

Abstract
GNE myopathy, previously known as hereditary inclusion body myopathy (HIBM), or Nonaka myopathy, is a rare autosomal recessive muscle disease characterized by progressive skeletal muscle atrophy. It has an estimated prevalence of 1 to 9:1,000,000. GNE myopathy is caused by mutations in the GNE gene which encodes the rate-limiting enzyme of sialic acid biosynthesis. The pathophysiology of the disease is not entirely understood, but hyposialylation of muscle glycans is thought to play an essential role. The typical presentation is bilateral foot drop caused by weakness of the anterior tibialis muscles with onset in early adulthood. The disease slowly progresses over the next decades to involve skeletal muscles throughout the body, with relative sparing of the quadriceps until late stages of the disease. The diagnosis of GNE myopathy should be considered in young adults presenting with bilateral foot drop. Histopathologic findings on muscle biopsies include fiber size variation, atrophic fibers, lack of inflammation, and the characteristic “rimmed” vacuoles on modified Gomori trichome staining. The diagnosis is confirmed by the presence of pathogenic (mostly missense) mutations in both alleles of the GNE gene. Although there is no approved therapy for this disease, preclinical and clinical studies of several potential therapies are underway, including substrate replacement and gene therapy-based strategies. However, developing therapies for GNE myopathy is complicated by several factors, including the rare incidence of disease, limited preclinical models, lack of reliable biomarkers, and slow disease progression.
Electronic supplementary material
The online version of this article (10.1007/s13311-018-0671-y) contains supplementary material, which is available to authorized users.
Key Words: GNE myopathy, genetics, rare diseases, gene therapy, ManNAc, sialic acid.
Introduction
GNE myopathy (OMIM 605820) is a rare genetic muscle disease characterized by progressive skeletal muscle atrophy due to mutations in GNE, the gene encoding the bifunctional enzyme UDP-N-acetylglucosamine (GlcNAc) 2-epimerase/N-acetylmannosamine (ManNAc) kinase (EC:3.2.1.183, EC:2.7.1.60) (Fig. 1).
Fig. 1.

Sialic acid biosynthesis pathway. The biosynthesis of sialic acid (Neu5Ac) is an intracellular process depicted above with enzymes shown in yellow and substrates in white. The initial steps of this pathway occur in the cytoplasm, with the substrate, UDP-GlcNAc, which is derived from glucose. In the rate-limiting step of the pathway, UDP-GlcNAc is converted into ManNAc by UDP-GlcNAc 2-epimerase, encoded by the epimerase domain of GNE. ManNAc is phosphorylated by ManNAc kinase encoded by the kinase domain of GNE. Sialic acid becomes “activated” by CMP-sialic acid synthetase in the cell nucleus. CMP-sialic acid acts as a sialic acid donor to sialylate glycans on nascent glycoproteins (sia) and glycolipids in the Golgi; it also acts as a cytoplasmic feedback inhibitor of the UDP-GlcNAc 2-epimerase enzyme by binding to its allosteric site. Sialylation-increasing therapies (ManNAc and sialic acid), and sialylated glycoproteins (siallylactose and IVIG) are boxed in white. Adapted with permission from Xu et al. [1]
GNE myopathy was initially reported by Nonaka et al. in Japan and Argov and Yarom in Israel. Nonaka et al. used the term distal myopathy with rimmed vacuoles (DMRV) to describe a familial myopathy with onset in early adulthood, predominantly affecting the anterior tibialis muscles, with mild serum creatine kinase (CK) elevation, and muscle histopathology characterized by fiber atrophy, rimmed vacuoles, lack of inflammation, and no evidence of regeneration [2, 3]. Argov and Yarom described autosomal recessive inheritance of several Iranian Jewish families with a rimmed vacuole myopathy that spared the quadriceps [4]; Argov et al. later introduced the term “hereditary inclusion body myopathy” (HIBM) to describe the disease [5]. Other historical names include Nonaka myopathy and inclusion body myopathy 2 (IBM2). In 2001 and 2002, mutations in the GNE gene were identified as the cause of HIBM and DMRV, respectively [6, 7], confirming a monogenic disorder and unifying various historical names into a single disease entity now commonly known as GNE myopathy [8]. Although initially identified in Japanese and Middle Eastern populations, patients with GNE myopathy have been reported worldwide. The prevalence of GNE myopathy is estimated at ~ 1 to 9/1,000,000 (Orphanet; http://www.orpha.net/).
Here, we provide an overview of the clinical and molecular aspects of GNE myopathy and describe the challenges in developing therapies for the disease.
Clinical Manifestations of GNE Myopathy
Patients with GNE myopathy typically present in early adulthood, between 20 and 40 years of age [9, 10] with manifestations of anterior tibialis weakness, such as tripping, gait disturbance, inability to lift toes, or foot drop [9–11], although some patients present to medical attention at later stages of the disease.
The clinical manifestations of the disease are a result of the progressive skeletal muscle atrophy and weakness with the subsequent impairment of function and quality of life. The specific muscle groups and functions impacted depend on the muscles involved a different stages of the disease. GNE myopathy is slowly progressing, and it typically takes decades for patients to progress from early to late stages of the disease. The disease progresses from the distal muscles of the lower extremities, followed by thigh muscles, with relative sparing of the quadriceps [4] (Fig. 2). The gait becomes progressively affected, resulting in steppage gait, increased risk of falls, and decreased balance. Patients may use assistive devices such as ankle-foot orthotics, cane, and walker, and it is estimated that patients may use a wheelchair ~ 10 to 20 years after the onset of symptoms [9, 11–13]. The upper extremities are affected 5–10 years after the onset of symptoms [9, 11] and do not necessarily follow the distal to proximal progression seen in the lower extremities. In advanced stages of the disease, neck muscles can also be affected [10]. Ultimately, disease progression may result in complete loss of skeletal muscle function and dependence on caregivers [11, 12]. The disease does not affect swallowing or facial muscles, and it does not impair cognition.
Fig. 2.

Progressive muscle involvement in GNE myopathy. Representative lower extremity muscle MRI images of patients with GNE myopathy, with advancing disease progression from left to right. Coronal T1-weighted muscle MRI images (upper panels). Axial T1-weighted images of the mid-femoral thigh (middle panels). Axial T1-weighted images of the lower leg (lower panels). Progressive muscle atrophy of the lower extremities is noted initially in the anterior tibialis muscle (B lower panel), followed by involvement of muscles in the calves (C lower panel) and posterior thigh muscles (B, C middle panel), and finally involvement of the quadriceps in advanced stages of the disease (D middle panel)
Disease progression has been analyzed prospectively in two studies. Twenty-four Japanese patients evaluated over a 1-year period showed a significant reduction from 36 to 33.2 (p < 0.001) in the summed manual muscle testing of 19 muscles (MMT). Smaller differences were noted in other measures of strength, including knee extension by hand-held dynamometry, pinch, and grip power [10]. Another study evaluating patients over a 2-year period using the GNE myopathy functional activity scale (GNEM-FAS) questionnaire, estimated an average annual rate of decline in function of − 9.6% and − 3.2% in 16 ambulatory and 6 non-ambulatory patients, respectively [9], as measured by this reported outcome with preliminary validity and reliability in GNE myopathy [14]. Further prospective studies are needed to understand disease progression.
The age of onset and the severity of the disease may vary significantly [10, 13], even among siblings [15–17]. The factors that affect progression are not well understood, but genetic modifiers and environmental factors are thought to play a role. Both extremes of muscle overuse or underuse (such as fractures or prolonged inactivity) may increase the rate of progression. Severe and prolonged muscle overuse, associated with intense pain, has been shown to cause significant weakness and atrophy of muscles involved in work-related repetitive muscle overuse in a patient with GNE myopathy [18]. A balanced physical activity, which includes regular exercise, as feasible and tolerated, should be encouraged in patients with GNE myopathy, with the possible exception of lifting weights. Further studies investigating the genetic and environmental modifiers of progression are warranted to understand disease variability in GNE myopathy.
Respiratory muscle involvement has been described at late stages of the disease. A retrospective study of patients in Japan showed decreased forced vital capacity (FVC < 80%) in 12 out of 39 of patients with GNE myopathy, 11 of which were non-ambulatory. Clinically significant involvement was only seen in 4 wheelchair-dependent patients, 2 of which required nocturnal non-invasive positive pressure ventilation [19]. Another study found no clinical respiratory dysfunction in 50 Bulgarian Roma patients, even those with advanced disease [20]. However, given that clinically significant abnormalities have been identified in a small proportion of patients, respiratory function should be evaluated particularly in non-ambulatory patients.
Cardiac involvement has been reported in a mouse model of GNE myopathy [21] but is not usually seen in patients. As part of the Japanese Registry of GNE myopathy, all 35 subjects who had an echocardiogram had normal cardiac function [22]. A cohort of Bulgarian Roma patients had cardiac abnormalities detected by electrocardiogram (EKG) (n = 12) or echocardiogram (n = 33), but most of these were subclinical or due to other predisposing factors [20]. As part of a survey, 2/137 respondents with GNE myopathy reported having cardiomyopathy, although it is unclear whether the cardiomyopathy was due to an alternative etiology. Furthermore, there is no description of whether patients who reported having no cardiac complications had a formal cardiac evaluation [9]. Dilated cardiomyopathy has been reported in a pair of siblings compound heterozygous for the Phe559Cys and Ala662Val mutations [23], and another pair of siblings with sudden death had vacuoles on cardiac tissue evaluated during autopsy [24], suggesting that predisposition may be familial or due to another underlying pathology. Further studies are needed to characterize cardiac involvement in patients with GNE myopathy and determine whether patients should be monitored for cardiac complications.
