In: Dickson DW, Weller RO, editors. Neurodegeneration: the molecular pathology of dementia and movement disorders. 2nd ed. Chichester, West Sussex: Willey-Blackwell; 2011. p. 354–363. [Google Scholar]
- [10].Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- [11].Parchi P, Strammiello R, Notari S, et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSC types: an updated classification. Acta Neuropathol. 2009;118:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Giese A, Brown DR, Groschup MH, et al. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998;8:449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Van EB, Dewulf E, Pals P, et al. The role of cytokines, astrocytes, microglia and apoptosis in Creutzfeldt-Jakob disease. Neurobiol Aging. 2002;23:59–64. [DOI] [PubMed] [Google Scholar]
- [14].Szpak GM, Lewandowska E, Lechowicz W, et al. The brain immune response in human prion diseases. Microglial activation and microglial disease. I. Sporadic Creutzfeldt-Jakob disease. Folia Neuropathol. 2006;44:202–213. [PubMed] [Google Scholar]
- [15].Llorens F, López-González I, Thüne K, et al. Subtype and regional-specific neuroinflammation in sporadic Creutzfeldt-Jakob disease. Front Aging Neurosci. 2014;6:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kovacs GG, Preusser M, Strohschneider M, et al. Subcellular localization of disease-associated prion protein in the human brain. Am J Pathol. 2005;166:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rodríguez A, Pérez-Gracia E, Espinosa JC, et al. Increased expression of water channel aquaporin 1 and aquaporin 4 in Creutzfeldt-Jakob disease and in bovine spongiform encephalopathy-infected bovine-PrP transgenic mice. Acta Neuropathol. 2006;112:573–585. [DOI] [PubMed] [Google Scholar]
- [18].Iwasaki Y, Mimuro M, Yoshida M, et al. Enhanced aquaporin-4 immunoreactivity in sporadic Creutzfeldt-Jakob disease. Neuropathology. 2007;27:314–323. [DOI] [PubMed] [Google Scholar]
- [19].Choi YP, Head MW, Ironside JW, et al. Uptake and degradation of protease-sensitive and -resistant forms of abnormal human prion protein aggregates by human astrocytes. Am J Pathol. 2014;184:3299–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Victoria GS, Arkhipenko A, Zhu S, et al. Astrocyte-to-neuron intercellular prion transfer is mediated by cell-cell contact. Sci Rep. 2016;6:20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Prinz M, Montrasio F, Furukawa H, et al. Intrinsic resistance of oligodendrocytes to prion infection. J Neurosci. 2004;24:5974–5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liberski PP, Yanagihara R, Wells GA, et al. Ultrastructural pathology of axons and myelin in experimental scrapie in hamsters and bovine spongiform encephalopathy in cattle and a comparison with the panencephalopathic type of Creutzfeldt-Jakob disease. J Comp Pathol. 1992;106:383–398. [DOI] [PubMed] [Google Scholar]
- [23].Liberski PP, Brown P, Cervenakova L, et al. Interactions between astrocytes and oligodendroglia in human and experimental Creutzfeldt-Jakob disease and scrapie. Exp Neurol. 1997;144:227–234. [DOI] [PubMed] [Google Scholar]
- [24].Shintaku M, Yutani C. Oligodendrocytes within astrocytes (“emperipolesis”) in the white matter in Creutzfeldt-Jakob disease. Acta Neuropathol. 2004;108:201–206. [DOI] [PubMed] [Google Scholar]
- [25].El Hachimi KH, Chaunu MP, Brown P, et al. Modifications of oligodendroglial cells in spongiform encephalopathies. Exp Neurol. 1998;154:23–30. [DOI] [PubMed] [Google Scholar]
- [26].Saab AS, Tzvetanova ID, Nave K-A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol. 2013;23:1065–1072. [DOI] [PubMed] [Google Scholar]
- [27].Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94:1–14. [DOI] [PubMed] [Google Scholar]
- [28].Rinholm JE, Hamilton NB, Kessaris N, et al. Regulation of oligodendrocyte development and myelination by glucose and lactate. J Neurosci. 2011;31:538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee Y, Morrison BM, Li Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Marinelli C, Bertalot T, Zusso M, et al. Systematic review of pharmacological properties of the oligodendrocyte lineage. Front Cell Neurosci. 2016;10:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dawson MR, Polito A, Levine JM, et al. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. [DOI] [PubMed] [Google Scholar]
- [32].Peters A. A fourth type of neuroglial cell in the adult central nervous system. J Neurocytol. 2004;33:345–357. [DOI] [PubMed] [Google Scholar]
- [33].Kursula P. The current status of structural studies on proteins of the myelin sheath. Int J Mol Med. 2001;8:475–479. [PubMed] [Google Scholar]
- [34].Harauz G, Ladizhansky V, Boggs JM. Structural polymorphism and multifunctionality of myelin basic protein. Biochemistry. 2009;48:8094–8104. [DOI] [PubMed] [Google Scholar]
- [35].Jahn O, Tenzer S, Werner HB. Myelin proteomics: molecular anatomy of an insulating sheath. Mol Neurobiol. 2009;40:55–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gow A, Southwood CM, Li JS, et al. CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell. 1999;99:649–659. [DOI] [PubMed] [Google Scholar]