Thrombocytopenia has been reported in a small number of patients with GNE myopathy [25, 26], but one study determined the occurrence of thrombocytopenia was similar to that of the general population in Japan [10, 22]. Congenital thrombocytopenia has also been reported in families with compound heterozygous GNE mutations, and although the etiology still needs to be determined, the authors suggested hyposialylation of platelets leading to increased platelet clearance as a possible mechanism [26, 27].
Etiology of GNE Myopathy
GNE Function
The GNE gene encodes for UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE), the bifunctional and rate-limiting enzyme in the biosynthesis of sialic acid [28, 29] and a regulator of cell surface sialylation [30]. The GNE enzyme has an N-terminal epimerase enzymatic domain, containing the allosteric site for CMP-sialic acid feedback inhibition, and a C-terminal kinase enzymatic domain (Fig. 3). The activities of the GNE enzymatic domains are interrelated; mutations in one domain also affect the enzyme activity of the other domain [32–35]. For example, modeling the effects of mutations on enzyme function has shown that the Middle Eastern founder mutation Met743Thr in the kinase domain likely affects GlcNAc binding of the epimerase domain [34].
Fig. 3.

GNE gene and protein structure. (A) Exon (boxes)-intron (lines) structures of the two major human GNE mRNA transcripts, formed by alternative splicing of N-terminal exons. mRNA variant 1 (the longest splice form) encodes the hGNE2 protein, and mRNA variant 2 encodes the hGNE1 protein. Colored boxes represent the open reading frame and functional domains (colors correspond with translated protein domains in (B)). White boxes represent untranslated regions (UTRs) and white dotted lined boxes the skipped exons of each transcript. (B) hGNE2 protein structure. Gray UF, unknown function; dark red, UDP-GlcNAc 2-epimerase enzymatic activity encoding domain; vertical lined, putative nuclear export signal (NES); dotted, experimental allosteric region (AR) for CMP-sialic acid binding; green, ManNAc kinase enzymatic activity encoding domain. Amino acid numbering of both hGNE1 and hGNE2 protein isoforms is indicated below the structure. (C) Location on the GNE protein of frequent GNE mutations associated (see Table 1 for details)
In GNE myopathy, decreased GNE function leading to hyposialylation of muscle glycans has been thought to mediate the disease process. However, apart from its role in sialic acid biosynthesis and regulation, there is evidence to suggest that GNE plays other roles in cellular function that may shed light on understanding disease mechanisms and developing novel therapies. The function of GNE appears to be critical during development since a Gne knock-out mouse is embryonic lethal [36]. GNE is present in the nucleus where it may play a role in muscle regeneration after injury [37, 38]; the GNE protein interacts with alpha-actinin [39, 40] and other proteins involved in cytoskeletal organization and development [41] and has been shown to play a role in survival–apoptosis signaling, proliferation, gene expression, and oxidative stress [42–45].
Sialic Acid Biosynthesis Pathway
Sialic acids are negatively charged sugars, and the terminal monosaccharides of most glycoproteins and glycolipids (glycans) found on circulating proteins and cell surfaces, where they mediate several biological functions and play essential roles in disease processes [30, 46, 47]. The most abundant mammalian sialic acid and the precursor of most other sialic acids, N-acetylneuraminic acid (Neu5Ac, generally referred to as “sialic acid”), is mainly produced through the sialic acid biosynthesis pathway [48].
As shown in Fig. 1, the biosynthesis of Neu5Ac in humans is an intracellular pathway initiated and regulated by the bifunctional enzyme encoded by the GNE gene, UDP-N-acetylglucosamine 2-epimerase (UDP-GlcNAc 2-epimerase; EC 5.1.3.14)/N-acetylmannosamine kinase (ManNAc kinase; EC 2.7.1.60). UDP-GlcNAc 2-epimerase catalyzes the conversion of UDP-GlcNAc into ManNAc, which is followed by phosphorylation into ManNAc-6P by ManNAc kinase (Fig. 1) [29, 30, 49]. Afterward, two additional enzymatic steps combine ManNAc-6P with phosphoenolpyruvate (PEP) to produce Neu5Ac (sialic acid). In the nucleus, Neu5Ac is activated into the nucleotide sugar donor cytidine-5′-monophosphate-N-acetylneuraminic acid (CMP-sialic acid) [50]. In the final step of the pathway, CMP-sialic acid is transported to the Golgi complex where sialylation occurs. CMP-sialic acid is the only substrate donor used by sialyltransferases to sialylate nascent glycoproteins and glycolipids (glycans) (Fig. 1). The pathway is regulated by cytoplasmic CMP-sialic acid that feedback-inhibits UDP-GlcNAc 2-epimerase activity by binding to its allosteric site [51, 52]. Either Neu5Ac or CMP-sialic acid levels are believed to induce the transcription of sialyltransferases in the nucleus [53].
Sialylated glycans (glycoproteins and glycolipids), including endocytosed extracellular glycans from food sources, are reduced inside lysosomes. Lysosomal neuraminidases release sialic acid (Neu5Ac) from individual glycans and the newly formed free sialic acid subsequently exits lysosomes into the cytoplasm through the sialic acid transporter sialin (SLC17A5). It is presumed that cytoplasmic free Neu5Ac can potentially be reused in the de novo sialic acid synthesis pathway. Extracellular free Neu5Ac enters cells through endocytotic vesicles (micropinocytosis) that fuse with lysosomes, from where free Neu5Ac gets released into the cytoplasm though the sialin transporter [54].
Sialylation of Muscle Glycans
Sialylation of cell surface and circulating glycans, the end-product of the sialic acid biosynthesis pathway, is crucial for many biological processes, including cell–cell interactions, adhesion, and processes during development and regeneration [46, 47, 55]. By affecting the activity of the rate-limiting enzyme in the pathway, GNE mutations presumably affect the sialylation of glycans [56, 57]. There is supporting evidence suggesting that hyposialylation of muscle cell surface glycans play a significant role in GNE myopathy. Although overall sialylation in GNE myopathy serum glycans and myoblasts appears normal [58, 59], organ-specific membrane hyposialylation was found in GNE-deficient mice [56], and specific skeletal muscle glycans including alpha-dystroglycan, NCAM, neprilysin, GM3 ganglioside, and O-linked glycans have been reported to be hyposialylated in GNE myopathy [58, 60–66]. Additional evidence comes from murine models of GNE myopathy; in one model, muscle atrophy and weakness could be prevented by treatment with oral sialic acid metabolites [67] and in another model, impaired muscle sialylation was restored with the administration of oral N-acetylmannosamine (ManNAc) [68].
Diagnosis
GNE myopathy should be considered as a potential diagnosis in patients presenting in early adulthood with bilateral foot drop. Currently, the diagnosis of GNE myopathy is based on clinical and muscle pathology findings and confirmed by identification of biallelic mutations in the GNE gene.
Several factors may delay the diagnosis of this rare disease. The clinical manifestations in early stages of the disease are nonspecific and the characteristic clinical sign, sparing of the quadriceps, is not evident until late in the disease. No routine laboratory test suggests the presence of GNE myopathy. Serum creatine phosphokinase (CPK) levels may be elevated (usually < 1000 mcg/L) or within the normal range [20, 22, 69]. Electromyograms (EMGs) and nerve conduction studies may be nonspecific and do not contribute significantly to the diagnosis [61, 70, 71]. Under certain circumstances, as described below, muscle biopsy may fail to show characteristic histopathological findings. When performed, T1-weighted muscle magnetic resonance imaging (MRI) shows fatty-fibrous replacement of affected muscles [72] (Fig. 2) and may aid in the identification of the site of muscle biopsy. Genetic testing is unlikely to be performed unless there is clinical suspicion, although more patients are being identified by neuromuscular genetic testing panels that include GNE or next-generation (including exome) sequencing [16, 73, 74].
Muscle Histology
Muscle biopsies of affected muscles show variation in muscle fiber size, atrophic fibers that are usually angular and clustered, the presence of characteristic “rimmed” vacuoles, and typically lack of inflammation (Fig. 4a-1) [2, 4]. Modified Gomori trichome staining (Fig. 4a-2) facilitates the identification of “rimmed vacuoles,” which may be missed with hematoxylin and eosin staining. These rimmed vacuoles appear empty but are, in fact, artificially created during the staining procedure by the detachment of red-colored granules, which are thought to be protein aggregates. These rimmed vacuoles are stained intensely with acid phosphatase (Fig. 4a-3), indicating increased acidity within the tissues. By electron microscopy, areas of rimmed vacuoles appear to be enriched with autophagosomal structures (Fig. 4a-4).
Fig. 4.
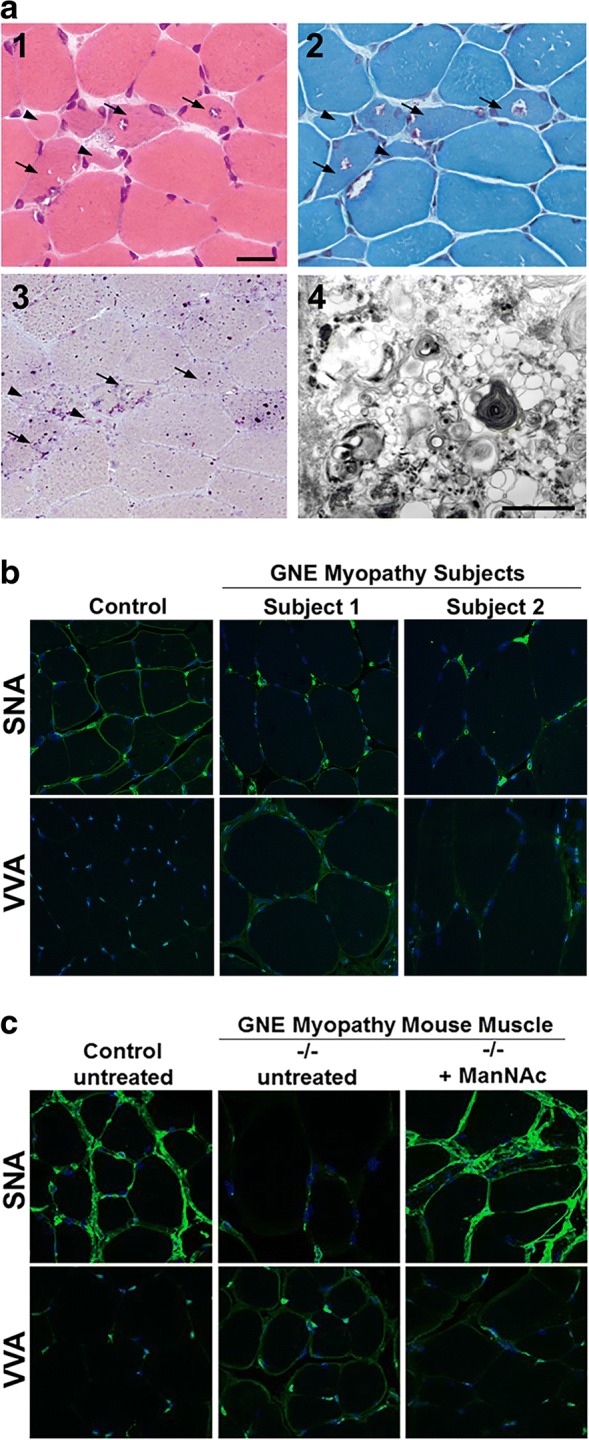
GNE Myopathy muscle histology and lectin histochemistry. (a) Stained cryosections of a muscle biopsy (biceps brachii) from a GNE myopathy patient. (1) H&E staining shows variation in muscle fiber size, atrophic fibers (arrowheads), and characteristic fibers with rimmed vacuoles (arrows). (2) Modified Gomori trichrome staining in which rimmed vacuoles appear as vacuoles rimmed by red granules. (3) Acid phosphatase staining showing intense lysosomal staining in regions with rimmed vacuoles, implying that these rimmed vacuolar areas maybe clusters of autophagic vacuoles. (4) Electron microscopy of a rimmed vacuole containing cytoplasmic region, confirming that structures are indeed composed of autophagic vacuoles and myeloid bodies, in addition to excessive cellular debris. Scale bar in (1) represents 50 μm in section images shown in (1, 2, 3). Scale bar in (4) denotes 1 nm. (b, c) Paraffin embedded skeletal muscle sections of control and GNE myopathy subjects (b) and mice (c) stained with the lectins (green) SNA (predominantly binding terminal α(2,6)-linked sialic acid on all glycans) or VVA (predominantly binding terminal GalNAc, without sialic acid attached, O-linked to serine or threonine residues of glycoproteins), costained with the nuclear dye DAPI (blue). GNE myopathy sections show hyposialylation, indicated by decreased staining of SNA and increased staining of VVA, compared to control sections. Muscle sections of GNE myopathy mutant mice (−/−) that received oral ManNAc therapy exhibited a sialylation status restored to the normal range. All confocal imaging was performed at the same microscope intensity settings per tissue and per lectin (with a 63× objective). All images are 1D projections of confocal Z-stacks. Images in (a): adapted with permission from Huizing et al., OMMBID [11]. Images in (b): adapted with permission from: Leoyklang et al. [65]. Images in (c): adapted with permission from Niethamer et al. [68]
Histopathologic findings may be absent in biopsies of unaffected muscle areas. Therefore, it is recommended to avoid biopsy of the quadriceps muscle, which is unaffected until late stages of the disease, and to perform muscle biopsy in a center with expertise in the diagnosis of GNE myopathy [75].
Lectin histochemistry of human and mouse GNE myopathy muscle shows hyposialylation of muscle surface glycoproteins (Fig. 4b, c) [11, 64, 66]. Since SNA predominantly recognizes terminal sialic acid, muscle histochemistry could be further developed, including testing sensitivity and specificity, to assist in the diagnosis and evaluation of patients with GNE myopathy [64–66, 68].
Genetics
GNE myopathy is an autosomal recessive inherited disease. There are ethnic GNE founder mutations found in Middle Eastern (Met743Thr), Japanese (Cys44Ser, Asp207Val, and Val603Leu), Roma Bulgarian (Ile618Thr) and Indian/Asian (Val727Met) populations (Table 1). Besides founder mutations, there are more than 200 GNE gene mutations reported in over 950 GNE myopathy patients worldwide, in either the epimerase or the kinase encoding domains of the enzyme [20, 31, 76, 77], several proven to result in decreased enzyme activity [31, 33, 57]. Most pathogenic mutations that cause GNE myopathy are missense, but nonsense mutations, insertion/deletions, and intronic (splice site) variants have also been described [76, 77]. Patients may be homozygous for a single mutation or compound heterozygote for two different mutations in either the same or different domains of the enzyme. Interestingly, no patients have been reported to have two null mutations (resulting in decay of GNE mRNA), supporting other evidence that absent GNE function is incompatible with life [36].
Table 1.
Frequently reported ethnic GNE variants associated with GNE myopathy
| Nucleotide substitution | Predicted amino acid substitution | |||
|---|---|---|---|---|
| Current nomenclature | Previous nomenclature | GNE prote in domain | Reported ethnicity | |
| c.131G>C | p.Cys44Ser | p.C13S | ep | Japanese, Korean, Chinese |
| c.620A>T | p.Asp207Val | p.D176V | ep | Japanese, Korean, Chinese |
| c.1225G>T | p.Asp409Tyr | p.D378Y | UF | Caucasian, Japanese |
| c.1616T>C | p.Leu539Ser | p.L508S | kin | Chinese |
| c.1807G>C | p.Val603Leu | p.V572L | kin | Japanese, Korean, Chinese |
| c.1853T>C | p.Ile618Thr | p.I587T | kin | Roma Bulgarian, Asian |
| c.1985C>T | p.Ala662Val | p.A631V | kin | Caucasian, Japanese, Korean |
| c.2179G>A | p.Val727Met | p.V696M | kin | Indian, Asian |
| c.2228T>C | p.Met743Thr | p.M712T | kin | Middle Eastern |
Nucleotide substitution are provided in the current nomenclature only (mRNA variant 1 NM_001128227.2). Predicted amino acid substitutions are provided in the current hGNE2 (NP_001121699.1) with the preferred 3-letter amino acid code and in the previously used hGNE1 (NP_005467.1) nomenclature [8], with 1-letter amino acid code for easy reference with previous reviews [31]. GNE protein domains: ep = UDP-GlcNAc 2-epimerase domain; UF = unknown function; kin = ManNAc kinase domain (Fig. 2). Ethnicities listed in order of prevalence, some variants are also sporadically reported in other ethnicities, which are not included in this table
A recent study estimated carrier rates and prevalence of GNE myopathy using allele frequencies of GNE gene variants (excluding frequent nondisease causing SNPs) in large-scale exome databases according to the Hardy–Weinberg principle of population genetics. The worldwide carrier rate of a GNE variant was estimated to average ~ 1/203 individuals, resulting in an estimated prevalence of GNE myopathy of ~ 6/1,000,000 [31], in the same range as 1 to 9/1,000,000 as previously predicted [Orphanet; http://www.orpha.net]. These predictions estimate the existence of at least ~ 40,000 GNE myopathy subjects worldwide. Since the disease is not symptomatic until early adulthood, it can be conservatively assumed that there are currently 20,000 subjects affected worldwide. Since there are only ~ 950 subjects reported, it is predicted that many patients with the disease remain misdiagnosed or undiagnosed. Indeed, recent sequencing studies on cohorts of patients with myopathy identified several GNE myopathy patients, many of which could have escaped diagnosis without access to advanced genetic testing [74, 78].
Genetic Testing
Genetic testing has become the test of choice to confirm the diagnosis of GNE myopathy. A diagnosis is established with the identification of pathogenic variants in both alleles of the GNE gene. Targeted genetic sequencing can be performed in siblings of affected patients and patients of a particular ancestry, such as Met743Thr in Middle Eastern, Val727Met in Asian/Indian or (Cys44Ser, Asp207Val, and Val603Leu) in Japanese/Asian patients, and other ethnicities with founder effects. Sequence analysis of the entire coding region should be performed in all other patients. Sequencing of the entire coding region may be followed by deletion/duplication analysis in patients with characteristic findings and a single pathogenic variant identified [76, 77]. In rare occasions, patients may have mutations in deep intronic regions or other regulatory regions that are not detected by analysis of the GNE coding region [76].
As several GNE myopathy patients have been identified by genetic panels or next-generation sequencing studies on patient cohorts with limb-girdle muscular dystrophy phenotype and other undiagnosed myopathies [74, 78, 79], the GNE gene should be included on neuromuscular or similar gene sequencing panels. Sanger sequencing should be used to confirm any variants identified by exome or whole genome analysis. We recommend that all forms of genetic testing described above, if possible, are performed by a Clinical Laboratory Improvement Amendments (CLIA) compliant laboratory and that the current ACMG Standards and Guidelines are used for interpretation of pathogenicity of sequence variants [80].
Once the diagnosis has been established, genetic counseling should be provided to patients and families. Carrier testing for relatives can be performed by targeted genetic sequencing of the pathogenic variants in the family. Carrier testing of unrelated individuals such as spouses requires sequencing of the entire coding region.