- [37].Morita K, Sasaki H, Fujimoto K, et al. Claudin-11/OSP-based tight junctions of myelin sheaths in brain and Sertoli cells in testis. J Cell Biol. 1999;145:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Suzuki K, Chen G. Chemical studies on Jakob-Creutzfeldt disease. J Neuropathol Exp Neurol. 1966;25:396–408. [DOI] [PubMed] [Google Scholar]
- [40].Bass NH, Hess HH, Pope A. Altered cell membranes in Creutzfeldt-Jakob disease. Microchemical Studies. Arch Neurol. 1974;31:174–182. [DOI] [PubMed] [Google Scholar]
- [41].Tamai Y, Kojima H, Ikuta F, et al. Alterations in the composition of brain lipids in patients with Creutzfeldt-Jakob disease. J Neurol Sci. 1978;35:59–76. [DOI] [PubMed] [Google Scholar]
- [42].Yu RK, Manuelidis EE. Ganglioside alterations in guinea pig brains at end stages of experimental Creutzfeldt-Jakob disease. J Neurol Sci. 1978;35:15–23. [DOI] [PubMed] [Google Scholar]
- [43].Bedner P, Steinhauser C, Theis M. Functional redundancy and compensation among members of gap junction protein families? Biochim Biophys Acta. 2012; 1818:1971–1984. [DOI] [PubMed] [Google Scholar]
- [44].Nualart-Marti A, Solsona C, Fields RD. Gap junction communication in myelinating glia. Biochim Biophys Acta. 2013;1828:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Orthmann-Murphy JL, Freidin M, Fischer E, et al. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J Neurosci. 2007;27:13949–13957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li Y, Han G, Ji Y, et al. Establishment of mitochondrial pyruvate carrier 1 (MPC1) gene knockout mice with preliminary gene function analyses. Oncotarget. 2016;7:79981–79994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kim-Han JS, Reichert SA, Quick KL, et al. BMCP1: a mitochondrial uncoupling protein in neurons which regulates mitochondrial function and oxidant production. J Neurochem. 2001;79:658–668. [DOI] [PubMed] [Google Scholar]
- [48].Shawahna R, Uchida Y, Declèves X, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8:1332–1341. [DOI] [PubMed] [Google Scholar]
- [49].Nakamura S, Muramatsu SI, Takino N, et al. Gene therapy for Glut1-deficient mouse using an adeno-associated virus vector with the human intrinsic GLUT1 promoter. J Gene Med. 2018;20:e3013. [DOI] [PubMed] [Google Scholar]
- [50].Smith JP, Drewes LR. Modulation of monocarboxylic acid transporter-1 kinetic function by the cAMP signaling pathway in rat brain endothelial cells. J Biol Chem. 2006;281:2053–2060. [DOI] [PubMed] [Google Scholar]
- [51].Daneman R, Zhou L, Agalliu D, et al. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PloS ONE. 2010;5:e13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu Z, Sneve M, Haroldson TA, et al. Regulation of monocarboxylic acid transporter 1 trafficking by the canonical Wnt/β-catenin pathway in rat brain endothelial cells requires cross-talk with Notch signaling. J Biol Chem. 2016;291:8059–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gerhart DZ, Enerson BE, Zhdankina OY, et al. Expression of monocarboxylate transporter MCT1 by brain endothelium and glia in adult and suckling rats. Am J Physiol. 1997;273:E207–213. [DOI] [PubMed] [Google Scholar]
- [54].Zhang M, Ma Z, Qin H, et al. Monocarboxylate transporter 1 in the medial prefrontal cortex developmentally expresses in oligodendrocytes and associates with neuronal amounts. Mol Neurobiol. 2017;54:2315–2326. [DOI] [PubMed] [Google Scholar]
- [55].Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci. 2006;7:194–206. [DOI] [PubMed] [Google Scholar]
- [56].Pekny M, Pekna M, Messing A, et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 2016;131:323–345. [DOI] [PubMed] [Google Scholar]
- [57].Tognata R, Miller RH. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology. 2016;110:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ettle B, Schlachetzki JCM, Winkler J. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol Neurobiol. 2016;53:3046–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ferrer I. Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathol. 2017;27:645–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Verkhratsky A, Zorec R, Parpura V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol. 2017;27:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Parchi P, de Boni L, Saverioni D, et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012;124:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jansen G, Head MW, Rozemuller AJ, et al. Panencephalopathic Creutzfeldt-Jakob disease in the Netherlands and the UK: clinical and pathological characteristics of nine patients. Neuropathol Appl Neurobiol. 2009;35:272–282. [DOI] [PubMed] [Google Scholar]
- [63].Barrachina M, Castaño E, Ferrer I. TaqMan PCR assay in the control of RNA normalization in human post-mortem brain tissue. Neurochem Int. 2006;49:276–284. [DOI] [PubMed] [Google Scholar]