Genetic Testing Results: GNE Nomenclature
There are at least six different GNE mRNA transcripts and protein isoforms described in humans [8, 81]. The mutation nomenclature currently in use is based on the longest GNE mRNA transcript (GenBank: NM_001128227; Ensembl: ENST00000396594), following the guidelines of the Human Genome Variation Society (http://www.hgvs.org). The NM_001128227 encodes 753 amino acids instead of 722 amino acids encoded by the previously used transcript (GenBank: NM_005476; Ensembl: ENST00000377902). Since the longer transcript has an extra partially coding exon at the N-terminus of the transcript, the nomenclature of the translated protein has been shifted by 31 amino acids; for example, the Middle Eastern mutation previously known as Met712Thr is now known as Met743Thr (Fig. 3) [8]. This nomenclature change should be taken into account by GNE mutations reported before ~ 2014, which did not always report the associated GenBank accession numbers. The most recent GNE mutation review published in 2014, lists all reported GNE variants at that time in both previous and current nomenclatures for reference and clarity [31].
Potential Therapies
There is no approved treatment available for GNE myopathy. However, understanding the underlying defect in GNE myopathy has allowed the development of targeted therapeutic strategies for the disease based on either providing precursors of the sialic acid biosynthetic pathway (sialylation-increasing therapies) or restoring UDP-GlcNAc 2-epimerase and ManNAc kinase enzymatic activities by gene or cell therapy approaches (Table 2). Studies on murine models have been conducted for sialylation-increasing therapies, including Neu5Ac, N-acetyl-D-mannosamine (ManNAc), peracetylated ManNAc, or sialyllactose [11, 67, 68, 82, 83], and gene therapy applications [84].
Table 2.
Potential therapies for GNE myopathy
| Potential therapy | Preclinical evidence | Clinical trials in GNE myopathy patients | ||
|---|---|---|---|---|
| Mouse model (year) | Trial design (ClinicalTrials.gov Identifier) | Dosing, trial duration (number of patients) | Sponsor year |
|
| Substrate replacement therapies | ||||
| ManNAc |
Gne Met743Thr knock-in (2007, 2012) [68, 91] Gne Asp207Val transgenic (2009) [67] |
Phase 1, randomized, double-blind, placebo-controlled (NCT01634750) [1] | Oral, single dose of 3 g, 6 g, and 10 g (n = 22) |
NHGRI/NCATS 2013–2014 |
| Phase 2, open label (NCT02346461) | Oral 6 g BID, 30 months (n = 12) |
NHGRI/NCATS 2015–2017 |
||
| Phase 3, randomized, double-blind, placebo-controlled | Oral 4 g TID, 24 months (tbd) | Planned 2018 | ||
| Neu5Ac | Gne Asp207Val transgenic (2009) [67] | Phase 1, open label (NCT01236898) | oral sialic acid, single and multiple doses, up to 800 mg TID for 5 days (n = 6) |
Tohoku University 2010–2011 |
| Phase 1, open label (NCT01359319) | Ace-ER single and repeat dosing (n = 26) |
Ultragenyx 2011–2012 |
||
| Phase 2, randomized, double-blind, placebo-controlled (NCT01517880) [94] | Ace-ER 3 or 6 g/day, 48 weeks (n = 47) |
Ultragenyx 2012–2013 |
||
| Phase 2, open label (NCT01830972)* | Ace-ER/SA-IR 3 g, 6 g, 12 g/day, 48 weeks (n = 59) |
Ultragenyx 2013–2017 |
||
| Phase 2, open label, safety (NCT02731690) | Ace-ER 6 g/day, 48 weeks (n = 45) |
Ultragenyx 2016–2018† |
||
| Phase 3, randomized, double-blind, placebo-controlled (NCT02377921) | Ace-ER 2 g TID, 48 weeks (n = 89) |
Ultragenyx 2015–2017 |
||
| Phase 3, open-label extension (NCT02736188) | Ace-ER 2 g TID, 24 months (n = 144) |
Ultragenyx 2016–2020† |
||
| Peracetylated ManNAc | Gne Asp207Val transgenic (2012) [82] | None | – | – |
| Sialylated glycan therapies | ||||
| Sialyllactose | Gne Asp207Val transgenic (2009, 2014) [67, 83] | None | – | – |
| IVIG | – | Phase 1, open label (NCT00195637) [92] | IVIG 1 g/kg, 400 mg/kg, 4 weeks (n = 4) |
NHGRI, NIH 2005–2006 |
| Gene-based therapies | ||||
| GNE cDNA-Lipoplex | BALB/c wild type [111] | Single patient “compassionate use” (not on ClinicalTrials.gov) [110] | Intramuscular, multiple doses GNE cDNA-lipoplex (n = 1) |
Gradalis Inc. 2008–2009 |
| Single patient “compassionate use” (not on ClinicalTrials.gov) [102] | Intravenous GNE cDNA-lipoplex, every 60 days, 7 doses total (n = 1) |
Gradalis Inc. 2009–2010 |
||
| AAV8-GNE cDNA |
C57BL/6 wild-type mice [84] GNE myopathy human myoblasts [84] |
– | – | – |
| AAV8 trans-splicing | GNE myopathy human myoblasts [104] | – | – | – |
Ace-ER = extended-release sialic acid; BID = twice daily; IVIG = intravenous immunoglobulin; NHGRI = National Human Genome Research Institute; NCATS = National Center for Advancing Translational Sciences; SA-IR = immediate-release sialic acid; TID = three times a day
*Results posted on ClinicalTrials.gov
†Studies ended prematurely
Sialylation-Increasing Therapies
The role of sialylation-increasing therapies in GNE myopathy assumes that the disease pathophysiology is primarily due to a defect in the sialic acid biosynthesis pathway. Sialylation-increasing therapies include pathway substrates, such as ManNAc and sialic acid, and highly sialylated glycoproteins, such as siallylactose and intravenous immunoglobulin (IVIG). Potential sialylation-increasing molecules should be able to reach the intracellular space and incorporated in the metabolic pathway to be efficacious (Fig. 1). ManNAc, a neutral molecule, enters cells through passive diffusion or a plasma membrane transporter, as shown by in vitro studies [85–88]. Neu5Ac and sialyllactose are negatively charged and are taken up into cells by an active transport endocytic lysosomal pathway [85, 86, 89]. Sialylated proteins, like sialyllactose, are degraded in lysosomes and release its Neu5Ac molecules through lysosomal neuraminidases [90].
Preclinical Studies
There is encouraging preclinical evidence that sialylation-increasing therapies, such as sialic acid, N-acetyl-D-mannosamine (ManNAc) [67, 68, 91], tetra-O-acetylated ManNAc (Ac4ManNAc) [82], and sialyllactose (a highly sialylated protein) [83], can prevent, arrest, or improve the development of the disease in mouse models of GNE myopathy and provide benefit for affected individuals.
A transgenic mouse model for a founder mutation in Japanese patients on a Gne null background (Gne−/−hGNEAsp207Val-Tg; originally Gne−/−hGNED176V-Tg) recapitulated the muscle phenotype of GNE myopathy, developing progressive muscle weakness starting at 10 weeks and by 40 weeks of age showing significant changes in muscle pathology and biochemistry similar to that of GNE myopathy patients. Prophylactic oral treatment of these mice with ManNAc, sialic acid, sialyllactose, or intravenous Ac4ManNAc resulted in prevention of muscle weakness and atrophy, reduced or absent rimmed vacuoles and intracellular amyloid inclusions, improved survival, and improved sialylation of membrane-bound glycans in muscle and other tissues compared to untreated affected littermates [67, 82]. The administration of oral sialyllactose after the onset of symptoms in the same mouse model ameliorated the myopathic phenotype of affected mice [83].
In a gene-targeted knock-in Gne mouse homozygous for the Middle Eastern founder mutation, Met743Thr (originally M712T), oral prophylactic administration of ManNAc to pregnant and nursing mice, conferred increased survival of mutant pups beyond postnatal day 3 [91]. ManNAc administration was also associated with increased enzymatic activity [91]. The therapeutic potential of ManNAc treatment to rescue muscle hyposialylation after its onset was studied in adult mutant mice (~ 6 months of age) orally treated with ManNAc (1 or 2 g/kg/day) for 12 weeks and showed marked improvement of muscle sialylation status into the normal range. These results support the therapeutic potential of ManNAc for treatment of GNE myopathy subjects after disease onset [68].
IVIG Therapy
In 2005, a pilot study of IVIG, a highly sialylated glycoprotein that is presumed to be metabolized in lysosomes, similar to sialyllactose, was given to four GNE myopathy patients at doses of 1 g/kg on 2 consecutive days followed by 3 doses of 400 mg/kg at weekly intervals (ClinicalTrials.gov: NCT00195637). The patients exhibited mild increase in muscle strength but no biochemical evidence of significantly increased glycans or sialylation on muscle biopsies [92].
Sialic Acid (Neu5Ac)
Neu5Ac (MW 309.3 Da) is a negatively charged acidic monosaccharide transported intracellularly through an active transport endocytic lysosomal pathway, which may be inefficient [85, 86, 89]. Oral free sialic acid (Neu5Ac) administered orally to rats, was almost entirely absorbed and, within hours, excreted mainly as free Neu5Ac via the kidneys [93]. The significant in vivo first pass effect of oral Neu5Ac and inefficient cellular uptake necessitated a different formulation or route of administration to obtain sufficient cellular uptake.
Ultragenyx Pharmaceutical developed an extended-release formulation of Neu5Ac (Ace-ER) and have conducted multiple studies to evaluate the safety, pharmacokinetics, and efficacy of Ace-ER in patients with GNE myopathy (Table 2).
In 2012, a phase 2, randomized, double-blind, placebo-controlled trial (ClinicalTrials.gov NCT01517880) conducted in 47 subjects with GNE myopathy treated patients with either Ace-ER 3 g/day, 6 g/day, or placebo for 24 weeks. After 24 weeks, subjects receiving placebo were assigned to receive Ace-ER at doses of either 3 g/day or 6 g/day. Results at 24 weeks showed maintenance of upper extremity strength (as measured by composite upper extremity strength obtained by dynamometry) in the 6 g/day group compared to placebo, especially in subjects able to walk > 200 m on the 6-min walk test (6MWT) at baseline. At 48 weeks, there was significant improvement on upper extremity composite strength in the combined 6 g/day group compared to the combined 3 g/day group (+ 3.46 kg, p = 0.0031), but no other significant effect was measured on other tested endpoints, including lower extremity composite score, 6MWT, or GNEM-FAS [94].
This study was followed by a phase 3, randomized, double-blind, placebo-controlled trial (ClinicalTrials.gov NCT02377921) in 89 patients, which failed to detect clinical efficacy. In August 2017, Ultragenyx Pharmaceutical discontinued the clinical development of Ace-ER for GNE myopathy.
N-Acetyl-D-mannosamine (ManNAc)
ManNAc is a naturally occurring uncharged monosaccharide, the first committed precursor for the biosynthesis of Neu5Ac (sialic acid), and a substrate of the GNE enzyme (Fig. 1). ManNAc is the only neutral molecule in the committed sialic acid biosynthesis pathway, facilitating its permeability across membranes. Several in vitro studies suggest that ManNAc reaches the intracellular space either through passive diffusion or an unidentified membrane transporter [85–88]. Moreover, assays of cells incubated with labeled ManNAc showed a half-life of the intracellular ManNAc pool longer than 2 h, which allows ManNAc to be incorporated into the metabolic pathway [86]. ManNAc bypasses the rate-limiting feedback inhibition enzymatic step catalyzed by UDP-GlcNAc 2-epimerase [42] and can be phosphorylated by other kinase enzymes, such as GlcNAc kinase if the enzymatic activity of ManNAc kinase is compromised [95].
The clinical development of ManNAc was pursued based on the in vitro and in vivo evidence that ManNAc restores sialic acid production and increases the sialylation of hyposialylated glycoproteins and glycolipids in disease models of GNE myopathy [67, 68, 91]. After IND approval by the FDA in 2012, a first-in-human phase 1, randomized, placebo-controlled, double-blind, single-ascending dose study (ClinicalTrials.gov NCT01634750; IND No.78,091) was conducted at the National Institutes of Health (NIH, Bethesda, USA). This study evaluated the safety, tolerability, and pharmacokinetics of single oral doses of ManNAc in subjects with GNE myopathy. Quantification of ManNAc and Neu5Ac in human plasma was performed using a validated liquid chromatography and tandem mass spectrometry (LC-MS/MS) method with an assay range of 10 to 5000 ng/mL [96]. Oral ManNAc was absorbed rapidly and exhibited a short half-life (~ 2.4 h) as expected [1].
Following administration of a single oral dose of ManNAc, there was a significant and sustained increase in plasma Neu5Ac levels up to 48 h post dose. Given that Neu5Ac is known to have a short half-life, the prolonged elevation of Neu5Ac after a single dose of ManNAc strongly suggested that intracellular biosynthesis of sialic acid was restored in subjects with GNE myopathy, including those homozygous for mutations in the kinase domain [1].
A phase 2 open-label clinical trial of ManNAc in GNE myopathy (ClinicalTrials.gov NCT02346461) was initiated in January 2015 and was recently completed. The objectives of this study were to assess the long-term safety, tolerability, pharmacokinetics, and biochemical efficacy of orally administered ManNAc in GNE myopathy subjects. A pivotal multicenter trial of ManNAc for GNE myopathy is being planned.
Gene and Cell Therapies
Gene therapy is a significant scientific development with the potential of effectively treating genetic diseases. Since several aspects of the disease pathophysiology are still being elucidated in GNE myopathy, gene therapy strategies offer the potential of delivering an unaffected copy of the gene to target muscle tissues with the aim of restoring or preventing the loss of muscle function.
Over the last several decades, several challenges have delayed the development of effective and safe gene therapies, such as ethical and safety concerns associated with the risk of life-threatening immune responses [97], and insertional mutagenesis that may result in malignancy [98]. For muscle diseases, the most significant hurdle in developing successful gene therapy strategies has been the ability to deliver the gene to affected muscles throughout the body effectively. Recombinant adeno-associated viruses (rAAV) are the most commonly used gene therapy vectors and the ones used for FDA-approved gene therapies [99]. The main issue with the use of rAAV is the presence of preexisting neutralizing antibodies against the rAAV capsid proteins which decrease the transduction of the gene to target tissues [100]. The AAV8 vector has shown to provide effective delivery of the GNE gene to murine and human-derived muscle cells in culture and safe systemic administration to healthy mice [84]. However, preexisting serum antibodies to a wide variety of rAAV serotypes, including rAAV8, were present in ~ 50% of GNE myopathy patients tested in a recent study [101].
A single subject with advanced stage GNE myopathy has been treated in a “compassionate” investigational new drug trial with intravenous [102] injections of human GNE gene therapy with a cytomegalovirus (CMV) promoter and a vector complexed with a cationic liposome for delivery (GNE-lipoplex). It was reported that there was significant muscle mRNA expression of the delivered GNE, increased sialylation of muscle glycoproteins, and apparent stabilization of muscle strength in this subject, without any severe adverse events [102]. Further gene therapy studies in preclinical models are necessary to optimize the vector, promoter, and mode of delivery to correct the underlying defect in skeletal muscles throughout the body in a safe manner.
A more novel technology, targeted genome editing, uses viral vectors to deliver a gene-editing system, such as the CRISPR technology, to replace mutations in genes with the correct nucleotides [103]. Such an approach can be considered for GNE ethnic founder mutations. A rAAV8-based trans-splicing vector was used in a recent in vitro study to correct cultured muscle cells containing the Middle Eastern Met743Thr GNE mutation [104].
Cell therapy strategies have been explored for myopathies [105, 106] and could be translated to GNE myopathy if proven successful in animal studies. However, several issues still need to be addressed, such as grafting and survival of muscle precursor cells before human studies are conducted [107].
Future Challenges and Approaches
Almost 20 years after the identification of the molecular basis of the disease, there is still no approved therapy for GNE myopathy, and several aspects of the pathophysiology remain a mystery. As our understanding of GNE myopathy increases, better diagnostic tools, novel biomarkers, and therapeutic strategies continue to be explored.
The diagnosis of GNE myopathy remains challenging, but continued efforts to increase awareness and identify biomarkers will facilitate the identification of patients. Lately, the majority of patients are identified through genetic testing, and currently, there are > 250 GNE variants reported in patients, some of which are of unknown significance. This raises a new challenge of novel sequence variants identified in the GNE gene that require proper evidence to determine pathogenicity to ensure that patients receive a proper diagnosis [80]. To determine the pathogenicity of novel sequence variants, development of GNE-specific assays are needed, such as an accessible enzymatic assay for GNE, development of functional GNE antibodies, or other cellular assays that assess sialylation or GNE interactions.
The two mouse models of GNE myopathy currently available have facilitated the understanding of disease mechanisms and identification of potential therapies [21, 91]. However, these preclinical models have limitations, as commonly seen in mouse models for human metabolic diseases [108]. The knock-in model develops severe glomerular disease, which is not part of the human phenotype, and, unless it is treated with ManNAc or other sialic acid-increasing molecules, develops neonatal lethality. The transgenic model on a Gne null background may be cumbersome to breed, and depending on the genetic background, the mutated transgene may be overexpressed or the phenotype not consistently replicated. Therefore, the creation of better murine models for GNE myopathy will be an invaluable tool for future preclinical research.
The wide variety in disease onset and severity among cases with the same GNE mutations and even among affected siblings is possibly due to disease modifiers. Such modifiers can be explored through large cohort studies with data-mining techniques such as whole genome sequencing, metabolomics, proteomics, epigenetic, or RNA analysis.
Studies of substrate replacement therapy are ongoing, but the termination of the clinical program for Ace-ER has left their efficacy in question. However, it is estimated that a significant proportion of clinical trials fail during drug development due to inappropriate study design or endpoint selection [1]. Unfortunately, this leads to the inability to distinguish whether negative results are due to lack of therapeutic effect or the inability to detect it due to poor protocol design. Therefore, potential therapies may be discarded without a proper understanding of their efficacy. Clinical trial designs can be improved to more confidently determine treatment effect when based on a detailed understanding of the disease. In the case of GNE myopathy, the limited knowledge on natural history, the small number of patients, and the slow progression of the disease pose a difficult challenge for designing clinical trials able to determine treatment effect confidently. A better understanding of disease progression and more sensitive endpoints are needed to inform the design and endpoint selection for clinical trials in GNE myopathy.
Gene and cell-based therapeutic approaches are promising but should be further investigated in preclinical models to determine successful approaches that are safe, are feasibly delivered, and achieve proper gene expression, and cell survival, in skeletal muscles affected in GNE myopathy [109].
Resources
The existence of patient organizations is extremely important for rare diseases. Patient organizations dedicated to GNE myopathy such as the Neuromuscular Disease Foundation (NDF; http://curehibm.org) and the Advancement of Research for Myopathies (ARM; https://www.hibm.org/arm/) in the USA, TREAT-NMD in Europe (http://www.treat-nmd.eu/gne/about), and Patient Association for Distal Myopathies (PADM) in Japan, as well as GNE Myopathy International (http://gne-myopathy.org), are available to provide support for individuals with GNE myopathy.
Information resources are available at the US National Library of Medicine, including the Genetics Home Reference page (https://ghr.nlm.nih.gov/condition/inclusion-body-myopathy-2). Finally, ClinicalTrials.gov (https://clinicaltrials.gov) provides information on clinical studies around the world that conform with human subject and ethics review regulations.
Electronic Supplementary Material
(PDF 498 kb)
Acknowledgments
This research was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
References
- 1.Xu X, Wang AQ, Latham LL, Celeste F, Ciccone C, Malicdan MC, et al. Safety, pharmacokinetics and sialic acid production after oral administration of N-acetylmannosamine (ManNAc) to subjects with GNE myopathy. Mol Genet Metab. 2017;122(1–2):126–134. doi: 10.1016/j.ymgme.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nonaka I, Sunohara N, Ishiura S, Satoyoshi E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci. 1981;51(1):141–155. doi: 10.1016/0022-510X(81)90067-8. [DOI] [PubMed] [Google Scholar]
- 3.Nonaka I, Sunohara N, Satoyoshi E, Terasawa K, Yonemoto K. Autosomal recessive distal muscular dystrophy: a comparative study with distal myopathy with rimmed vacuole formation. Ann Neurol. 1985;17(1):51–59. doi: 10.1002/ana.410170113. [DOI] [PubMed] [Google Scholar]
- 4.Argov Z, Yarom R. “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. 1984;64(1):33–43. doi: 10.1016/0022-510X(84)90053-4. [DOI] [PubMed] [Google Scholar]
- 5.Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet. 1996;5(1):159–163. doi: 10.1093/hmg/5.1.159. [DOI] [PubMed] [Google Scholar]
- 6.Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nature genetics. 2001;29(1):83–87. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 7.Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59(11):1689–1693. doi: 10.1212/01.WNL.0000041631.28557.C6. [DOI] [PubMed] [Google Scholar]
- 8.Huizing M, Carrillo-Carrasco N, Malicdan MC, Noguchi S, Gahl WA, Mitrani-Rosenbaum S, et al. GNE myopathy: new name and new mutation nomenclature. Neuromuscul Disord. 2014;24(5):387–389. doi: 10.1016/j.nmd.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pogoryelova O, Cammish P, Mansbach H, Argov Z, Nishino I, Skrinar A, et al. Phenotypic stratification and genotype-phenotype correlation in a heterogeneous, international cohort of GNE myopathy patients: first report from the GNE myopathy Disease Monitoring Program, registry portion. Neuromuscul Disord. 2018;28(2):158–168. doi: 10.1016/j.nmd.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mori-Yoshimura M, Oya Y, Yajima H, Yonemoto N, Kobayashi Y, Hayashi YK, et al. GNE myopathy: a prospective natural history study of disease progression. Neuromuscul Disord. 2014;24(5):380–386. doi: 10.1016/j.nmd.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Huizing M, Malicdan MV, Krasnewich DM, Manoli I, Carrillo-Carrasco N. GNE Myopathy. In: Scriver CR, Childs B, Sly WS, Valle D, Beaudet AL, Vogelstein B, Kinsler KW, editors. Scriver’s online metabolic and molecular bases of inherited disease. http://www.ommbid.com. Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, Ballabio A, et al., editors. New York: McGraw-Hill; 2014.
- 12.Slota C, Bevans M, Yang L, Shrader J, Joe G, Carrillo N. Patient reported outcomes in GNE myopathy: incorporating a valid assessment of physical function in a rare disease. Disabil Rehabil. 2017:1–8. [DOI] [PMC free article] [PubMed]
- 13.Cho A, Hayashi YK, Monma K, Oya Y, Noguchi S, Nonaka I, et al. Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy) J Neurol Neurosurg Psychiatry. 2014;85(8):914–917. doi: 10.1136/jnnp-2013-305587. [DOI] [PubMed] [Google Scholar]
- 14.Mayhew J, Bonner N, Arbuckle R, Turnbull A, Bowden A, Skrinar A. Development and preliminary evidence of the psychometric properties of the GNE myopathy functional activity scale. J Comp Eff Res. 2018;7(4):381–395. doi: 10.2217/cer-2017-0062. [DOI] [PubMed] [Google Scholar]
- 15.Boyden SE, Duncan AR, Estrella EA, Lidov HG, Mahoney LJ, Katz JS, et al. Molecular diagnosis of hereditary inclusion body myopathy by linkage analysis and identification of a novel splice site mutation in GNE. BMC Med Genet. 2011;12:87. doi: 10.1186/1471-2350-12-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dotti MT, Malandrini A, Lornage X, Mignarri A, Cantisani TA, Bohm J, et al. Discordant manifestations in Italian brothers with GNE myopathy. J Neurol Sci. 2018;386:1–3. doi: 10.1016/j.jns.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Ro LS, Lee-Chen GJ, Wu YR, Lee M, Hsu PY, Chen CM. Phenotypic variability in a Chinese family with rimmed vacuolar distal myopathy. J Neurol Neurosurg Psychiatry. 2005;76(5):752–755. doi: 10.1136/jnnp.2004.048876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Dios JK, Shrader JA, Joe GO, McClean JC, Williams K, Evers R, et al. Atypical presentation of GNE myopathy with asymmetric hand weakness. Neuromuscul Disord. 2014;24(12):1063–1067. doi: 10.1016/j.nmd.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mori-Yoshimura M, Oya Y, Hayashi YK, Noguchi S, Nishino I, Murata M. Respiratory dysfunction in patients severely affected by GNE myopathy (distal myopathy with rimmed vacuoles) Neuromuscular disorders : NMD. 2013;23(1):84–88. doi: 10.1016/j.nmd.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Chamova T, Guergueltcheva V, Gospodinova M, Krause S, Cirak S, Kaprelyan A, et al. GNE myopathy in Roma patients homozygous for the p.I618T founder mutation. Neuromuscul Disord. 2015;25(9):713–718. doi: 10.1016/j.nmd.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 21.Malicdan MC, Noguchi S, Nonaka I, Hayashi YK, Nishino I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. 2007;16(22):2669–2682. doi: 10.1093/hmg/ddm220. [DOI] [PubMed] [Google Scholar]
- 22.Mori-Yoshimura M, Hayashi YK, Yonemoto N, Nakamura H, Murata M, Takeda S, et al. Nationwide patient registry for GNE myopathy in Japan. Orphanet J Rare Dis. 2014;9:150. doi: 10.1186/s13023-014-0150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chai Y, Bertorini TE, McGrew FA. Hereditary inclusion-body myopathy associated with cardiomyopathy: report of two siblings. Muscle & nerve. 2011;43(1):133–136. doi: 10.1002/mus.21839. [DOI] [PubMed] [Google Scholar]
- 24.Kimpara T, Imamura T, Tsuda T, Sato K, Tsuburaya K. Distal myopathy with rimmed vacuoles and sudden death—report of two siblings. Rinsho Shinkeigaku. 1993;33(8):886–890. [PubMed] [Google Scholar]
- 25.Zhen C, Guo F, Fang X, Liu Y, Wang X. A family with distal myopathy with rimmed vacuoles associated with thrombocytopenia. Neurol Sci. 2014;35(9):1479–1481. doi: 10.1007/s10072-014-1790-9. [DOI] [PubMed] [Google Scholar]
- 26.Izumi R, Niihori T, Suzuki N, Sasahara Y, Rikiishi T, Nishiyama A, et al. GNE myopathy associated with congenital thrombocytopenia: a report of two siblings. Neuromuscul Disord. 2014;24(12):1068–1072. doi: 10.1016/j.nmd.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 27.Futterer J, Dalby A, Lowe GC, Johnson B, Simpson MA, Motwani J, et al. Mutation in GNE is associated with a severe form of congenital thrombocytopenia. Blood. 2018. [DOI] [PMC free article] [PubMed]
- 28.Hinderlich S, Weidemann W, Yardeni T, Horstkorte R, Huizing M. UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE): a master regulator of sialic acid synthesis. Top Curr Chem. 2015;366:97–137. doi: 10.1007/128_2013_464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinderlich S, Stasche R, Zeitler R, Reutter W. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. 1997;272(39):24313–24318. doi: 10.1074/jbc.272.39.24313. [DOI] [PubMed] [Google Scholar]
- 30.Keppler OT, Hinderlich S, Langner J, Schwartz-Albiez R, Reutter W, Pawlita M. UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science. 1999;284(5418):1372–1376. doi: 10.1126/science.284.5418.1372. [DOI] [PubMed] [Google Scholar]
- 31.Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MC, Leoyklang P, et al. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat. 2014;35(8):915–926. doi: 10.1002/humu.22583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen SC, Huang CH, Lai SJ, Yang CS, Hsiao TH, Lin CH, et al. Mechanism and inhibition of human UDP-GlcNAc 2-epimerase, the key enzyme in sialic acid biosynthesis. Sci Rep. 2016;6:23274. doi: 10.1038/srep23274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sparks SE, Ciccone C, Lalor M, Orvisky E, Klootwijk R, Savelkoul PJ, et al. Use of a cell-free system to determine UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities in human hereditary inclusion body myopathy. Glycobiology. 2005;15(11):1102–1110. doi: 10.1093/glycob/cwi100. [DOI] [PubMed] [Google Scholar]
- 34.Kurochkina N, Yardeni T, Huizing M. Molecular modeling of the bifunctional enzyme UDP-GlcNAc 2-epimerase/ManNAc kinase and predictions of structural effects of mutations associated with HIBM and sialuria. Glycobiology. 2010;20(3):322–337. doi: 10.1093/glycob/cwp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tong Y, Tempel W, Nedyalkova L, Mackenzie F, Park HW. Crystal structure of the N-acetylmannosamine kinase domain of GNE. PLoS One. 2009;4(10):e7165. doi: 10.1371/journal.pone.0007165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarzkopf M, Knobeloch KP, Rohde E, Hinderlich S, Wiechens N, Lucka L, et al. Sialylation is essential for early development in mice. Proc Natl Acad Sci U S A. 2002;99(8):5267–5270. doi: 10.1073/pnas.072066199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krause S, Hinderlich S, Amsili S, Horstkorte R, Wiendl H, Argov Z, et al. Localization of UDP-GlcNAc 2-epimerase/ManAc kinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp Cell Res. 2005;304(2):365–379. doi: 10.1016/j.yexcr.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura K, Tsukamoto Y, Hijiya N, Higuchi Y, Yano S, Yokoyama S, et al. Induction of GNE in myofibers after muscle injury. Pathobiology. 2010;77(4):191–199. doi: 10.1159/000292652. [DOI] [PubMed] [Google Scholar]
- 39.Amsili S, Zer H, Hinderlich S, Krause S, Becker-Cohen M, MacArthur DG, et al. UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) binds to alpha-actinin 1: novel pathways in skeletal muscle? PLoS One. 2008;3(6):e2477. doi: 10.1371/journal.pone.0002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harazi A, Becker-Cohen M, Zer H, Moshel O, Hinderlich S, Mitrani-Rosenbaum S. The interaction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) and alpha-actinin 2 is altered in GNE myopathy M743T mutant. Mol Neurobiol. 2017;54(4):2928–2938. doi: 10.1007/s12035-016-9862-x. [DOI] [PubMed] [Google Scholar]
- 41.Weidemann W, Stelzl U, Lisewski U, Bork K, Wanker EE, Hinderlich S, et al. The collapsin response mediator protein 1 (CRMP-1) and the promyelocytic leukemia zinc finger protein (PLZF) bind to UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE), the key enzyme of sialic acid biosynthesis. FEBS Lett. 2006;580(28–29):6649–6654. doi: 10.1016/j.febslet.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 42.Weidemann W, Klukas C, Klein A, Simm A, Schreiber F, Horstkorte R. Lessons from GNE-deficient embryonic stem cells: sialic acid biosynthesis is involved in proliferation and gene expression. Glycobiology. 2010;20(1):107–117. doi: 10.1093/glycob/cwp153. [DOI] [PubMed] [Google Scholar]
- 43.Harazi A, Chaouat M, Shlomai Z, Levitzki R, Becker-Cohen M, Sadeh M, et al. Survival-apoptosis associated signaling in GNE myopathy-cultured myoblasts. J Recept Signal Transduct Res. 2015;35(4):249–257. doi: 10.3109/10799893.2014.956755. [DOI] [PubMed] [Google Scholar]
- 44.Chanana P, Padhy G, Bhargava K, Arya R. Mutation in GNE downregulates peroxiredoxin IV altering ER redox homeostasis. Neuromolecular Med. 2017;19(4):525–540. doi: 10.1007/s12017-017-8467-5. [DOI] [PubMed] [Google Scholar]
- 45.Cho A, Christine M, Malicdan V, Miyakawa M, Nonaka I, Nishino I, et al. Sialic acid deficiency is associated with oxidative stress leading to muscle atrophy and weakness in GNE myopathy. Hum Mol Genet. 2017;26(16):3081–3093. doi: 10.1093/hmg/ddx192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schauer R. Sialic acids as regulators of molecular and cellular interactions. Curr Opin Struct Biol. 2009;19(5):507–514. doi: 10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varki A. Sialic acids in human health and disease. Trends Mol Med. 2008;14(8):351–360. doi: 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Varki A. Diversity in the sialic acids. Glycobiology. 1992;2(1):25–40. doi: 10.1093/glycob/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stasche R, Hinderlich S, Weise C, Effertz K, Lucka L, Moormann P, et al. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Molecular cloning and functional expression of UDP-N-acetyl-glucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. 1997;272(39):24319–24324. doi: 10.1074/jbc.272.39.24319. [DOI] [PubMed] [Google Scholar]
- 50.Kean EL, Munster-Kuhnel AK, Gerardy-Schahn R. CMP-sialic acid synthetase of the nucleus. Biochim Biophys Acta. 2004;1673(1–2):56–65. doi: 10.1016/j.bbagen.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 51.Leroy JG, Seppala R, Huizing M, Dacremont G, De Simpel H, Van Coster RN, et al. Dominant inheritance of sialuria, an inborn error of feedback inhibition. Am J Hum Genet. 2001;68(6):1419–1427. doi: 10.1086/320598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seppala R, Lehto VP, Gahl WA. Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am J Hum Genet. 1999;64(6):1563–1569. doi: 10.1086/302411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bork K, Weidemann W, Berneck B, Kuchta M, Bennmann D, Thate A, et al. The expression of sialyltransferases is regulated by the bioavailability and biosynthesis of sialic acids. Gene Expr Patterns. 2017;23-24:52–58. doi: 10.1016/j.gep.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 54.Varki A CR, Esko J, et al., editors. . Essentials of glycobiology. NY: Cold Spring Harbor Laboratory Press; 1999. [PubMed]
- 55.Reinke SO, Lehmer G, Hinderlich S, Reutter W. Regulation and pathophysiological implications of UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE) as the key enzyme of sialic acid biosynthesis. Biol Chem. 2009;390(7):591–599. doi: 10.1515/BC.2009.073. [DOI] [PubMed] [Google Scholar]
- 56.Gagiannis D, Orthmann A, Danssmann I, Schwarzkopf M, Weidemann W, Horstkorte R. Reduced sialylation status in UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE)-deficient mice. Glycoconj J. 2007;24(2–3):125–130. doi: 10.1007/s10719-006-9019-7. [DOI] [PubMed] [Google Scholar]
- 57.Noguchi S, Keira Y, Murayama K, Ogawa M, Fujita M, Kawahara G, et al. Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J Biol Chem. 2004;279(12):11402–11407. doi: 10.1074/jbc.M313171200. [DOI] [PubMed] [Google Scholar]
- 58.Savelkoul PJ, Manoli I, Sparks SE, Ciccone C, Gahl WA, Krasnewich DM, et al. Normal sialylation of serum N-linked and O-GalNAc-linked glycans in hereditary inclusion-body myopathy. Mol Genet Metab. 2006;88(4):389–390. doi: 10.1016/j.ymgme.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 59.Salama I, Hinderlich S, Shlomai Z, Eisenberg I, Krause S, Yarema K, et al. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNE mutation. Biochem Biophys Res Commun. 2005;328(1):221–226. doi: 10.1016/j.bbrc.2004.12.157. [DOI] [PubMed] [Google Scholar]
- 60.Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, et al. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Molecular Genetics and Metabolism. 2004;81(3):196–202. doi: 10.1016/j.ymgme.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 61.Ricci E, Broccolini A, Gidaro T, Morosetti R, Gliubizzi C, Frusciante R, et al. NCAM is hyposialylated in hereditary inclusion body myopathy due to GNE mutations. Neurology. 2006;66(5):755–758. doi: 10.1212/01.wnl.0000200956.76449.3f. [DOI] [PubMed] [Google Scholar]
- 62.Broccolini A, Gidaro T, De Cristofaro R, Morosetti R, Gliubizzi C, Ricci E, et al. Hyposialylation of neprilysin possibly affects its expression and enzymatic activity in hereditary inclusion-body myopathy muscle. J Neurochem. 2008;105(3):971–981. doi: 10.1111/j.1471-4159.2007.05208.x. [DOI] [PubMed] [Google Scholar]
- 63.Paccalet T, Coulombe Z, Tremblay JP. Ganglioside GM3 levels are altered in a mouse model of HIBM: GM3 as a cellular marker of the disease. PLoS One. 2010;5(4):e10055. doi: 10.1371/journal.pone.0010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tajima Y, Uyama E, Go S, Sato C, Tao N, Kotani M, et al. Distal myopathy with rimmed vacuoles: impaired O-glycan formation in muscular glycoproteins. Am J Pathol. 2005;166(4):1121–1130. doi: 10.1016/S0002-9440(10)62332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leoyklang P, Malicdan MC, Yardeni T, Celeste F, Ciccone C, Li X, et al. Sialylation of Thomsen-Friedenreich antigen is a noninvasive blood-based biomarker for GNE myopathy. Biomark Med. 2014;8(5):641–652. doi: 10.2217/bmm.14.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leoyklang P, Class B, Noguchi S, Gahl WA, Carrillo N, Nishino I, et al. Quantification of lectin fluorescence in GNE myopathy muscle biopsies. Muscle Nerve. 2018;58(2):286–292. doi: 10.1002/mus.26135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Malicdan MC, Noguchi S, Hayashi YK, Nonaka I, Nishino I. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. 2009;15(6):690–695. doi: 10.1038/nm.1956. [DOI] [PubMed] [Google Scholar]
- 68.Niethamer TK, Yardeni T, Leoyklang P, Ciccone C, Astiz-Martinez A, Jacobs K, et al. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab. 2012;107(4):748–755. doi: 10.1016/j.ymgme.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Chen Q, Liu F, Zhang X, Liu T, Li W, et al. Clinical and molecular genetic analysis in Chinese patients with distal myopathy with rimmed vacuoles. J Hum Genet. 2011;56(4):335–338. doi: 10.1038/jhg.2011.15. [DOI] [PubMed] [Google Scholar]
- 70.Saechao C, Valles-Ayoub Y, Esfandiarifard S, Haghighatgoo A, No D, Shook S, et al. Novel GNE mutations in hereditary inclusion body myopathy patients of non-Middle Eastern descent. Genet Test Mol Biomarkers. 2010;14(2):157–162. doi: 10.1089/gtmb.2009.0157. [DOI] [PubMed] [Google Scholar]
- 71.Voermans NC, Guillard M, Doedee R, Lammens M, Huizing M, Padberg GW, et al. Clinical features, lectin staining, and a novel GNE frameshift mutation in hereditary inclusion body myopathy. Clin Neuropathol. 2010;29(2):71–77. [PMC free article] [PubMed] [Google Scholar]
- 72.Tasca G, Ricci E, Monforte M, Laschena F, Ottaviani P, Rodolico C, et al. Muscle imaging findings in GNE myopathy. J Neurol. 2012;259(7):1358–1365. doi: 10.1007/s00415-011-6357-6. [DOI] [PubMed] [Google Scholar]
- 73.Khadilkar SV, Nallamilli BRR, Bhutada A, Hegde M, Gandhi K, Faldu HD, et al. A report on GNE myopathy: individuals of Rajasthan ancestry share the Roma gene. J Neurol Sci. 2017;375:239–240. doi: 10.1016/j.jns.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 74.Park HJ, Jang H, Kim JH, Lee JH, Shin HY, Kim SM, et al. Discovery of pathogenic variants in a large Korean cohort of inherited muscular disorders. Clinical genetics. 2017;91(3):403–410. doi: 10.1111/cge.12826. [DOI] [PubMed] [Google Scholar]
- 75.Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry. 2015;86(4):385–392. doi: 10.1136/jnnp-2013-307051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu W, Mitsuhashi S, Yonekawa T, Noguchi S, Huei JC, Nalini A, et al. Missing genetic variations in GNE myopathy: rearrangement hotspots encompassing 5′UTR and founder allele. J Hum Genet. 2017;62(2):159–166. doi: 10.1038/jhg.2016.134. [DOI] [PubMed] [Google Scholar]
- 77.Garland J, Stephen J, Class B, Gruber A, Ciccone C, Poliak A, et al. Identification of an Alu element-mediated deletion in the promoter region of GNE in siblings with GNE myopathy. Mol Genet Genomic Med. 2017;5(4):410–417. doi: 10.1002/mgg3.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Monies D, Alhindi HN, Almuhaizea MA, Abouelhoda M, Alazami AM, Goljan E, et al. A first-line diagnostic assay for limb-girdle muscular dystrophy and other myopathies. Hum Genomics. 2016;10(1):32. doi: 10.1186/s40246-016-0089-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khadilkar SV, Faldu HD, Patil SB, Singh R. Limb-girdle muscular dystrophies in India: a review. Ann Indian Acad Neurol. 2017;20(2):87–95. doi: 10.4103/aian.AIAN_81_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yardeni T, Choekyi T, Jacobs K, Ciccone C, Patzel K, Anikster Y, et al. Identification, tissue distribution, and molecular modeling of novel human isoforms of the key enzyme in sialic acid synthesis, UDP-GlcNAc 2-epimerase/ManNAc kinase. Biochemistry. 2011;50(41):8914–8925. doi: 10.1021/bi201050u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malicdan MC, Noguchi S, Tokutomi T, Goto Y, Nonaka I, Hayashi YK, et al. Peracetylated N-acetylmannosamine, a synthetic sugar molecule, efficiently rescues muscle phenotype and biochemical defects in mouse model of sialic acid-deficient myopathy. J Biol Chem. 2012;287(4):2689–2705. doi: 10.1074/jbc.M111.297051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yonekawa T, Malicdan MC, Cho A, Hayashi YK, Nonaka I, Mine T, et al. Sialyllactose ameliorates myopathic phenotypes in symptomatic GNE myopathy model mice. Brain. 2014;137(Pt 10):2670–2679. doi: 10.1093/brain/awu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mitrani-Rosenbaum S, Yakovlev L, Becker Cohen M, Telem M, Elbaz M, Yanay N, et al. Sustained expression and safety of human GNE in normal mice after gene transfer based on AAV8 systemic delivery. Neuromuscul Disord. 2012;22(11):1015–1024. doi: 10.1016/j.nmd.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 85.Bardor M, Nguyen DH, Diaz S, Varki A. Mechanism of uptake and incorporation of the non-human sialic acid N-glycolylneuraminic acid into human cells. J Biol Chem. 2005;280(6):4228–4237. doi: 10.1074/jbc.M412040200. [DOI] [PubMed] [Google Scholar]
- 86.Diaz S, Varki A. Metabolic labeling of sialic acids in tissue culture cell lines: methods to identify substituted and modified radioactive neuraminic acids. Anal Biochem. 1985;150(1):32–46. doi: 10.1016/0003-2697(85)90438-5. [DOI] [PubMed] [Google Scholar]
- 87.Jones MB, Teng H, Rhee JK, Lahar N, Baskaran G, Yarema KJ. Characterization of the cellular uptake and metabolic conversion of acetylated N-acetylmannosamine (ManNAc) analogues to sialic acids. Biotechnol Bioeng. 2004;85(4):394–405. doi: 10.1002/bit.10901. [DOI] [PubMed] [Google Scholar]
- 88.Gilormini PA, Lion C, Vicogne D, Levade T, Potelle S, Mariller C, et al. A sequential bioorthogonal dual strategy: ManNAl and SiaNAl as distinct tools to unravel sialic acid metabolic pathways. Chem Commun (Camb). 2016;52(11):2318–2321. doi: 10.1039/C5CC08838K. [DOI] [PubMed] [Google Scholar]
- 89.Hirschberg CB, Goodman SR, Green C. Sialic acid uptake by fibroblasts. Biochemistry. 1976;15(16):3591–3599. doi: 10.1021/bi00661a029. [DOI] [PubMed] [Google Scholar]
- 90.Nohle U, Schauer R. Metabolism of sialic acids from exogenously administered sialyllactose and mucin in mouse and rat. Hoppe Seylers Z Physiol Chem. 1984;365(12):1457–1467. doi: 10.1515/bchm2.1984.365.2.1457. [DOI] [PubMed] [Google Scholar]
- 91.Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117(6):1585–1594. doi: 10.1172/JCI30954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sparks S, Rakocevic G, Joe G, Manoli I, Shrader J, Harris-Love M, et al. Intravenous immune globulin in hereditary inclusion body myopathy: a pilot study. BMC Neurol. 2007;7:3. doi: 10.1186/1471-2377-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Witt W, von Nicolai H, Zilliken F. Uptake and distribution of orally applied N-acetyl-(14C)neuraminosyl-lactose and N-acetyl-(14C)neuraminic acid in the organs of newborn rats. Nutr Metab. 1979;23(1):51–61. doi: 10.1159/000176241. [DOI] [PubMed] [Google Scholar]
- 94.Argov Z, Caraco Y, Lau H, Pestronk A, Shieh P, Skrinar A, et al. Aceneuramic acid extended release administration maintains upper limb muscle strength in a 48-week study of subjects with GNE myopathy: results from a phase 2, randomized, controlled study. Journal of Neuromuscular Diseases. 2016;3(1):49–66. doi: 10.3233/JND-159900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hinderlich S, Berger M, Keppler OT, Pawlita M, Reutter W. Biosynthesis of N-acetylneuraminic acid in cells lacking UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. Biol Chem. 2001;382(2):291–297. doi: 10.1515/BC.2001.036. [DOI] [PubMed] [Google Scholar]
- 96.Shi Y, Xu X, Fang M, Zhang M, Li Y, Gillespie B, et al. Quantitative hydrophilic interaction chromatography-mass spectrometry analysis of N-acetylneuraminic acid and N-acetylmannosamine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;1000:105–111. doi: 10.1016/j.jchromb.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Somia N, Verma IM. Gene therapy: trials and tribulations. Nat Rev Genet. 2000;1(2):91–99. doi: 10.1038/35038533. [DOI] [PubMed] [Google Scholar]
- 98.Collins FS, Gottlieb S. The next phase of human gene-therapy oversight. N Engl J Med. 2018. [DOI] [PMC free article] [PubMed]
- 99.Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, Sadelain M. Gene therapy comes of age. Science. 2018;359(6372). [DOI] [PubMed]
- 101.Zygmunt DA, Crowe KE, Flanigan KM, Martin PT. Comparison of serum rAAV serotype-specific antibodies in patients with Duchenne muscular dystrophy, Becker muscular dystrophy, inclusion body myositis, or GNE myopathy. Hum Gene Ther. 2017;28(9):737–746. doi: 10.1089/hum.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nemunaitis G, Jay CM, Maples PB, Gahl WA, Huizing M, Yardeni T, et al. Hereditary inclusion body myopathy: single patient response to intravenous dosing of GNE gene lipoplex. Hum Gene Ther. 2011;22(11):1331–1341. doi: 10.1089/hum.2010.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tal-Goldberg T, Lorain S, Mitrani-Rosenbaum S. Correction of the Middle Eastern M712T mutation causing GNE myopathy by trans-splicing. Neuromolecular Med. 2014;16(2):322-31. [DOI] [PubMed]
- 105.Wilschut KJ, Ling VB, Bernstein HS. Concise review: stem cell therapy for muscular dystrophies. Stem Cells Transl Med. 2012;1(11):833–842. doi: 10.5966/sctm.2012-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tremblay JP, Skuk D. Another new “super muscle stem cell” leaves unaddressed the real problems of cell therapy for duchenne muscular dystrophy. Mol Ther. 2008;16(12):1907–1909. doi: 10.1038/mt.2008.243. [DOI] [PubMed] [Google Scholar]
- 107.Skuk D, Tremblay JP. Cell therapy in myology: dynamics of muscle precursor cell death after intramuscular administration in non-human primates. Mol Ther Methods Clin Dev. 2017;5:232–240. doi: 10.1016/j.omtm.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thiel C, Korner C. Mouse models for congenital disorders of glycosylation. J Inherit Metab Dis. 2011;34(4):879–889. doi: 10.1007/s10545-011-9295-7. [DOI] [PubMed] [Google Scholar]
- 109.Cheever TR, Berkley D, Braun S, Brown RH, Byrne BJ, Chamberlain JS, et al. Perspectives on best practices for gene therapy programs. Hum Gene Ther. 2015;26(3):127–133. doi: 10.1089/hum.2014.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nemunaitis G, Maples PB, Jay C, Gahl WA, Huizing M, Poling J, et al. Hereditary inclusion body myopathy: single patient response to GNE gene Lipoplex therapy. J Gene Med. 2010;12(5):403-12. [DOI] [PubMed]
- 111.Phadke AP, Jay C, Chen SJ, Haddock C, Wang Z, Yu Y, et al. Safety and in vivo expression of a GNE-transgene: a novel treatment approach for hereditary inclusion body myopathy-2. Gene Regul Syst Bio. 2009;3:89-101. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 498 kb)